Determination of the Bridging Ligand in the Active Site of Tyrosinase
by

Congming Zou
1,†, 
Wei Huang
1,†,
Gaokun Zhao
1,
Xiao Wan
2,
Xiaodong Hu
1,
Yan Jin
1,
Junying Li
1 and
Junjun Liu
2,* 1
Yunnan Academy of Tobacco Agricultural Sciences, 33 Yuantong Street, Kunming 650021, China
2
School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, 13 Hangkong Road, Wuhan 430030, China
*
Author to whom correspondence should be addressed.
†
These authors contribute equally to this work.
Molecules 2017, 22(11), 1836; https://doi.org/10.3390/molecules22111836
Submission received: 29 September 2017
/
Revised: 23 October 2017
/
Accepted: 25 October 2017
/
Published: 28 October 2017
(This article belongs to the Section Computational and Theoretical Chemistry)
Abstract
:Tyrosinase is a type-3 copper enzyme that is widely distributed in plants, fungi, insects, and mammals. Developing high potent inhibitors against tyrosinase is of great interest in diverse fields including tobacco curing, food processing, bio-insecticides development, cosmetic development, and human healthcare-related research. In the crystal structure of Agaricus bisporus mushroom tyrosinase, there is an oxygen atom bridging the two copper ions in the active site. It is unclear whether the identity of this bridging oxygen is a water molecule or a hydroxide anion. In the present study, we theoretically determine the identity of this critical bridging oxygen by performing first-principles hybrid quantum mechanics/molecular mechanics/Poisson-Boltzmann-surface area (QM/MM-PBSA) calculations along with a thermodynamic cycle that aim to improve the accuracy. Our results show that the binding with water molecule is energy favored and the QM/MM-optimized structure is very close to the crystal structure, whereas the binding with hydroxide anions causes the increase of energy and significant structural changes of the active site, indicating that the identity of the bridging oxygen must be a water molecule rather than a hydroxide anion. The different binding behavior between water and hydroxide anions may explain why molecules with a carboxyl group or too many negative charges have lower inhibitory activity. In light of this, the design of high potent active inhibitors against tyrosinase should satisfy both the affinity to the copper ions and the charge neutrality of the entire molecule.
1. Introduction
Tyrosinase (Enzyme Commission, EC 1.14.18.1) is a type-3 copper enzyme that is widely distributed in plants, fungi, insects, and mammals [1,2,3]. It catalyzes both the hydroxylation of monophenols and the oxidation of o-diphenols, and thus is directly associated with plant browning [4], cuticle formation [3], and skin pigmentation [1,5]. Tyrosinase is also involved in Parkinson’s disease, due to its ability to oxidize dopamine to form melanin in the brain [6]. As a result, considerable efforts have been invested in developing safe and high-potential tyrosinase inhibitors in diverse fields including tobacco curing [7], food processing [8], bio-insecticides development [3], cosmetic development [9], and human healthcare-related research [10,11].
The active site of tyrosinase contains six conserved histidine residues coordinating two copper ions [12], which are critical for both catalytic activities of tyrosinase. Shown in Figure 1 is the active site of the crystal structure of Agaricus bisporus mushroom tyrosinase. As we can see, an oxygen was determined at a location bridging the two copper ions, and it is uncertain whether this bridging oxygen is a water molecule or a hydroxide anion since hydrogen atoms cannot be determined by X-ray diffraction techniques [12].
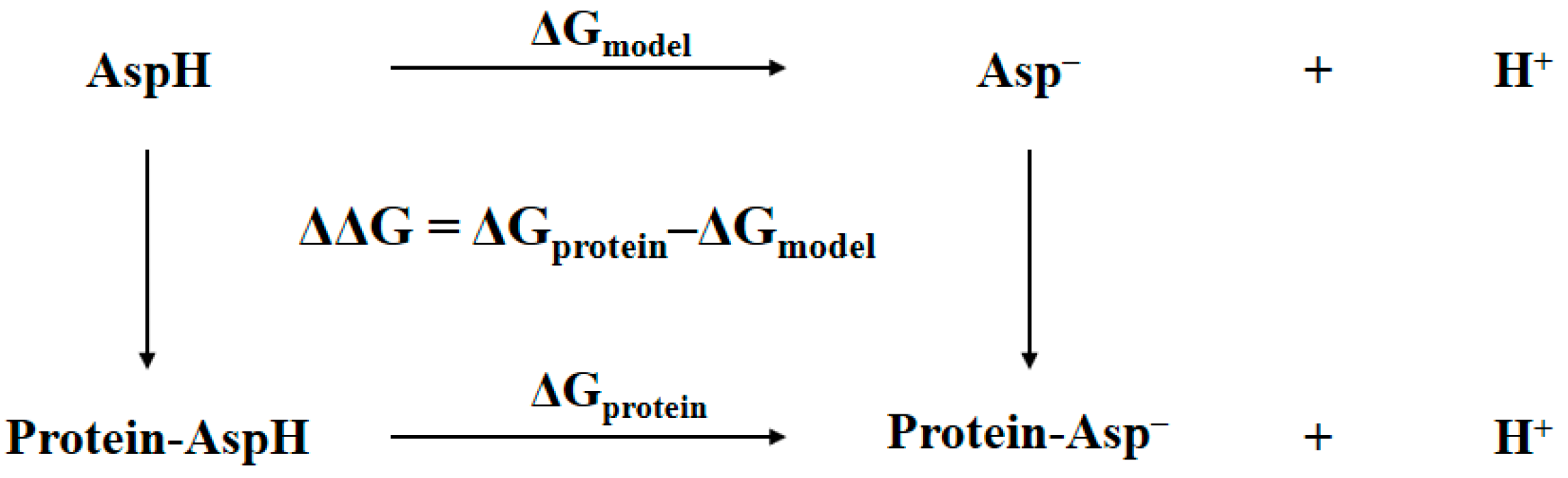
In general, the determination of the identity of the bridging oxygen can be achieved by comparing the Gibbs free energy of the protein with bridging water to that of the protein with bridging hydroxide anion plus proton in aqueous solution. The accuracy of this comparison can be improved by using a thermodynamic cycle where a model system is needed [13,14]. Shown in Figure 2 is an example of such a thermodynamic cycle that had been successfully used in pKa prediction or protonation state determination for an aspartic residue in protein [13]. The ΔGmodel is the free energy difference of the model system, which is the aspartic acid in aqueous solution, changing from the protonated state to the deprotonated state. The ΔGprotein is the free energy difference of the corresponding aspartic residue in protein changing from the protonated state to the deprotonated state. The double free energy difference ΔΔG is then equivalent to the pKa shifts of aspartic acid migrating from aqueous solution to protein, i.e., pKa,protein − pKa,model = ΔΔG/RTln(10). This equation can be re-written as follows:
The ΔGprotein(expt) can be used to determine the pKa,protein of aspartic residue in protein and to tell which protonation state is more favorable. As long as the model system has the experimentally determined pKa,model available and is structurally very close to its counterpart in protein, this thermodynamic cycle along with Equation (1) is highly efficient in pKa prediction or protonation state determination, because the systematic error caused by the theoretical method can be largely eliminated. In addition, the absolute free energy of the proton in aqueous solution is no longer necessary, and therefore the calculations of ΔGprotein(calc) and ΔGmodel(calc) are basically the free energy difference between the deprotonated and protonated states of the molecules. These two values could be estimated by the so-called QM/MM-PBSA method, in which the QM/MM refers to the hybrid quantum mechanical/molecular mechanics and PBSA refers to the solvation energy evaluated by solving Poisson–Boltzmann (PB) equation, and by estimating the nonpolar contribution in terms of the change in solvent-accessible surface area (SA). The QM/MM-PBSA method has been successfully applied to many protein systems, including reactions in proteins [15], binding mode analysis [16,17], inhibition constant evaluation [18], and protonation state determination [19]. By incorporating the abovementioned thermodynamic cycle, it should produce a reliable result for the values of ΔGprotein(calc) and ΔGmodel(calc).
