
Synthetic Assembly of Mannose Moieties Using Polymer Chemistry and the Biological Evaluation of Its Interaction towards Concanavalin A


Abstract
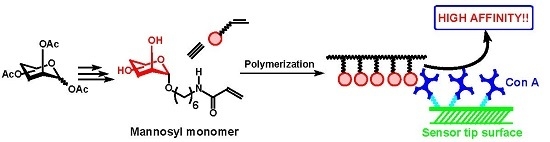
:
1. Introduction
2. Results and Discussion
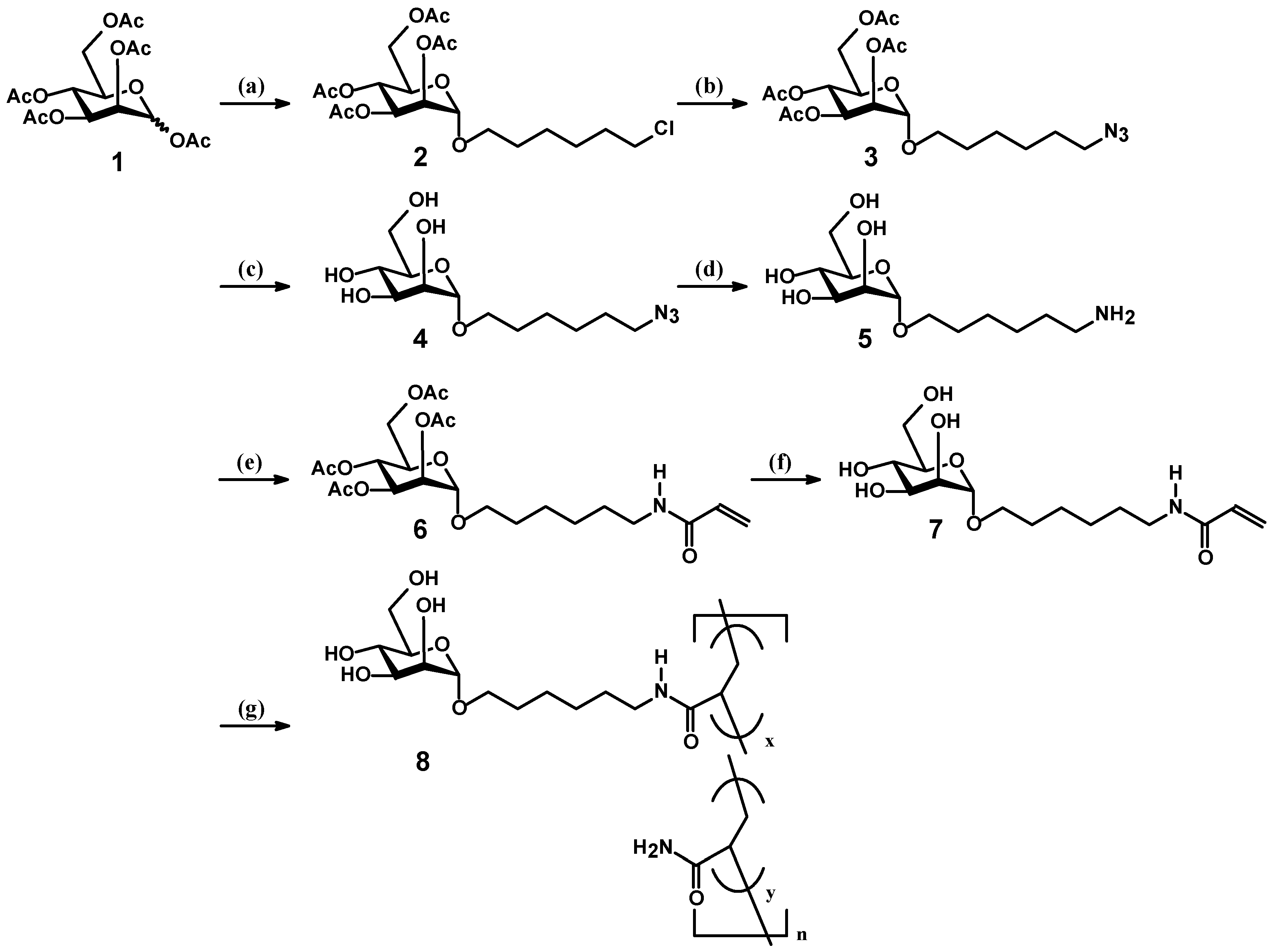
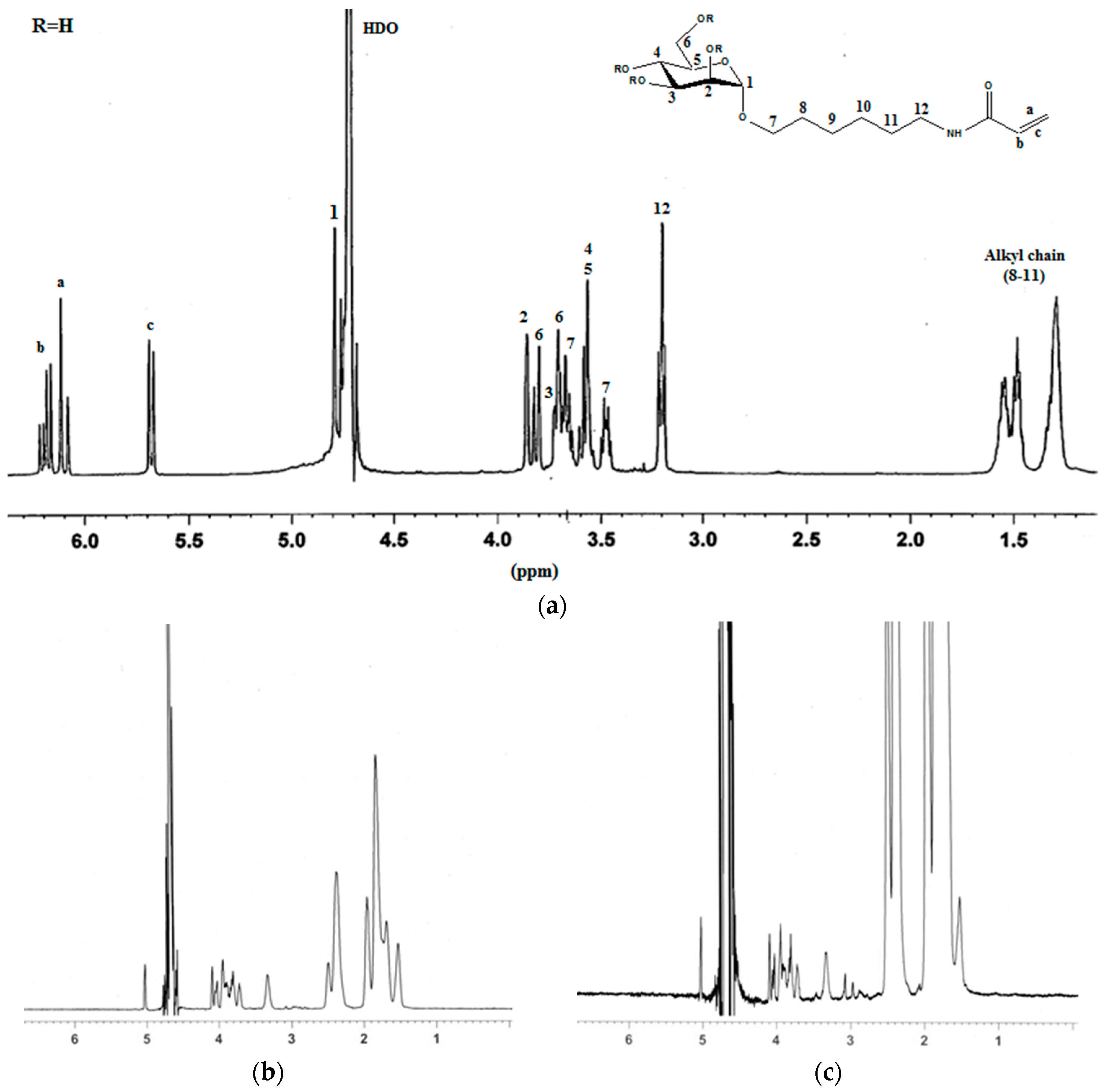
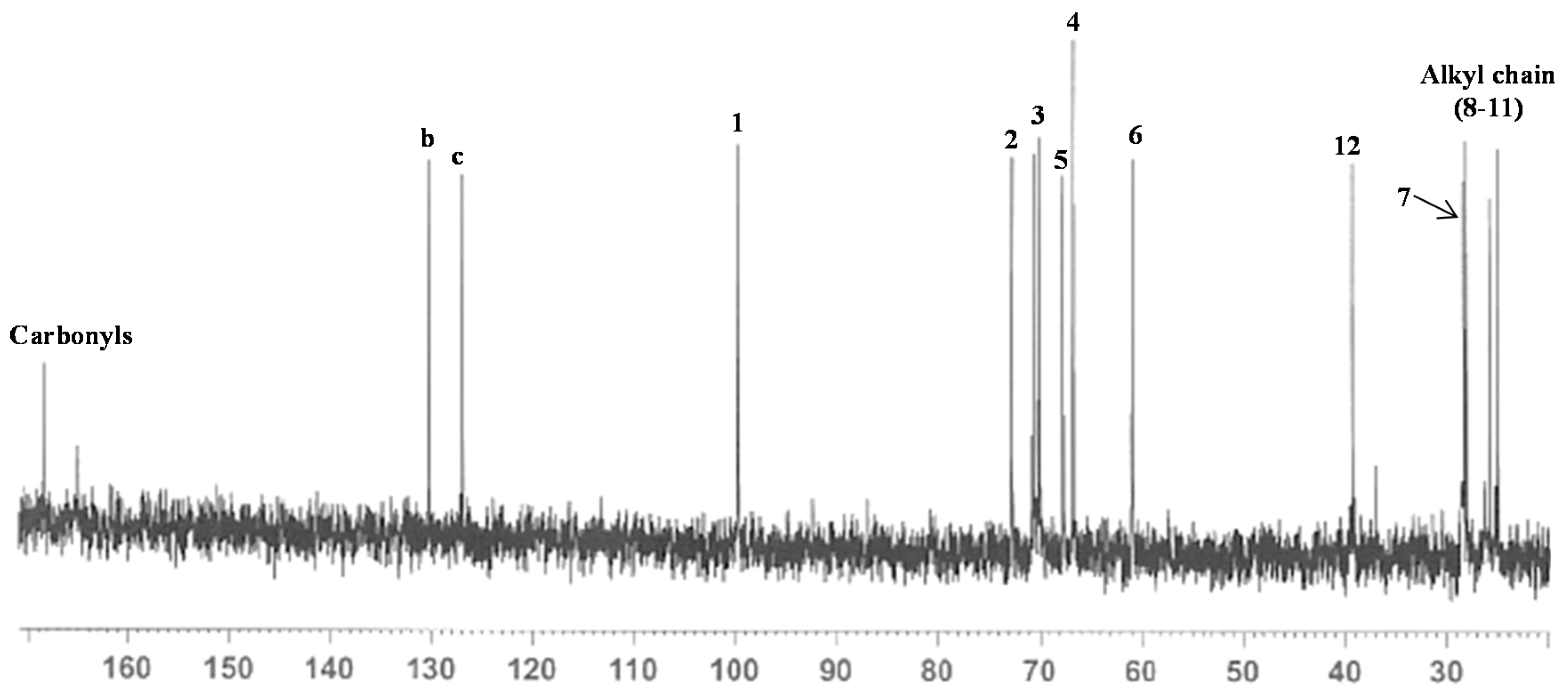
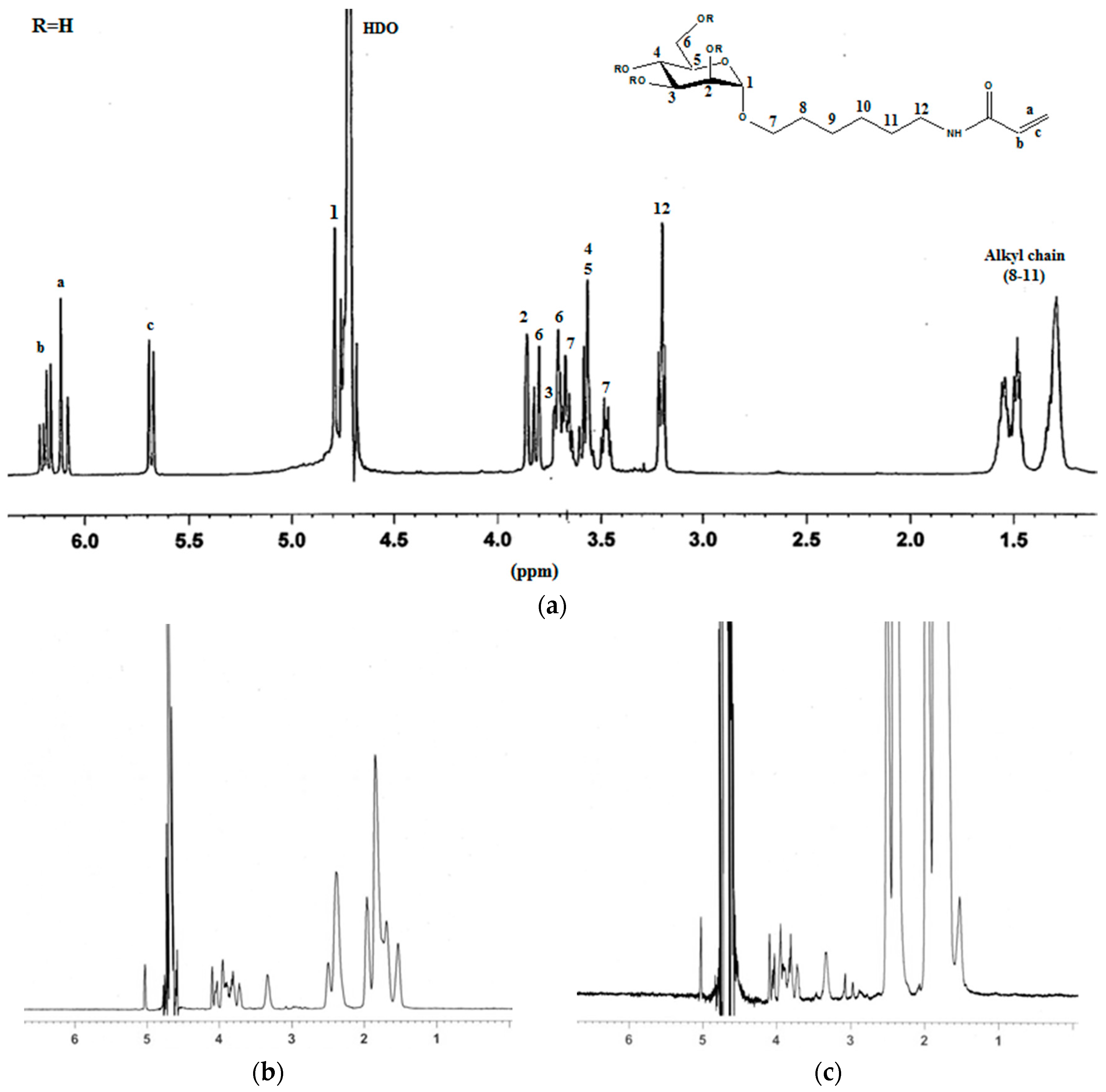
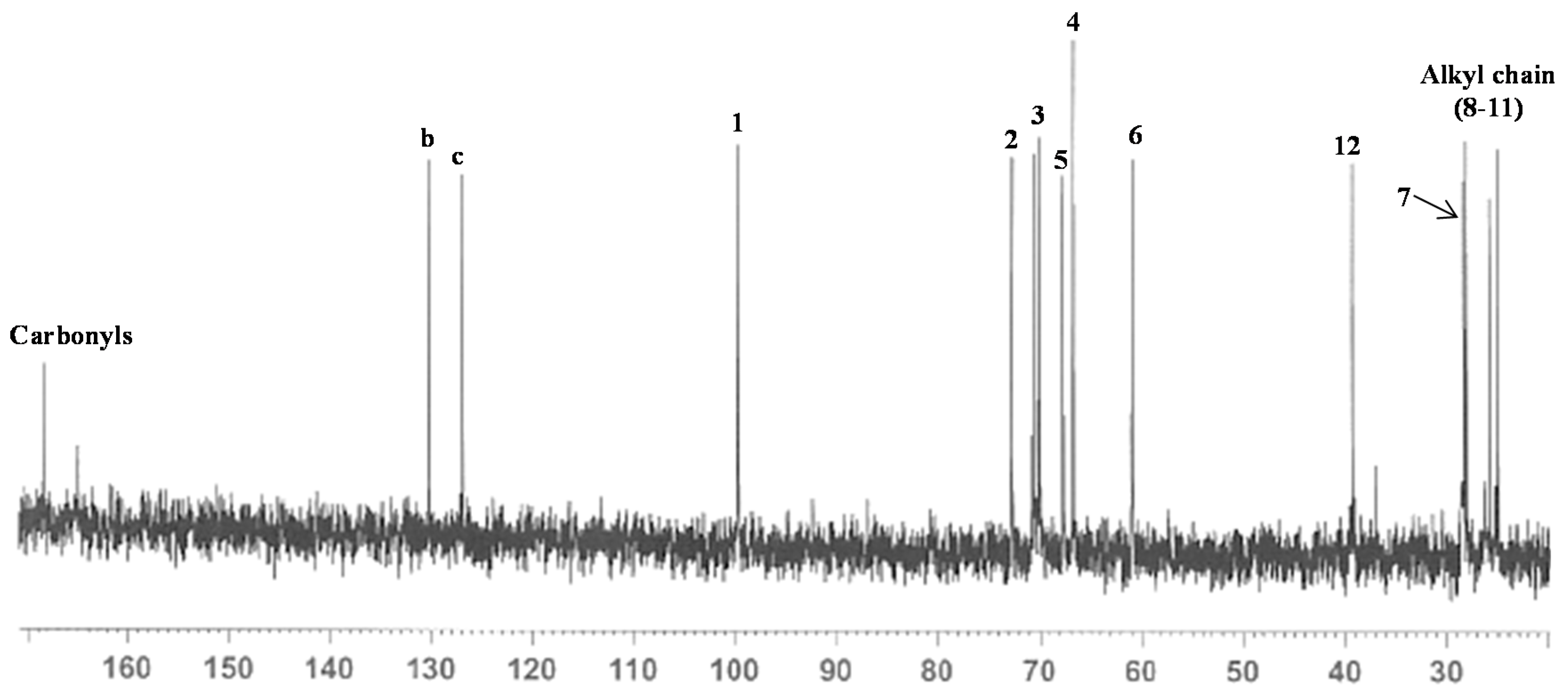
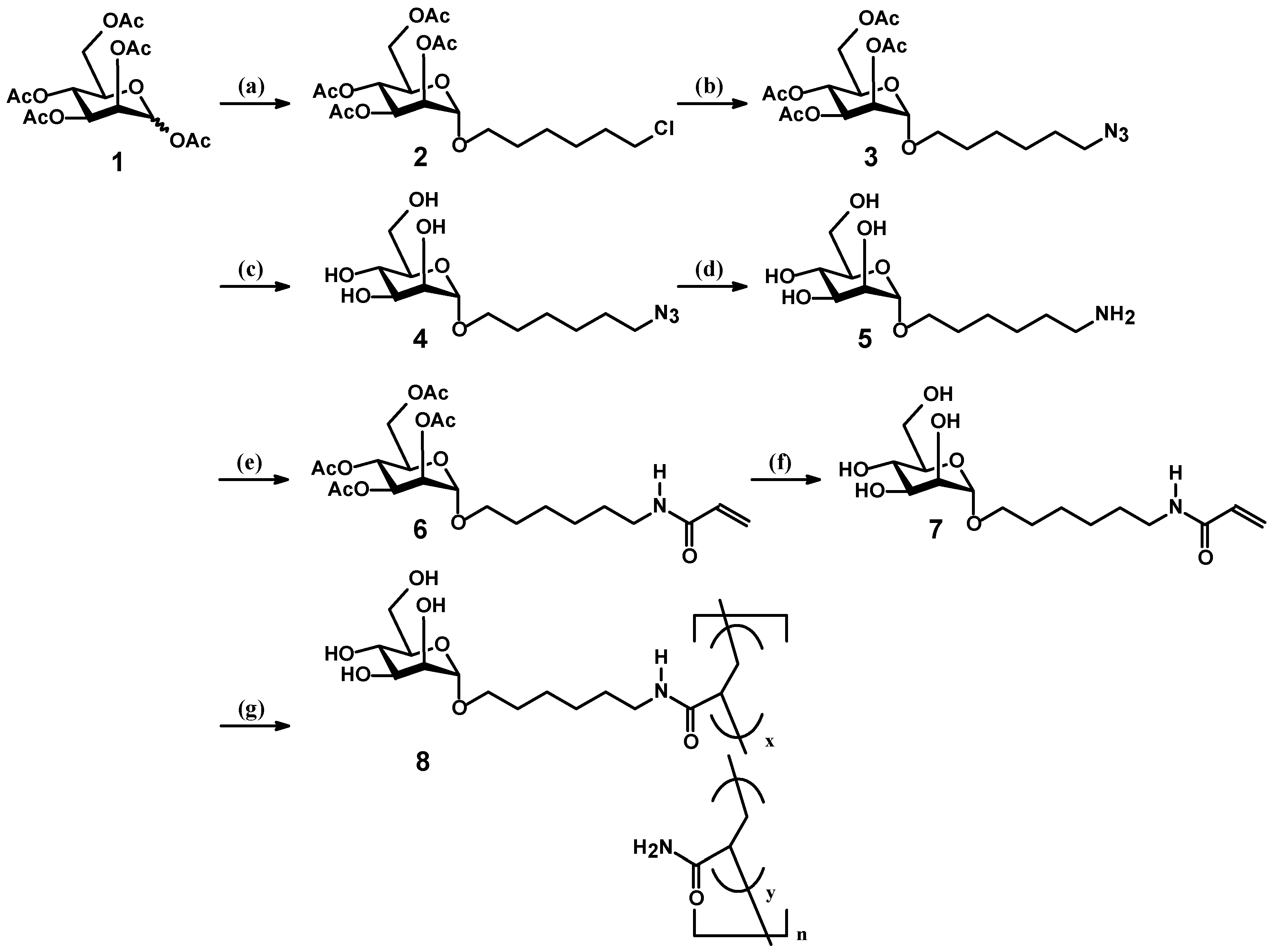
2.1. Synthesis of Mannosyl Glycomonomer
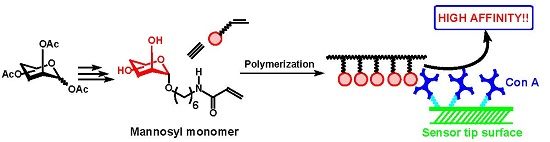
2.2. Polymerization of Mannosyl Glycomonomer
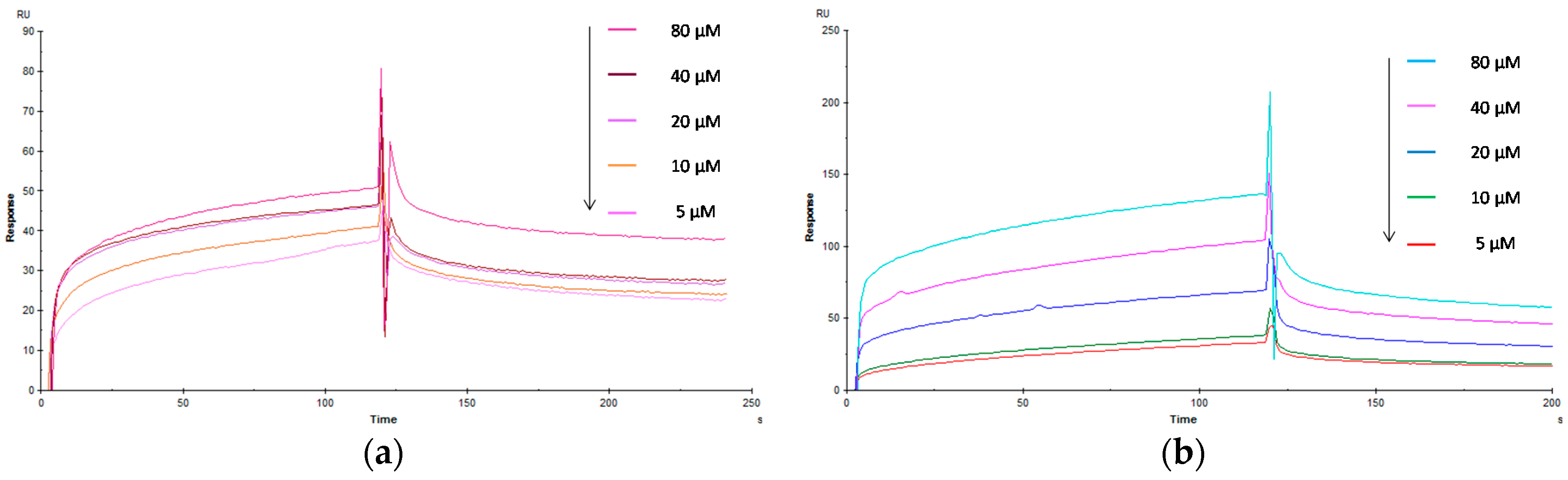
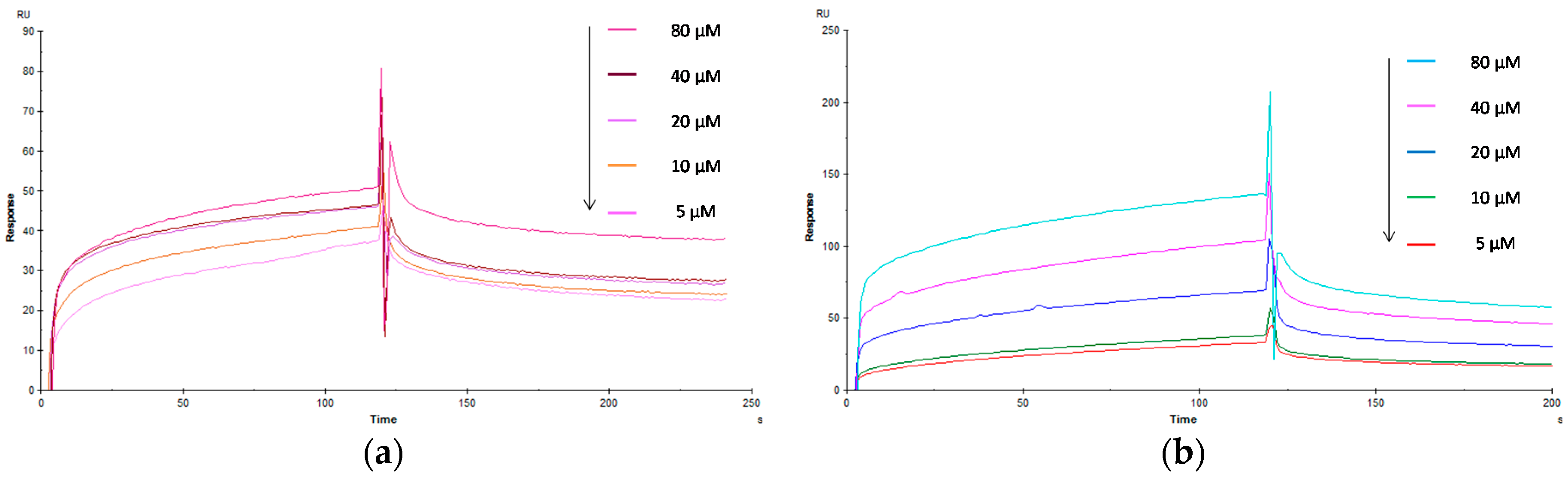
2.3. Biological Evaluation of Glycopolymers for Con A
3. Experimental Section
3.1. General Procedures
3.2. Synthesis of Compounds
3.3. Evaluation of the Interaction Kinetics of Glycopolymers for Lectin
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Holgersson, J.; Gustafsson, A.; Breimer, M.E. Characteristics of protein–carbohydrate interactions as a basis for developing novel carbohydrate-based antirejection therapies. Immunol. Cell Biol. 2005, 83, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Künzler, M. Hitting The sweet spot—Glycans as targets of fungal defense effector proteins. Molecules 2015, 20, 8144–8167. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Lee, R.T. Carbohydrate-protein interactions: Basis of glycobiology. Acc. Chem. Res. 1995, 28, 321–327. [Google Scholar] [CrossRef]
- Piotukh, K.; Serra, V.; Borriss, R.; Planas, A. Protein–carbohydrate interactions defining substrate specificity in bacillus 1,3-1,4-β-d-glucan 4-glucanohydrolases as dissected by mutational analysis. Biochemistry 1999, 38, 16092–16104. [Google Scholar] [CrossRef] [PubMed]
- Cairo, C.W.; Gestwicki, J.E.; Kanai, M.; Kiessling, L.L. Control of multivalent interactions by binding epitope density. J. Am. Chem. Soc. 2002, 124, 1615–1619. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, P.H.; Haase, W.C. Solid-phase oligosaccharide synthesis and combinatorial carbohydrate libraries. Chem. Rev. 2000, 100, 4349–4394. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Tao, L.; Mantovani, G.; Geng, J.; Nyström, D.; Haddleton, D.M. A Modular click approach to glycosylated polymeric beads: Design, synthesis and preliminary lectin recognition studies. Macromolecules 2007, 40, 7513–7520. [Google Scholar] [CrossRef]
- Kiessling, L.L.; Gestwicki, J.E.; Strong, L.E. Synthetic multivalent ligands as probes of signal transduction. Angew. Chem. Int. Ed. 2006, 45, 2348–2368. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.H.; Bertozzi, C.R. Glycans in cancer and inflammation-potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Spain, S.G.; Cameron, N.R. A spoonful of sugar: The application of glycopolymers in therapeutics. Polym. Chem. 2011, 2, 60–68. [Google Scholar] [CrossRef]
- Zhang, Q.; Collins, J.; Anastasaki, A.; Wallis, R.; Mitchell, D.A.; Becer, C.R.; Haddleton, D.M. Sequence-controlled multi-block glycopolymers to inhibit DC-SIGN-gp120 binding. Angew. Chem. Int. Ed. 2013, 52, 4435–4439. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Goshu, Y.; Takezawa, Y.; Mori, T.; Sakamoto, J.I.; Yamada, A.; Onaga, T.; Koyama, T.; Hatano, K.; Snyder, P.W.; et al. Practical synthesis of fully protected globotriaose and its glycopolymers. Carbohydr. Polym. 2007, 69, 326–335. [Google Scholar] [CrossRef]
