4. Experimental Section
The glassware used in air- and/or moisture-sensitive reactions was flame-dried and reactions were carried out under dry argon. Unless otherwise noted, chemicals were commercially available and used without further purification. Solvents were distilled before use. Dichloromethane was distilled from phosphorus pentoxide. Nuclear magnetic resonance spectra were recorded with a Bruker AM-500 MHz spectrometer. The 1H NMR spectra are referenced with respect to the residual CHCl3 proton of the solvent CDCl3 at δ = 7.26 ppm. Coupling constants are reported in Hz. 13C NMR spectra were fully decoupled and are referenced to the middle peak of the solvent CDCl3 at δ = 77.0 ppm. 31P NMR spectra are referenced with respect to the peak of 85% H3PO4 as external reference. For comparative purposes, all NMR spectra acquired in D2O for free bisphosphonic acids were performed at the same conditions. Splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quadruplet; dd, double doublet, etc. Melting points were determined with a Fisher–Johns apparatus and are uncorrected. High-resolution mass spectra were obtained using a Bruker micrOTOF-Q II spectrometer, which is a hybrid quadrupole time of flight mass spectrometer with MS/MS capability. Analytical TLC was performed on commercial 0.2 mm aluminum-coated silica gel plates (F254) and visualized by 254 nm UV or immersion in an aqueous solution of (NH4)6Mo7O24·4H2O (0.04 M), Ce(SO4)2 (0.003 M) in concentrated H2SO4 (10%).
As judged from the homogeneity of the 1H, 13C, 31P NMR spectra and HPLC analyses of the title compounds employing a Beckmann Ultrasphere ODS-2 column 5 μM, 250 × 10 mm eluting with water–acetonitrile (9:1) at 3.00 mL/min with a refractive index detector indicated a purity >97%.
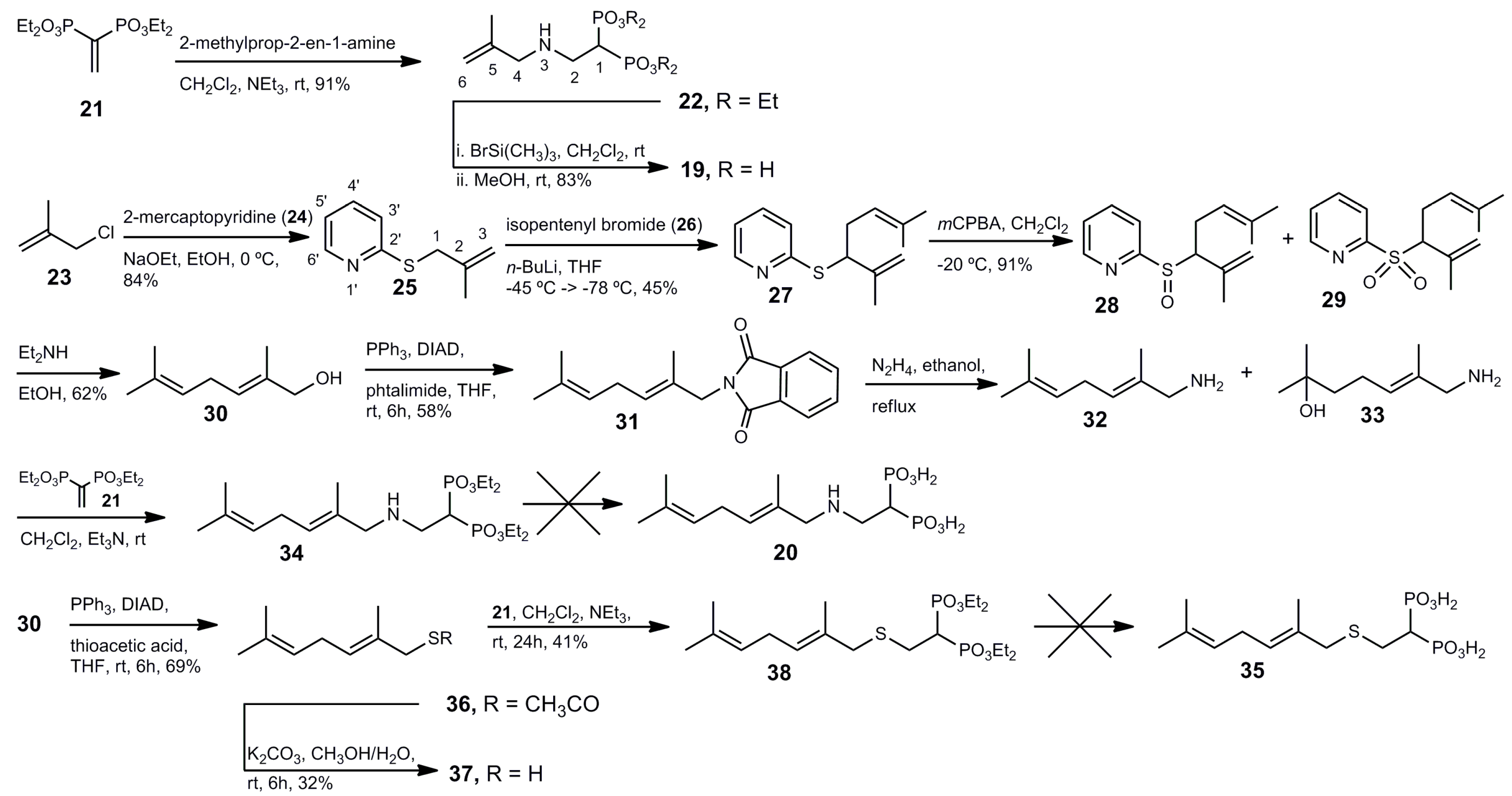
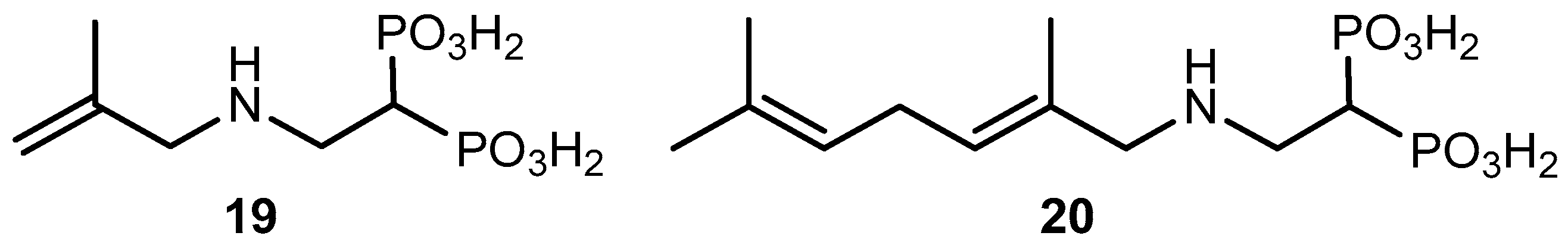
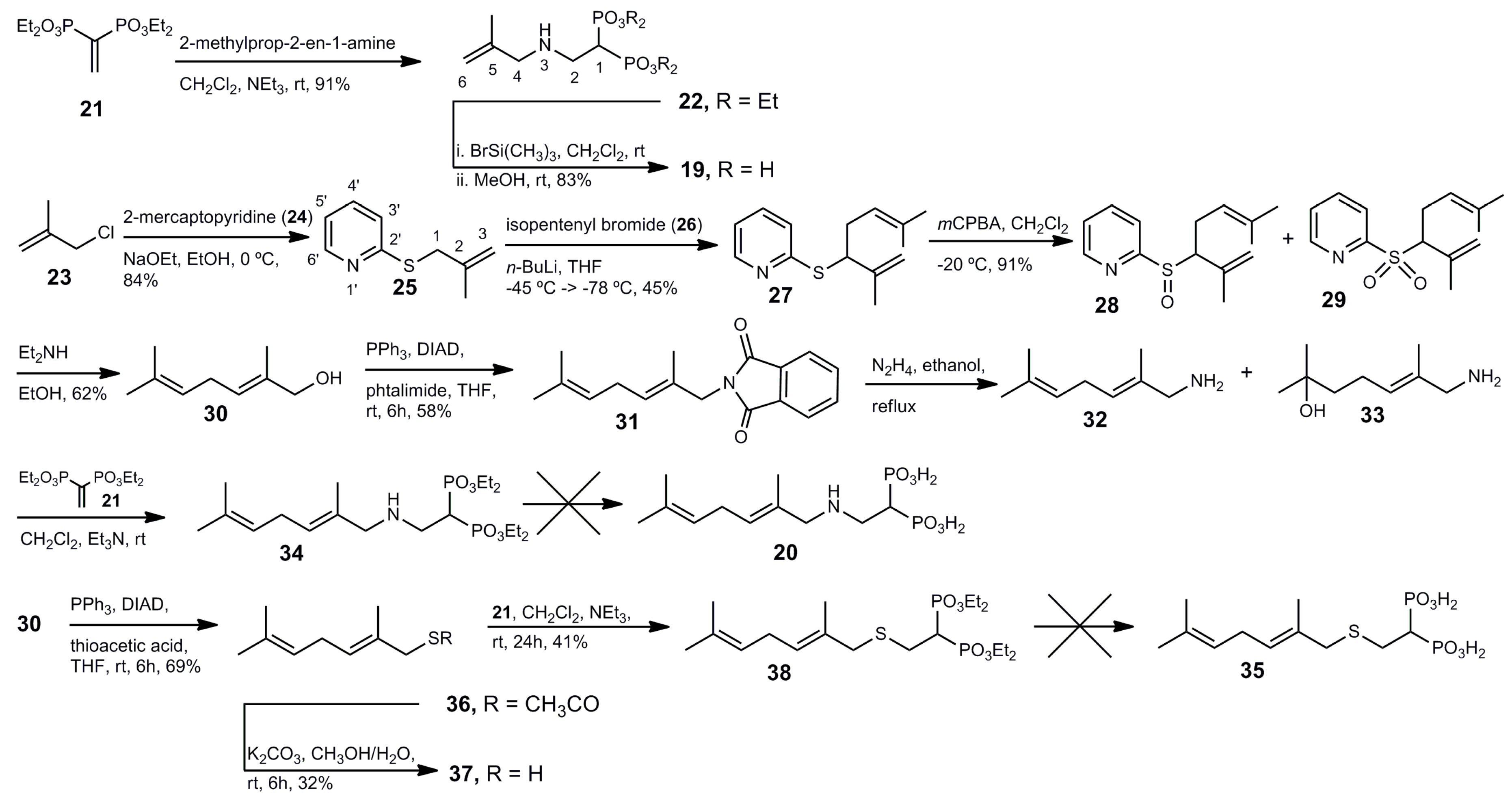
Tetraethyl 1-[(2-methylall-1-ylamino)ethyl] 1,1-bisphosphonate (22). A solution of compound 21 (300 mg, 1.0 mmol) in anhydrous methylene chloride (10 mL) was treated with triethylamine (121.2 mg, 168 μL, 1.2 mmol) and 2-methylallylamine hydrochloride (129 mg, 1.2 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was purified by column chromatography (silica gel) employing hexane–EtOAc (17:3) as eluent to produce 340 mg (91% yield) of 22 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.34 (t, J = 7.1 Hz, 12H, H-2′), 1.75 (s, 3H, CH3 at C-5), 2.66 (tt, J = 23.5, 5.8 Hz, 1H, H-1), 3.11 (dt, J = 16.9, 5.8 Hz, 2H, H-2), 3.17 (s, 2H, H-4), 4.19 (m, 8H, H-1′), 4.83 (br s, 1H, H-6a), 4.89 (br s, 1H, H-6b); 13C NMR (125.77 MHz, CDCl3) δ 16.4 (dd, J = 6.4, 2.6 Hz, C-2′), 20.6 (CH3 at C-5), 37.7 (t, J = 132.5 Hz, C-1), 44.9 (t, J = 4.3 Hz, C-2), 55.1 (C-4), 62.6 (dd, J = 32.6, 6.5 Hz, C-1′), 111.0 (C-6) 143.3 (C-5); 31P NMR (202.46 MHz, CDCl3) δ 22.73. HRMS (ESI) calcd. for (C14H32O6NP2) [M + H]+ 372.1707; found 372.1712.
1-[(2-Methylall-1-ylamino)ethyl] 1,1-bisphosphonic Acid (19). To a solution of the tetraethyl ester 22 (320 mg, 0.86 mmol) in anhydrous methylene chloride was added dropwise trimethylsilyl bromide (1.276 g, 1.10 mL, 8.6 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature for 48 h. After cooling at 0 °C, anhydrous methanol (10 mL) was added, and the resulting mixture was allowed to reach room temperature and stirred for 24 h. The solution was then concentrated under reduced pressure. The residue was dissolved in dry methanol (10 mL) and subsequently concentrated under reduced pressure twice. The solvent was evaporated and the residue was crystallized from ethanol–water to yield 183 mg (83% yield) of pure 19 as a white solid: 1H NMR (500.13 MHz, D2O) δ 1.74 (s, 3H, CH3 at C-5), 2.44 (m, 1H, H-1), 3.36 m, 2H, H-2), 3.59 (s, 2H, H-4), 5.03 (s, 1H, H-6a), 5.12 (s, 1H, H-6b); 13C NMR (125.77 MHz, D2O) δ 19.5 (CH3 at C-5), 35.7 (t, J = 128.8 Hz, C-1), 44.3 (C-2), 52.5 (C-4), 117.4 (C-6) 135.9 (C-5); 31P NMR (202.46 MHz, D2O) δ 16.96. HRMS (ESI) calcd. for (C6H15O6NP2Na) [M + Na]+ 282.0272; found 282.0268.
