Predictive QSAR Models for the Toxicity of Disinfection Byproducts
by
 ,
,
Litang Qin
1,2,3, 
Xin Zhang
1,
Yuhan Chen
1,
Lingyun Mo
2,3,*,
Honghu Zeng
1,2,3 and
Yanpeng Liang
1,2,3 1
College of Environmental Science and Engineering, Guilin University of Technology, Guilin 541004, China
2
Guangxi Key Laboratory of Environmental Pollution Control Theory and Technology, Guilin University of Technology, Guilin 541004, China
3
Collaborative Innovation Center for Water Pollution Control and Water Safety in Karst Area, Guilin University of Technology, Guilin 541004, China
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(10), 1671; https://doi.org/10.3390/molecules22101671
Submission received: 26 September 2017
/
Revised: 30 September 2017
/
Accepted: 1 October 2017
/
Published: 9 October 2017
(This article belongs to the Section Molecular Diversity)
Abstract
:Several hundred disinfection byproducts (DBPs) in drinking water have been identified, and are known to have potentially adverse health effects. There are toxicological data gaps for most DBPs, and the predictive method may provide an effective way to address this. The development of an in-silico model of toxicology endpoints of DBPs is rarely studied. The main aim of the present study is to develop predictive quantitative structure–activity relationship (QSAR) models for the reactive toxicities of 50 DBPs in the five bioassays of X-Microtox, GSH+, GSH−, DNA+ and DNA−. All-subset regression was used to select the optimal descriptors, and multiple linear-regression models were built. The developed QSAR models for five endpoints satisfied the internal and external validation criteria: coefficient of determination (R2) > 0.7, explained variance in leave-one-out prediction (Q2LOO) and in leave-many-out prediction (Q2LMO) > 0.6, variance explained in external prediction (Q2F1, Q2F2, and Q2F3) > 0.7, and concordance correlation coefficient (CCC) > 0.85. The application domains and the meaning of the selective descriptors for the QSAR models were discussed. The obtained QSAR models can be used in predicting the toxicities of the 50 DBPs.
1. Introduction
Disinfection byproducts (DBPs) have raised concerns since the first DBPs of trihalomethane (THM) compounds were identified in the 1970s [1]. The DBPs may result from reactions between disinfectants and dissolved organic matter present in source waters [2,3]. The THMs are the most common DBPs present in the typical chlorinated drinking water. Approximately 700 DBPs have been identified, as the increasingly employed disinfectants such as ozone, chlorine dioxide and chloramines in drinking water result in reactivity with organic compounds [2]. Research has demonstrated that several known DBPs are considered to be potent cytotoxins, genotoxins and carcinogens [4], which indicates that many DBPs may exert more toxicity to humans than THMs [3]. However, there are toxicological data gaps for most of the DBPs, and more in vitro bioassays or chronic in vivo bioassays need to be carried out. Compared with the experimental test for the toxicological data, the in-silico approach provides an effective way for predicting the toxicities of chemicals. Quantitative structure–activity relationship (QSAR) seems to be a useful method to study the toxicities of DBPs.
The QSAR method provides a promising, faster way of predicting the activity of chemicals using the structural information of the compound. A limited number of QSARs have been proposed for DBP studies. A QSAR for benchmark concentrations of cranial neural tube dysmorphogenesis was established for 10 halogenated derivatives (HAAs) [5]. The mutagenicity in the Salmonlla typhimurium strain TA100 of 42 HAAs was predicted by a QSAR model based on geometrical-, radial distribution function (RDF)-, weighted holistic invariant molecular (WHIM)-, eigenvalue- and 2D-autocorrelation-based methods, as well as information descriptors [6]. The eight-variable model was internally validated by leave-one-out (LOO) cross-validation, the bootstrapping test and Y-scrambling. Tang and Wang [7] developed a QSAR model for finding the energy of the highest occupied molecular orbital (EHOMO) of 36 DBPs, and the model was validated by LOO cross-validation and K-fold cross-validation. An empirical QSAR model based on liposome–water partition coefficients (logKlipw) was proposed for the 50% effective concentration (EC50) of five DBPs (1,1-dichloroethene, bromoethane, chloroform, dichloromethane and bromoform) on Vibrio fischeri [8]. Another empirical model was built for the first-order rate constants of photodegradation of six iodinated trihalomethanes (ITHMs) and three brominated THMs (BTHMs) [9]. Yang and Zhang [10] predicted the developmental toxicity of 19 DBPs to Platynereis dumerilii embryos using an oil–water partition coefficient (logP), protein kinase A (pK(a)), EHOMO and lowest unoccupied molecular orbital energy (ELUMO). All the aforementioned QSAR models for DBPs lack both strictly internal and external validation, which may not guarantee the real predictive ability of the models. Only two studies [8,10] were related to the toxicities of DBPs with a limited number of compounds (no more than 19 samples). The main reasons for the lack of QSAR study on DBPs is that only a small fraction of the DBPs identified (out of a total of ~700) have been tested for toxicity so far. It is implied that QSAR techniques remain underutilized by DBP researchers [2].
In the present study, we aim to develop QSAR models using multiple linear regression (MLR) to predict five toxicity endpoints of DBPs. The developed QSAR models were strictly internally validated by LOO and leave-many-out (LMO) cross-validation and Y-scrambling, and externally validated by several metrics, including variance explained in external prediction Q2F1 [11], Q2F2 [12], and Q2F3 [13], concordance correlation coefficient (CCC) [14,15], and metrics based on the correlation of the observed and predicted response data with and without the intercept [16,17], and the criteria recommended by Golbraikh and Tropsha [18].
2. Materials and Methods
2.1. Experimental Toxicity Data
The five endpoints of 50 drinking water DBPs are precisely explained (Table 1). Experimental reactive toxicity data for five endpoints (X-Microtox, GSH+, GSH−, DNA+ and DNA−) of 50 drinking water DBPs were obtained from the literature (Table 2) [4]. The 50 DBPs comprise a wide range of different chemical groups, 47 commonly detected drinking water DBPs together with 1,1-dichloroethene (1,1-DCE), dichloromethane (DCM) and bromochloromethane. The negative logarithm observed effect concentrations (pEC50 for Microtox and pECIR1.5 for the other assays, M (mol/L)) are listed in Table 2.
2.2. Molecular Structure Descriptors
The molecular structure descriptors of the chemicals were calculated by the Dragon software (version 6.0, Talete SRL, Milano, Italy). The original descriptors generated from the Dragon software were refined by the following principles [19,20]: (1) the descriptors with standard deviation less than 0.0001 were excluded; (2) the descriptors with at least one missing value were deleted; (3) the descriptors with (abs)pair correlation larger than or equal to 0.8 were excluded; and (4) the descriptors that Pearson correlation coefficients (|r|) between descriptors and toxicities of DBPs lower than 0.3 were deleted. The remaining descriptors were used for the further analysis.
2.3. Data Splits and Model Development
The whole dataset was randomly split into several training and test sets. It was recommended that analysis of the models should be obtained from various splits into the training set and test set [21]. For each toxicity endpoint of DBPs, we randomly split the whole dataset into five training sets and five test sets.
All-subset regression for the whole dataset was performed with the QSARINS software [22,23]. The four-variable multiple linear regression (MLR) models with the highest coefficients of determination (R2) and explained variance in leave-one-out (Q2LOO) prediction were selected for the whole dataset. The MLR models for the training sets based on the same descriptors derived from the whole dataset were developed, and the test sets were used to validate the external predictive abilities of the models.
2.4. Model Validation
The statistical parameters for modeling, internal and external validation metrics were adopted to evaluate the fit, stability and predicative power of the QSAR model. The quality parameters include R2, adjusted coefficient of determination (), root mean square error in fitting (RMSEtr) and F-value (F). The internal validations were performed by the LOO and LMO cross-validation (Q2LOO and Q2LMO) and the Y-scrambling test (R2Yscr and Q2Yscr). The external validation was evaluated by a test set. The parameters Q2F1 [11], Q2F2 [12], Q2F3 [13], CCC [14,15], average of () and the difference between () [16,17] were used as the measures of the predictive power of a QSAR model. The proposed parameters by Golbraikh and Tropsha were also applied for the external validation criteria [18]: slope of the regression line over external data (k and k’), coefficients of determination between predicted and observed activities () and coefficients of determination between observed and predicted activities ().
