
Dysregulation of Cell Death and Its Epigenetic Mechanisms in Systemic Lupus Erythematosus

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Dysregulated Apoptosis in SLE
1.2. Epigenetic Modification-Mediated Increased Apoptosis in SLE
1.2.1. miRNAs-Mediated Apoptosis in SLE
1.2.2. DNA Methylation and Apoptosis
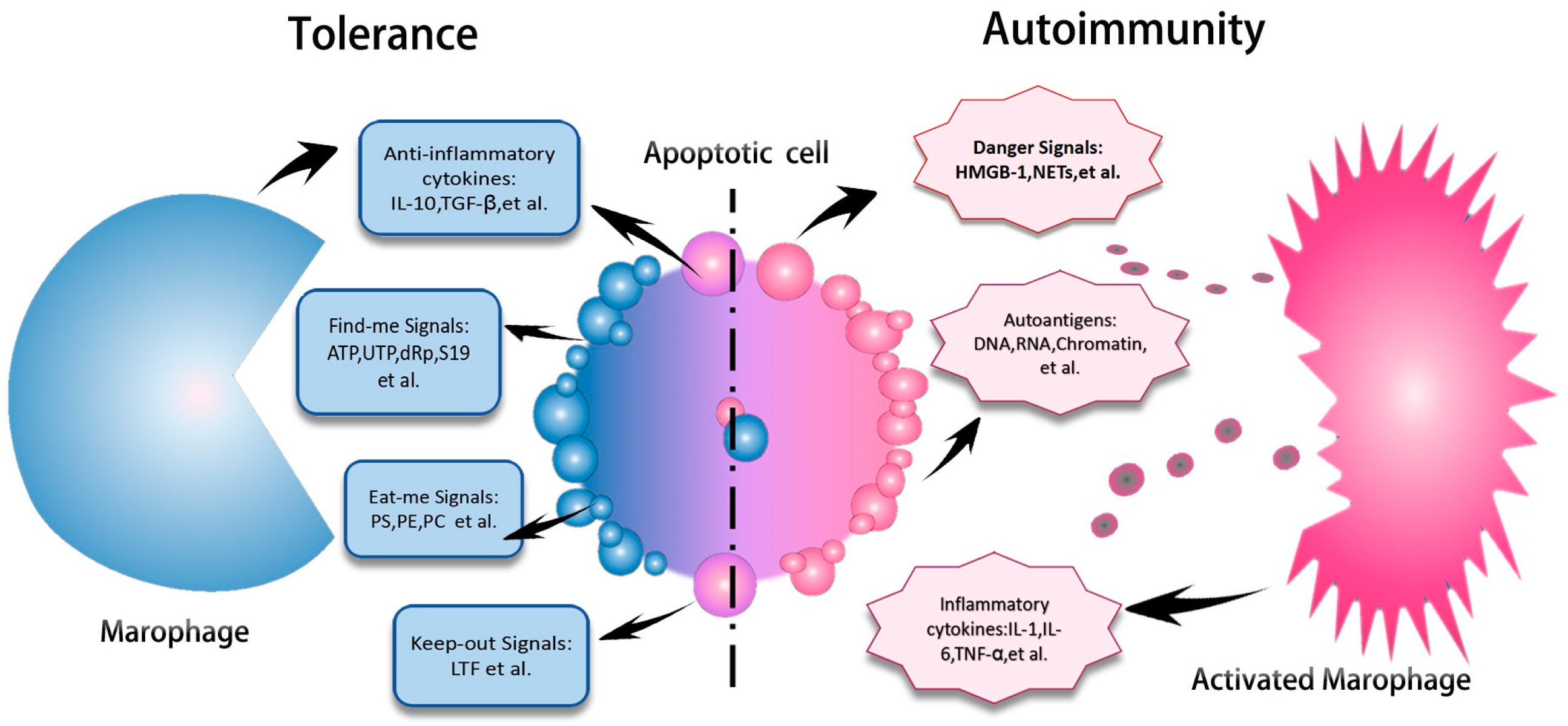
1.3. Impaired Clearance of Apoptotic Materials in SLE
Epigenetic Mechanisms Underlying Impaired Phagocytic Function of Macrophages
1.4. NETosis and SLE
1.5. Autophagy and SLE
1.6. Necroptosis and SLE
2. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- D’Cruz, D.P.; Khamashta, M.A.; Hughes, G.R. Systemic lupus erythematosus. Lancet 2007, 369, 587–596. [Google Scholar] [CrossRef]
- Tan, E.M.; Kunkel, H.G. Characteristics of a soluble nuclear antigen precipitating with sera of patients with systemic lupus erythematosus. J. Immunol. 1966, 96, 464–471. [Google Scholar] [PubMed]
- Takeno, M.; Nagafuchi, H.; Kaneko, S.; Wakisaka, S.; Oneda, K.; Takeba, Y.; Yamashita, N.; Suzuki, N.; Kaneoka, H.; Sakane, T. Autoreactive T cell clones from patients with systemic lupus erythematosus support polyclonal autoantibody production. J. Immunol. 1997, 158, 3529–3538. [Google Scholar] [PubMed]
- Santulli-Marotto, S.; Retter, M.W.; Gee, R.; Mamula, M.J.; Clarke, S.H. Autoreactive B cell regulation: Peripheral induction of developmental arrest by lupus-associated autoantigens. Immunity 1998, 8, 209–219. [Google Scholar] [CrossRef]
- Gatto, M.; Zen, M.; Ghirardello, A.; Bettio, S.; Bassi, N.; Iaccarino, L.; Punzi, L.; Doria, A. Emerging and critical issues in the pathogenesis of lupus. Autoimmun. Rev. 2013, 12, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Joseph, F.G.; Lammie, G.A.; Scolding, N.J. CNS lupus: A study of 41 patients. Neurology 2007, 69, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.; Ruland, V.; Bonsmann, G. Cutaneous lupus erythematosus: Update of therapeutic options part II. J. Am. Acad. Dermatol. 2011, 65, e195–e213. [Google Scholar] [CrossRef] [PubMed]
- Gabba, A.; Piga, M.; Vacca, A.; Porru, G.; Garau, P.; Cauli, A.; Mathieu, A. Joint and tendon involvement in systemic lupus erythematosus: An ultrasound study of hands and wrists in 108 patients. Rheumatology (Oxf.) 2012, 51, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Wanitpongpun, C.; Teawtrakul, N.; Mahakkanukrauh, A.; Siritunyaporn, S.; Sirijerachai, C.; Chansung, K. Bone marrow abnormalities in systemic lupus erythematosus with peripheral cytopenia. Clin. Exp. Rheumatol. 2012, 30, 825–829. [Google Scholar] [PubMed]
- Bouts, Y.M.; Wolthuis, D.F.; Dirkx, M.F.; Pieterse, E.; Simons, E.M.; van Boekel, A.M.; Dieker, J.W.; van der Vlag, J. Apoptosis and NET formation in the pathogenesis of SLE. AutoImmunity 2012, 45, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Corral, C.I.; Vega-Memije, M.E.; Martinez-Luna, E.; Cuevas-Gonzalez, J.C.; Rodriguez-Carreon, A.A.; de la Rosa, J.J.; Fde, J.D.; Avalos-Diaz, E. Apoptosis in chronic cutaneous lupus erythematosus, discoid lupus, and lupus profundus. Int. J. Clin. Exp. Pathol. 2015, 8, 7260–7265. [Google Scholar] [PubMed]