2-((2-Methylallyl)thio)pyridine (25). To a solution of sodium ethoxide (prepared from sodium (620 mg, 27.0 mmol) and anhydrous ethanol (20 mL)) was added 2-mercaptopyridine (24; 3.00 g, 27.0 mmol) at 0 °C. The mixture was stirred for 15 min. Then, methallyl chloride (23; 3.06 mL, 2.81 g, 31.0 mmol) was added and the mixture was stirred at 0 °C for 3 h. The solvent was evaporated and the residue was partitioned between water (50 mL) and ethyl ether (50 mL). The organic phase was washed with an aqueous 5% solution of sodium hydroxide (2 × 30 mL) and water (2 × 30 mL). The organic layer was dried (MgSO4) and the solvent was evaporated to produce 3.743 g (84% yield) of 25 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.85 (s, 3H, CH3 at C-2), 3.86 (s, 2H, H-1), 4.86 (t, J = 1.4 Hz, 1H, H-3a), 5.02 (q, J = 0.9 Hz, 1H, H-3b), 6.97 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H, H-5′), 7.18 (dt, J = 8.1, 0.9 Hz, 1H, H-3′), 7.47 (ddd, J = 7.9, 7.5, 1.8 Hz, 1H, H-4′), 8.42 (ddd, J = 4.9, 1.7, 0.9 Hz, 1H, H-6′); 13C NMR (125.77 MHz, CDCl3) δ 21.5 (CH3 at C-2), 37.1 (C-3), 113.7 (C-1), 119.5 (C-5′), 122.3 (C-3′),135.9 (C-4′), 141.3 (C-2), 149.4 (C-6′), 159.0 (C-2′).
2-((2,6-Dimethylhepta-1,5-dien-3-yl)thio)pyridine (27). A solution of 25 (3.74 g, 22.7 mmol) in anhydrous tetrahydrofuran (50 mL) cooled at −50 °C was treated dropwise with an 1.1 M solution of n-butyllithium in hexane (22 mL). The reaction mixture turned deep-red and was cooled to −78 °C. Then, to the resulting mixture a solution of isopentenyl bromide (26; 2.8 mL, 3.61 g, 24.2 mmol) in anhydrous tetrahydrofuran (10 mL) was added dropwise. The reaction mixture was stirred at −78 °C for 1 h. Then, the mixture was allowed to reach room temperature gradually. The reaction was quenched by addition of a saturated solution of ammonium chloride (50 mL). The organic layer was separated and the aqueous phase was extracted with ethyl ether (50 mL). The combined organic layers were dried (MgSO4) and the solvent was evaporated to give 2.384 g of 27 (45% yield) as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.64 (s, 3H, CH3 at C-6), 1.68 (s, 3H, H-7), 1.82 (s, 3H, CH3 at C-2), 2.43 (m, 1H, H-4a), 2.56 (m, 1H, H-4b), 4.40 (dd, J = 8.6, 6.6 Hz, 1H, H-3), 4.85 (br s, 1H, H-1a), 4.98 (br s, 1H, H-1b), 5.15 (tt, J = 7.0, 1.2 Hz, 1H, H-5), 6.92 (ddt, J = 7.3, 5.1, 1.0 Hz, 1H, H-5′), 7.17 (ddd, J = 8.0, 1.9, 1.0 Hz, 1H, H-3′), 7.46 (m, 1H, H-4′), 8.42 (ddd, J = 4.0, 1.8, 0.9 Hz, 1H, H-6′); 13C NMR (125.77 MHz, CDCl3) δ 18.0 (CH3 at C-6), 19.5 (CH3 at C-2), 25.8 (C-7), 32.1 (C-4), 51.2 (C-3), 113.3 (C-1), 119.6 (C-5′), 121.2 (C-3′), 123.2 (C-5), 133.5 (C-6), 135.9 (C-4′), 144.2 (C-2), 149.4 (C-6′), 159.1 (C-2′). HRMS (ESI) calcd. for C14H20NS [M + H]+ 234.1316; found 234.1319.
2-((2,6-Dimethylhepta-1,5-dien-3-yl)sulfinyl)pyridine (28) and 2-((2,6-dimethylhepta-1,5-dien-3-yl)sulfonyl)pyridine (29). To a solution of 27 (5.91 mg, 25.3 mmol) in methylene chloride (100 mL) cooled at −25 °C under an argon atmosphere was added dropwise a solution of m-chloroperbenzoic acid (77% purity, 5.61 g, 26.6 mmol) in methylene chloride (100 mL). The mixture was stirred at −25 °C for 1 h. Then, the temperature was allowed to reach room temperature. Then, the mixture was extracted with a saturated solution of potassium carbonate (2 × 75 mL) and water (2 × 75 mL). The organic phase was dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography (silica gel) eluting was hexane–EtOAc (19.1) to give 5.68 g of 28 (91% yield) and 403 mg of 29 (6% yield) as colorless oils: Compound 28 (two diastereomers): 1H NMR (500.13 MHz, CDCl3) δ 1.42 (s, 3H, CH3 at C-6), 1.55 (s, 3H, CH3* at C-6), 1.50 (s, 3H, H-7), 1.70 (s, 3H, H-7*), 1.73 (s, 3H, CH3 at C-2), 1.88 (s, 3H, CH3* at C-2), 2.25 (m, 1H, H-4a), 2.60 (m, 1H, H-4a*), 2.47 (m, 1H, H-4b), 2.85 (m, 1H, H-4b*), 3.46 (dd, J = 8.2, 7.9 Hz, 1H, H-3), 3.57 (dd, J = 10.3, 4.9 Hz, 1H, H-3*), 4.48 (br s, 1H, H-1a), 4.77 (p, J = 1.5 Hz, 1H, H-1a*), 4.83 (tp, J = 7.0, 1.4 Hz, 1H, H-3), 5.03 (br s, 1H, H-1b), 5.13 (p, J = 1.5 Hz, 1H, H-1b*), 5.19 (m, 1H, H-3*), 7.31 (m, 1H, aromatic proton), 7.88–7.97 (m, 2H, aromatic proton), 8.60–8.64 (m, 1H, aromatic proton); compound 29 1H NMR (500.13 MHz, CDCl3) δ 1.57 (s, 3H, CH3 at C-6), 1.67 (s, 3H, H-7), 1.82 (br s, 3H, CH3 at C-2), 2.48 (m, 1H, H-4a), 2.72 (m, 1H, H-4b), 4.21 (dd, J = 11.2, 4.2 Hz, 1H, H-3), 4.84 (br s, 1H, H-1a), 4.88 (t, J = 7.0 Hz, 1H, H-5), 5.02 (br s, 1H, H-1b), 7.51 (ddd, J = 7.7, 4.7, 1.1 Hz, 1H, H-5′), 7.91 (dt, J = 7.7, 1.7 Hz, 1H, H-4′), 8.02 (d, J = 7.9 Hz, 1H, H-3′), 8.74 (ddd, J = 4.6, 1.6, 0.8 Hz, 1H, H-6′); 13C NMR (125.77 MHz, CDCl3) δ 17.9 (CH3 at C-6), 20.2 (CH3 at C-2), 24.6 (C-7), 25.7 (C-4), 67.7 (C-3), 118.4 (C-1), 120.8 (C-5′), 123.4 (C-3′), 127.1 (C-5), 135.1 (C-6), 136.4 (C-2), 137.7 (C-4′), 150.1 (C-6′), 156.6 (C-2′).
(E)-2,6-Dimethylhepta-2,5-dien-1-ol (30). A solution of 28 (5.65 g, 22.7 mmol) in methanol (100 mL) was treated with diethylamine (100 mL) and the resulting mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was partitioned between ethyl ether (100 mL) and water (100 mL). The aqueous phase was extracted with ethyl ether (2 × 50 mL). The combined organic layers were washed with 5% hydrochloric acid (2 × 30 mL), a saturated solution of sodium bicarbonate (2 × 30 mL), brine (3 × 30 mL). The organic layer was dried (MgSO4) and the solvent was evaporated. The product was purified by column chromatography (silica gel) eluting with a mixture of hexane EtOAc (19:1) to yield 1.973 g (62% yield) of alcohol 30 as a colorless oil: Rf = 0.51 (hexane–EtOAc, 1:1): 1H NMR (500.13 MHz, CDCl3) δ 1.64 (s, 3H, C-7), 1.70 (s, 6H, CH3 at C-2, CH3 at C-6), 2.73 (t, J = 7.2 Hz, 2H, H-4), 4.00 (s, 2H, H-1), 5.11 (tp, J = 7.0, 1.3 Hz, 1H, H-3), 5.39 (tq, J = 7.3, 1.3 Hz, 1H, H-5); 13C NMR (125.77 MHz, CDCl3) δ 13.6 (CH3 at C-2), 17.7 (CH3 at C-6), 25.7 (C-7), 26.7 (C-4), 69.0 (C-1), 122.4 (C-3), 125.1 (C-5), 132.0 (C-6), 134.5 (C-2).
(E)-2-(2,6-Dimethylhepta-2,5-dien-1-yl)isoindoline-1,3-dione (31). To a mixture of alcohol 30 (406 mg, 2.9 mmol), triphenylphosphine (911.3 mg, 3.5 mmol), phtalimide (511.2 mg, 3.5 mmol) in anhydrous tetrahydrofuran (10 mL) cooled at 0 °C was added a solution of diisopropyl azodicarboxylate (702.6 mg, 3.5 mmol). The reaction mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was purified by column chromatography (silica gel) eluting with a mixture of hexane–EtOAc (19:1) to give 453.0 mg (58% yield) of pure 31 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.60 (s, 3H, C-7), 1.67 (s, 6H, CH3 at C-2, CH3 at C-6), 2.70 (t, J = 7.3 Hz, 2H, H-4), 4.20 (s, 2H, H-1), 5.05 (tp, J = 7.0, 1.4 Hz, 1H, H-3), 5.36 (tq, J = 7.2, 1.3 Hz, 1H, H-5); 13C NMR (125.77 MHz, CDCl3) δ 14.6 (CH3 at C-2), 17.7 (CH3 at C-6), 25.6 (C-7), 26.9 (C-4), 45.0 (C-1), 122.3 (C-3), 123.3 (C-2′), 126.6 (C-5), 129.0(C-1′), 132.0 (C-6), 132.1 (C-2), 133.9 (C-3′), 168.3 (CO). HRMS (ESI) calcd. for C17H19O2NNa [M + Na]+ 292.1313; found 292.1315.
(E)-2,6-Dimethylhepta-2,5-dien-1-amine (32) and (E)-7-amino-2,6-dimethylhept-5-en-2-ol (33). A solution of 31 (400 mg, 1.5 mmol) in ethanol (10 mL) was treated with a solution of 60% hidrazine in water (0.1 mL). The reaction mixture was refluxed for 3 h. The mixture was allowed to cool to room temperature and was filtered. The solvent was evaporated. The residue was treated with a 20% aqueous solution of hydrochloric acid (10 mL) and the mixture was filtered. The pH of the solution was adjusted to 11 by addition of a 1.0 M solution of sodium hydroxide. The aqueous solution was extracted with methylene chloride (3 × 30 mL), dried (MgSO4) and the solvent was evaporated. The residue was purified by column chromatography (silica gel) employing a mixture of hexane–EtOAc (4:1) to give 175.4 mg of 32 (84% yield) and 21.2 mg of 33 (9% yield): Compound 32: 1H NMR (500.13 MHz, CDCl3) δ 1.64 (s, 3H, C-7), 1.66 (CH3 at C-2), 1.70 (CH3 at C-6), 2.72 (t, J = 7.1 Hz, 2H, H-4), 3.17 (s, 2H, H-1), 5.11 (tq, J = 7.1, 1.2 Hz, 1H, H-3), 5.26 (tt, J = 7.1, 1.0 Hz, 1H, H-5); 13C NMR (125.77 MHz, CDCl3) δ 14.6 (CH3 at C-2), 17.7 (CH3 at C-6), 25.7 (C-7), 26.8 (C-4), 49.9 (C-1), 122.6 (C-3), 122.9 (C-5), 131.7 (C-6), 136.3 (C-2). Compound 33: 1H NMR (500.13 MHz, CDCl3) δ 1.24 (s, 6H, H-7, CH3 at C-6), 1.36 (s, 2H, NH2), 1.55 (m, 2H, H-5), 1.66 (s, 3H, CH3 at C-2), 2.11 (m, 2H, H-4), 3.17 (s, 2H, H-1), 5.30 (tq, J = 7.1, 1.2 Hz, 1H, H-3); 13C NMR (125.77 MHz, CDCl3) δ 14.5 (CH3 at C-2), 22.7 (C-4), 29.2 (C-7, CH3 at C-6), 43.5 (C-5), 49.9 (C-1), 70.8 (C-6), 123.7 (C-3), 136.6 (C-2). HRMS (ESI) calcd. for C9H19ONNa [M + Na]+ 180.1364; found 180.1353.