The validation criteria thresholds for the parameters mentioned above are: (1) R2 > 0.7, Q2LOO and Q2LMO > 0.6, Q2F1, Q2F2 and Q2F3 > 0.7, difference between R2 and Q2LOO smaller than 0.1 [15]; (2) > 0.65; (3) CCC > 0.85 [15]; and (4) criteria recommended by Golbraikh and Tropsha [18]: or , 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k’ ≤ 1.15.
The descriptors included in the whole dataset, training set and the test set should satisfy the following conditions [24]: (1) the Pearson correlation coefficients for the complete (rc), training (rt) and test (re) sets are equal to or greater than 0.3: |rc| and |rt| ≥ 0.3; (2) the normalized regression coefficient of the descriptor for the complete (βc) and the training (βt) sets are equal to or greater than 0.001: |βc| and |βt| ≥ 0.001; and (3) absence of the sign-change problem: sign(rc) = sign(rt) = sign(re); sign(rc) = sign(βc) = sign(βt)
Models that have acceptable validation criteria thresholds for all conditions were considered as the final models. These models are robust and able to make good internal and external predictions.
2.5. Applicability Domain
The application domain of the QSAR model was defined by the leverage approach from the hat matrix (hi in Equation (1)), which is calculated from the descriptors of chemicals [19,25], and by identification of chemicals with LOO cross-validated standardized residuals greater than 2.0 standard deviation units. An outlier in the QSAR model is defined as hi value larger than the warning leverage h* and LOO standardized residuals greater than 2.0, which is graphically depicted in the Williams plot. The warning leverage h* is fixed at (3k)/n, where k is the number of model parameters and n is the number of the objects used to calculate the model.
where xi is the descriptor row vector of the query compound; X is the n × k matrix of k model descriptor values for n training set compounds and the superscript T refers to the transpose of the matrix/vector.
3. Results and Discussion
3.1. Selected Descriptors
For each five endpoints of X-Microtox, GSH+, GSH−, DNA+ and DNA− of the selected drinking-water DBPs, four descriptors were selected by all-subset regression for the whole dataset, which was performed by the QSARINS software [22,23]. The selected descriptors for X-Microtox were the spectral diameter from Burden matrix weighted by mass (SpDiam_B(m)), average vertex sum from Burden matrix weighted by van der Waals volume (AVS_B(v)), eigenvalue no. 5 from augmented edge adjacency mat weighted by dipole moment (Eig05_AEA(dm)) and sum of ddsN E-states (SddsN). For the endpoints of GSH+ and GSH−, the four selected descriptors were percentage of C atoms (C%), SpDiam_B(m), P_VSA-like on LogP bin 8 (P_VSA_LogP_8) and sum of topological distances between N..Br (T(N..Br)). The selected descriptors for DNA+ were P_VSA-like on LogP, bin 7 (P_VSA_LogP_7), signal 04/weighted by I-state (Mor04s), T(N..Br) and sum of topological distances between N..I (T(N..I)). For the endpoints of DNA−, the four selected descriptors were sum of atomic van der Waals volumes (Sv), P_VSA_LogP_7, signal 03/weighted by I-state (Mor03s) and T(N..I). There was no high correlation between the selected descriptors, and these descriptors were used as inputs for the training set.
3.2. Models Development and Validation