- Souliotis, V.L.; Sfikakis, P.P. Increased DNA double-strand breaks and enhanced apoptosis in patients with lupus nephritis. Lupus 2015, 24, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sun, B.; Wang, H.; Yin, C.; Wang, X.; Ji, X. Increased serum IL-10 in lupus patients promotes apoptosis of T cell subsets via the caspase 8 pathway initiated by Fas signaling. J. Biomed. Res. 2015, 29, 232–240. [Google Scholar] [PubMed]
- Colonna, L.; Lood, C.; Elkon, K.B. Beyond apoptosis in lupus. Curr. Opin. Rheumatol. 2014, 26, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, A.A.; Gullstrand, B.; Truedsson, L.; Sturfelt, G. SLE serum induces classical caspase-dependent apoptosis independent of death receptors. Clin. Immunol. 2008, 126, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Mecklenbrauker, I.; Saijo, K.; Zheng, N.Y.; Leitges, M.; Tarakhovsky, A. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature 2002, 416, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Silvestris, F.; Williams, R.C.; Calvani, N.; Grinello, D.; Tucci, M.; Cafforio, P.; Dammacco, F. Serum elevations of soluble Fas (CD95/apo-I) concur in deregulating T cell apoptosis during active lupus disease. Clin. Exp. Med. 2002, 2, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Dwivedi, A.; Mujtaba, S.F.; Singh, K.P.; Pal, M.K.; Chopra, D.; Goyal, S.; Srivastav, A.K.; Dubey, D.; Gupta, S.K.; et al. Ambient UV-B exposure reduces the binding of ofloxacin with bacterial DNA gyrase and induces DNA damage mediated apoptosis. Int. J. Biochem. Cell Biol. 2016, 73, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Ishikawa, O.; Miyachi, Y.; Kanai, Y. In vitro spontaneous and UVB-induced lymphocyte apoptosis are not specific to SLE. Photodermatol. Photoimmunol. Photomed. 1997, 13, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Fransen, J.H.; van der Vlag, J.; Ruben, J.; Adema, G.J.; Berden, J.H.; Hilbrands, L.B. The role of dendritic cells in the pathogenesis of systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, M.A.; Sawalha, A.H. Epigenetics in systemic lupus erythematosus: Leading the way for specific therapeutic agents. Int. J. Clin. Rheumtol. 2011, 6, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, A.L.; Schetter, A.J.; Nielsen, C.T.; Lood, C.; Knudsen, S.; Voss, A.; Harris, C.C.; Hellmark, T.; Segelmark, M.; Jacobsen, S.; et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheumatol. 2013, 65, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Wu, J.; Zhao, J.; Wang, H.; Liu, Y.; Chen, T.; Kan, X.; Tao, Q.; Shen, X.; Yan, K.; et al. miR-29b and miR-29c are involved in Toll-like receptor control of glucocorticoid-induced apoptosis in human plasmacytoid dendritic cells. PLoS ONE 2013, 8, e69926. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhu, X.; Liang, J.; Wu, J.; Yang, Y.; Wang, S.; Shi, W.; Xu, J. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J. Dermatol. Sci. 2013, 69, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Wang, P.; Wang, T.; Qi, J.; Wei, M.; Wang, S.; Fan, T.; Johnson, D.; Wan, X.; Shi, W.; et al. MicroRNA-21 regulates T-cell apoptosis by directly targeting the tumor suppressor gene Tipe2. Cell Death Dis. 2014, 5, e1095. [Google Scholar] [CrossRef] [PubMed]