Tetraethyl (E)-2,6-dimethylhepta-2,5-dien-1-ylamino ethyl-1,1-bisphosphonate (34). A solution of 21 (327.8 mg, 1.1 mmol) in anhydrous methylene chloride (10 mL) was treated with triethylamine (133.1 mg, 185 μL, 1.3 mmol) and 32 (152.0 mg, 1.1 mmol) as described for the preparation of 22. Evaporation of the solvent yielded 483.2 mg (100% yield) of pure 34 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.34 (t, J = 7.1 Hz, 12H, CH2CH3), 1.63 (s, 3H, C-10), 1.66 (CH3 at C-5), 1.68 (CH3 at C-9), 2.64 (tt, J = 23.7, 5.9 Hz, 1H, H-1), 2.71 (t, J = 6.8 Hz, 2H, H-7), 3.08 (dt, J = 16.8, 5.9 Hz, 2H, H-2), 3.12 (s, 2H, H-4), 4.18 (m, 8H, CH2CH3), 5.09 (tt, J = 7.1, 1.3 Hz, 1H, H-6), 5.29 (t, J = 7.2 Hz, 1H, H-8); 13C NMR (125.77 MHz, CDCl3) δ 14.6 (CH3 at C-5), 16.3 (d, J = 3.0 Hz, CH2CH3), 16.4 (d, J = 2.7 Hz, CH2C′H3), 18.7 (CH3 at C-9), 24.7 (C-10), 26.9 (C-7), 37.6 (t, J = 132.4 Hz, C-1), 44.8 (t, J = 4.3 Hz, C-2), 57.0 (C-4), 62.4 (d, J = 6.6 Hz, CH2CH3), 62.7 (d, J = 6.6 Hz, C′H2CH3), 122.8 (C-6), 125.1 (C-8), 131.5 (C-9), 132.8 (C-5); 31P NMR (202.46 MHz, CDCl3) δ 22.82 ppm.
(E)-S-(2,6-Dimethylhepta-2,5-dien-1-yl) ethanethioate (36). To a mixture of alcohol 30 (471.0 mg, 3.4 mmol), triphenylphosphine (1.32 g, 5.04 mmol), thioacetic acid (255.7 mg, 3.4 mmol) in anhydrous tetrahydrofuran (10 mL) cooled at 0 °C was added a solution of diisopropyl azodicarboxylate (1.02 g, 5.04 mmol) as depicted for the preparation of 31. The product was purified by column chromatography (silica gel) eluting with a mixture of hexane–EtOAc (49:1) to give 459.0 mg (69% yield) of 36 as a colorless oil: Rf 0.37 (hexane–EtOAc, 19:1); 1H NMR (500.13 MHz, CDCl3) δ 1.62 (s, 3H, C-7), 1.65 (m, 3H, CH3 at C-2), 1.68 (d, J = 1.1 Hz, 3H, CH3 at C-6), 2.33 (s, 3H, CH3C(O)), 2.68 (t, J = 6.9 Hz, 2H, H-4), 3.55 (q, J = 0.9 Hz, 2H, H-1), 5.11 (tq, J = 7.1, 1.2 Hz, 1H, H-3), 5.38 (tq, J = 7.3, 1.2 Hz, 1H, H-5); 13C NMR (125.77 MHz, CDCl3) δ 15.2 (CH3 at C-2), 17.7 (CH3 at C-6), 25.7 (C-7), 27.2 (C-4), 30.5 (CH3C(O)), 38.2 (C-1), 122.2 (C-3), 127.9 (C-5), 130.0 (C-2), 132.1 (C-6), 195.1 (CH3C(O)).
(E)-2,6-Dimethylhepta-2,5-dien-1-thiol (37). To a solution of 36 (353.0 mg, 1.78 mmol) in 20.0 mL of a mixture of methanol–water (5:3) was added potassium carbonate (100 mg, 0.72 mmol). The reaction mixture was stirred at room temperature for 6 h. Then, water (50 mL) was added and the mixture was extracted with methylene chloride (3 × 30 mL). The combined organic layers were washed with brine (2 × 30 mL), dried (MgSO4) and the solvent was evaporated. The residue was purified by column chromatography (silica gel) eluting with hexane to yield 89 mg (32% yield) of pure 37 as a colorless oil: Rf 0.57 (hexane–EtOAc, 19:1); 1H NMR (300.18 MHz, CDCl3) δ 1.40 (t, J = 7.8 Hz, 1H, SH), 1.63 (s, 3H, C-7), 1.69 (s, 3H, CH3 at C-2), 1.75 (q, J = 1.0 Hz, 3H, CH3 at C-6), 2.71 (q, J = 7.9 Hz, 2H, H-4), 3.12 (d, J = 7.8 Hz, 2H, H-1), 5.08 (tp, J = 7.1, 1.4 Hz, 1H, H-3), 5.31 (tq, J = 7.2, 1.0 Hz, 1H, H-5).
Tetraethyl (E)-2,6-dimethylhepta-2,5-dien-1-ylthioethyl-1,1-bisphosphonate (38). A solution of 21 (430.3 mg, 1.43 mmol) in anhydrous methylene chloride (15 mL) was treated with triethylamine (147.1 mg, 205 μL, 1.43 mmol) and 37 (224.1 mg, 1.43 mmol) as described for the preparation of 22. The solvent was evaporated and the residue was purified by column chromatography (silica gel) employing hexane–EtOAc (4:1) as eluent to give 268 mg (41% yield) of 38 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.366 (t, J = 7.0 Hz, 6H, CH2CH3), 1.374 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.64 (s, 3H, C-10), 1.70 (s, 3H, CH3 at C-5), 1.74 (m, 3H, CH3 at C-9), 2.61 (tt, J = 23.8, 6.2 Hz, 1H, H-1), 2.74 (t, J = 7.6 Hz, 2H, H-7), 2.94 (dt, J = 16.2, 6.2 Hz, 2H, H-2), 3.15 (s, 2H, H-4), 4.21 (m, 8H, CH2CH3), 5.11 (tp, J = 7.0, 1.6 Hz, 1H, H-6), 5.31 (t, J = 7.1 Hz, 1H, H-8); 13C NMR (125.77 MHz, CDCl3) δ 14.9 (CH3 at C-5), 16.2 (t, J = 3.3 Hz, CH2CH3), 16.4 (dd, J = 6.5, 1.3 Hz, CH2C′H3), 17.7 (CH3 at C-9), 25.6 (C-10), 26.6 (t, J = 5.0 Hz, C-2), 27.2 (C-7), 38.7 (t, J = 131.7 Hz, C-1), 42.4.0 (C-4), 62.7 (d, J = 4.8 Hz, CH2CH3), 62.8 (d, J = 6.7 Hz, C′H2CH3), 122.2 (C-6), 127.9 (C-8), 130.0 (C-4), 132.1 (C-9); 31P NMR (202.47 MHz, CDCl3) δ 21.77 ppm.
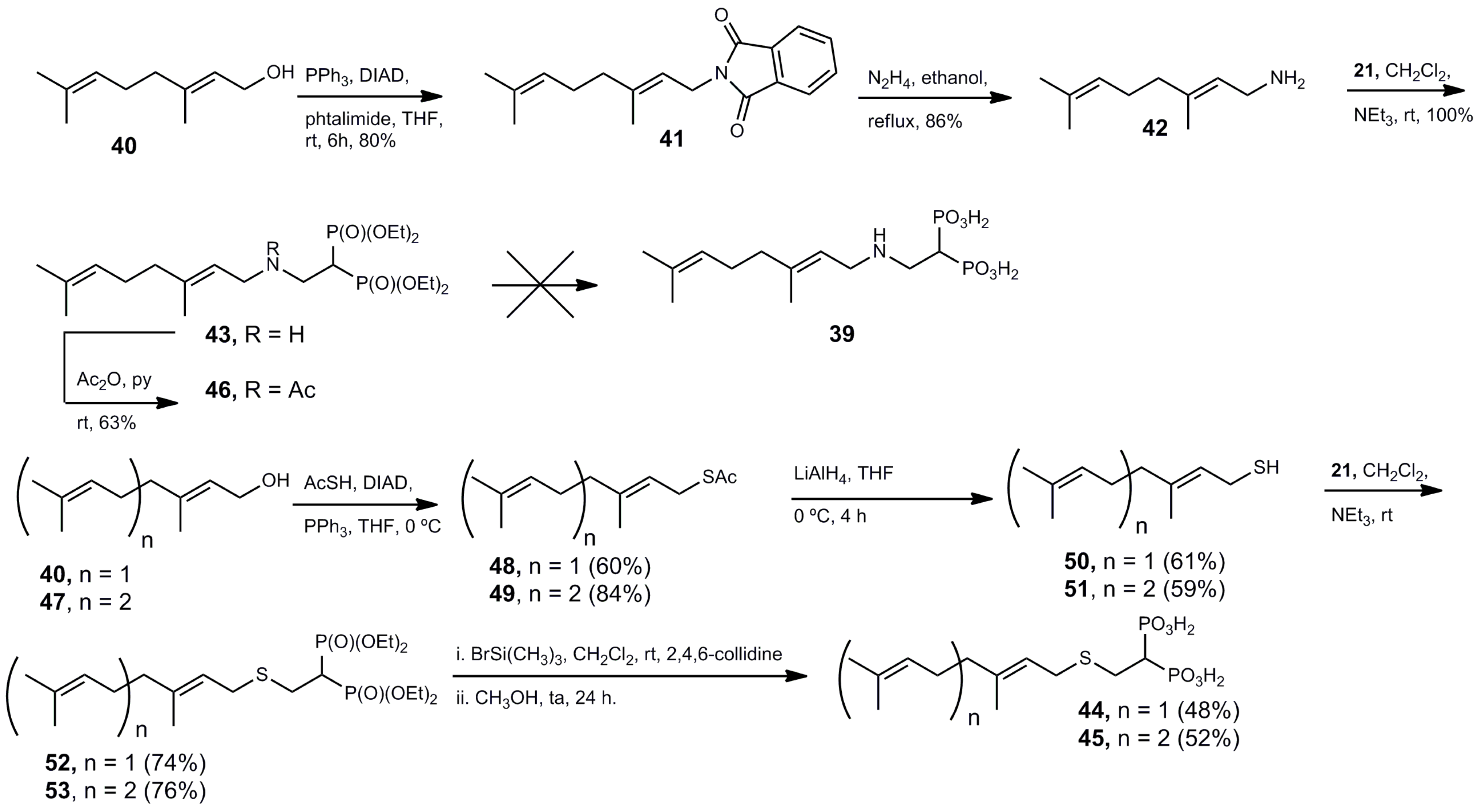
(E)-2-(3,7-Dimethylocta-2,6-dien-1-yl)isoindoline-1,3-dione (41). To a mixture of geraniol 40 (1.00 g, 6.5 mmol), triphenylphosphine (1.70 g, 6.5 mmol), phtalimide (953.3 mg, 6.5 mmol) in anhydrous tetrahydrofuran (15 mL) cooled at 0 °C was added a solution of diisopropyl azodicarboxylate (1.38 g, 6.5 mmol) as described for the preparation of 31. The residue was purified by column chromatography (silica gel) eluting with hexane–EtOAc (19:1) to give 1.47 g (80 yield) of pure 41 as a colorless oil: Rf 0.52 (hexane–EtOAc, 4:1); 1H NMR (200 MHz, CDCl3) δ 1.56 (s, 3H, H-8), 1.63 (s, 3H, CH3 at C-7), 1.82 (s, 3H, CH3 at C-3), 2.00 (m, 2H, H-5), 2.08 (p, J = 7.0 Hz, 2H, H-4), 4.27 (d, J = 7.2 Hz, 2H, H-1), 5.05 (m, 1H, H-6), 5.27 (t, J = 7.4 Hz, 1H, H-2).
(E)-3,7-Dimethylocta-2,6-dien-1-amine (42). A solution of 41 (900 mg, 3.2 mmol) in ethanol (13 mL) was treated with a solution of 60% hidrazine in water (150 µL, 101 mg, 3.2 mmol). The reaction mixture was refluxed for 3 h and was treated as described for the preparation of 32 to yield 422 mg (86% yield) as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.60 (s, 3H, H-8), 1.63 (s, 3H, CH3 at C-7), 1.69 (s, 3H, CH3 at C-3), 2.00 (m, 2H, H-5), 2.08 (p, J = 7.0 Hz, 2H, H-4), 3.27 (d, J = 6.8 Hz, 2H, H-1), 5.10 (tp, J = 6.7, 1.3 Hz, 1H, H-6), 5.26 (tp, J = 6.9, 1.2 Hz, 1H, H-2); 13C NMR (125.77 MHz, CDCl3) δ 16.1 (CH3 at C-3), 17.7 (CH3 at C-7), 25.7 (C-8), 26.5 (C-5), 39.5 (C-4), 39.6 (C-1), 124.1 (C-2), 125.9 (C-6), 131.5 (C-7), 136.3 (C-3).