- Matsuoka, K.; Oka, H.; Koyama, T.; Hatano, K. Synthetic assembly of bifluorescence-labeled glycopolymers as substrates for assaying α-amylase by resonance energy transfer. ACS Macro Lett. 2012, 1, 266–269. [Google Scholar] [CrossRef]
- Matsuoka, K.; Kurita, A.; Koyama, T.; Hatano, K. Use of chloromethylstyrene as a supporter for convenient preparation of carbohydrate monomer and glycopolymers. Carbohydr. Polym. 2014, 107, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Papp, I.; Dernedde, J.; Enders, S.; Riese, S.B.; Shiao, T.C.; Roy, R.; Haag, R. Multivalent presentation of mannose on hyperbranched polyglycerol and their interaction with concanavalin A lectin. Chembiochem 2011, 12, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.-I.; Matsuoka, K.; Furuike, T.; Ishii, S.; Kurita, K.; Nishimura, K.M. Synthetic glycoconjugates. 2. n-pentenyl glycosides as convenient mediators for the syntheses of new types of glycoprotein models. Macromolecules 1991, 24, 4236–4241. [Google Scholar]
- Toshima, K.; Tatsuta, K. Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem. Rev. 1993, 93, 1503–1531. [Google Scholar] [CrossRef]
- Takano, T.; Nakatsubo, F.; Murakami, K. A facile allyl β-glycosylation in the presence of a benzyl protecting group using boron trifluoride etherate. Carbohydr. Res. 1990, 203, 341–342. [Google Scholar] [CrossRef]
- Nagahori, N.; Nishimura, S.-I. Tailored glycopolymers: Controlling the carbohydrate-protein interaction based on template effect. Biomacromolecules 2001, 2, 21–24. [Google Scholar] [CrossRef]
- Matsuoka, K.; Nishimura, S.-I. Glycoconjugates. 5. Polymeric sugar ligands available for determining the binding-specificity of lectins. Macromolecules 1995, 28, 2961–2968. [Google Scholar] [CrossRef]
- Lis, H.; Sharon, N. Lectins: Carbohydrate-specific proteins that mediate cellular recognition. Chem. Rev. 1998, 98, 637–674. [Google Scholar] [CrossRef] [PubMed]
- Kogelberg, H.; Feizi, T. New structural insights into lectin-type proteins of the immune system. Curr. Opin. Struct. Biol. 2001, 11, 635–643. [Google Scholar] [CrossRef]
- De la Fuente, J.M.; Eaton, P.; Barrientos, A.G.; Menedez, M.; Penades, S. Thermodynamic evidence for Ca2+-mediated self-aggregation of Lewis X gold glyconanoparticles. A model for cell adhesion via carbohydrate-carbohydrate interaction. J. Am. Chem. Soc. 2005, 127, 6192–6197. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, P.V.; Basu, A. Studies of the carbohydrate-carbohydrate interaction between lactose and GM3 using langmuir monolayers and glycolipid micelles. Glycoconj. J. 2004, 21, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Myc, A.; Silpe, J.E.; Sumit, M.; Wong, P.T.; McCarthy, K.; Desai, A.M.; Thomas, T.P.; Kotlyar, A.; Holl, M.M.B.; et al. Dendrimer-based multivalent vancomycin nanoplatform for targeting the drug-resistant bacterial surface. ACS Nano 2013, 7, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hatano, K.; Matsuoka, K.; Esumi, Y.; Toone, E.J.; Terunuma, D. Synthesis of carbosilane dendrimers having peripheral mannose and mannobiose. Tetrahedron 2005, 61, 2751–2760. [Google Scholar] [CrossRef]
- O’Brien, M.J., II; Brueck, S.R.J.; Perez-Luna, V.H.; Tender, L.M.; Lopez, G.P. SPR biosensors: Simultaneously removing thermal & bulk-composition effects. Biosens. Bioelectron. 1999, 14, 145–154. [Google Scholar]
- Myszka, D.G. Improving biosensor analysis. J. Mol. Recognit. 1999, 12, 279–284. [Google Scholar] [CrossRef]
- Haseley, S.R.; Talaga, P.; Kamerling, J.P.; Vliegenthart, J.F.G. Characterization of carbohydrate binding specificity and kinetic parameters of lectins by surface plasmon resonance. Anal. Biochem. 1999, 274, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.A.; Kanai, M.; Maly, D.J.; Kiessling, L.L. Probing low affinity and multivalent interactions with surface plasmon resonance: Ligands for concanavalin A. J. Am. Chem. Soc. 1998, 120, 10575–10582. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 7, 8a, and 8b are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Monomer Ratio a | Total Yield b (%) | Polymer Composition c | Sugar Content (wt %) | d (kDa) | |||
|---|---|---|---|---|---|---|---|---|
| 8 | x | y | n | |||||
| a | 1:10 | 71 | 1 | 12 | 532 | 23.8 | 178 | 1.1 |
| b | 1:20 | 90 | 1 | 130 | 310 | 3.6 | 124 | 1.0 |
| Compound | Working Concentrations (μM) | Flow Rate (μL/min) | Contact Time (min) | Dissociation Time (min) | ||||
|---|---|---|---|---|---|---|---|---|
| 8a | 5 | 10 | 20 | 40 | 80 | 30 | 1 | 1 |
| 8b | 5 | 10 | 20 | 60 | 80 | 30 | 2 | 10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diwan, D.; Shinkai, K.; Tetsuka, T.; Cao, B.; Arai, H.; Koyama, T.; Hatano, K.; Matsuoka, K. Synthetic