The whole dataset for each endpoint was randomly split into training and test sets by five iterations (splits 1–5) for the same size of training and test sets. Of the chemicals in the dataset, 80% were selected for the training set and the remaining 20% were considered as the test set. Five QSAR models based on the same size of training sets were built for five endpoints of X-Microtox, GSH+, GSH−, DNA+ and DNA−. The statistical parameters of modeling, internal and external validations were calculated for each model. We have examined five splits into the training and test sets. The realistic reliability of the QSAR model was estimated by the result of the analysis of five splits into the training and test sets. The statistical characteristics of QSAR models of five splits for five endpoints are given in Supplementary data. It can be found that all QSAR models presented high predictive power, as those models satisfy the internal and external validation criteria: R2 > 0.7, Q2LOO and Q2LMO > 0.6, Q2F1, Q2F2 and Q2F3 > 0.7, CCC > 0.85, and > 0.65.
In order to validate whether the descriptors presented in the QSAR models were real or not before model validation and interpretation, we checked the sign-change-problem correlation coefficients and regression coefficients of a descriptor in the MLR model regressions, before and after the data split [24]. It was found that all descriptors in the five QSAR models satisfy the conditions [24]: |rc| and |rt| ≥ 0.3, |βc| and |βt| ≥ 0.001, sign(rc) = sign(rt) = sign(re), and sign(rc) = sign(βc) = sign(βt). Thus, the selected descriptors are considered to be real variables.
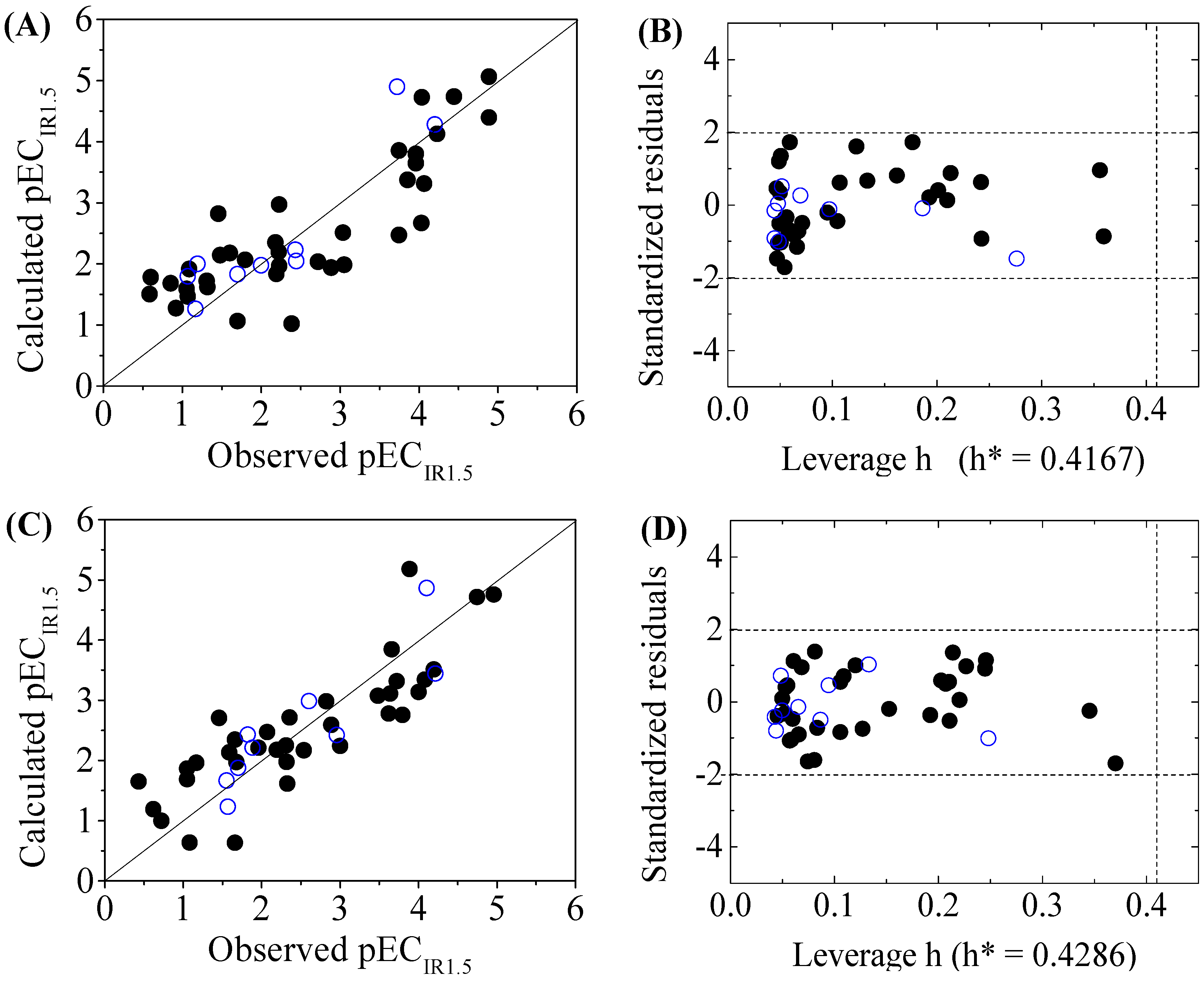
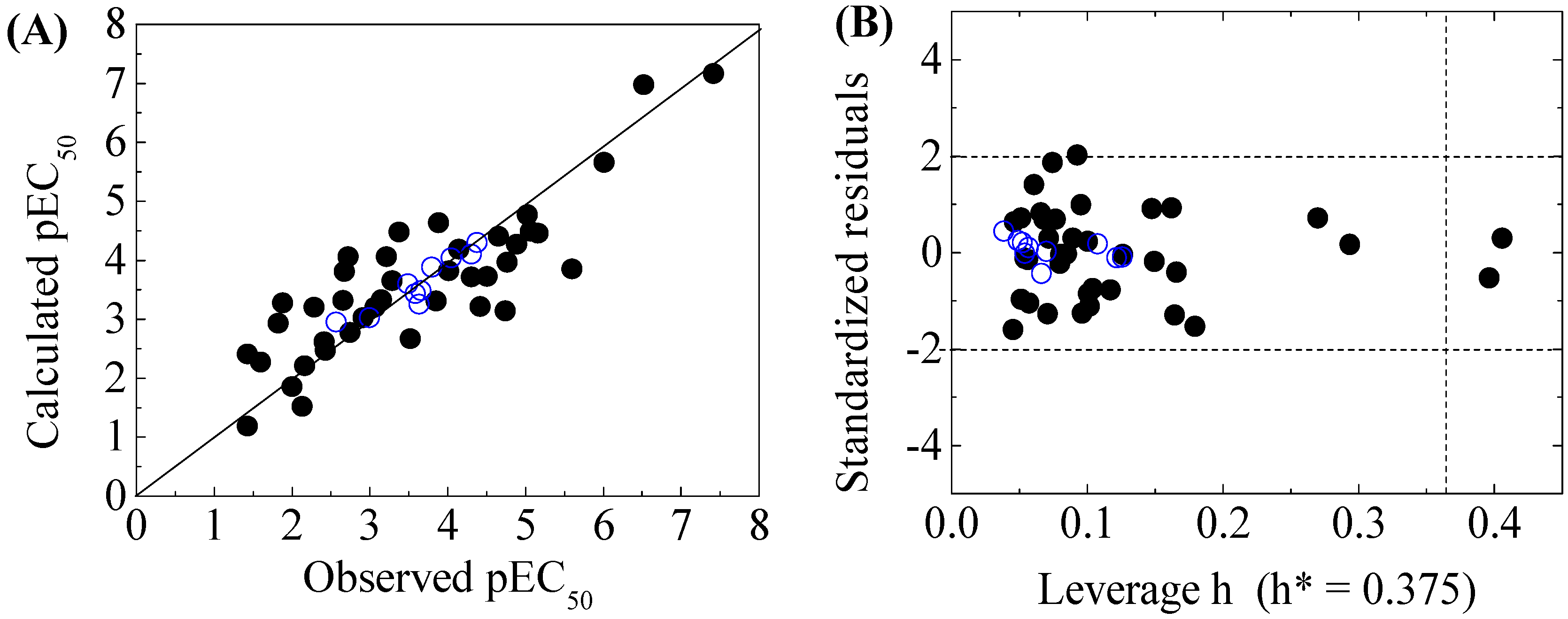
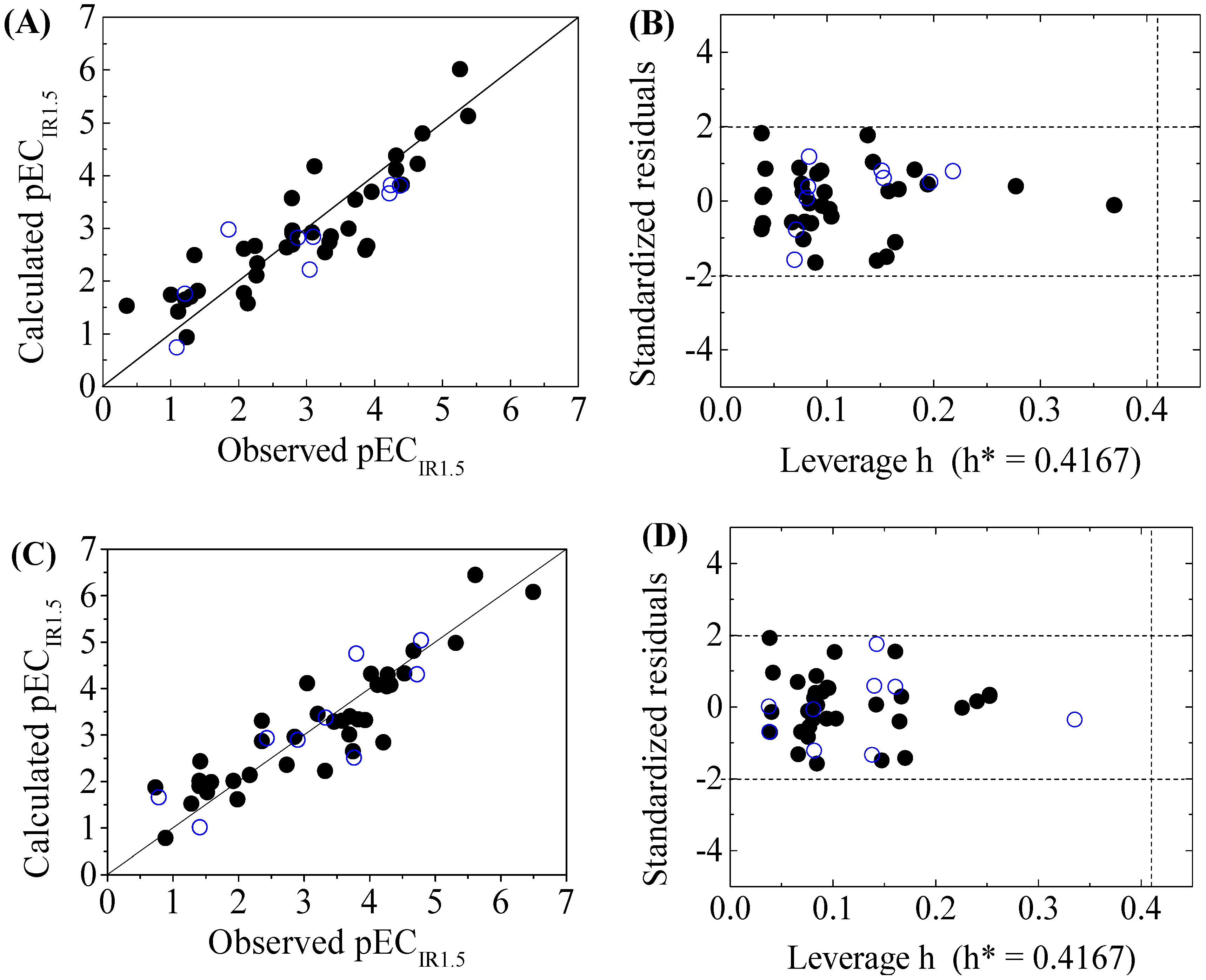
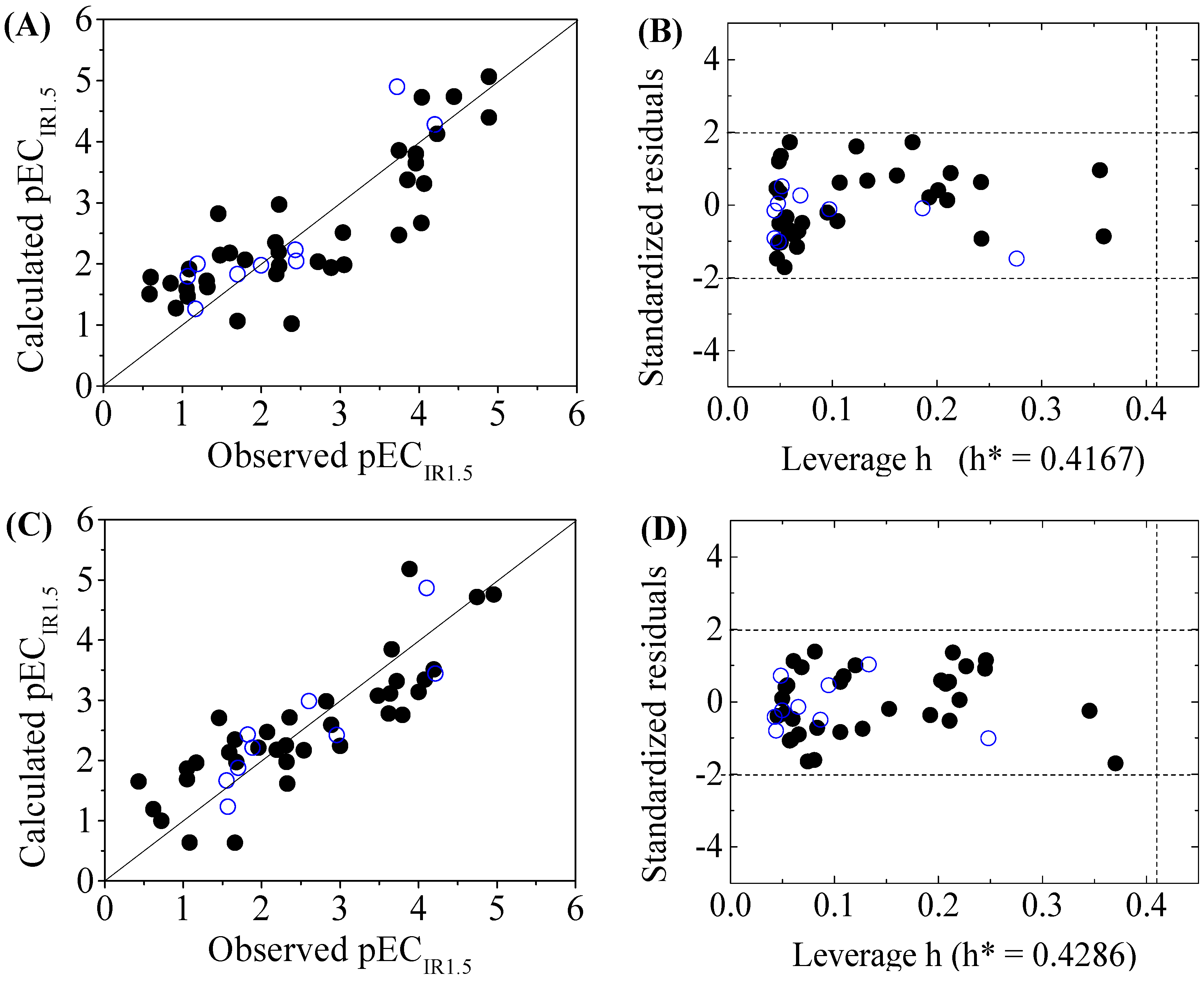
The four-variable QSAR models for the first split and its statistical parameters for five toxicity endpoints are listed in Table 3. All five QSAR models for toxicity bioassays of X-Microtox, GSH+, GSH−, DNA+ and DNA− are satisfactory, according to all conditions of R2 > 0.7, Q2LOO and Q2LMO > 0.6, Q2F1, Q2F2 and Q2F3 > 0.7, CCC > 0.85 [15]; > 0.65; or , and 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k’ ≤ 1.15 [18]. Figure 1, Figure 2 and Figure 3 present the correlations between experimental and calculated –logEC50 or –logECIR1.5 (pEC50 or pECIR1.5) values for the five models. The pEC50 or pECIR1.5 values calculated from the QSAR models are listed in Table 2.
3.3. Domain of Applicability
The criteria for an outlier are expressed as hi > h* and LOO standardized residuals greater than 2.0. For the first split (split 1), the Williams plot of the five QSAR models for toxicity bioassays of X-Microtox, GSH+, GSH−, DNA+ and DNA− are shown in Figure 1, Figure 2 and Figure 3. All 50 DBPs in the training and test sets satisfy the outlier criteria, and the QSAR models lead to reliably predicted data. The outlier was also examined for the splits 2–5 (the statistical parameters are listed in Supplementary data). There were no outliers in the training and test sets.
3.4. Explanation of Descriptors