Tetraethyl 1-[((E)-3,7-dimethylocta-2,6-diene-1-amino)ethyl] 1,1-bisphosphonate (43). A solution of 21 (400.1 mg, 1.33 mmol) in anhydrous methylene chloride (10 mL) was treated with amine 42 (204.0 mg, 1.33 mmol) as described for the preparation of 22. Evaporation of the solvent gave 611.1 mg (100% yield) of pure 43 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.34 (t, J = 7.1 Hz, 12H, CH2CH3), 1.63 (s, 3H, H-11), 1.66 (s, 3H, CH3 at C-10), 1.68 (s, 3H, CH3 at C-6), 2.71 (t, J = 6.8 Hz, 4H, H-7, H-8), 2.65 (tt, J = 23.7, 5.9 Hz, 1H, H-1), 3.08 (dt, J = 16.8, 5.9 Hz, 2H, H-2), 3.13 (br s, 2H, H-4), 5.09 (tt, J = 7.1, 1.3 Hz, 1H, H-9), 5.29 (t, J = 7.2 Hz, 1H, H-5); 13C NMR (125.77 MHz, CDCl3) δ 14.6 (CH3 at C-6), 16.3 (d, J = 3.0 Hz, CH2CH3), 16.4 (d, J = 2.7 Hz, CH2CH3′), 17.7 (CH3 at C-10), 25.6 (C-11), 26.9 (C-8), 37.6 (t, J = 132.4 Hz, C-1), 44.8 (t, J = 4.3 Hz, C-2), 57.0 (C-4), 62.4 (d, J = 6.8 Hz, CH2CH3), 62.7 (d, J = 6.8 Hz, CH2CH3′), 122.8 (C-5), 125.1 (C-9), 131.5 (C-10), 132.8 (C-6); 31P NMR (202.46 MHz, CDCl3) δ 22.82 ppm.
Tetraethyl 1-[((E)-3,7-dimethylocta-2,6-diene-1-acetamido)ethyl] 1,1-bisphosphonate (46). A solution of 43 (227 mg, 0.5 mmol) in anhydrous pyridine (3.0 mL) was treated with acetic anhydride (3.0 mL) and the mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was purified by column chromatography (silica gel) eluting with a mixture of CH2Cl2–methanol (19:1) to give 312.2 mg (63% yield) of pure 46 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.34 (t, J = 7.0 Hz, 6H, CH2CH3), 1.35 (t, J = 7.1 Hz, 6H, CH2CH3′), 1.61 (s, 3H, H-11), 1.68 (s, 3H, CH3 at C-10), 1.69 (s, 3H, CH3 at C-6), 2.00–2.11 (m, 4H, H-7, H-8), 2.07 (s, 3H, C(O)CH3), 3.38 (tt, J = 22.7, 5.5 Hz, 1H, H-1), 3.71 (ddd, J = 14.7, 11.1, 7.4 Hz, 2H, H-2), 4.04 (d, J = 6.4 Hz, 2H, H-4), 4.18 (m, 8H, CH2CH3), 5.07 (m, 2H, H-5, H-9); 13C NMR (125.77 MHz, CDCl3) δ 16.3 (CH3 at C-6), 16.34 (d, J = 6.7 Hz, CH2CH3), 16.37 (d, J = 6.6 Hz, CH2CH3′), 17.7 (CH3 at C-10), 21.9 (C(O)CH3), 25.7 (C-11), 26.3 (C-8), 34.4 (t, J = 129.9 Hz, C-1), 39.5 (C-7), 44.4 (t, J = 3.5 Hz, C-2), 48.8 (C-4), 62.4 (d, J = 6.8 Hz, CH2CH3), 62.7 (d, J = 6.8 Hz, CH2CH3′), 119.7 (C-5), 123.6 (C-9), 131.9 (C-10), 139.4 (C-6), 171.2 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.49 ppm.
(E)-S-(3,7-Dimethylocta-2,6-dien-1-yl) ethanethioate (48). To a mixture of geraniol 40 (1.00 g, 6.5 mmol), triphenylphosphine (2.55 g, 9.7 mmol), thioacetic acid (0.55 mL, 592.2 mg, 7.8 mmol) in anhydrous tetrahydrofuran (20 mL) cooled at 0 °C was added a solution of diisopropyl azodicarboxylate (1.62 mL, 1.66 g, 9.7 mmol) as depicted for the preparation of 31. The product was purified by column chromatography (silica gel) eluting with hexane–EtOAc (9:1) to give 828.2 mg (60% yield) of 48 as a colorless oil: Rf 0.39 (hexane–EtOAc, 4:1); 1H NMR (500.13 MHz, CDCl3) δ 1.59 (s, 3H, H-8), 1.68 (s, 6H, CH3 at C-3, CH3 at C-7), 2.00 (m, 2H, H-4), 2.06 (m, 2H, H-4), 2.32 (s, 3H, SC(O)CH3), 3.54 (d, J = 7.9 Hz, 2H, H-1), 5.06 (tp, J = 6.8, 1.4 Hz, 1H, H-6), 5.21 (tp, J = 7.9, 1.3 Hz, 1H, H-2); 13C NMR (125.77 MHz, CDCl3) δ 16.2 (CH3 at C-2), 17.7 (CH3 at C-6), 25.6 (C-1), 26.4 (C-8), 27.3 (C-5), 30.4 (SC(O)CH3), 39.5 (C-4), 118.4 (C-2), 123.8 (C-5), 131.7 (C-7), 140.1 (C-3), 196.1 (SC(O)CH3). HRMS (ESI) calcd. for C12H20OS [M + Na]+ 235.1133; found 235.1138.
(E)-3,7-Dimethylocta-2,6-diene-1-thiol (50). A solution of 48 (846 mg, 3.98 mmol) in anhydrous tetrahydrofuran (10 mL) cooled at 0 °C was treated with portionwise with lithium aluminum hydride (181 mg, 4.74 mmol). The mixture was stirred at 0 °C for 4 h. The reaction was quenched by addition of ethyl acetate (2.0 mL). Then, the mixture was partitioned between methylene chloride (50 mL) and an aqueous saturated solution of potassium tartrate (50 mL). The organic phase was washed with water (2 × 30 mL), dried (MgSO4), and the solvent was evaporated. The product was purified by column chromatography (silica gel) eluting with hexane to give 411 mg (61% yield) of 50 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.40 (t, J = 7.1 Hz, 1H, SH), 1.60 (s, 3H, H-8), 1.66 (d, J = 1.2 Hz, CH3 at C-7), 1.68 (d, J = 1.2 Hz, CH3 at C-3), 2.00 (m, 2H, H-4), 2.07 (m, 2H, H-4), 3.16 (ddd, J = 7.6, 7.3, 0.4 Hz, 2H, H-1), 5.08 (tq, J = 6.9, 1.4 Hz, 1H, H-6), 5.34 (tsxt, J = 7.8, 1.3 Hz, 1H, H-2); 13C NMR (125.77 MHz, CDCl3) δ 15.8 (CH3 at C-2), 17.7 (CH3 at C-6), 22.1 (C-1), 25.7 (C-8), 26.4 (C-5), 39.4 (C-4), 123.3 (C-2), 123.6 (C-5), 131.7 (C-7), 137.5 (C-3). HRMS (ESI) calcd. for C10H19S [M + H]+ 171.1207; found 171.1194.
Tetraethyl 1-[((E)-3,7-dimethylocta-2,6-diene-1-thio)ethyl] 1,1-bisphosphonate (52). A solution of 21 (1.00 mg, 3.34 mmol) in anhydrous methylene chloride (15 mL) was treated with triethylamine (337 mg, 470 μL, 3.34 mmol) and 50 (580.9 mg, 3.34 mmol) as described for the preparation of 22. The solvent was evaporated and the residue was purified by column chromatography (silica gel) employing EtOAc as eluent to give 1.163 g (74% yield) of 52 as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.35 (t, J = 7.1 Hz, 12H, CH2CH3), 1.60 (s, 3H, H-11), 1.67 (s, 3H, CH3 at C-10), 1.68 (s, 3H, CH3 at C-6), 2.02 (m, 2H, H-7), 2.07 (m, 2H, H-8), 2.61 (tt, J = 23.8, 6.1 Hz, 1H, H-1), 3.02 (dt, J = 16.2, 6.1 Hz, 2H, H-2), 3.22 (d, J = 7.7 Hz, 2H, H-4), 5.08 (m, 1H, H-9), 5.26 (t, J = 7.7 Hz, 1H, H-5); 13C NMR (125.77 MHz, CDCl3) δ 16.2 (CH3 at C-6), 16.4 (d, J = 6.1 Hz, CH2CH3), 17.7 (CH3 at C-10), 25.7 (C-11), 26.5 (C-8), 27.3 (t, J = 5.0 Hz, C-2), 30.7 (C-4), 38.9 (t, J = 131.6 Hz, C-1), 39.6 (C-4), 119.9 (C-5), 123.9 (C-9), 131.7 (C-10), 139.4 (C-6); 31P NMR (202.46 MHz, CDCl3) δ 21.72 ppm. HRMS (ESI) calcd. for C20H40O6SP2 [M + Na]+ 493.1919; found 493.1929.
1-[((E)-3,7-Dimethylocta-2,6-diene-1-thio)ethyl] 1,1-bisphosphonic Acid (44). A solution of the Michael adduct 52 (997 mg, 2.1 mmol) and 2,4,6-collidine (2.53 g, 2.76 mL, 21.0 mmol) in anhydrous methylene chloride (20 mL) was treated with bromotrimethylsilane (3.23 g, 2.73 mL, 21.1 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature for 48 h. Then, methanol (1.0 mL) was added and the solvent was evaporated. The residue was dissolved in methanol (10 mL) and the mixture was stirred at room temperature for 24 h. The solvent was evaporated and the residue redissolved/evaporated in methanol four times. The product was purified by column chromatography on reverse phase eluting with a mixture of water–acetonitrile (3:2) to give 361.2 mg (48% yield) of 44 as an amorphous solid: 1H NMR (500.13 MHz, D2O) δ 1.54 (s, 3H, H-11), 1.61 (s, 3H, CH3 at C-10), 1.62 (s, 3H, CH3 at C-6), 1.94–2.08 (m, 5H, H-1, H-7, H-8), 2.93 (ddd, J = 15.1, 14.0, 7.3 Hz, 2H, H-2), 3.20 (d, J = 7.7 Hz, 2H, H-4), 5.11 (t, J = 7.0 Hz, 1H, H-9), 5.30 (t, J = 7.5 Hz, 1H, H-5); 13C NMR (125.77 MHz, D2O) δ 15.3 (CH3 at C-6), 16.9 (CH3 at C-10), 24.8 (C-11), 25.7 (C-8), 28.7 (t, J = 3.7 Hz, C-2), 29.9 (C-4), 40.1 (t, J = 109.3 Hz, C-1), 38.7 (C-4), 119.8 (C-5), 124.2 (C-9), 133.6 (C-10), 140.2 (C-6); 31P NMR (202.46 MHz, D2O) δ 15.85 ppm. HRMS (ESI) calcd. for C12H22O6SP2Na3 [M + H]+ 425.0305; found 425.0298.
S-((2E,6E)-3,7,11-Trimethyldodeca-2,6,10-trien-1-yl) ethanethioate (49). To a mixture of farnesol 47 (1.00 g, 4.5 mmol), triphenylphosphine (1.77 g, 6.7 mmol), thioacetic acid (0.38 mL, 410.1 mg, 5.4 mmol) in anhydrous tetrahydrofuran (20 mL) cooled at 0 °C was added a solution of diisopropyl azodicarboxylate (1.13 mL, 1.15 g, 6.7 mmol) as depicted for the preparation of 31. The product was purified by column chromatography (silica gel) eluting with hexane–EtOAc (9:1) to give 840.0 mg (84% yield) of 49 as a colorless oil: Rf 0.38 (hexane–EtOAc, 4:1); 1H NMR (500.13 MHz, CDCl3) δ 1.59 (s, 3H, C-12), 1.60 (s, 3H, CH3 at C-7), 1.68 (s, 6H, CH3 at C-3CH3 at C-11), 1.95–2.10 (m, 8H, H-4, H-5, H-8, H-9), 2.32 (s, 3H, SC(O)CH3), 3.54 (t, J = 7.9 Hz, 2H, H-1), 5.08 (m, 2H, H-6, H-10), 5.21 (tq, J = 7.9, 1.1 Hz, 1H, H-2); 13C NMR (125.77 MHz, CDCl3) δ 16.0 (CH3 at C-3), 16.2 (CH3 at C-7), 17.7 (CH3 at C-11), 25.9 (C-12), 26.3 (C-9), 26.7 (C-5), 27.3 (C-1), 30.4 (SC(O)CH3), 39.5 (C-4), 39.7 (C-8), 118.4 (C-2), 123.7 (C-10), 124.3 (C-6), 131.3 (C-11), 135.3 (C-7), 140.2 (C-3), 196.1 (SC(O)CH3). HRMS (ESI) calcd. for C17H28OS [M + Na]+ 303.1759; found 303.1757.