- Stagakis, E.; Bertsias, G.; Verginis, P.; Nakou, M.; Hatziapostolou, M.; Kritikos, H.; Iliopoulos, D.; Boumpas, D.T. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: MiR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann. Rheum. Dis. 2011, 70, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.M.; Fang, M.; Wang, J.P.; Cai, P.C.; Wang, P.; Hu, L.H. Clinical relevance of plasma miR-21 in new-onset systemic lupus erythematosus patients. J. Clin. Lab. Anal. 2014, 28, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Amatya, V.J.; Naumann, U.; Weller, M.; Ohgaki, H. TP53 promoter methylation in human gliomas. Acta Neuropathol. 2005, 110, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Lima, E.M.; Leal, M.F.; Burbano, R.R.; Khayat, A.S.; Assumpcao, P.P.; Bello, M.J.; Rey, J.A.; Smith, M.A.; Casartelli, C. Methylation status of ANAPC1, CDKN2A and TP53 promoter genes in individuals with gastric cancer. Braz. J. Med. Biol. Res. 2008, 41, 539–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreb, D.B.; Desman, G.T. Expression of p53 and apoptosis in discoid lupus erythematosus. Croat. Med. J. 2005, 46, 998. [Google Scholar] [PubMed]
- Chauhan, R.; Handa, R.; Das, T.P.; Pati, U. Over-expression of TATA binding protein (TBP) and p53 and autoantibodies to these antigens are features of systemic sclerosis, systemic lupus erythematosus and overlap syndromes. Clin. Exp. Immunol. 2004, 136, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Javierre, B.M.; Fernandez, A.F.; Richter, J.; Al-Shahrour, F.; Martin-Subero, J.I.; Rodriguez-Ubreva, J.; Berdasco, M.; Fraga, M.F.; O’Hanlon, T.P.; Rider, L.G.; et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, S.; Luo, S.; Wu, H.; Tang, M.; Cheng, W.; Zhang, Q.; Zhang, P.; Yu, X.; Xia, Y.; et al. DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J. Autoimmun. 2014, 54, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Serra, L.; Esteller, M. Proteins that bind methylated DNA and human cancer: Reading the wrong words. Br. J. Cancer 2008, 98, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.X.; Hackett, J.A.; Nestor, C.; Dunican, D.S.; Madej, M.; Reddington, J.P.; Pennings, S.; Harrison, D.J.; Meehan, R.R. Apoptosis and DNA methylation. Cancers (Basel) 2011, 3, 1798–1820. [Google Scholar] [CrossRef] [PubMed]
- Balada, E.; Ordi-Ros, J.; Serrano-Acedo, S.; Martinez-Lostao, L.; Vilardell-Tarres, M. Transcript overexpression of the MBD2 and MBD4 genes in CD4+ T cells from systemic lupus erythematosus patients. J. Leukoc. Biol. 2007, 81, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Thaler, R.; Karlic, H.; Spitzer, S.; Klaushofer, K.; Varga, F. Extra-cellular matrix suppresses expression of the apoptosis mediator Fas by epigenetic DNA methylation. Apoptosis 