The five developed QSAR models allow for mechanical interpretation of the toxicities of DBPs to X-Microtox, GSH+, GSH−, DNA+ and DNA−. Four descriptors for five models selected by stepwise MLR helped to improve the understanding of DBPs. A total of 12 descriptors were included in five four-variable models. The selected descriptors are SpDiam_B(m), AVS_B(v), Eig05_AEA(dm), SddsN, Sv, C%, P_VSA_LogP_7, P_VSA_LogP_8, Mor03s, Mor04s, T(N..Br) and T(N..I). These structural features are related to DBP toxicity. The positive values of the regression coefficients indicate increasing toxicity with increasing descriptor values, while the negative values indicate decreasing toxicity with increasing descriptor values. The 12 descriptors belong to different groups of descriptors: 2D matrix-based descriptors (SpDiam_B(m) and AVS_B(v)), edge adjacency indices (Eig05_AEA(dm)), atom-type E-state indices (SddsN), constitutional indices (Sv and C%), P_VSA-like descriptors (P_VSA_LogP_7 and P_VSA_LogP_8), 3D-MoRSE descriptors (Mor03s and Mor04s) and 2D atom pairs (T(N..Br) and T(N..I)).
For the QSAR model based on the toxicity of the X-Microtox bioassay, the standard regression coefficients of AVS_B(v) and SddsN were higher than the other two descriptors. AVS_B(v) and SddsN were the main factors affecting the toxicity of DBPs to X-Microtox. The descriptor SddsN indicated that −N(=)= (nitro) (where “=” represents a double bond and “−” represents a single bond) is one of the main factors that affected the toxicity of DBPs to X-Microtox. For the toxicities of DBPs toward GSH+ and GSH−, the same descriptors were selected in the QSAR models, which indicates the similar toxicity mechanism of the two endpoints. SpDiam_B(m) and T(N..Br) were the main positive contributors to the toxicity, as their standard regression coefficients were higher than C% and P_VSA_LogP_8. T(N..Br), the heteroatom between N and Br, was one of the main factors affecting the toxicity of DBPs toward GSH+ and GSH−. There were two descriptors (P_VSA_LogP_7 and T(N..I)) in the QSAR model for DNA+ and DNA−, where P_VSA_LogP_7 made a positive contribution to toxicity while T(N..I) made a negative contribution to toxicity. T(N..I), the heteroatom between N and I, was one of the main factors affecting the toxicity of DBPs toward DNA+ and DNA−.
4. Conclusions
All five considered QSAR models, resulting from the random split of the whole dataset intro training and test sets, satisfied the validation criteria. The application domain was clearly defined and the mechanism was interpreted. The reliability of the five QSAR models met the Organization for Economic Co-operation and Development (OECD) principles [26]: (1) a defined endpoint; (2) an unambiguous algorithm; (3) a defined domain of applicability; (4) appropriate measures of goodness-of-fit, robustness and predictivity; and (5) a mechanistic interpretation, if possible.
The QSAR method was used to develop several predictive models for the toxicities of DBPs toward X-Microtox, GSH+, GSH−, DNA+, and DNA−. Using the selected descriptors, which can be easily generated from the Dragon software, all the developed QSAR models with a good predictive performance were used for estimating toxicities of DBPs. It is expected that the proposed QSAR models could be used to predict the toxicities of DBPs.
Supplementary Materials
Supplementary Materials are available online.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 21407032, 51578171, and 21667013), Science Research and Technology Development Project of Guilin (2016012505), Science Research and Technology Development Project of Guangxi (Guikehe1599005-2-2), and the Project of High-level Innovation Team and Outstanding Scholar in Guangxi Colleges and Universities (002401013001).