(2E,6E)-3,7,11-Trimethyldodeca-2,6,10-triene-1-thiol (51). A solution of 49 (840.3 mg, 3.0 mmol) in anhydrous tetrahydrofuran (10 mL) cooled at 0 °C was treated with portionwise with lithium aluminum hydride (170 mg, 4.5 mmol) as described for the preparation of 50 to obtain 421.8 mg of 51 (59% yield) as a colorless oil: 1H NMR (500.13 MHz, CDCl3) δ 1.42 (t, J = 7.1 Hz, 1H, SH), 1.62 (s, 6H, C-12, CH3 at C-3), 1.68 (s, 3H, CH3 at C-7), 1.71 (s, 3H, CH3 at C-11), 1.99–2.14 (m, 8H, H-4, H-5, H-8, H-9), 3.18 (t, J = 7.2 Hz, 2H, H-1), 5.12 (m, 2H, H-6, H-10), 5.37 (tq, J = 7.8, 1.2 Hz, 1H, H-2); 13C NMR (125.77 MHz, CDCl3) δ 15.8 (CH3 at C-3), 16.0 (CH3 at C-7), 17.7 (CH3 at C-11), 22.1 (C-1), 25.7 (C-12), 26.3 (C-9), 26.7 (C-5), 39.4 (C-4), 39.7 (C-8), 123.3 (C-2), 123.7 (C-10), 124.3 (C-6), 131.3 (C-11), 135.3 (C-7), 137.5 (C-3). HRMS (ESI) calcd. for C15H27S [M + H]+ 239.1833; found 239.1820.
Tetraethyl 1-[((2E,6E)-3,7,11-trimethyldodeca-2,6,10-triene-1-thio)ethyl]-1,1-bisphosphonate (53). A solution of 21 (303 mg, 1.0 mmol) in anhydrous methylene chloride (10 mL) was treated with triethylamine (112 mg, 154 μL, 1.1 mmol) and 51 (241 mg, 1.0 mmol) as described for the preparation of 22. The solvent was evaporated and the residue was purified by column chromatography (silica gel) employing EtOAc as eluent to yield 412.3 mg (76% yield) of 53 as a colorless oil: Rf 0.50 (EtOAc–methanol, 9:1); 1H NMR (500.13 MHz, CDCl3) δ 1.35 (t, J = 7.1 Hz, 12H, CH2CH3), 1.36 (t, J = 7.0 Hz, 12H, CH2CH′3), 1.60 (s, 6H, C-15, CH3 at C-6), 1.68 (s, 6H, CH3 at C-10, CH3 at C-14), 1.96–2.10 (m, 8H, H-7, H-8, H-11, H-12), 2.61 (tt, J = 23.8, 6.1 Hz, 1H, H-1), 3.02 (dt, J = 18.2, 6.1 Hz, 2H, H-2), 3.22 (d, J = 7.7 Hz, 2H, H-4), 4.20 (m, 8H, CH2CH3), 5.10 (m, 2H, H-9, H-13), 5.26 (tq, J = 7.6, 1.0 Hz, 1H, H-5); 13C NMR (125.77 MHz, CDCl3) δ 16.2 (CH3 at C-6), 16.3 (CH3 at C-10), 16.4 (d, J = 6.1 Hz, CH2CH3), 17.7 (CH3 at C-14), 25.7 (C-15), 27.3 (t, J = 5.0 Hz, C-2), 26.5 (C-12), 26.7 (C-8), 30.7 (C-4), 38.9 (t, J = 131.6 Hz, C-1), 39.6 (C-7), 39.7 (C-11), 62.7 (d, J = 6.7 Hz, CH2CH3), 62.8 (d, J = 6.6 Hz, C′H2CH3), 119.8 (C-5), 123.8 (C-13), 124.3 (C-9), 131.3 (C-14), 135.3 (C-10), 139.5 (C-6); 31P NMR (202.46 MHz, CDCl3) δ 21.47. HRMS (ESI) calcd. for C25H48O6SP2 [M + Na]+ 561.2545; found 561.2534.
1-[((2E,6E)-3,7,11-Trimethyldodeca-2,6,10-triene-1-thio)ethyl]-1,1-bisphosphonic Acid (45). A solution of 53 (375 mg, 0.7 mmol), 2,4,6-collidine (843 mg, 920 µL, 7.0 mmol) in anhydrous methylene chloride (10 mL) was treated with bromotrimethylsilane (1.07 g, 0.9 mL, 7.0 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature for 48 h. Then, methanol (1.0 mL) was added and the reaction mixture was treated as depicted for the preparation of 44. The residue was purified by reversed-phase column chromatography eluting with a mixture of water–acetonitrile (2:1) to give 170 mg (52% yield) of 45 as an amorphous solid: 1H NMR (500.13 MHz, D2O-CD3OD) δ 1.61 (s, 6H, C-15, CH3 at C-6), 1.69 (s, 3H, CH3 at C-10) 1.72 (s, 3H, CH3 at C-14), 1.96–2.17 (m, 9H, H-1, H-7, H-8, H-11, H-12), 3.06 (dt, J = 14.6, 7.1 Hz, 2H, H-2), 3.31 (d, J = 7.9 Hz, 2H, H-4), 5.14 (m, 2H, H-9, H-13), 5.26 (tq, J = 7.8, 1.0 Hz, 1H, H-5); 13C NMR (125.77 MHz, D2O-CD3OD) δ 16.1 (CH3 at C-6), 16.3 (CH3 at C-10), 17.9 (CH3 at C-14), 24.3 (C-15), 25.9 (C-12), 30.2 (t, J = 3.7 Hz, C-2), 26.9 (C-12), 27.0 (C-8), 31.4 (C-4), 41.4 (t, J = 109.4 Hz, C-1), 40.09 (C-7), 40.11 (C-11), 120.9 (C-5), 125.23 (C-13), 125.22 (C-9), 133.4 (C-14), 136.7 (C-10), 140.6 (C-6); 31P NMR (202.46 MHz, D2O-CD3OD) δ 18.16 ppm. HRMS (ESI) calcd. for C17H29O6SP2Na4 [M + H]+ 515.0751; found 515.0737.
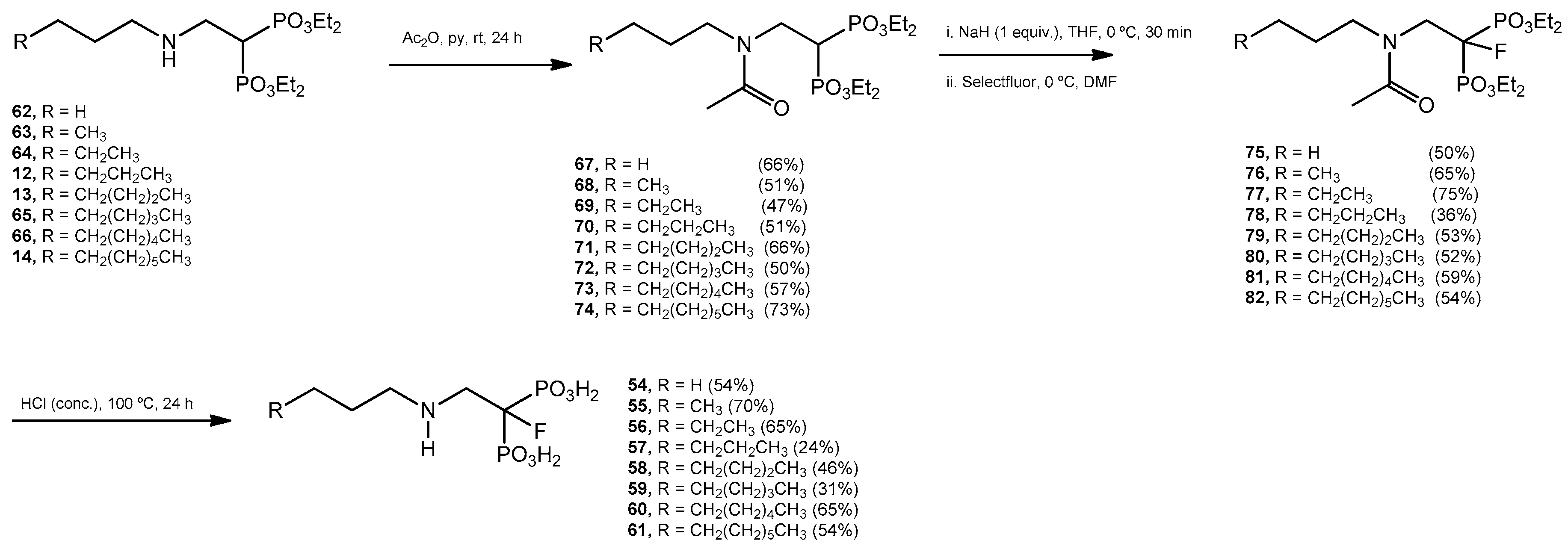
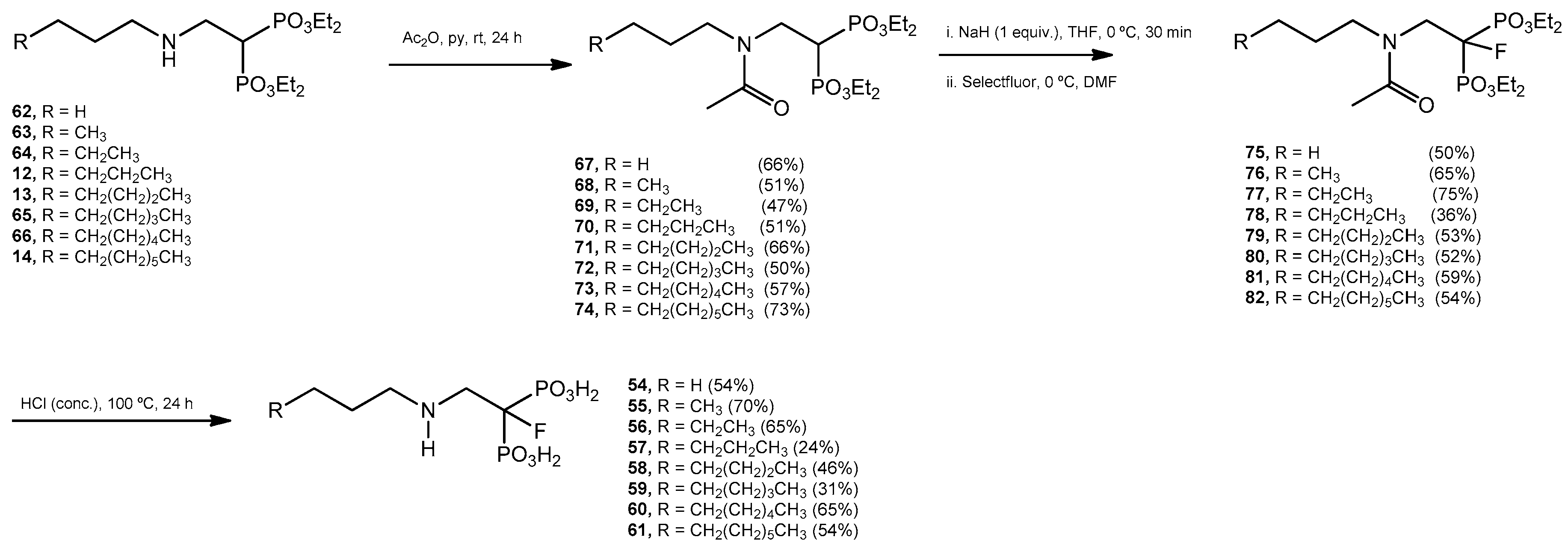
Acetylation Reaction. General Procedure. Tetraethyl 2-(N-n-alkylamino)ethyl bisphosphonates 12–14, 61–65 (15 mmol) were treated individually with acetic anhydride (3.0 mL) and pyridine (3.0 mL). The mixture was stirred at room temperature overnight. The solvent was evaporated and each residue was purified by column chromatography (silica gel) eluting with CH2Cl2–methanol (99:1) to produce the acetylated products 67–74.