To determine the identity of the critical bridging oxygen in the active site of tyrosinase, we may employ the QM/MM-PBSA method, incorporating a similar thermodynamic cycle as that in Figure 2. The most appropriate model system should be a small cluster representing the active site of tyrosinase, and practically could be the atoms in the stick and sphere representations shown in Figure 1. It contains the side chains of six histidine residues coordinating two copper ions, in which the two copper ions are connected by a bridging water or a bridging hydroxide anion. Unfortunately, this model system does not have the experimental pKa value available, causing the lack of ΔGmodel(expt), and consequently prevents the use of Equation (1) to estimate ΔGprotein(expt). However, it is possible to accurately estimate ΔGmodel(expt) by using first-principles quantum mechanical calculations with the continuum solvation model. The pure continuum solvation model is well-suited to describe the long-range electrostatic interactions between solute and the bulk solvent, but only partially considers the short-range electrostatic interactions, e.g., the electrostatics between the solute and first solvation shell [20,21,22]. As a result, the inclusion of water molecules in the solvation shell around the solute to form a solute-water cluster can largely improve the calculation accuracy for ions where solvation effects are significant [23,24]. For example, the absolute hydration free energy of the proton was determined in such a way by including the water clusters in the first and second solvation shells [25]. In addition, a recent study showed that the average error for pKa prediction on molecules with hydroxyl and hydroperoxyl functional groups was about 0.02 pKa units by employing the SMD solvation model [26] at the B3LYP/6-311++G** level, in which the continuum/cluster model was used with three explicit water molecules in the first solvation shell [27]. Accordingly, the ΔGmodel(expt) for the model systems here could be calculated by using the same method.
In this study, we theoretically determine the identity of the critical bridging oxygen by performing first-principles QM/MM-PBSA calculations along with a thermodynamic cycle that aim to improve the accuracy. By analyzing the QM/MM-optimized geometries, the keys to design high potent active inhibitors against tyrosinase are discussed.
2. Methods
2.1. Thermodynamic Cycle
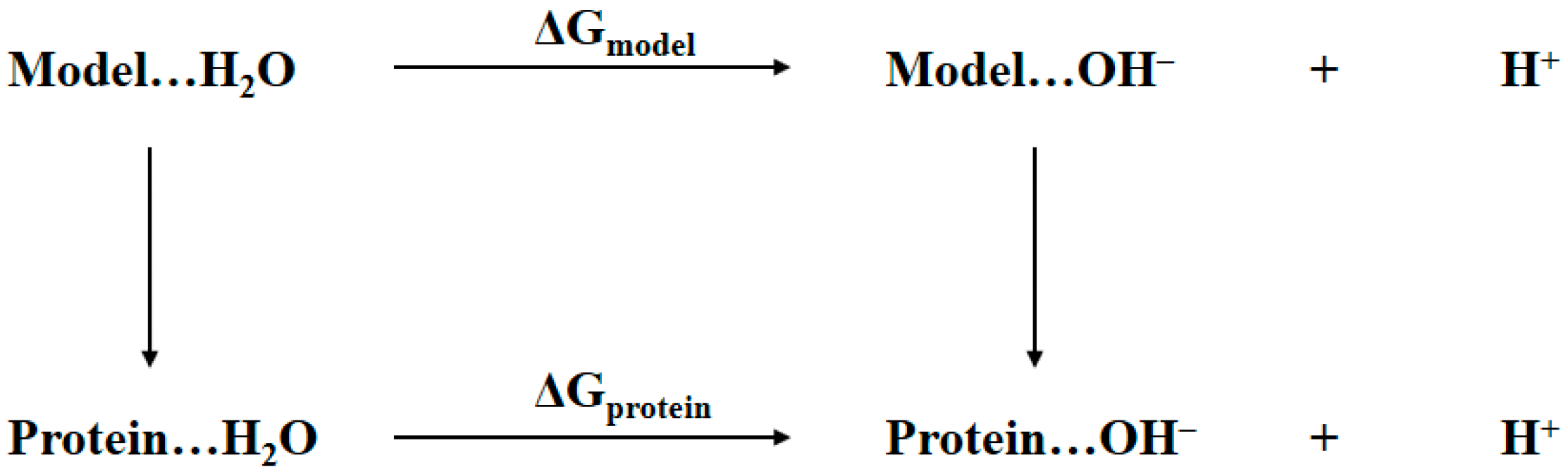
The determination of the bridging ligand in the active site of tyrosinase to be water or hydroxide anion is essentially the same as determining the protonation state of aspartic residue in protein shown in Figure 2. Thus, a similar thermodynamic cycle is built in Figure 3. The notations Protein…H2O and Protein…OH− refer to the two tyrosinase systems of which the bridging ligand is water or hydroxide anion, respectively. The Model refers to a cluster model system that is structurally as close as possible to the active site of tyrosinase, namely the atoms rendered in stick and sphere representations in Figure 1. The ΔGprotein and ΔGmodel are the Gibbs free energy changes from bridging water to bridging hydroxide anion for tyrosinase and the model systems, respectively. In the present study, these two values are obtained by using the QM/MM-PBSA calculations. Considering the fact that the pKa for the model system is determined by first-principles QM calculations, the notations of terms in Equation (1) shall be revised in a more appropriate form:
in which the value of ΔG can be used to determine the identity of the bridging ligand.
2.2. QM/MM-PBSA Calculation
In the QM/MM-PBSA method, the free energy is calculated by using the following equation:
where EQM/MM is the QM/MM potential energy; GPBSA is the solvation energy that consists of the polar solvation energy estimated by solving the Poisson-Boltzmann equation and the nonpolar solvation energy estimated from the solvent-accessible surface area of the molecule; T is the room temperature; and SQM/MM is the entropy of the molecule, which could be estimated as:
G = EQM/MM + GPBSA − TSQM/MM
SQM/MM = SQM + SMM + SQM-MM
SQM and SMM are the entropy terms of QM and MM systems, respectively. SQM-MM is the coupling term between QM and MM systems.