Author Contributions
Litang Qin: The writer of the manuscript, provide the main research ideas. Xin Zhang: Data collecion; data analysis; draw pictures and tables. Yuhan Chen: Data collection. Lingyun Mo: Build QSAR model. Honghu Zeng : Data analysis.Yanpeng Liang: Calculation of molecular descriptors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rook, J.J. Formation of haloforms during chlorination of natural waters. Water Treat. Exam. 1974, 23, 234–243. [Google Scholar]
- Chen, B.; Zhang, T.; Bond, T.; Gan, Y. Development of quantitative structure activity relationship (QSAR) model for disinfection byproduct (DBP) research: A review of methods and resources. J. Hazard. Mater. 2015, 299, 260–279. [Google Scholar] [PubMed]
- Grellier, J.; Rushton, L.; Briggs, D.J.; Nieuwenhuijsen, M.J. Assessing the human health impacts of exposure to disinfection by-products—A critical review of concepts and methods. Environ. Int. 2015, 78, 61–81. [Google Scholar] [PubMed]
- Stalter, D.; O’Malley, E.; von Gunten, U.; Escher, B.I. Fingerprinting the reactive toxicity pathways of 50 drinking water disinfection by-products. Water Res. 2016, 91, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hunter, E.S.; Rogers, E.; Blanton, M.; Richard, A.; Chernoff, N. Bromochloro-haloacetic acids: Effects on mouse embryos in vitro and QSAR considerations. Reprod. Toxicol. 2006, 21, 260–266. [Google Scholar] [PubMed]
- Perez-Garrido, A.; Gonzalez, M.P.; Escudero, A.G. Halogenated derivatives QSAR model using spectral moments to predict haloacetic acids (HAA) mutagenicity. Bioorg. Med. Chem. 2008, 16, 5720–5732. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.Z.; Wang, F. Quantitative structure activity relationship (QSAR) of chlorine effects on E-LUMO of disinfection by-product: Chlorinated alkanes. Chemosphere 2010, 78, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Stalter, D.; Dutt, M.; Escher, B.I. Headspace-Free setup of in vitro bioassays for the evaluation of volatile disinfection by-products. Chem. Res. Toxicol. 2013, 26, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.J.; Fan, R.L.; Zhang, L.F.; Yue, J.Q.; Webster, R.D.; Lim, T.T. Photodegradation of iodinated trihalomethanes in aqueous solution by UV 254 irradiation. Water Res. 2014, 49, 275–285. [Google Scholar] [PubMed]
- Yang, M.T.; Zhang, X.R. Comparative developmental toxicity of new aromatic halogenated DBPs in a chlorinated saline sewage effluent to the marine polychaete Platynereis dumerilii. Environ. Sci. Technol. 2013, 47, 10868–10876. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.M.; Fang, H.; Tong, W.; Wu, J.; Perkins, R.; Blair, R.M.; Branham, W.S.; Dial, S.L.; Moland, C.L.; Sheehan, D.M. QSAR models using a large diverse set of estrogens. J. Chem. Inf. Comput. Sci. 2001, 41, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Schüürmann, G.; Ebert, R.U.; Chen, J.W.; Wang, B.; Kuhne, R. External validation and prediction employing the predictive squared correlation coefficient - test set activity mean vs training set activity mean. J. Chem. Inf. Model. 2008, 48, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Ballabio, D.; Todeschini, R. Evaluation of model predictive ability by external validation techniques. J. Chemom. 2010, 24, 194–201. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate it? comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative studies on some metrics for external validation of QSPR models. J. Chem. Inf. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Ojha, P.K.; Mitra, I.; Das, R.N.; Roy, K. Further exploring R2M metrics for validation of QSPR models. Chemom. Intell. Lab. Syst. 2011, 107, 194–205. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Mode.l 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Qin, L.T.; Liu, S.S.; Chen, F.; Wu, Q.S. Development of validated quantitative structure–retention relationship models for retention indices of plant essential oils. J. Sep. Sci. 2013, 36, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.T.; Liu, S.S.; Chen, F.; Xiao, Q.F.; Wu, Q.S. Chemometric model for predicting retention indices of constituents of essential oils. Chemosphere 2013, 90, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Toropova, A.P.; Toropov, A.A.; Benfenati, E.; Leszczynska, D.; Leszczynski, J. QSAR model as a random event: A case of rat toxicity. Bioorg. Med. Chem. 2015, 23, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Kiralj, R.; Ferreira, M.M.C. Is your QSAR/QSPR descriptor real or trash? J. Chemom. 2010, 24, 681–693. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Organisation for Economic Co-operation and Development. Guidance Document on the Validation of (quanTitative) Structure-Activity Relationship [(Q)SAR] Models; ENV/JM/MONO: Paris, France, 2007; pp. 1–154. [Google Scholar]
Sample Availability: Not available. |
Figure 1.
Scatter plot of observed versus calculated pEC50 (A), and the Williams plot of the final model (B) for 50 disinfection byproducts to X-Microtox. “●”: training set, “○”: test set. h*( warning leverage).
Figure 1.
Scatter plot of observed versus calculated pEC50 (A), and the Williams plot of the final model (B) for 50 disinfection byproducts to X-Microtox. “●”: training set, “○”: test set. h*( warning leverage).
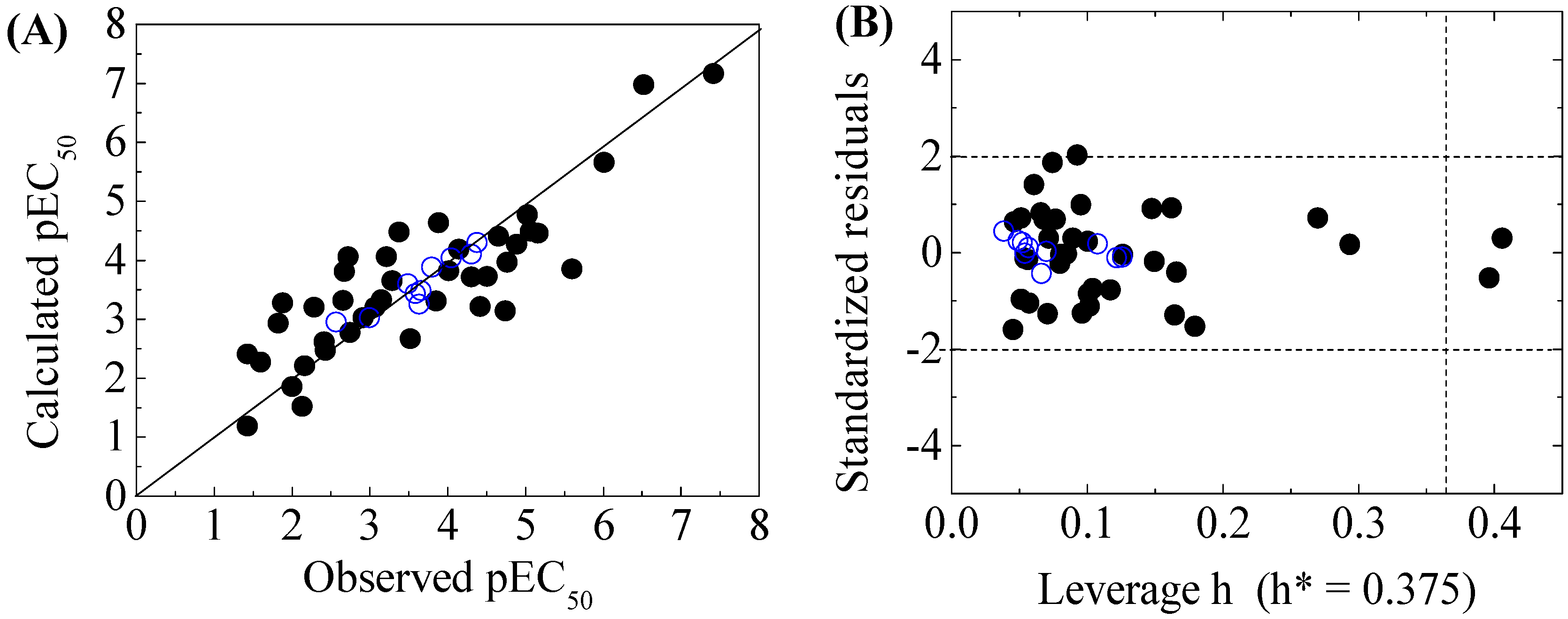
Figure 2.