Tetraethyl 2-(N-n-propylacetamido)ethyl-1,1-bisphosphonate (67): 66% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.90 (t, J = 7.4 Hz, 3H, H-6), 1.33 (t, J = 7.0 Hz, 6H, CH2CH3), 1.34 (t, J = 7.0 Hz, 6H, CH2CH′3), 1.55 (p, J = 7.1 Hz, 2H, H-5), 2.06 (s, 3H, C(O)CH3), 3.38 (t, J = 7.7 Hz, 1H, H-4), 3.46 (tt, J = 22.6, 7.5 Hz, 1H, H-1), 3.74 (ddd, J = 14.7, 11.2, 7.4 Hz, 2H, H-2), 4.17 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 11.2 (C-6), 16.30 (d, J = 4.1 Hz, CH2CH3), 16.36 (d, J = 4.1 Hz, CH2CH3), 21.6 ((C(O)CH3), 22.2 (C-5), 34.1 (t, J = 130.1 Hz, C-1), 44.6 (t, J = 3.3 Hz, C-2), 52.7 (C-4), 62.4 (d, J = 6.8 Hz, CH2CH3), 63.0 (d, J = 6.7 Hz, CH2CH3), 171.1 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.48.
Tetraethyl 2-(N-n-butylacetamido)ethyl-1,1-bisphosphonate (68): 51% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.95 (t, J = 7.3 Hz, 3H, H-7), 1.34 (t, J = 7.0 Hz, 6H, CH2CH3), 1.35 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.53 (p, J = 7.1 Hz, 2H, H-5), 2.07 (s, 3H, C(O)CH3), 3.41 (t, J = 7.8 Hz, 1H, H-4), 3.44 (tt, J = 22.8, 7.4 Hz, 1H, H-1), 3.74 (ddd, J = 14.7, 11.1, 7.4 Hz, 2H, H-2), 4.19 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 13.8 (C-7), 16.27 (d, J = 4.1 Hz, CH2CH3), 16.33 (d, J = 4.1 Hz, CH2CH3), 20.0 (C-6), 21.6 ((C(O)CH3), 31.1 (C-5), 34.1 (t, J = 130.1 Hz, C-1), 44.6 (t, J = 3.3 Hz, C-2), 50.6 (C-4), 62.4 (d, J = 6.8 Hz, CH2CH3), 62.9 (d, J = 6.8 Hz, CH2CH3), 171.0 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.47.
Tetraethyl 2-(N-n-penylacetamido)ethyl-1,1-bisphosphonate (69): 47% yield, colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.91 (t, J = 7.2 Hz, 3H, H-8), 1.26 (m, 2H, CH2), 1.34 (t, J = 7.1 Hz, 6H, CH2CH3), 1.35 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.53 (p, J = 7.7 Hz, 2H, H-5), 2.07 (s, 3H, C(O)CH3), 3.41 (t, J = 7.7 Hz, 1H, H-4), 3.44 (tt, J = 22.8, 7.4 Hz, 1H, H-1), 3.74 (ddd, J = 14.7, 11.1, 7.4 Hz, 2H, H-2), 4.19 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 13.9 (C-8), 16.3 (d, J = 4.2 Hz, CH2CH3), 16.4 (d, J = 4.2 Hz, CH2CH3), 21.6 ((C(O)CH3), 22.4 (C-7), 28.7 (C-6), 28.9 (C-5), 34.1 (t, J = 130.0 Hz, C-1), 44.6 (t, J = 3.4 Hz, C-2), 50.9 (C-4), 62.4 (d, J = 6.8 Hz, CH2CH3), 62.9 (d, J = 6.7 Hz, CH2CH3), 171.0 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.47.
Tetraethyl 2-(N-n-hexylacetamido)ethyl-1,1-bisphosphonate (70): 51% yield, colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.87 (t, J = 6.9 Hz, 3H, H-9), 1.29 (m, 6H, CH2), 1.34 (t, J = 7.0 Hz, 6H, CH2CH3), 1.35 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.52 (m, 2H, H-5), 2.07 (s, 3H, C(O)CH3), 3.41 (t, J = 7.7 Hz, 1H, H-4), 3.44 (tt, J = 22.8, 7.4 Hz, 1H, H-1), 3.74 (ddd, J = 14.7, 11.1, 7.4 Hz, 2H, H-2), 4.18 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-9), 16.2 (d, J = 4.3 Hz, CH2CH3), 16.4 (d, J = 4.1 Hz, CH2CH3), 21.6 ((C(O)CH3), 22.6 (C-8), 26.5 (C-6), 29.0 (C-5), 31.5 (C-7), 34.1 (t, J = 130.0 Hz, C-1), 44.6 (t, J = 3.2 Hz, C-2), 50.9 (C-4), 62.4 (d, J = 6.8 Hz, CH2CH3), 62.9 (d, J = 6.7 Hz, CH2CH3), 171.0 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.48.
Tetraethyl 2-(N-n-heptylacetamido)ethyl-1,1-bisphosphonate (71): 66% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.89 (t, J = 7.1 Hz, 3H, H-10), 1.29 (m, 8H, CH2), 1.34 (t, J = 7.1 Hz, 6H, CH2CH3), 1.35 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.52 (p, J = 7.5 Hz, 2H, H-5), 2.07 (s, 3H, C(O)CH3), 3.41 (t, J = 7.7 Hz, 1H, H-4), 3.44 (tt, J = 23.1, 7.4 Hz, 1H, H-1), 3.74 (ddd, J = 14.7, 11.1, 7.4 Hz, 2H, H-2), 4.18 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-10), 16.3 (d, J = 4.4 Hz, CH2CH3), 16.4 (d, J = 4.1 Hz, CH2CH3), 21.6 ((C(O)CH3), 22.6 (C-9), 26.8 (C-6), 29.03 (C-5), 29.04 (C-7), 31.5 (C-8), 34.1 (t, J = 130.0 Hz, C-1), 44.6 (C-2), 50.9 (C-4), 62.4 (d, J = 6.6 Hz, CH2CH3), 62.9 (d, J = 6.8 Hz, CH2CH3), 171.0 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.49.
Tetraethyl 2-(N-n-octylacetamido)ethyl-1,1-bisphosphonate (72): 50% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.89 (t, J = 7.0 Hz, 3H, H-11), 1.29 (m, 10H, CH2), 1.34 (t, J = 7.1 Hz, 6H, CH2CH3), 1.35 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.52 (p, J = 7.4 Hz, 2H, H-5), 2.07 (s, 3H, C(O)CH3), 3.41 (t, J = 7.8 Hz, 1H, H-4), 3.44 (tt, J = 22.9, 7.4 Hz, 1H, H-1), 3.74 (ddd, J = 14.7, 11.1, 7.4 Hz, 2H, H-2), 4.19 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-11), 16.2 (d, J = 3.9 Hz, CH2CH3), 16.3 (d, J = 3.9 Hz, CH2CH3), 21.5 ((C(O)CH3), 22.5 (C-10), 26.7 (C-6), 28.9 (C-8), 29.1 (C-5), 29.2 (C-7), 31.6 (C-9), 34.0 (t, J = 130.0 Hz, C-1), 44.5 (t, J = 3.2 Hz, C-2), 50.8 (C-4), 62.3 (d, J = 6.8 Hz, CH2CH3), 62.8 (d, J = 6.6 Hz, CH2CH3), 170.9 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.44.
Tetraethyl 2-(N-n-nonylacetamido)ethyl-1,1-bisphosphonate (73): 57% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.84 (t, J = 6.9 Hz, 3H, H-12), 1.23 (m, 12H, CH2), 1.30 (t, J = 7.1 Hz, 6H, CH2CH3), 1.31 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.49 (p, J = 7.1 Hz, 2H, H-5), 2.03 (s, 3H, C(O)CH3), 3.37 (t, J = 7.8 Hz, 1H, H-4), 3.40 (tt, J = 23.0, 7.4 Hz, 1H, H-1), 3.70 (ddd, J = 14.8, 11.2, 7.4 Hz, 2H, H-2), 4.15 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-12), 16.2 (d, J = 4.0 Hz, CH2CH3), 16.3 (d, J = 3.9 Hz, CH2CH3), 21.5 ((C(O)CH3), 22.5 (C-11), 26.7 (C-6), 29.0 (C-8), 29.1 (C-5), 29.3 (C-7), 29.4 (C-9), 31.8 (C-10), 34.0 (t, J = 130.1 Hz, C-1), 44.5 (t, J = 3.3 Hz, C-2), 50.9 (C-4), 62.3 (d, J = 6.9 Hz, CH2CH3), 62.9 (d, J = 6.7 Hz, CH2CH3), 170.9 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.45.
Tetraethyl 2-(N-n-decylacetamido)ethyl-1,1-bisphosphonate (74): 73% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.85 (t, J = 6.9 Hz, 3H, H-13), 1.23 (m, 14H, CH2), 1.31 (t, J = 7.0 Hz, 6H, CH2CH3), 1.32 (t, J = 7.1 Hz, 6H, CH2CH′3), 1.49 (p, J = 7.1 Hz, 2H, H-5), 2.04 (s, 3H, C(O)CH3), 3.38 (t, J = 7.5 Hz, 1H, H-4), 3.44 (tt, J = 23.2, 7.6 Hz, 1H, H-1), 3.71 (ddd, J = 14.6, 11.2, 7.5 Hz, 2H, H-2), 4.15 (m, 8H, CH2CH3); 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-13), 16.27 (d, J = 6.8 Hz, CH2CH3), 16.30 (d, J = 6.6 Hz, CH2CH3), 21.6 ((C(O)CH3), 22.6 (C-12), 26.8 (C-6), 29.0 (C-8), 29.2 (C-5), 29.3 (C-7), 29.4 (C-9), 29.5 (C-10), 31.8 (C-11), 34.0 (t, J = 130.0 Hz, C-1), 44.5 (t, J = 3.3 Hz, C-2), 50.9 (C-4), 62.4 (d, J = 6.9 Hz, CH2CH3), 62.9 (d, J = 6.7 Hz, CH2CH3), 171.0 (C(O)CH3); 31P NMR (202.46 MHz, CDCl3) δ 21.46.
Fluorination Reaction. General Procedure. Sodium hydride (10.5 mmol, 60% in mineral oil) was washed with anhydrous hexane under an argon atmosphere, and then anhydrous tetrahydrofuran (10 mL) was added. The mixture, cooled at 0 °C, was treated with the corresponding tetraethyl 2-(N-n-alkylacetamido)ethyl-1,1-bisphosphonate such as 67–74 (10.0 mmol) in anhydrous tetrahydrofuran (10 mL). The solution was stirred at 0 °C for 15 min, then, the mixture was allowed to reach room temperature and stirred for additional 60 min. The mixture was cooled at 0 °C and Selectfluor (12.5 mmol) was added followed by addition of N,N-dimethylformamide (5.0 mL). The reaction mixture was allowed to reach room temperature and stirred for 4 h. The mixture was partitioned between methylene chloride (30 mL) and brine (30 mL). The aqueous phase was extracted with methylene chloride (2 × 30 mL). The combined organic layers were washed with brine (5 × 30 mL), water (2 × 25 mL), dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography eluting with a mixture of EtOAc–methanol (49:1) to produce the corresponding 1-fluoro tetraethyl esters 75–82.
Tetraethyl 2-(N-n-propylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (75): 50% yield; 1H NMR (500.13 MHz, CDCl3) δ 0.88 (t, J = 7.3 Hz, 3H, H-6), 0.91 (t, J = 7.4 Hz, 3H, H-6′), 1.38 (t, J = 7.0 Hz, 12H, CH2CH3), 1.39 (dt, J = 6.9, 3.3 Hz, 12H, CH2CH′3), 1.53 (sext, J = 7.6 Hz, 2H, H-5), 1.60 (sext, J = 7.5 Hz, 2H, H-5′), 2.14 (s, 3H, C(O)CH3), 2.21 (s, 3H, C(O)CH3), 3.45 (t, J = 7.8 Hz, 2H, H-4), 3.47 (t, J = 7.9 Hz, 2H, H-4′), 4.10 (dd, J = 10.1, 8.8 Hz, 2H, H-2), 4.15 (dd, J = 10.2, 8.8 Hz, 2H, H-2′), 4.27 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 11.1 (C-6), 11.3 (C-6′), 16.3 (t, J = 3.3 Hz, CH2CH3), 16.4 (t, J = 2.3 Hz, CH2CH′3), 19.8 (C-5), 21.2 (C-5′), 21.4 (C(O)CH3), 21.9 (d, J = 3.5 Hz, C(O)CH′3), 44.9 (d, J = 17.9 Hz, C-2), 48.4 (d, J = 3.5 Hz, C-4), 48.9 (d, J = 18.3 Hz, C-2′), 50.9 (d, J = 4.2 Hz, C-2′), 64.2 (m, CH2CH3), 64.7 (t, J = 3.4 Hz, CH2CH3), 171.3 (C(O)CH3), 171.6 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.70 (d, J = 70.2 Hz), 11.90 (d, J = 77.4 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.55 (t, J = 77.4 Hz), −192.80 (t, J = 71.3 Hz), two rotamers.