The crystal structure 2Y9W was used for the preparation of the initial structure for QM/MM calculations. The QM system is depicted as the atoms that are rendered as sticks and spheres in Figure 1. It consists of the two copper ions, the side chains of histidine residues 61, 85, 94, 259, 263, and 296 that form around the copper ions, and the bridging ligand which is a water molecule or a hydroxide anion. The rest of the atoms are defined as the MM system. The QM/MM interface was described by the link-atom method where the boundary atoms are capped by hydrogen atoms. [28]. The QM/MM calculations were carried out by using an interface program implemented in Amber16 [29] at the B3LYP/6-31+G*:AMBER level, in which the QM system was described at the B3LYP/6-31+G* level of theory by using Gaussian 09 program [30] and the MM system was described with the Amber14SB force field by using the sander module in Amber16. In all QM/MM calculations, atoms within 15 Å of the two copper ions were allowed to move while all the other atoms outside this range were kept frozen. The geometry optimizations were performed by using the Limited-memory Broyden-Fletcher-Goldfarb-Shanno (LBFGS) algorithm [31] implemented in the sander program. The convergence criterion is the root-mean-square (RMS) deviation of the energy gradient being less than 0.01 kcal·mol−1·Å−1. At the convergence of the geometry optimization, the RESP (restrained electrostatic potential) charges [32] of the QM system along with the MM charges obtained from the QM/MM calculations were applied in the PBSA calculations by using the PBSA program in Amber16.
The QM/MM and PBSA calculations generate the first and second terms in Equation (3). For the entropy term that is described with details in Equation (4), we ignored the term SMM because it is unlikely that the entropy of the protein is significantly different for the two states. Likewise, the contributions from bonded and van der Waals QM-MM interactions to the term SQM-MM were also ignored, because SQM-MM majorly comprises the contribution from QM-MM electrostatic interactions. Technically, the entropy term was calculated by using Gaussian 09 program with normal mode analysis of harmonic frequencies for the QM system, in which MM point charges were taken as background charges. The QM/MM-PBSA calculations were performed on both protein and model systems in order to cancel the systematic errors from the theoretical method. Since the model system is defined exactly the same as that for the QM system, the calculations for the model system are basically the QM/MM-PBSA calculations on an isolated QM system that does not have MM background charges.
2.3. QM Calculation for pKa Determination
The pKa could be accurately determined by first-principles QM calculations employing the continuum/cluster model. For example, it was found that the SMD solvation method incorporating three explicit water molecules in the first solvation shell produced accurate pKa for molecules with hydroxyl and hydroperoxyl functional groups [27]. In the present study, the oxygen of the bridging ligand binds with the two copper ions, leaving the hydrogens of the bridging ligand pointing towards the outside of the active site. Thus, there are two water molecules in the first solvation shell for bridging water, and one water molecule in the first solvation shell for bridging hydroxide anion. In order for the computed energies to be reliable as well as comparable, the water molecules in the first and second solvation shells are explicitly included, resulting in three explicit water molecules for both bridging water and bridging hydroxide anion states of model system. We carried out constrained molecular dynamics (MD) simulations on the model system to obtain the three explicit water molecules at reasonable locations. Since the model system is defined as the same as that for the QM system, the RESP charges for the QM system were used here for the model system in the constrained MD simulation. The model system was solvated in a rectangular box of TIP3P water molecules with a minimum solute wall distance of 8 Å. Prior to MD simulation, the energy minimization was carried out to remove possible bad contacts. Then, the equilibration was performed for 40 ps with the temperature gradually increasing from 10 K to 300 K, followed by a 50-ps simulation at 300 K in NPT ensemble, i.e., isothermal–isobaric ensemble. The time step in the MD simulation was 2 fs. It is worth noting that the purpose of the MD simulation here was to obtain the three explicit water molecules at reasonable locations, and the geometry of the model system constructed from the crystal structure should not be altered by the molecular force field, in case the molecular force field may not be appropriate for describing the bi-nuclear copper system. Therefore, in the energy-minimization and the MD simulation, only water molecules and the hydrogen atoms were allowed to move, and all remaining atoms were kept fixed at their crystal structure positions. The three water molecules that had closest contacts with the bridging ligand were selected from the last snapshot of the MD simulations. After that, the two states of the model system, i.e., the bridging ligand as water molecule or hydroxide anion, respectively, were subjected to the geometry optimizations at the B3LYP/6-31+G* level of theory with the SMD solvation method by using Gaussian 09 program. The optimized geometries were verified by harmonic normal mode calculations. The energies of the two states were further refined by single-point calculations at the B3LYP/6-311++G** level of theory. The free energy of proton in aqueous solution was calculated as:
1 M and 1atm refer to the standard states for the aqueous solution and gas phase, respectively. Ggas,H+ is the gas-phase free energy of the proton with the value of −6.28 kcal/mol at the 1 atm standard state and 298.15 K [27]. ΔG1atm→1 M is the free energy change from the standard state of 1 atm to 1 M, of which the value is 1.89 kcal/mol [33]. ΔGsolv,H+ is the hydration energy of proton at the 1 M standard state and 298.15 K, of which the value is −265.9 kcal/mol [25,33]. Therefore, the free energy of the proton in aqueous solution at the standard state of 1 M and 298.15 K is −270.3 kcal/mol. Finally, the free energy difference of the model system changing from bridging water to bridging hydroxide anion is calculated by Equation (6), which can be converted to pKa by Equation (7):
ΔGmodel(QM) = Gmodel(hydroxide anion) + Gaq,H+ − Gmodel(water)
3. Results and Discussion
3.1. Charge States of Copper Ions
The two copper ions in tyrosinase may take two possible charge states [11], i.e., the +1 formal charge corresponding to the singlet state, and the +2 formal charge corresponding to the triplet state. The two charge states of copper ions are denoted by Cu(I) and Cu(II), respectively. By combining these two charge states with the two possibilities of bridging ligands, we have four possible structures for the active site of tyrosinase, which were subjected to QM/MM optimizations at the B3LYP/6-31+G*:AMBER level of theory. Shown in Figure 4 are the comparisons of the four QM/MM-optimized structures to the crystal structure 2Y9W. We can see from Figure 4A that the bridging water molecule is unable to bind with the two Cu(I) ions. If the bridging ligand is hydroxide anion, as shown in Figure 4B, the bridging ligand may keep the binding with two Cu(I) ions. However, the distance between the right side copper ion and the epsilon nitrogen of His263 changes from 2.1 Å in the crystal structure to 3.3 Å in the QM/MM-optimized structure, indicating that the coordination of copper ion to His263 is lost. This is further confirmed by the fact that His295 forms a hydrogen bond with His263. Clearly, no matter what the bridging ligand is, the coordination of copper ions cannot be maintained when the two copper ions are at the singlet state, suggesting that the copper ions in the crystal structure 2Y9W are not Cu(I). On the other hand, as we can see from Figure 4C,D, the coordination of copper ions are maintained on both the bridging water and bridging hydroxide anion systems when the copper ions are at the triplet state. In addition, the hydrogen bond formed between side chains of His263 and His295 in Figure 4B, which is an indication that the coordination of the right side Cu(I) is disrupted, is not observed in Figure 4D, where copper ions are Cu(II). As a result, the copper in crystal structure 2Y9W should be Cu(II).