Scatter plot of observed versus calculated pECIR1.5 ((A) for GSH+ and (C) for GSH− ) and the Williams plot ((B) for GSH+ and (D) for GSH− ) of the final model for 45 disinfection byproducts. “●”: training set, “○”: test set.
Figure 2.
Scatter plot of observed versus calculated pECIR1.5 ((A) for GSH+ and (C) for GSH− ) and the Williams plot ((B) for GSH+ and (D) for GSH− ) of the final model for 45 disinfection byproducts. “●”: training set, “○”: test set.
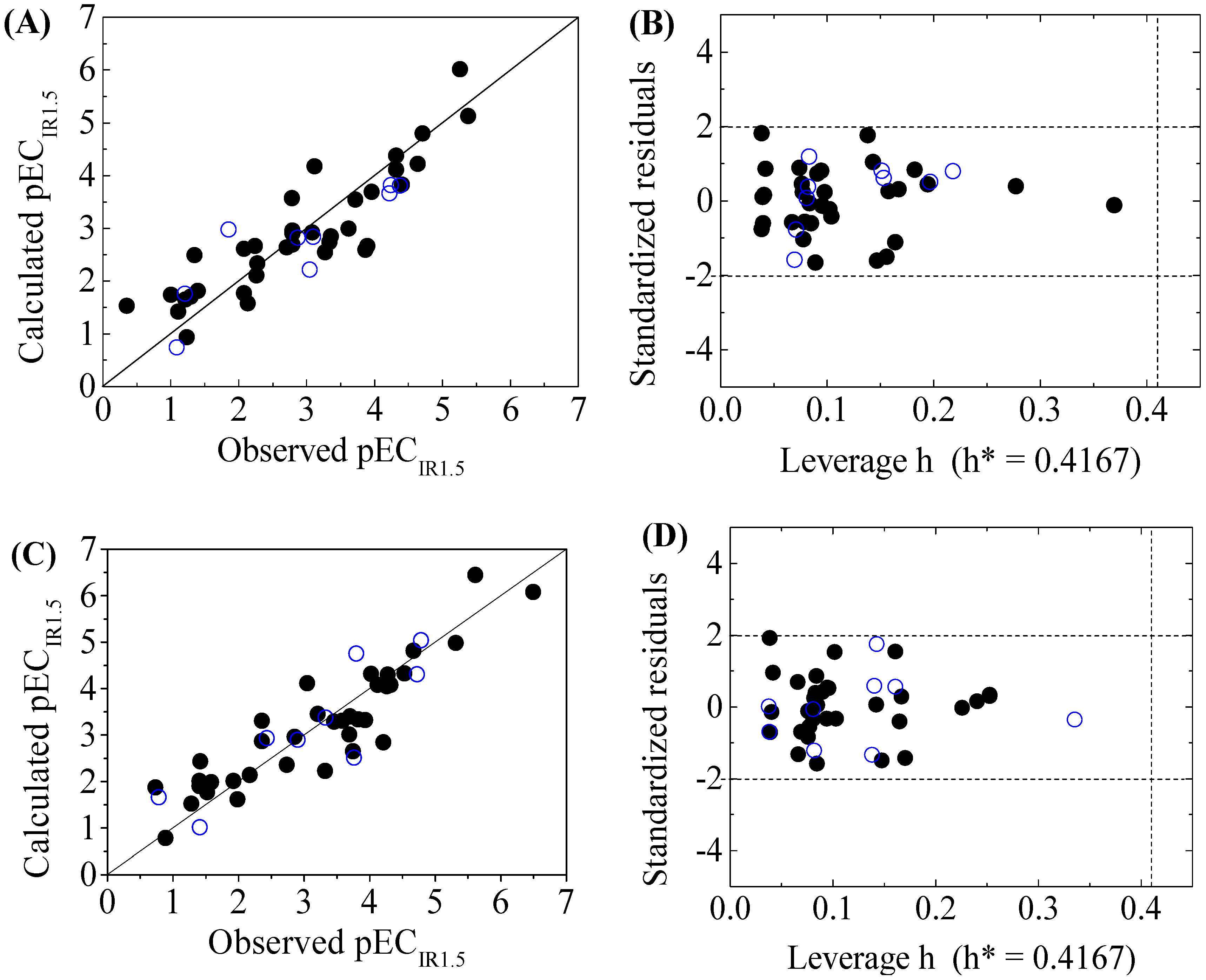
Figure 3.
Scatter plot of observed versus calculated pECIR1.5 ((A) for DNA+ and (C) for DNA−) and the Williams plot ((B) for DNA+ and (D) for DNA−) of the final model for 45 disinfection byproducts. “●”: training set, “○”: test set.
Figure 3.
Scatter plot of observed versus calculated pECIR1.5 ((A) for DNA+ and (C) for DNA−) and the Williams plot ((B) for DNA+ and (D) for DNA−) of the final model for 45 disinfection byproducts. “●”: training set, “○”: test set.

{kind=link}
{kind=link}
{kind=link}
Table 1.
The five endpoints of the 50 drinking water disinfection byproducts (DBPs).
| Bioassay | Test Species (Strain/Cell Line) a | Endpoint | Detected Signal |
|---|---|---|---|
| Microtox | Aliivibrio fischeri | Cytotoxicity | Bioluminescence as indicator for cell viability |
| E. coli ± GSH | Escherichia coli MJF335 (GSH−) and MJF276 (GSH+) | Interaction with proteins/peptides | OD at 600 nm as indicator for cell density and descriptor of cell growth |
| E. coli ± DNA | Escherichia coli MV4108 (DNA−) and MV1161 (DNA+) | Interaction with DNA | OD at 600 nm as indicator for cell density and descriptor of cell growth |
a: GSH+: EC50 of E. coli strain; MJF276: capable to produce glutathione (GSH); GSH−: EC50 of E. coli strain; MJF335: not capable to produce GSH and hence more susceptible to compounds which react with proteins (i.e., soft electrophiles); DNA+: EC50 of E. coli strain; MV1161: capable of repairing DNA damage; DNA−: EC50 of E. coli strain; MV4108: not capable of repairing DNA damage and hence more susceptible to compounds which react with DNA (i.e., hard electrophiles).
Table 2.
Observed and calculated effect concentrations (pEC50 (negative logarithm of 50% effective concentration) for X-Microtox and pECIR1.5 (negative logarithm of 1.5 induction ratio effective concentration) for the other assays, mol/L) of disinfection byproducts.
Table 2.
Observed and calculated effect concentrations (pEC50 (negative logarithm of 50% effective concentration) for X-Microtox and pECIR1.5 (negative logarithm of 1.5 induction ratio effective concentration) for the other assays, mol/L) of disinfection byproducts.