Tetraethyl 2-(N-n-butylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (76): 65% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.91 (t, J = 7.3 Hz, 3H, H-7), 0.94 (t, J = 7.4 Hz, 3H, H-7′), 1.30 (m, 2H, H-6), 1.37 (t, J = 6.8 Hz, 12H, CH2CH3), 1.38 (dt, J = 6.8, 3.3 Hz, 12H, CH2CH′3), 1.49 (p, J = 7.7 Hz, 2H, H-5), 1.54 (p, J = 7.8 Hz, 2H, H-5′), 2.12 (s, 3H, C(O)CH3), 2.20 (s, 3H, C(O)CH3), 3.46 (t, J = 7.9 Hz, 2H, H-4), 3.49 (t, J = 7.9 Hz, 2H, H-4′), 4.10 (dd, J = 10.5, 8.5 Hz, 2H, H-2), 4.14 (dd, J = 10.3, 8.9 Hz, 2H, H-2′), 4.27 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 13.75 (C-7), 13.84 (C-7′), 16.39 (t, J = 3.2 Hz, CH2CH3), 16.44 (t, J = 2.6 Hz, CH2CH′3), 20.0 (C-6), 20.1 (C-6′), 21.5 (C(O)CH3), 22.0 (d, J = 3.6 Hz, C(O)CH′3), 28.7 (C-5), 30.1 (C-5′), 44.9 (d, J = 18.1 Hz, C-2), 46.5 (d, J = 3.3 Hz, C-4), 49.1 (d, J = 4.2 Hz, C-4′), 49.9 (d, J = 18.1 Hz, C-2′), 64.2 (m, CH2CH3), 64.7 (m, CH2CH3), 171.2 (C(O)CH3), 171.6 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.70 (d, J = 70.2 Hz), 11.90 (d, J = 77.2 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.46 (t, J = 77.5 Hz), −192.81 (t, J = 71.4 Hz), two rotamers. HRMS (ESI) calcd. for (C16H34O7NFP2Na) [M + Na]+ 456.1692; found 456.1700.
Tetraethyl 2-(N-n-pentylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (77): 75% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.89 (t, J = 7.5 Hz, 3H, H-8), 0.90 (t, J = 7.5 Hz, 3H, H-8′), 1.24 (m, 2H, CH2), 1.37 (t, J = 6.7 Hz, 12H, CH2CH3), 1.38 (dt, J = 6.5, 2.6 Hz, 12H, CH2CH′3), 1.51 (p, J = 7.6 Hz, 2H, H-5), 1.55 (p, J = 7.7 Hz, 2H, H-5′), 2.12 (s, 3H, C(O)CH3), 2.20 (s, 3H, C(O)CH3), 3.47 (t, J = 7.9 Hz, 2H, H-4), 4.10 (t, J = 9.6 Hz, 2H, H-2), 4.15 (t, J = 9.1 Hz, 2H, H-2′), 4.27 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 13.86 (C-8), 13.90 (C-8′), 16.36 (m, CH2CH3), 21.4 (C(O)CH3), 21.88 (d. J = 3.5 Hz, C(O)CH′3), 22.3 (C-7), 22.4 (C-7′), 26.1 (C-6), 27.6 (C-6′), 28.8 (C-5), 29.0 (C-5′), 44.8 (d, J = 18.5 Hz, C-2), 46.7 (d, J = 3.3 Hz, C-4), 49.2 (d, J = 4.2 Hz, C-4′), 49.8 (d, J = 18.2 Hz, C-2′), 64.1 (m, CH2CH3), 64.6 (m, CH2CH3), 95.8 (dt, J = 153.2, 109.3 Hz, 1H, H-1), 97.3 (dt, J = 153.0, 103.1 Hz, 1H, H-1′), 171.1 (C(O)CH3), 171.4 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.66 (d, J = 71.8 Hz), 11.87 (d, J = 78.0 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.56 (t, J = 77.4 Hz), −192.82 (t, J = 71.2 Hz); two rotamers.
Tetraethyl 2-(N-n-hexylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (78): 36% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.87 (t, J = 6.9 Hz, 3H, H-9), 0.89 (t, J = 6.9 Hz, 3H, H-9′), 1.29 (m, 6H, CH2), 1.37 (t, J = 6.9 Hz, 12H, CH2CH3), 1.38 (dt, J = 6.9, 2.7 Hz, 12H, CH2CH′3), 1.49 (p, J = 7.5 Hz, 2H, H-5), 1.54 (p, J = 7.8 Hz, 2H, H-5′), 2.12 (s, 3H, C(O)CH3), 2.19 (s, 3H, C(O)CH3), 3.46 (t, J = 7.9 Hz, 2H, H-4), 4.09 (t, J = 9.4 Hz, 2H, H-2), 4.09 (t, J = 9.4 Hz, 2H, H-2′), 4.26 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 13.95 (C-9), 14.00 (C-9′), 16.39 (t, J = 3.2 Hz, CH2CH3), 16.44 (t, J = 2.6 Hz, CH2CH′3), 21.5 (C(O)CH3), 22.0 (d, J = 3.5 Hz, C(O)CH′3), 22.5 (C-8), 22.6 (C-8′), 26.56 (C-6), 26.58 (C-6′), 28.0 (C-5), 31.5 (C-7), 31.6 (C-7′), 44.9 (d, J = 18.2 Hz, C-2), 46.8 (d, J = 3.2 Hz, C-4), 49.3 (d, J = 4.3 Hz, C-4′), 49.9 (d, J = 18.4 Hz, C-2′), 62.4 (d, J = 6.8 Hz, CH2CH3), 62.9 (d, J = 6.7 Hz, CH2CH3), 171.1 (C(O)CH3), 171.5 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.71 (d, J = 69.7 Hz), 11.91 (d, J = 77.1 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.50 (t, J = 77.6 Hz), −192.78 (t, J = 71.2 Hz); two rotamers. HRMS (ESI) calcd. for (C18H39O7NFP2) [M + H]+ 462.2188; found 462.2197.
Tetraethyl 2-(N-n-heptylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (79): 53% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.87 (t, J = 6.4 Hz, 3H, H-10), 0.88 (t, J = 6.9 Hz, 3H, H-10′), 1.27 (m, 8H, CH2), 1.36 (t, J = 7.0 Hz, 12H, CH2CH3), 1.38 (dt, J = 6.8, 3.4 Hz, 12H, CH2CH′3), 1.49 (p, J = 7.6 Hz, 2H, H-5), 1.54 (p, J = 7.8 Hz, 2H, H-5′), 2.11 (s, 3H, C(O)CH3), 2.19 (s, 3H, C(O)CH3), 3.46 (t, J = 7.9 Hz, 2H, H-4), 4.09 (dd, J = 10.3, 8.6 Hz, 2H, H-2), 4.13 (t, J = 10.2, 8.7 Hz, 2H, H-2′), 4.27 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 13.99 (C-10), 14.02 (C-10′), 16.3 (t, J = 3.2 Hz, CH2CH3), 16.4 (t, J = 2.4 Hz, CH2CH′3), 21.5 (C(O)CH3), 21.9 (d, J = 3.5 Hz, C(O)CH′3), 22.50 (C-9), 22.53 (C-9′), 26.6 (C-6), 26.7 (C-6′), 26.9 (C-7), 28.0 (C-5), 31.69 (C-8), 31.74 (C-8′), 44.9 (d, J = 17.9 Hz, C-2), 46.8 (d, J = 3.2 Hz, C-4), 49.3 (d, J = 4.3 Hz, C-4′), 49.9 (d, J = 18.3 Hz, C-2′), 64.2 (m, CH2CH3), 95.9 (dt, J = 153.1, 102.8 Hz, 1H, H-1), 97.4 (dt, J = 153.1, 102.8 Hz, 1H, H-1′), 171.2 (C(O)CH3), 171.5 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.69 (d, J = 72.1 Hz), 11.89 (d, J = 78.4 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.52 (t, J = 77.5 Hz), −192.79 (t, J = 71.3 Hz); two rotamers.
Tetraethyl 2-(N-n-octylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (80): 52% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.87 (t, J = 7.1 Hz, 3H, H-11), 0.88 (t, J = 6.9 Hz, 3H, H-11′), 1.27 (m, 10H, CH2), 1.37 (t, J = 7.0 Hz, 12H, CH2CH3), 1.38 (dt, J = 6.8, 3.4 Hz, 12H, CH2CH′3), 1.50 (p, J = 7.6 Hz, 2H, H-5), 1.54 (p, J = 7.9 Hz, 2H, H-5′), 2.12 (s, 3H, C(O)CH3), 2.19 (s, 3H, C(O)CH3), 3.46 (t, J = 8.2 Hz, 2H, H-4), 4.09 (dd, J = 12.0, 7.2 Hz, 2H, H-2), 4.15 (t, J = 10.2, 8.8 Hz, 2H, H-2′), 4.27 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 14.01 (C-11), 14.02 (C-11′), 16.4 (m, CH2CH3), 21.5 (C(O)CH3), 21.9 (d, J = 3.5 Hz, C(O)CH′3), 22.55 (C-10), 22.58 (C-10′), 26.6 (C-6), 26.8 (C-6′), 26.9 (C-7), 28.0 (C-5), 29.1 (C-8), 29.3 (C-8′), 31.74 (C-9), 31.79 (C-9′), 44.9 (d, J = 18.0 Hz, C-2), 46.8 (d, J = 3.3 Hz, C-4), 49.3 (d, J = 4.2 Hz, C-4′), 49.9 (d, J = 18.4 Hz, C-2′), 64.2 (m, CH2CH3), 95.9 (dt, J = 153.1, 109.2 Hz, 1H, H-1), 97.4 (dt, J = 153.1, 103.3 Hz, 1H, H-1′), 171.2 (C(O)CH3), 171.5 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.69 (d, J = 72.1 Hz), 11.89 (d, J = 79.6 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.51 (t, J = 77.5 Hz), −192.78 (t, J = 71.3 Hz); two rotamers.
Tetraethyl 2-(N-n-nonylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (81): 59% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.875 (t, J = 7.0 Hz, 3H, H-11), 0.882 (t, J = 6.9 Hz, 3H, H-11′), 1.26 (m, 12H, CH2), 1.37 (t, J = 7.0 Hz, 12H, CH2CH3), 1.38 (dt, J = 6.9, 3.3 Hz, 12H, CH2CH′3), 1.50 (p, J = 7.6 Hz, 2H, H-5), 1.54 (p, J = 7.7 Hz, 2H, H-5′), 2.12 (s, 3H, C(O)CH3), 2.19 (s, 3H, C(O)CH3), 3.46 (t, J = 7.9 Hz, 2H, H-4), 3.48 (t, J = 8.5 Hz, 2H, H-4′), 4.09 (dd, J = 10.0, 8.9 Hz, 2H, H-2), 4.15 (t, J = 10.2, 8.8 Hz, 2H, H-2′), 4.27 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-12), 14.1 (C-12′), 16.4 (m, CH2CH3), 21.5 (C(O)CH3), 21.9 (d, J = 3.6 Hz, C(O)CH′3), 22.59 (C-11), 22.61 (C-11′), 26.6 (C-6), 26.8 (C-6′), 26.9 (C-7), 28.0 (C-5), 29.16 (C-8), 29.21 (C-8′), 29.28 (C-7′) 29.39 (C-5′), 29.46 (C-9), 29.50 (C-9′), 31.78 (C-10), 31.81 (C-10′), 44.9 (d, J = 17.9 Hz, C-2), 46.8 (d, J = 3.4 Hz, C-4), 49.3 (d, J = 4.3 Hz, C-4′), 49.9 (d, J = 18.2 Hz, C-2′), 64.2 (m, CH2CH3), 64.7 (t, J = 3.2 Hz, CH2CH3), 95.9 (dt, J = 153.0, 109.0 Hz, 1H, H-1), 97.4 (dt, J = 153.0, 103.0 Hz, 1H, H-1′), 171.2 (C(O)CH3), 171.5 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.70 (d, J = 77.9 Hz), 11.91 (d, J = 77.9 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.50 (t, J = 77.4 Hz), −192.77 (t, J = 71.3 Hz); two rotamers.