2010, 15, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Orskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Gronbaek, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ke, S.; Wang, W.; Ran, H.; Chen, M.; Zhang, F.; Qiu, X.; Jiang, M.; Zou, C.; Zhang, R.; et al. A single fas gene mutation changes lupus onset, severity, location, and molecular abnormalities in mice. Curr. Mol. Med. 2015, 15, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Siwiec, A.; Majdan, M. The role of the PD-1 protein in pathogenesis of autoimmune diseases, with particular consideration of rheumatoid arthritis and systemic lupus erythematosus. Postepy Hig. Med. Dosw. (Online) 2015, 69, 534–542. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.C.; Konkel, J.E.; Prendergast, C.T.; Thomson, J.P.; Ottaviano, R.; Leech, M.D.; Kay, O.; Zandee, S.E.; Sweenie, C.H.; Wraith, D.C.; et al. Epigenetic modification of the PD-1 (Pdcd1) promoter in effector CD4(+) T cells tolerized by peptide immunotherapy. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Navratil, J.S.; Ahearn, J.M. Apoptosis, clearance mechanisms, and the development of systemic lupus erythematosus. Curr. Rheumatol. Rep. 2001, 3, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Tas, S.W.; Quartier, P.; Botto, M.; Fossati-Jimack, L. Macrophages from patients with SLE and rheumatoid arthritis have defective adhesion in vitro, while only SLE macrophages have impaired uptake of apoptotic cells. Ann. Rheum. Dis. 2006, 65, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Voll, R.E.; Zoller, O.M.; Hagenhofer, M.; Ponner, B.B.; Kalden, J.R. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheumatol. 1998, 41, 1241–1250. [Google Scholar] [CrossRef]
- Baumann, I.; Kolowos, W.; Voll, R.E.; Manger, B.; Gaipl, U.; Neuhuber, W.L.; Kirchner, T.; Kalden, J.R.; Herrmann, M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheumatol. 2002, 46, 191–201. [Google Scholar] [CrossRef]
- Ren, Y.; Tang, J.; Mok, M.Y.; Chan, A.W.; Wu, A.; Lau, C.S. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheumatol. 2003, 48, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Kruse, K.; Janko, C.; Urbonaviciute, V.; Mierke, C.T.; Winkler, T.H.; Voll, R.E.; Schett, G.; Munoz, L.E.; Herrmann, M. Inefficient clearance of dying cells in patients with SLE: Anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis 2010, 15, 1098–1113. [Google Scholar] [CrossRef] [PubMed]
- Church, L.D.; Cook, G.P.; McDermott, M.F. Primer: Inflammasomes and interleukin 1beta in inflammatory disorders. Nat. Clin. Pract. Rheumatol. 2008, 4, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yue, Y.; Zhu, Y.; Xiong, S. Extracellular, but not intracellular HMGB1, facilitates self-DNA induced macrophage activation via promoting DNA accumulation in endosomes and contributes to the pathogenesis of lupus nephritis. Mol. Immunol. 2015, 65, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yu, S.; Xu, W.; Gao, B.; Xiong, S. HMGB1 Promotes Systemic Lupus Erythematosus by Enhancing Macrophage Inflammatory Response. J. Immunol. Res. 2015, 2015, 946748. [Google Scholar] [CrossRef] [PubMed]