| No. | Name | X-Microtox | GSH+ | GSH− | DNA+ | DNA− | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Observed pEC50 | Calculated pEC50 | Observed pECIR1.5 | Calculated pECIR1.5 | Observed pECIR1.5 | Calculated pECIR1.5 | Observed pECIR1.5 | Calculated pECIR1.5 | Observed pECIR1.5 | Calculated pECIR1.5 | ||
| Halomethanes | |||||||||||
| 1 | 1,1-dichloroethene | 3.1549 | 3.3214 | 1.3516 | 2.4847 | 1.4145 | 2.4357 | 1.0706 * | 1.7985 * | 0.4318 | 1.6512 |
| 2 | dichloromethane | 2.2840 | 3.1963 | 1.0888 * | 0.7391 * | 0.8861 | 0.7818 | 0.8539 | 1.6828 | 0.7212 | 0.9997 |
| 3 | bromochloromethane | 5.0706 | 4.4813 | 1.2182 | 1.6474 | 0.7328 | 1.8741 | 0.6021 | 1.7808 | - | - |
| 4 | chloroform | 2.4318 | 2.4675 | 1.2366 | 0.9330 | 1.4089 * | 1.0149 * | 1.1675 * | 1.2621 * | 1.0809 | 0.6385 |
| 5 | bromodichloromethane | 5.0269 | 4.7723 | 1.2111 * | 1.7614 * | 1.4034 | 1.9032 | 1.3188 | 1.6196 | 1.5686 * | 1.2324 * |
| 6 | bromoform | 3.6383 * | 3.2549 * | 1.0066 | 1.7407 | 1.5850 | 1.9863 | 1.0862 | 1.9166 | 1.1675 | 1.9629 |
| 7 | dibromochloromethane | 3.0000 * | 3.0193 * | 2.0809 | 1.7617 | 1.9208 | 2.0116 | 1.6990 * | 1.8321 * | 1.5528 * | 1.6647 * |
| 8 | dichloroiodomethane | 3.4949 * | 3.5960 * | 2.7959 | 2.9585 | 3.2076 | 3.4508 | 2.1938 | 1.8353 | 2.3279 | 1.6163 |
| 9 | bromochloroiodomethane | 1.6021 | 2.2717 | 2.7959 | 2.8967 | 3.3279 * | 3.3766 * | 2.2291 | 2.9723 | 2.3188 | 1.9777 |
| 10 | dibromoiodomethane | 4.0506 * | 4.0353 * | 2.8697 * | 2.8209 * | 3.4559 | 3.2854 | 2.0000 * | 1.9755 * | 1.9586 | 2.2126 |
| 11 | chlorodiiodomethane | 4.6576 | 4.4046 | 3.0809 | 2.9161 | 3.6990 | 3.3999 | 2.4437 * | 2.0457 * | 2.3098 | 2.2495 |
| 12 | bromodiiodomethane | 5.6021 | 3.8512 | 3.0969 * | 2.8369 * | 3.5850 | 3.3046 | 3.0506 | 1.9836 | 2.9586 * | 2.4210 * |
| 13 | triiodomethane | 2.4202 | 2.6149 | 3.3615 | 2.8486 | 3.9337 | 3.3188 | 2.8861 | 1.9398 | 2.8861 | 2.5894 |
| Halonitromethanes | |||||||||||
| 14 | trichloronitromethane | 4.3098 | 3.7132 | 4.6383 | 4.2152 | 5.3143 | 4.9812 | 4.2007 * | 4.2819 * | 4.0809 | 3.3428 |
| 15 | tribromonitromethane | 2.7447 | 2.7705 | 5.3820 | 5.1283 | 6.4949 | 6.0793 | 4.8861 | 5.0640 | 4.7447 | 4.7139 |
| Haloacetonitriles | |||||||||||
| 16 | dichloroacetonitrile | 4.5086 | 3.7184 | 3.2757 | 2.5481 | 3.7632 * | 2.5119 * | 3.0362 | 2.5123 | 2.8239 | 2.9808 |
| 17 | trichloroacetonitrile | 4.8861 | 4.2672 | 3.8979 | 2.6617 | 3.7447 | 2.6486 | 3.7447 | 3.8560 | 3.4815 | 3.0753 |
| 18 | bromochloroacetonitrile | 4.0132 | 3.8159 | 4.3188 | 4.1067 | 4.2757 | 4.2982 | 3.8539 | 3.3736 | 3.7212 | 3.3159 |
| 19 | dibromoacetonitrile | 4.7696 | 3.9655 | 4.7100 | 4.7981 | 4.7825 * | 5.0416 * | 4.2291 | 4.1282 | 4.1938 | 3.5113 |
| Haloketones | |||||||||||
| 20 | 1,1-dichloropropanone | 2.7212 | 4.0555 | 3.0506 * | 2.2187 * | 3.3188 | 2.2263 | 2.4318 * | 2.2303 * | 2.3565 | 2.7129 |
| 21 | 1,1,1-trichloropropanone | 3.6576 * | 3.4857 * | 2.2803 | 2.3311 | 2.7364 | 2.3615 | 2.3872 | 1.0225 | 3.0000 | 2.2423 |
| Haloacetic acids | |||||||||||
| 22 | chloroacetic acid | 6.0088 | 5.6592 | 2.1367 | 1.5737 | 1.9851 | 1.6179 | 1.6990 | 1.0652 | 1.6576 | 2.3448 |
| 23 | bromoacetic acid | 1.8861 | 3.2782 | 3.8697 | 2.5921 | 4.2111 | 2.8428 | 4.0655 | 3.3127 | 4.0000 | 3.1346 |
| 24 | iodoacetic acid | 2.6778 | 3.8068 | 4.3768 * | 3.8079 * | 4.7212 * | 4.305 * | 3.7447 | 2.4730 | 3.6576 | 3.8483 |
| 25 | dichloroacetic acid | 3.2147 | 4.0622 | 1.2967 | 1.6975 | 1.5229 | 1.7669 | 0.9208 | 1.2745 | 0.6198 | 1.1912 |
| 26 | bromochloroacetic acid | 5.1612 | 4.4508 | 2.0783 | 2.6122 | 2.3565 | 2.8669 | 1.1938 * | 1.9983 * | 1.6990 * | 1.8777 * |
| 27 | dibromoacetic acid | 4.1487 | 4.1865 | 2.2403 | 2.6637 | 2.4318 * | 2.9289 * | 1.6021 | 2.1776 | 1.8861 * | 2.2108 * |
| 28 | chloroiodoacetic acid | 1.8239 | 2.9267 | 4.4034 | 3.8242 | 4.5302 | 4.3246 | 4.0362 | 4.7239 | 4.1024 * | 4.8634 * |
| 29 | bromoiodoacetic acid | 3.7959 * | 3.8762 * | 4.2403 * | 3.8168 * | 4.0200 | 4.3157 | 3.7212 * | 4.8968 * | 3.8861 | 5.1786 |
| 30 | trichloroacetic acid | 3.2924 | 3.6489 | 1.4034 | 1.8108 | 1.4034 | 2.0112 | 1.0555 | 1.5905 | 1.0506 | 1.6881 |
| 31 | bromodichloroacetic acid | 1.4318 | 2.4029 | 2.7100 | 2.6367 | 2.9031 * | 2.8964 * | 1.7959 | 2.0658 | 1.6576 | 0.6340 |
| 32 | dibromochloroacetic acid | 3.5229 | 2.6718 | 2.7959 | 2.6882 | 2.8539 | 2.9584 | 1.4815 | 2.1417 | 1.4559 | 2.7074 |
| 33 | tribromoacetic acid | 4.4202 | 3.2073 | 3.3372 | 2.7322 | 3.6882 | 3.0113 | 2.1805 | 2.3490 | 2.6021 * | 2.9859 * |
| Haloacetaldehyde | |||||||||||
| 34 | chloral hydrate | 2.1675 | 2.2067 | 2.2636 | 2.1046 | 2.1707 | 2.1359 | 1.3098 | 1.7185 | 1.6778 | 1.9750 |
| Haloacetamides | |||||||||||
| 35 | dichloracetamide | 6.5229 | 6.9769 | 1.1135 | 1.4161 | 1.2798 | 1.5222 | 0.5850 | 1.5056 | 1.0506 | 1.6881 |
| 36 | bromochloroacetamide | 2.5686 * | 2.9516 * | 1.8539 * | 2.9712 * | 2.3565 | 3.3043 | 1.4559 | 2.8215 | 1.8239 * | 2.4295 * |
| 37 | dibromoacetamide | 3.0706 | 3.2043 | 4.2218 * | 3.6626 * | 4.2596 | 4.0477 | 3.9586 | 3.6467 | 3.6198 | 2.7807 |
| 38 | chloroiodoacetamide | 2.6576 | 3.3142 | 3.7212 | 3.5437 | 4.1192 | 4.0810 | 2.7212 | 2.0333 | 2.5376 | 2.1670 |
| 39 | bromoiodoacetamide | 3.3768 | 4.4729 | 3.1163 | 4.1762 | 3.7959 * | 4.7536 * | 2.2291 | 1.9651 | 2.0706 | 2.4718 |
| 40 | diiodoacetamide | 1.4318 | 1.1768 | 2.7825 | 3.5724 | 3.0482 | 4.1155 | 2.2218 | 2.1941 | 2.1938 | 2.1785 |
| 41 | trichloroacetamide | 2.0000 | 1.8559 | 0.3565 | 1.5288 | 0.7825 * | 1.6577 * | 1.0706 | 1.4651 | 1.5850 | 2.1329 |
| 42 | bromodichloroacetamide | 4.3098 * | 4.099 * | 3.6198 | 2.9951 | 3.8239 | 3.3331 | 4.0315 | 2.6693 | 3.7959 | 2.7569 |
| 43 | dibromochloroacetamide | 4.3768 * | 4.2953 * | 3.9566 | 3.6865 | 4.3188 | 4.0764 | 3.9586 | 3.8000 | 3.6383 | 3.1107 |
| 44 | tribromoacetamide | 2.1308 | 1.5148 | 4.3233 | 4.3703 | 4.6676 | 4.8106 | 4.4437 | 4.7359 | 4.2147 * | 3.4421 * |
| Nitrosamines | |||||||||||
| 45 | n-nitrosodimethylamine | 2.9208 | 3.0246 | - | - | - | - | - | - | - | - |
| 46 | n-nitrosodiethylamine | 7.4202 | 7.1686 | - | - | - | - | - | - | - | - |
| 47 | n-nitrosopiperidine | 3.8861 | 4.6292 | - | - | - | - | - | - | - | - |
| 48 | n-nitrosomorpholine | 3.8539 | 3.3096 | - | - | - | - | - | - | - | - |
| 49 | nitrosodi-n-butylamine | 3.5850 * | 3.4285 * | - | - | - | - | - | - | - | - |
| Furanone | |||||||||||
| 50 | 3-chloro-4-(dichloromethyl)-5- | 4.7447 | 3.1323 | 5.2596 | 6.0139 | 5.6108 | 6.4454 | 4.8861 | 4.3949 | 4.9586 | 4.7578 |
* The chemical included in the test set.