3.2. Determination of Bridging Ligand Identity
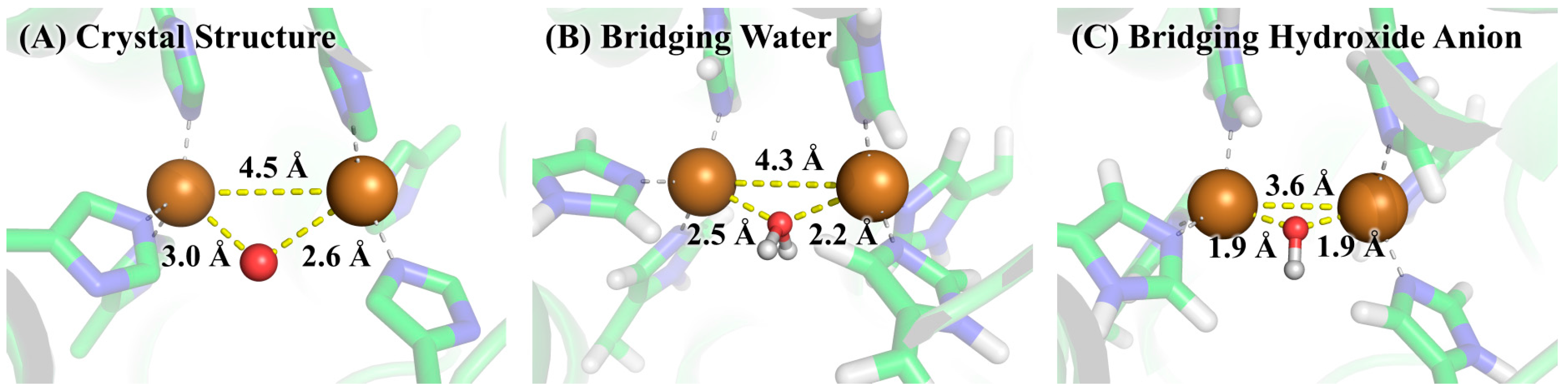
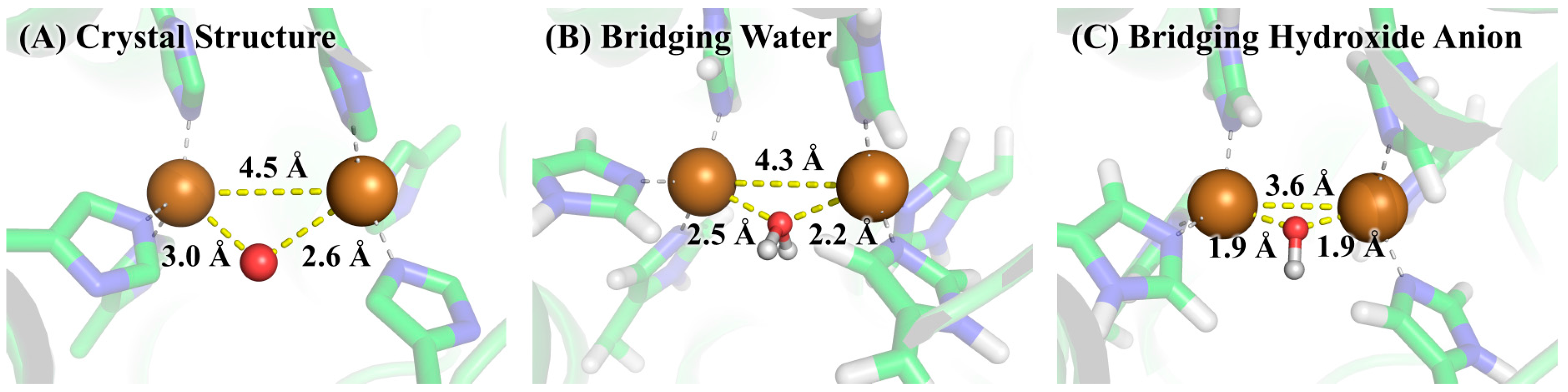
The free energy difference for the model system changing from the bridging water state to the bridging hydroxide state was calculated by the SMD solvation model with three explicit water molecules in the first and second solvation shells at the B3LYP/6-311++G**//B3LYP/6-31+G* level of theory. The optimized geometries of the two states of the model system are given in Figure 5. As can be seen from Table 1, the calculated ΔGmodel(QM) is 2.8 kcal/mol and the corresponding pKa is 2.1, indicating that the bridging ligand in the model system is hydroxide anion at a neutral pH condition. To determine the identity of the bridging oxygen in protein, we still need the values of ΔGprotein(QM/MM-PBSA) and ΔGmodel(QM/MM-PBSA) in terms of Equation (2). For this purpose, QM/MM-PBSA calculations were performed on both the protein and model systems at the B3LYP/6-311++G**//B3LYP/6-31+G* level of theory. Given in Table 2 are the calculated results for these two terms, whose values are 295.3 and 260.9 kcal/mol, respectively. According to Equation (2), the ΔG for determining the bridging ligand identity should be 37.1 kcal/mol, indicating that the identity of the bridging oxygen in protein must be a water molecule rather than a hydroxide anion. This conclusion can be further confirmed by the comparison between the structures in crystal structure and those in the QM/MM-optimized geometries, as shown in Figure 6. In crystal structure 2Y9W, the distance between the two copper ions is 4.5 Å. This distance is barely changed with the bridging ligand being a water molecule. However, if the bridging ligand is a hydroxide anion, the distance between the copper ions dramatically changes to 3.6 Å. In addition, the distances between the ions and the bridging oxygen are slightly altered from 3.0 and 2.6 Å to 2.6 and 2.3 Å, respectively, if the bridging ligand is a water molecule, whereas these two distances both significantly decrease to 1.9 Å if the bridging ligand is a hydroxide anion. Clearly, the QM/MM-optimized geometry of the active site with the bridging water is very close to that in the crystal structure, which is also consistent with the result from our QM/MM-PBSA free energy evaluations. As a result, the identity of the bridging ligand in the active site of tyrosinase is determined to be a water molecule.