Assembly of Mannose Moieties Using Polymer Chemistry and the Biological Evaluation of Its Interaction towards Concanavalin A. Molecules 2017, 22, 157. https://doi.org/10.3390/molecules22010157
Diwan D, Shinkai K, Tetsuka T, Cao B, Arai H, Koyama T, Hatano K, Matsuoka K. Synthetic Assembly of Mannose Moieties Using Polymer Chemistry and the Biological Evaluation of Its Interaction towards Concanavalin A. Molecules. 2017; 22(1):157. https://doi.org/10.3390/molecules22010157
Chicago/Turabian StyleDiwan, Deepti, Kohei Shinkai, Toshihiro Tetsuka, Bin Cao, Hidenao Arai, Tetsuo Koyama, Ken Hatano, and Koji Matsuoka. 2017. "Synthetic Assembly of Mannose Moieties Using Polymer Chemistry and the Biological Evaluation of Its Interaction towards Concanavalin A" Molecules 22, no. 1: 157. https://doi.org/10.3390/molecules22010157
APA StyleDiwan, D., Shinkai, K., Tetsuka, T., Cao, B., Arai, H., Koyama, T., Hatano, K., & Matsuoka, K. (2017). Synthetic Assembly of Mannose Moieties Using Polymer Chemistry and the Biological Evaluation of Its Interaction towards Concanavalin A. Molecules, 22(1), 157. https://doi.org/10.3390/molecules22010157