Table 3.
QSAR(quantitative structure–activity relationship) model and statistical parameters for five endpoints of disinfection byproduct (training set = 80% of whole dataset, test set = 20% of whole dataset).
Table 3.
QSAR(quantitative structure–activity relationship) model and statistical parameters for five endpoints of disinfection byproduct (training set = 80% of whole dataset, test set = 20% of whole dataset).
| Endpoint | Equation a | Modeling b | Internal Validation c | External Validation d | Golbraikh & Tropsha e |
|---|---|---|---|---|---|
| X-Microtox | pEC50 = −11.8502 + 0.1230 SpDiam_B(m) + 4.9744 AVS_B(v) + 0.8805 Eig05_AEA(dm) − 3.3986 SddsN | ntr = 40, R2 = 0.7152, = 0.6826, RMSEtr = 0.7682, F = 21.9717 | = 0.6374, RMSEcv = 0.8668, = 0.6216, = 0.1034, = −0.2452 | ntest = 10, RMSEext = 0.2040, = 0.8660, = 0.8508, = 0.8496, = 0.9799, CCC = 0.9115 = 0.7185, = 0.1439 | k = 1.0136, k’ = 0.9837, = 0.8018, = 0.8584 |
| GSH+ | pECIR1.5 = −2.4744 + 0.1022C% + 0.3184SpDiam_B(m) + 0.0725 P_VSA_LogP_8+ 0.2132 T(N..Br) | ntr = 36, R2 = 0.7837, = 0.7558, RMSEtr = 0.5927, F = 28.0843 | = 0.6956, RMSEcv = 0.7032, = 0.6644, = 0.1121, = −0.2323 | ntest = 9, RMSEext = 0.6010, = 0.7715, = 0.7502, = 0.7502, = 0.7776, CCC = 0.8500, = 0.6558, = 0.1915 | k = 1.0596, k’ = 0.9119, = 0.6964, = 0.7709 |
| GSH- | pECIR1.5 = −2.4133 + 0.0894 C% + 0.3829SpDiam_B(m) + 0.0835 P_VSA_LogP_8 + 0.2270 T(N..Br) | ntr = 36, R2 = 0.8166, = 0.7929, RMSEtr = 0.5936, F = 34.5096 | = 0.7332, RMSEcv = 0.7160, = 0.6634, = 0.1140, = −0.2349 | ntest = 9, RMSEext = 0.6578, = 0.7593, = 0.7436, = 0.7430, = 0.7748, CCC = 0.8703, = 0.6688, = 0.0426 | k = 0.9659, k’ = 0.9969, = 0.7376, = 0.7510 |
| DNA+ | pECIR1.5 = 1.8732 + 0.0493 P_VSA_LogP_7 − 0.2258 Mor04s + 0.2798 T(N..Br) − 0.8971 T(N..I) | ntr = 36, R2 = 0.7019, = 0.6635, RMSEtr = 0.7113, F = 18.2520 | = 0.6287, RMSEcv = 0.7940, = 0.6338, = 0.1139, = −0.2471 | ntest = 9, RMSEext = 0.5570, = 0.8232, = 0.7482, = 0.7228, = 0.8173, CCC = 0.8781, = 0.7541, = 0.0264 | k = 0.8805, k ’= 1.0974, = 0.8132, = 0.8186 |
| DNA- | pECIR1.5 = 0.9105 + 0.3091Sv + 0.0493 P_VSA_LogP_7 + 0.2008 Mor03s − 1.0911 T(N..I) | ntr = 36, R2 = 0.7164, = 0.6786, RMSEtr = 0.6540, F = 18.9496 | = 0.6221, RMSEcv = 0.7550, = 0.5291, = 0.1200, = −0.2504 | ntest = 9, RMSEext = 0.4991, = 0.7774, = 0.7505, = 0.7500, = 0.8348, CCC = 0.8787, = 0.6920, = 0.0076 | k = 0.9538, k’ = 1.0145, = 0.7643, = 0.7664 |
a SpDiam_B(m): spectral diameter from Burden matrix weighted by mass; AVS_B(v): average vertex sum from Burden matrix weighted by van der Waals volume; Eig05_AEA(dm): eigenvalue no. 5 from augmented edge adjacency mat weighted by dipole moment; SddsN: sum of ddsN E-states; Sv: sum of atomic van der Waals volumes (scaled on carbon atom); C%: percentage of C atoms; P_VSA_LogP_7: P_VSA-like on LogP, bin 7; P_VSA_LogP_8: P_VSA-like on LogP, bin 8; T(N..Br): sum of topological distances between N..Br; T(N..I): sum of topological distances between N..I; Mor04s: signal 04/weighted by I-state; Mor03s: signal 03/weighted by I-state; b ntr: the number of samples in training set; R2: coefficient of determination; : adjusted R2; RMSEtr: root mean square error in fitting; F: F-value; c : explained variance in leave-one-out prediction; RMSEcv: root mean square error in cross-validation prediction; : explained variance in leave-many-out prediction; and : R2 and Q2 in Y-scrambling, respectively; d ntest: the number of samples in test set; RMSEext: root mean square error in test set; : external determination coefficient; , and : variance explained in test set; CCC: concordance correlation coefficient; and : average and delta values of Roy criteria, respectively; e k and k’: slopes of the regression line over external data; and : R2 values in Golbraikh & Tropsha criteria.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Qin, L.; Zhang, X.; Chen, Y.; Mo, L.; Zeng, H.; Liang, Y. Predictive QSAR Models for the Toxicity of Disinfection Byproducts. Molecules 2017, 22, 1671. https://doi.org/10.3390/molecules22101671
AMA Style
Qin L, Zhang X, Chen Y, Mo L, Zeng H, Liang Y. Predictive QSAR Models for the Toxicity of Disinfection Byproducts. Molecules. 2017; 22(10):1671. https://doi.org/10.3390/molecules22101671
Chicago/Turabian StyleQin, Litang, Xin Zhang, Yuhan Chen, Lingyun Mo, Honghu Zeng, and Yanpeng Liang. 2017. "Predictive QSAR Models for the Toxicity of Disinfection Byproducts" Molecules 22, no. 10: 1671. https://doi.org/10.3390/molecules22101671