3.3. Implication for the Design of Tyrosinase Inhibitors
A recent virtual screening study [34] showed that a tetrazole group was indispensable for the inhibitory activity against tyrosinase and one of the molecules with a tetrazole group exhibited the strongest inhibitory activity, although several other molecules with a carboxyl group were also subjected to the activity tests. This is a very interesting discovery, since the positive charged copper ions are also the binding sites to inhibitors and are supposed to have higher affinity to the negatively charged carboxyl group than to the neutral tetrazol group. As shown in Figure 6, after the bridging water in Figure 6B lost a hydrogen to become the hydroxide anion in Figure 6C, the bridging oxygen moves much closer to the copper ions, indicating much stronger electrostatic interactions between copper ions and hydroxide anion. This strong interaction also makes the two copper ions move dramatically closer to each other, and consequently generates an active site that is distinct from the crystal structure, which turns out to be energy unfavorable. Obviously, although molecules with negative charges may bind geometrically tighter with the copper ions, it causes the increase of the overall energy and thus is not favorable for the binding. Therefore, molecules with a carboxyl group should have lower binding affinity when compared to their neutral counterparts. Another example is of phthalic acid (PA) and cinnamic acid (CA), whose charges are −2 and −1 at pH 7 condition, respectively. Kinetic data on these two molecules show that PA has much lower inhibitory activity against tyrosinase than that of CA [35], suggesting that too many negative charges decrease the inhibitory activity. As a result, to design highly potent active inhibitors against tyrosinase, the molecule should have affinity to the copper ions as well as a neutral charge.
4. Conclusions
The present results obtained from QM/MM-PBSA calculations strongly support the conclusion that the bridging ligand in the active site of the crystal structure of Agaricus bisporus mushroom tyrosinase is a water molecule rather than a hydroxide anion. Although hydroxide anion binds geometrically tighter with the copper ions, it significantly changes the geometry of the active site of tyrosinase and causes an increase of the overall energy. The different binding behavior between water and hydroxide anion may explain why molecules with a carboxyl group or too many negative charges have lower inhibitory activity. In light of this, the design of highly potent active inhibitors against tyrosinase should satisfy both the affinity to the copper ions and the charge neutrality of the entire molecule.
Acknowledgments
This work was financially supported in part by the Yunnan Provincial Tobacco Monopoly Bureau Grants (2016YN28, 2017YN09), the Natural Science Foundation of Hubei Province of China Grants (2015CFB587), the Fundamental Research Funds for the Central Universities (2015QN160), the Yunnan Applied Basic Research Project (2017FB074), and the National Natural Science Foundation of China Grants (41601330, 21102050).
Author Contributions
J.L. conceived and designed the experiments; C.Z., W.H., G.Z., and X.W. performed the experiments; C.Z., W.H., and J.L. analyzed the data; X.H., Y.J., and J.Y.L. contributed materials preparation; J.L. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Olivares, C.; Solano, F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigment Cell Melanoma Res. 2009, 22, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M. Polyphenol oxidases in plants and fungi: Going places? A review. Phytochemistry 2006, 67, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Rosell, G. Biorational Approaches for Insect Control by Enzymatic Inhibition. Curr. Med. Chem. 2005, 12, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Gandia-Herrero, F.; Jimenez, M.; Cabanes, J.; Garcia-Carmona, F.; Escribano, J. Tyrosinase inhibitory activity of cucumber compounds: Enzymes responsible for browning in cucumber. J. Agric. Food. Chem. 2003, 51, 7764–7769. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Chaki, M.; Sengupta, M. Tyrosinase and ocular diseases: Some novel thoughts on the molecular basis of oculocutaneous albinism type 1. Prog. Retin. Eye Res. 2007, 26, 323–358. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Ni-hang, L.; Hong, L.; Huan-wen, Y. The Influence of Polyphenol Oxidase Inhibitors to the Fluecured Tobacco Polyphenol During Curing. J. Yunnan Agric. Univ. 2007, 1, 015. [Google Scholar]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Berthon, J.Y. Resveratrol: An original mechanism on tyrosinase inhibition. Int. J. Cosmet. Sci. 2000, 22, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Casanola-Martin, G.; Le-Thi-Thu, H.; Marrero-Ponce, Y.; Castillo-Garit, J.; Torrens, F.; Rescigno, A.; Abad, C.; Khan, M. Tyrosinase Enzyme: 1. An Overview on a Pharmacological Target. Curr. Top. Med. Chem. 2014, 14, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonson, T.; Carlsson, J.; Case, D.A. Proton binding to proteins: pKa calculations with explicit and implicit solvent models. J. Am. Chem. Soc. 2004, 126, 4167–4180. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sun, B.; Yao, Y.; Liu, J. Fast and Reliable Thermodynamic Approach for Determining the Protonation State of the Asp Dyad. J. Chem. Inf. Model. 2017, 57, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, M.; Soderhjelm, P.; Heimdal, J.; Ryde, U. QM/MM-PBSA method to estimate free energies for reactions in proteins. J. Phys. Chem. B 2008, 112, 12537–12548. [Google Scholar] [CrossRef] [PubMed]
- Tsitsanou, K.E.; Hayes, J.M.; Keramioti, M.; Mamais, M.; Oikonomakos, N.G.; Kato, A.; Leonidas, D.D.; Zographos, S.E. Sourcing the affinity of flavonoids for the glycogen phosphorylase inhibitor site via crystallography, kinetics and QM/MM-PBSA binding studies: Comparison of chrysin and flavopiridol. Food Chem. Toxicol. 2013, 61, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Manta, S.; Xipnitou, A.; Kiritsis, C.; Kantsadi, A.L.; Hayes, J.M.; Skamnaki, V.T.; Lamprakis, C.; Kontou, M.; Zoumpoulakis, P.; Zographos, S.E. 3′-Axial CH2OH Substitution on Glucopyranose does not Increase Glycogen Phosphorylase Inhibitory Potency. QM/MM-PBSA Calculations Suggest Why. Chem. Biol. Drug. Des. 2012, 79, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Ryde, U.; Soderhjelm, P. Ligand-Binding Affinity Estimates Supported by Quantum-Mechanical Methods. Chem. Rev. 2016, 116, 5520–5566. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Caldararu, O.; Ryde, U. Protonation States of Homocitrate and Nearby Residues in Nitrogenase Studied by Computational Methods and Quantum Refinement. J. Phys. Chem. B 2017, 121, 8242–8262. [Google Scholar] [CrossRef] [PubMed]
- Chipman, D.M.; Chen, F.W. Cation electric field is related to hydration energy. J. Chem. Phys. 2006, 124. [Google Scholar] [CrossRef] [PubMed]
- Chipman, D.M. Anion electric field is related to hydration energy. J. Chem. Phys. 2003, 118, 9937–9942. [Google Scholar] [CrossRef]
- Liu, J.; Kelly, C.P.; Goren, A.C.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G.; Zhan, C.-G. Free Energies of Solvation with Surface, Volume, and Local Electrostatic Effects and Atomic Surface Tensions to Represent the First Solvation Shell. J. Chem. Theory Comput. 2010, 6, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Aqueous solvation free energies of ions and ion-water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton. J. Phys. Chem. B 2006, 110, 16066–16081. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. SM6: A Density Functional Theory Continuum Solvation Model for Calculating Aqueous Solvation Free Energies of Neutrals, Ions, and Solute-Water Clusters. J. Chem. Theory Comput. 2005, 1, 1133–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.G.; Dixon, D.A. Absolute hydration free energy of the proton from first-principles electronic structure calculations. J. Phys. Chem. A 2001, 105, 11534–11540. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Thapa, B.; Schlegel, H.B. Improved pKa Prediction of Substituted Alcohols, Phenols, and Hydroperoxides in Aqueous Medium Using Density Functional Theory and a Cluster-Continuum Solvation Model. J. Phys. Chem. A 2017, 121, 4698–4706. [Google Scholar] [CrossRef] [PubMed]
- Senn, H.M.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. Engl. 2009, 48, 1198–1229. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Betz, R.M.; Botello-Smith, W.; Cerutti, D.S.; Cheatham, I.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Liu, D.C.; Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef]
- Cieplak, P.; Cornell, W.D.; Bayly, C.; Kollman, P.A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. [Google Scholar] [CrossRef]
- Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A. Calculation of solvation free energies of charged solutes using mixed cluster/continuum models. J. Phys. Chem. B 2008, 112, 9709–9719. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Choi, K.-E.; Park, S.J.; Kim, S.Y.; Jee, J.-G. Ensemble-Based Virtual Screening Led to the Discovery of New Classes of Potent Tyrosinase Inhibitors. J. Chem. Inf. Model. 2016, 56, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Hassani, S.; Haghbeen, K.; Fazli, M. Non-specific binding sites help to explain mixed inhibition in mushroom tyrosinase activities. Eur. J. Med. Chem. 2016, 122, 138–148. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
Figure 1.