- Radic, M.; Herrmann, M.; van der Vlag, J.; Rekvig, O.P. Regulatory and pathogenetic mechanisms of autoantibodies in SLE. AutoImmunity 2011, 44, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Barnawi, J.; Jersmann, H.; Haberberger, R.; Hodge, S.; Meech, R. Reduced DNA methylation of sphingosine-1 phosphate receptor 5 in alveolar macrophages in COPD: A potential link to failed efferocytosis. Respirology 2016. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.; Tullus, K.; Marks, S.D.; Holt, R.C.; Pilkington, C.; Beresford, M.W. Increased serum concentration of sphingosine-1-phosphate in juvenile-onset systemic lupus erythematosus. J. Clin. Immunol. 2012, 32, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Wallner, S.; Schroder, C.; Leitao, E.; Berulava, T.; Haak, C.; Beisser, D.; Rahmann, S.; Richter, A.S.; Manke, T.; Bonisch, U.; et al. Epigenetic dynamics of monocyte-to-macrophage differentiation. Epigenet. Chromatin 2016, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Bosch, X. Systemic lupus erythematosus and the neutrophil. N. Engl. J. Med. 2011, 365, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.L.; Kuan, W.P.; Wong, C.K.; Li, E.K.; Tam, L.S. Immunopathological roles of cytokines, chemokines, signaling molecules, and pattern-recognition receptors in systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 2012, 715190. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Radic, M. Clearance of Apoptotic Bodies, NETs, and Biofilm DNA: Implications for Autoimmunity. Front. Immunol. 2014, 5, 365. [Google Scholar] [CrossRef] [PubMed]
- Spengler, J.; Lugonja, B.; Ytterberg, A.J.; Zubarev, R.A.; Creese, A.J.; Pearson, M.J.; Grant, M.M.; Milward, M.; Lundberg, K.; Buckley, C.D.; et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can. Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol. 2015, 67, 3135–3145. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Muller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012, 14, R25. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, C.; Barbati, C.; Vacirca, D.; Piscopo, P.; Confaloni, A.; Sanchez, M.; Maselli, A.; Colasanti, T.; Conti, F.; Truglia, S.; et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J. 2012, 26, 4722–4732. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.L.; Kuballa, P.; Khor, B.; Zhang, M.; Shi, H.N.; Virgin, H.W.; Xavier, R.J. ATG5 regulates plasma cell differentiation. Autophagy 2013, 9, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, G. Lysosomes, Autoimmune Phenomena, and Diseases of Connective Tissue. Lancet 1964, 2, 1373–1375. [Google Scholar] [CrossRef]