Tetraethyl 2-(N-n-decylacetamido)ethyl-1-fluoro-1,1-bisphosphonate (82): 54% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.877 (t, J = 7.0 Hz, 3H, H-13), 0.881 (t, J = 6.8 Hz, 3H, H-13′), 1.26 (m, 14H, CH2), 1.37 (t, J = 7.0 Hz, 12H, CH2CH3), 1.38 (dt, J = 6.9, 3.1 Hz, 12H, CH2CH′3), 1.50 (p, J = 7.3 Hz, 2H, H-5), 1.54 (p, J = 7.6 Hz, 2H, H-5′), 2.12 (s, 3H, C(O)CH3), 2.19 (s, 3H, C(O)CH3), 3.46 (t, J = 7.9 Hz, 2H, H-4), 3.48 (t, J = 8.2 Hz, 2H, H-4′), 4.09 (dd, J = 10.4, 8.6 Hz, 2H, H-2), 4.15 (t, J = 10.1, 8.8 Hz, 2H, H-2′), 4.27 (m, 8H, CH2CH3), two rotamers; 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-13), 16.4 (m, CH2CH3), 21.5 (C(O)CH3), 21.9 (d, J = 3.9 Hz, C(O)CH′3), 22.61 (C-12), 22.61 (C-12′), 26.6 (C-6), 26.8 (C-6′), 26.9 (C-7), 28.0 (C-5), 29.23 (C-8), 29.25 (C-8′), 29.27 (C-7′) 29.39 (C-5′), 29.45 (C-9), 29.50 (C-10), 29.54 (C-10′), 31.81 (C-11), 31.83 (C-11′), 44.9 (d, J = 18.6 Hz, C-2), 46.8 (d, J = 3.4 Hz, C-4), 49.3 (d, J = 4.5 Hz, C-4′), 49.9 (d, J = 18.4 Hz, C-2′), 64.2 (m, CH2CH3), 64.7 (t, J = 3.2 Hz, CH2CH3), 95.9 (dt, J = 153.0, 109.0 Hz, 1H, H-1), 97.4 (dt, J = 153.0, 103.1 Hz, 1H, H-1′), 171.2 (C(O)CH3), 171.5 (C(O)′CH3), two rotamers; 31P NMR (202.46 MHz, CDCl3) δ 11.70 (d, J = 71.2 Hz), 11.90 (d, J = 77.7 Hz), two rotamers; 19F NMR (470.59 MHz, CDCl3) δ −192.50 (t, J = 77.5 Hz), −192.78 (t, J = 71.2 Hz); two rotamers.
Hydrolysys. General Procedure. Esters 75–82 were treated, in independent experiments, with a concentrated aqueous solution of hydrochloric acid (2.0 mL). The resulting mixtures were refluxed for 24 h. The solvent was evaporated and the residues were crystallized from water–ethanol.
1-[(n-Prop-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (54): 54% yield; white solid; m.p. 161 °C; 1H NMR (500.13 MHz, D2O) δ 0.91 (t, J = 7.5 Hz, 3H, H-6), 1.65 (sext, J = 7.4 Hz, 2H, H-5), 3.04 (t, J = 7.3 Hz, 2H, H-4), 3.60 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 10.4 (C-6), 19.5 (C-5), 49.6 (d, J = 16.1, C-2), 50.2 (C-4); 31P NMR (202.46 MHz, D2O) δ 8.89 (d, J = 65.4 Hz); 19F NMR (470.59 MHz, D2O) δ −193.32 (t, J = 65.5 Hz). HRMS (ESI) calcd. for (C5H15O6NFP2) [M + H]+ 266.0359; found 266.0359.
1-[(n-But-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (55): 70% yield; white solid; m.p. 153–154 °C; 1H NMR (500.13 MHz, D2O) δ 0.85 (t, J = 7.4 Hz, 3H, H-7), 1.32 (sext, J = 7.5 Hz, 2H, H-6), 1.61 (p, J = 7.5 Hz, 2H, H-5), 3.07 (t, J = 7.4 Hz, 2H, H-4), 3.59 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 12.7 (C-7), 19.0 (C-6), 27.5 (C-5), 48.1 (C-4), 49.1 (dt, J = 18.1, 2.3 Hz, C-2); 31P NMR (202.46 MHz, D2O) δ 8.84 (d, J = 65.5 Hz); 19F NMR (470.59 MHz, D2O) δ −193.39 (t, J = 65.6 Hz). HRMS (ESI) calcd. for (C6H17O6NFP2) [M + H]+ 280.0515; found 280.0514.
1-[(n-Pent-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (56): 65% yield; white solid; m.p. 164–165 °C; 1H NMR (500.13 MHz, D2O) δ 0.81 (t, J = 7.1 Hz, 3H, H-8), 1.28 (m, 4H, CH2), 1.63 (p, J = 7.3 Hz, 2H, H-5), 3.06 (t, J = 7.4 Hz, 2H, H-4), 3.59 (p, J = 10.1 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.0 (C-8), 21.4 (C-7), 25.1 (C-5), 27.7 (C-7), 48.3 (C-4), 49.1 (d, J = 18.3 Hz, C-2); 31P NMR (202.46 MHz, D2O) δ 8.93 (d, J = 47.3 Hz); 19F NMR (470.59 MHz, D2O) δ −193.32 (t, J = 65.5 Hz). HRMS (ESI) calcd. for (C7H19O6NFP2) [M + H]+ 294.0672; found 294.0667.
1-[(n-Hex-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (57): 34% yield; white solid; 1H NMR (500.13 MHz, D2O) δ 0.77 (t, J = 7.1 Hz, 3H, H-9), 1.22 (m, 4H, CH2), 1.28 (m, 2H, CH2), 1.61 (p, J = 7.5 Hz, 2H, H-5), 3.05 (t, J = 7.5 Hz, 2H, H-4), 3.58 (p, J = 10.6 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.1 (C-9), 21.6 (C-8), 25.2 (C-6), 25.4 (C-5), 30.3 (C-7), 49.1 (dt, J = 18.1, 2.3 Hz, C-2), 92.2 (dt, J = 184.1, 139.4 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 8.91 (d, J = 59.7 Hz); 19F NMR (470.59 MHz, D2O) δ −193.54 (t, J = 65.9 Hz). HRMS (ESI) calcd. for (C8H21O6NFP2) [M + H]+ 308.0823; found 308.0822.
1-[(n-Hept-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (58): 56% yield; white solid; m.p. 135–136 °C; 1H NMR (500.13 MHz, D2O) δ 0.76 (t, J = 7.1 Hz, 3H, H-10), 1.20 (m, 4H, CH2), 1.27 (m, 4H, CH2), 1.61 (p, J = 7.4 Hz, 2H, H-5), 3.06 (t, J = 7.7 Hz, 2H, H-4), 3.59 (p, J = 10.7 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.2 (C-10), 21.8 (C-9), 25.4 (C-6), 25.5 (C-7), 27.8 (C-5), 30.7 (C-8), 48.4 (C-4), 49.0 (dt, J = 17.9, 2.3 Hz, C-2), 92.1 (dt, J = 184.2 Hz, 139.6 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 8.90 (d, J = 60.8 Hz); 19F NMR (470.59 MHz, D2O) −193.65 (t, J = 66.0 Hz). HRMS (ESI) calcd. for (C9H23O6NFP2) [M + H]+ 322.0985; found 322.0969.
1-[(n-Oct-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (59): 31% yield; white solid; m.p. >270 °C; 1H NMR (500.13 MHz, D2O) δ 0.74 (t, J = 6.9 Hz, 3H, H-11), 1.19 (m, 10H, CH2), 1.59 (p, J = 7.4 Hz, 2H, H-5), 3.02 (t, J = 7.6 Hz, 2H, H-4), 3.57 (p, J = 10.2 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.4 (C-11), 22.0 (C-10), 25.4 (C-6), 25.6 (C-7), 28.1 (C-8), 28.2 (C-5), 31.0 (C-9), 48.2 (C-4), 48.9 (d, J = 18.1 Hz, C-2), 92.1 (dt, J = 184.6 Hz, 139.8 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 8.87 ppm (d, J = 61.0 Hz); 19F NMR (470.59 MHz, D2O) δ −193.71 (t, J = 66.1 Hz).
1-[(n-Non-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (60). White solid; m.p. 147 °C; 1H NMR (500.13 MHz, D2O) δ 0.77 (t, J = 7.1 Hz, 3H, H-12), 1.19 (m, 12H, CH2), 1.62 (p, J = 6.67 Hz, 2H, H-5), 3.06 (t, J = 7.1 Hz, 2H, H-4), 3.59 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.4 (C-12), 22.0 (C-11), 25.4 (C-6), 25.5 (C-7), 28.1 (C-9), 28.3 (C-8), 28.4 (C-5), 31.1 (C-10), 48.4 (C-4), 49.1 (d, J = 18.1 Hz, C-2), 92.3 (dt, J = 184.9 Hz, 139.4 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 8.83 ppm (d, J = 65.7 Hz); 19F NMR (470.59 MHz, D2O) δ −193.34 (t, J = 65.6 Hz). HRMS (ESI) calcd. for (C11H27O6NFP2) [M + H]+ 350.1298; found 350.1306.
1-[(n-Dec-1-ylamino)ethyl]-1-fluoro-1,1-bisphosphonic Acid (61): 54% yield; White solid; m.p. 151 °C; 1H NMR (500.13 MHz, D2O) δ 0.78 (t, J = 6.7 Hz, 3H, H-13), 1.20 (m, 10H, CH2), 1.27 (m, 4H, CH2), 1.64 (p, J = 7.3 Hz, 2H, H-5), 3.06 (t, J = 7.0 Hz, 2H, H-4), 3.62 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.4 (C-13), 22.0 (C-12), 25.4 (C-6), 25.5 (C-7), 28.1 (C-10), 28.4 (C-8,C-9), 28.6 (C-5), 31.1 (C-11), 48.4 (C-4), 49.1 (dt, J = 18.1, 2.3 Hz, C-2); 31P NMR (202.46 MHz, CDCl3) δ 8.75 ppm (d, J = 65.6 Hz); 19F NMR (470.59 MHz, D2O) δ −199.16 (t, J = 65.8 Hz). HRMS (ESI) calcd. for (C12H23O6NFP2) [M + H]+ 364.1454; found 364.1443.



 ,
, 




{kind=link}
{kind=link}
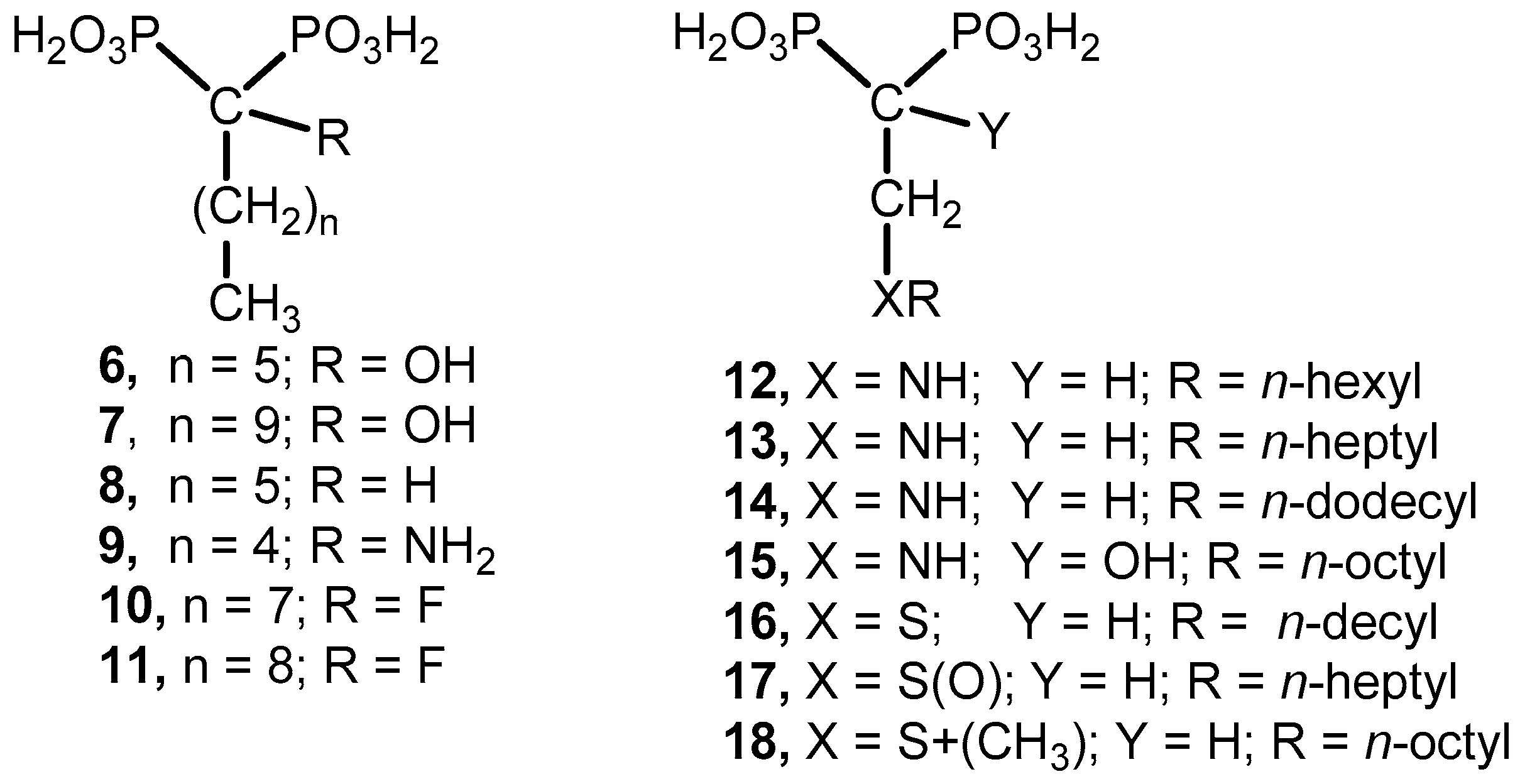{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}