The active site of the crystal structure of Agaricus bisporus mushroom tyrosinase (Protein Data Bank, PDB ID 2Y9W). The side chains of histidine residues are rendered as sticks, while copper ions and bridging oxygen are rendered as spheres. The mode system in the present study is defined by the atoms in stick and sphere representations. The carbon, nitrogen, oxygen, and copper atoms are colored with green, blue, red, and brown, respectively.
Figure 1.
The active site of the crystal structure of Agaricus bisporus mushroom tyrosinase (Protein Data Bank, PDB ID 2Y9W). The side chains of histidine residues are rendered as sticks, while copper ions and bridging oxygen are rendered as spheres. The mode system in the present study is defined by the atoms in stick and sphere representations. The carbon, nitrogen, oxygen, and copper atoms are colored with green, blue, red, and brown, respectively.
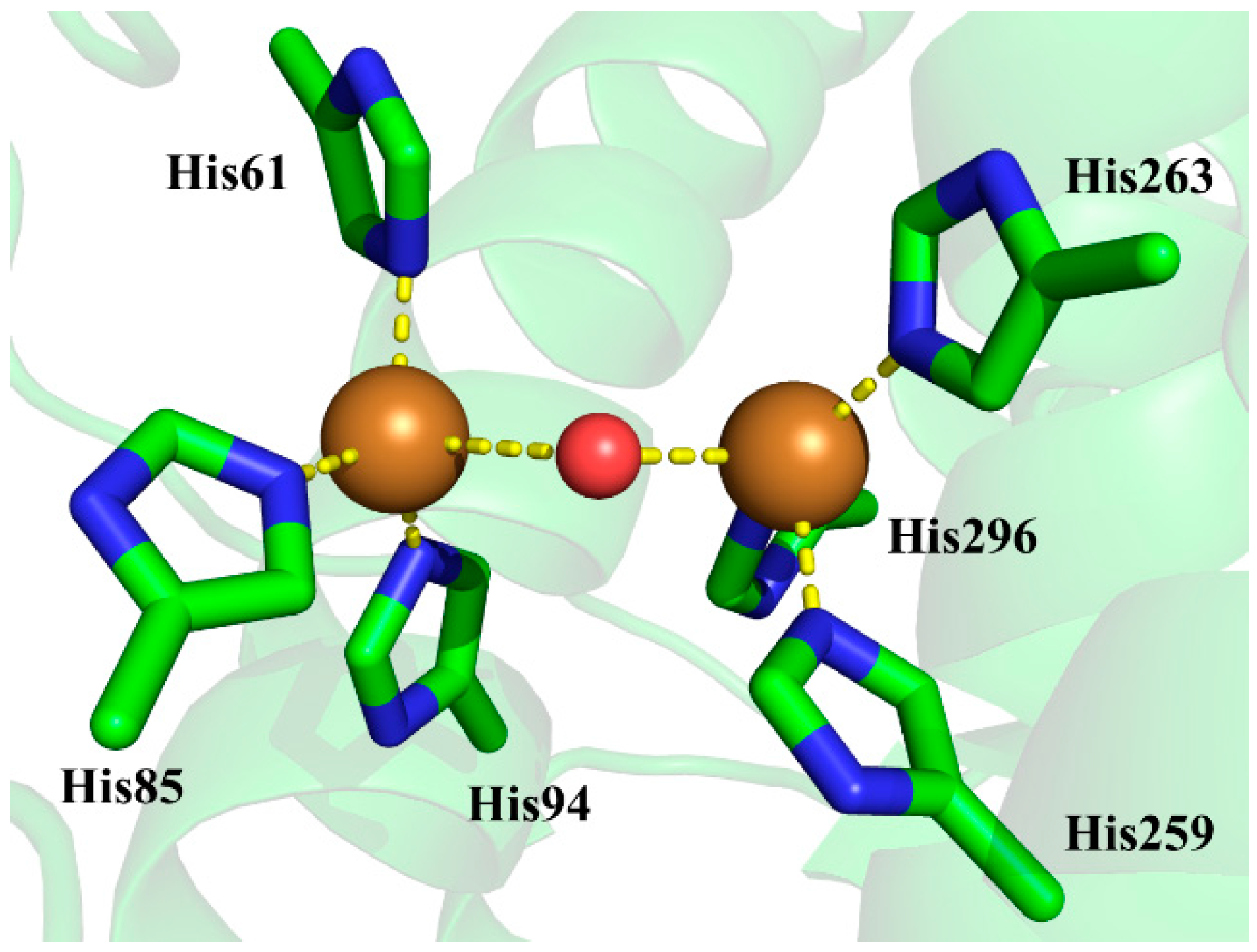
Figure 2.
Thermodynamic cycle for determining the pKa of aspartic residue in protein. The double free energy difference ΔΔG is for pKa shifts analysis.
Figure 2.
Thermodynamic cycle for determining the pKa of aspartic residue in protein. The double free energy difference ΔΔG is for pKa shifts analysis.