- International Consortium for Systemic Lupus Erythematosus Genetics; Harley, J.B.; Alarcon-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D.; et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Lu, X.L.; Lv, J.C.; Yang, H.Z.; Qin, L.X.; Zhao, M.H.; Su, Y.; Li, Z.G.; Zhang, H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann. Rheum. Dis. 2011, 70, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; Vyse, T.J. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Gao, C.; Chen, Y.; Feng, Y.; Liu, W.J.; Liu, H.F. Update on the role of autophagy in systemic lupus erythematosus: A novel therapeutic target. Biomed. Pharmacother. 2015, 71, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Harr, M.W.; McColl, K.S.; Zhong, F.; Molitoris, J.K.; Distelhorst, C.W. Glucocorticoids downregulate Fyn and inhibit IP(3)-mediated calcium signaling to promote autophagy in T lymphocytes. Autophagy 2010, 6, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Sun, J. Vitamin D, vitamin D receptor, and macroautophagy in inflammation and infection. Discov. Med. 2011, 11, 325–335. [Google Scholar] [PubMed]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Renaudineau, Y.; Youinou, P. Epigenetics and autoimmunity, with special emphasis on methylation. Keio J. Med. 2011, 60, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, N.; Long, H.; Zhao, M.; Yin, H.; Lu, Q. Aberrant expression pattern of histone acetylation modifiers and mitigation of lupus by SIRT1-siRNA in MRL/lpr mice. Scand. J. Rheumatol. 2009, 38, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; McComb, S.; Mulligan, R.; Dudani, R.; Krishnan, L.; Sad, S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 2012, 13, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Liu, F.; Dong, G.; Ren, D.; Xu, Y.; Dou, J.; Wang, T.; Sun, L.; Hou, Y. Activation-induced necroptosis contributes to B-cell lymphopenia in active systemic lupus erythematosus. Cell Death Dis. 2014, 5, e1416. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Izquierdo, M.C.; Sanchez-Nino, M.D.; Ucero, A.C.; Egido, J.; Ruiz-Ortega, M.; Ramos, A.M.; Putterman, C.; Ortiz, A. TWEAK and the progression of renal disease: Clinical translation. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 1), i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Yim, L.Y.; Tam, R.C.; Chan, A.; Lu, L.; Lau, C.S.; Chan, V.S. MicroRNA-155 Mediates Augmented CD40 Expression in Bone Marrow Derived Plasmacytoid Dendritic Cells in Symptomatic Lupus-Prone NZB/W F1 Mice. Int. J. Mol. Sci. 2016, 17, 1282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xiong, L.; Wang, Y.; Ding, L.; Hu, S.; Zhao, M.; Zhou, L. Treatment of murine lupus with PD-LIg. Clin. Immunol. 2016, 162, 1–8. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Fu, S.; Zhao, M.; Lu, L.; Lu, Q. Dysregulation of Cell Death and Its Epigenetic Mechanisms in Systemic Lupus Erythematosus. Molecules 2017, 22, 30. https://doi.org/10.3390/molecules22010030
Wu H, Fu S, Zhao M, Lu L, Lu Q. Dysregulation of Cell Death and Its Epigenetic Mechanisms in Systemic Lupus Erythematosus. Molecules. 2017; 22(1):30. https://doi.org/10.3390/molecules22010030
Chicago/Turabian StyleWu, Haijing, Siqi Fu, Ming Zhao, Liwei Lu, and Qianjin Lu. 2017. "Dysregulation of Cell Death and Its Epigenetic Mechanisms in Systemic Lupus Erythematosus" Molecules 22, no. 1: 30. https://doi.org/10.3390/molecules22010030