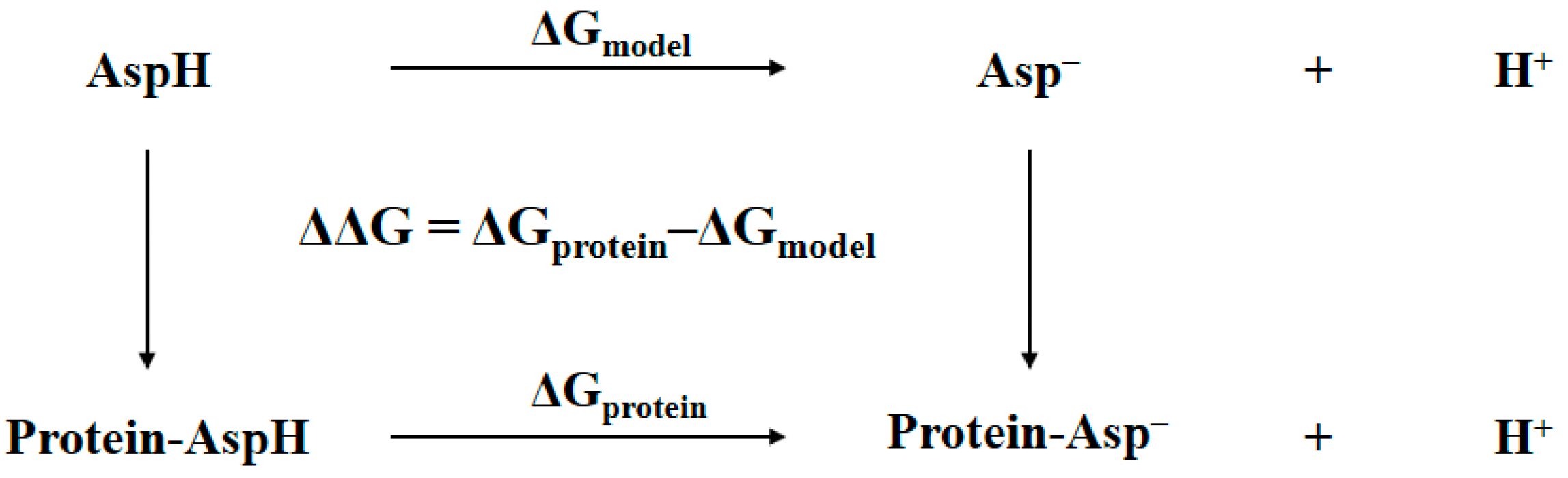
Figure 3.
Thermodynamic cycle for determining the identity of bridging oxygen, i.e., water or hydroxide anion, in the active site of tyrosinase.
Figure 3.
Thermodynamic cycle for determining the identity of bridging oxygen, i.e., water or hydroxide anion, in the active site of tyrosinase.
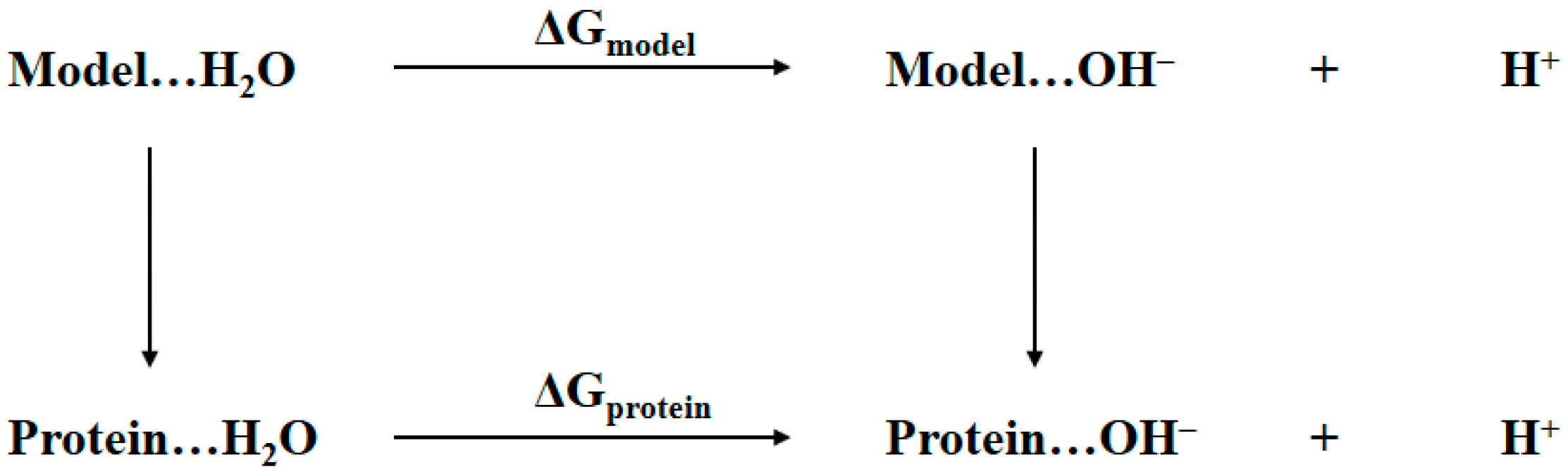
Figure 4.
The comparison of four possible QM/MM-optimized active site structures to that of crystal structure 2Y9W. The bridging ligand is water in (A,C); and is hydroxide anion in (B,D). The copper ions are Cu(I) in (A,B); and are Cu(II) in (C,D). For crystal structure 2Y9W, the carbon, nitrogen, oxygen, and copper atoms are colored with green, blue, red, and brown, respectively, whereas for the QM/MM-optimized structures these atoms are colored with magenta, blue, cyan, and yellow, respectively. For clarity, the copper ions and the bridging oxygen are scaled to small spheres.
Figure 4.
The comparison of four possible QM/MM-optimized active site structures to that of crystal structure 2Y9W. The bridging ligand is water in (A,C); and is hydroxide anion in (B,D). The copper ions are Cu(I) in (A,B); and are Cu(II) in (C,D). For crystal structure 2Y9W, the carbon, nitrogen, oxygen, and copper atoms are colored with green, blue, red, and brown, respectively, whereas for the QM/MM-optimized structures these atoms are colored with magenta, blue, cyan, and yellow, respectively. For clarity, the copper ions and the bridging oxygen are scaled to small spheres.

Figure 5.
The optimized geometries for the two states of the model system at the B3LYP/6-31+G* level of theory. (A) The bridging ligand is water; (B) The bridging ligand is hydroxide anion. Three water molecules in the first and second solvation shells of the bridging ligand were added explicitly for reliable solvation effect evaluation as well as for obtaining comparable energies.
Figure 5.
The optimized geometries for the two states of the model system at the B3LYP/6-31+G* level of theory. (A) The bridging ligand is water; (B) The bridging ligand is hydroxide anion. Three water molecules in the first and second solvation shells of the bridging ligand were added explicitly for reliable solvation effect evaluation as well as for obtaining comparable energies.
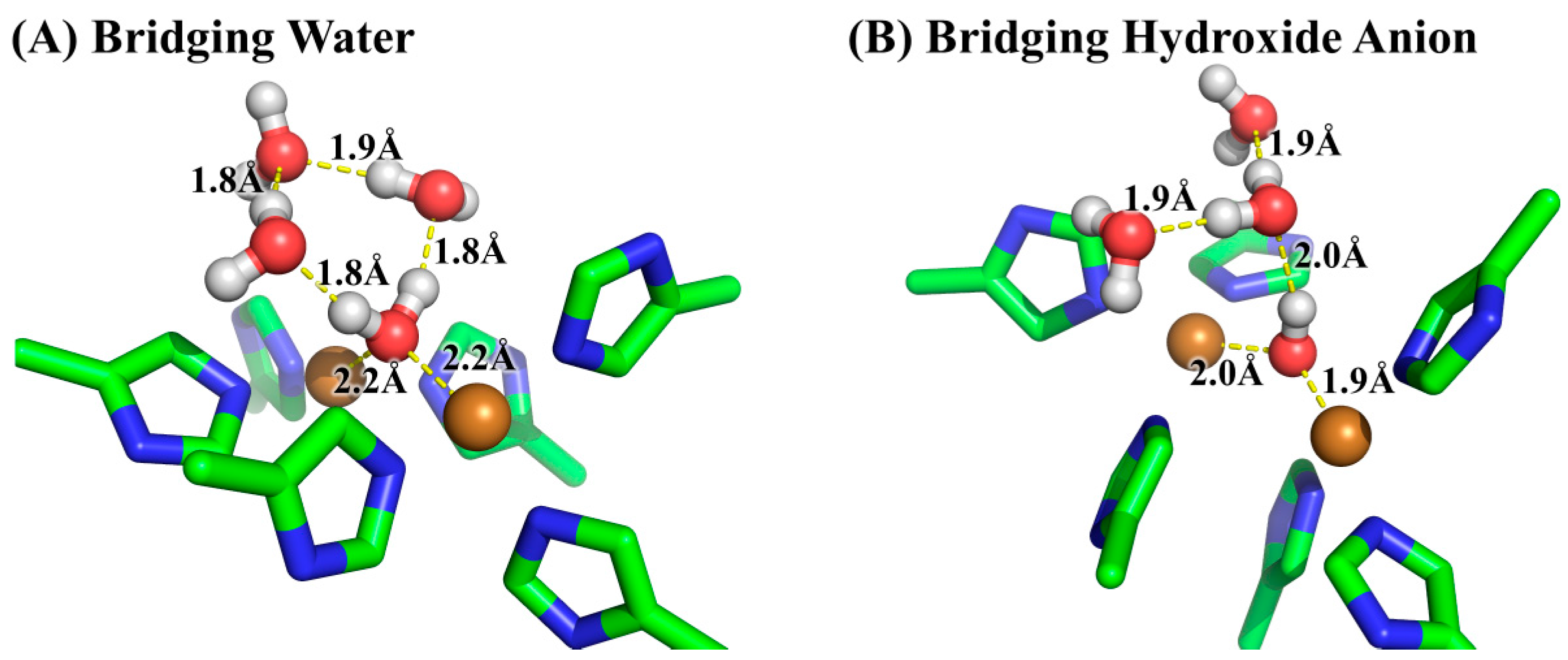
Figure 6.
The active site of tyrosinase in (A) crystal structure 2Y9W; (B) QM/MM-optimized geometry with bridging water; and (C) QM/MM-optimized geometry with bridging hydroxide anion. The geometry optimizations were performed by using the QM/MM method at the B3LYP/6-31+G*: AMBER level of theory.
Figure 6.
The active site of tyrosinase in (A) crystal structure 2Y9W; (B) QM/MM-optimized geometry with bridging water; and (C) QM/MM-optimized geometry with bridging hydroxide anion. The geometry optimizations were performed by using the QM/MM method at the B3LYP/6-31+G*: AMBER level of theory.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated value of ΔGmodel(QM) a.
| EQM b | dGQM c | GQM d | |
|---|---|---|---|
| water | −3,250,650.3 | 389.7 | −3,250,260.6 |
| hydroxide anion | −3,250,365.7 | 378.2 | −3,249,985.3 |
| proton | N/A | N/A | −270.3 e |
a ΔGmodel(QM) = Gmodel(hydroxide anion) + Gaq,H+ − Gmodel(water) = 2.8 kcal/mol. All energies are in the unit of kcal/mol and are calculated at the B3LYP/6-311++G**//B3LYP/6-31+G* level of theory; b EQM is the electronic energy; c dGQM is the thermal correction to Gibbs free energy; d GQM is the Gibbs free energy of each species and is calculated as the sum of EQM and dGQM; e the Gibbs free energy of the proton in aqueous solution is calculated according to Equation (5).
Table 2.
Calculated values of ΔGprotein(QM/MM-PBSA) and ΔGmodel(QM/MM-PBSA) a.
| EQM/MM b | dGQM/MM c | EPBSA d | GQM/MM-PBSA e | ΔG(QM/MM-PBSA) f | ||
|---|---|---|---|---|---|---|
| protein | water | −3,111,906.7 | 354.9 | −4967.8 | −3,116,519.6 | 295.3 |
| hydroxide anion | −3,111,396.4 | 357.4 | −5185.3 | −3,116,224.3 | ||
| model | water | −3,105,710.4 | 350.1 | −463.6 | −3,105,823.9 | 260.9 |
| hydroxide anion | −3,105,646.3 | 344.3 | −260.9 | −3,105,563.0 |
a all energies are in the unit of kcal/mol and are calculated at the B3LYP/6-31+G*:AMBER level of theory; b EQM/MM is the electronic energy; c dGQM is the thermal correction to Gibbs free energy for the QM system with embed MM background charges; d EPBSA is the solvation energy calculated by the PBSA method; e GQM/MM-PBSA is the Gibbs free energy of each species and is calculated as the sum of EQM/MM, dGQM/MM, and EPBSA; f ΔG(QM/MM-PBSA) is calculated as Ghydroxide anion(QM/MM-PBSA) − Gwater(QM/MM-PBSA).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zou, C.; Huang, W.; Zhao, G.; Wan, X.; Hu, X.; Jin, Y.; Li, J.; Liu, J. Determination of the Bridging Ligand in the Active Site of Tyrosinase. Molecules 2017, 22, 1836. https://doi.org/10.3390/molecules22111836
AMA Style
Zou C, Huang W, Zhao G, Wan X, Hu X, Jin Y, Li J, Liu J. Determination of the Bridging Ligand in the Active Site of Tyrosinase. Molecules. 2017; 22(11):1836. https://doi.org/10.3390/molecules22111836
Chicago/Turabian StyleZou, Congming, Wei Huang, Gaokun Zhao, Xiao Wan, Xiaodong Hu, Yan Jin, Junying Li, and Junjun Liu. 2017. "Determination of the Bridging Ligand in the Active Site of Tyrosinase" Molecules 22, no. 11: 1836. https://doi.org/10.3390/molecules22111836