
Preparation of Photoirradiation Molecular Imprinting Polymer for Selective Separation of Branched Cyclodextrins

Abstract
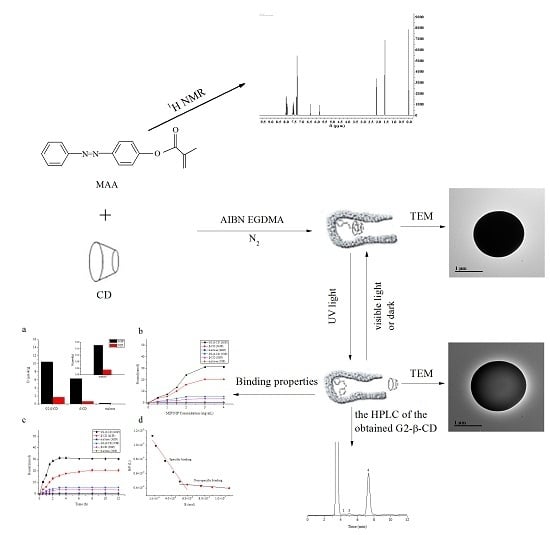
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Characterization of the 4-(Phenyldiazenyl)phenol and MAA
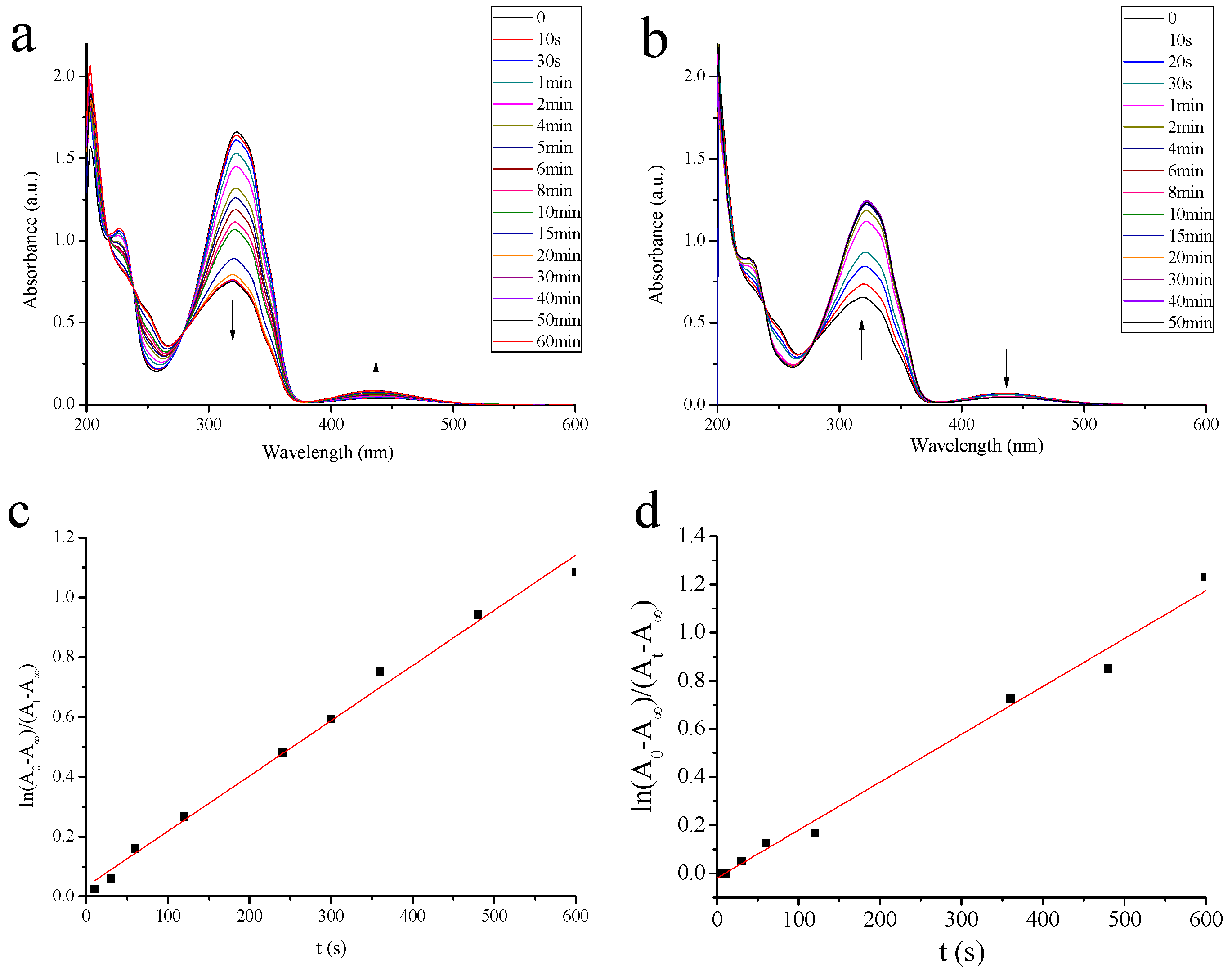
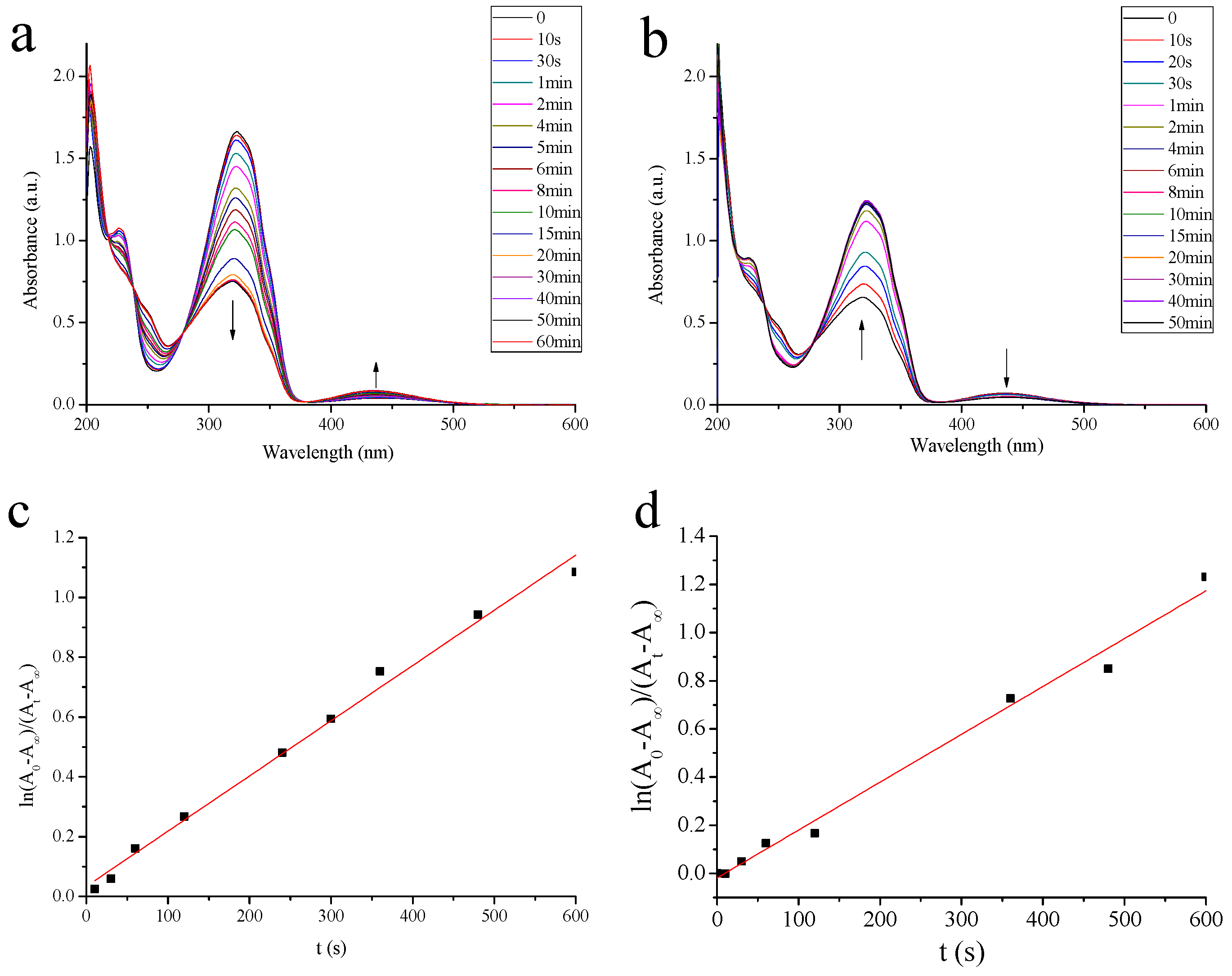
2.1.1. Photoisomerization Properties of MAA
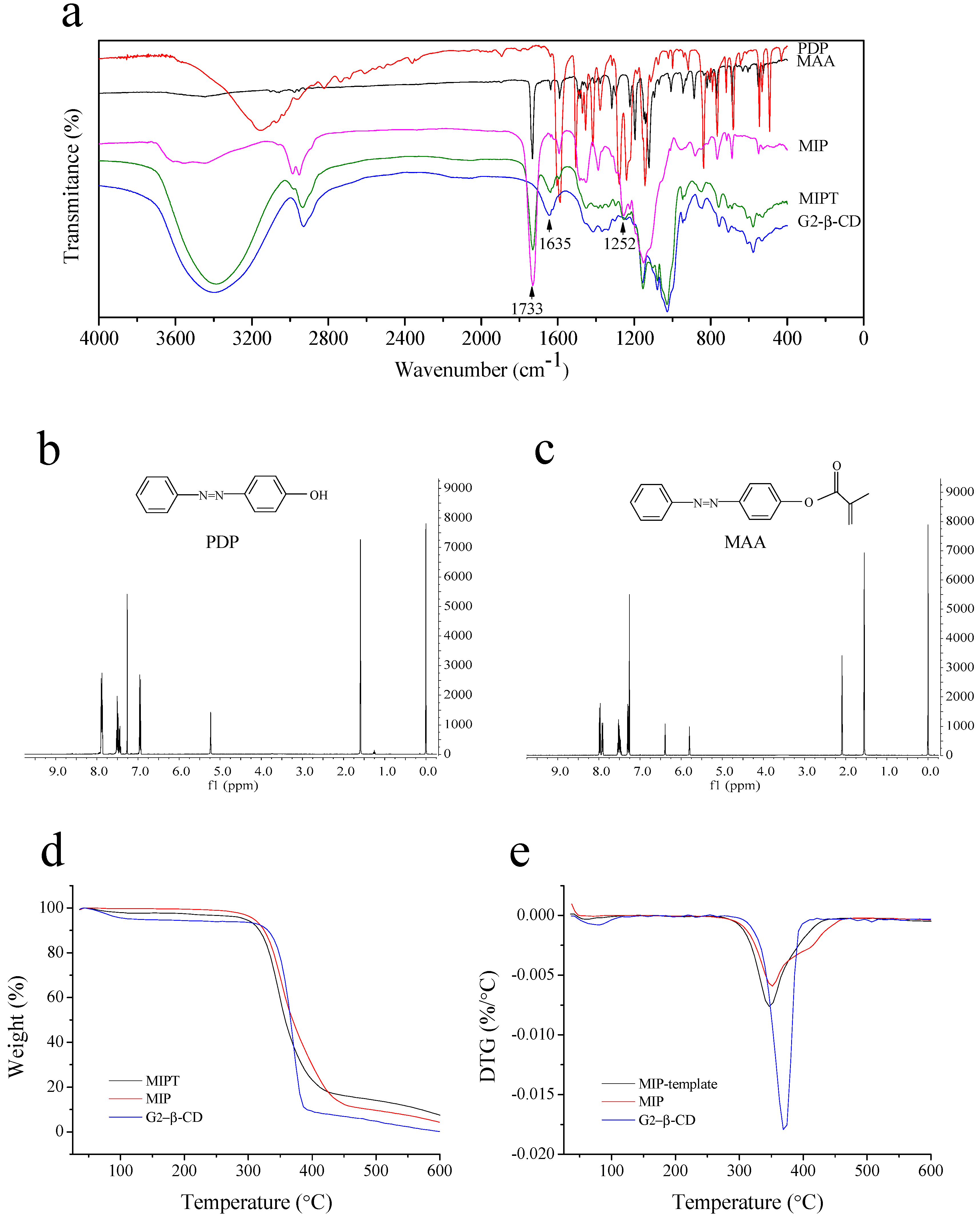
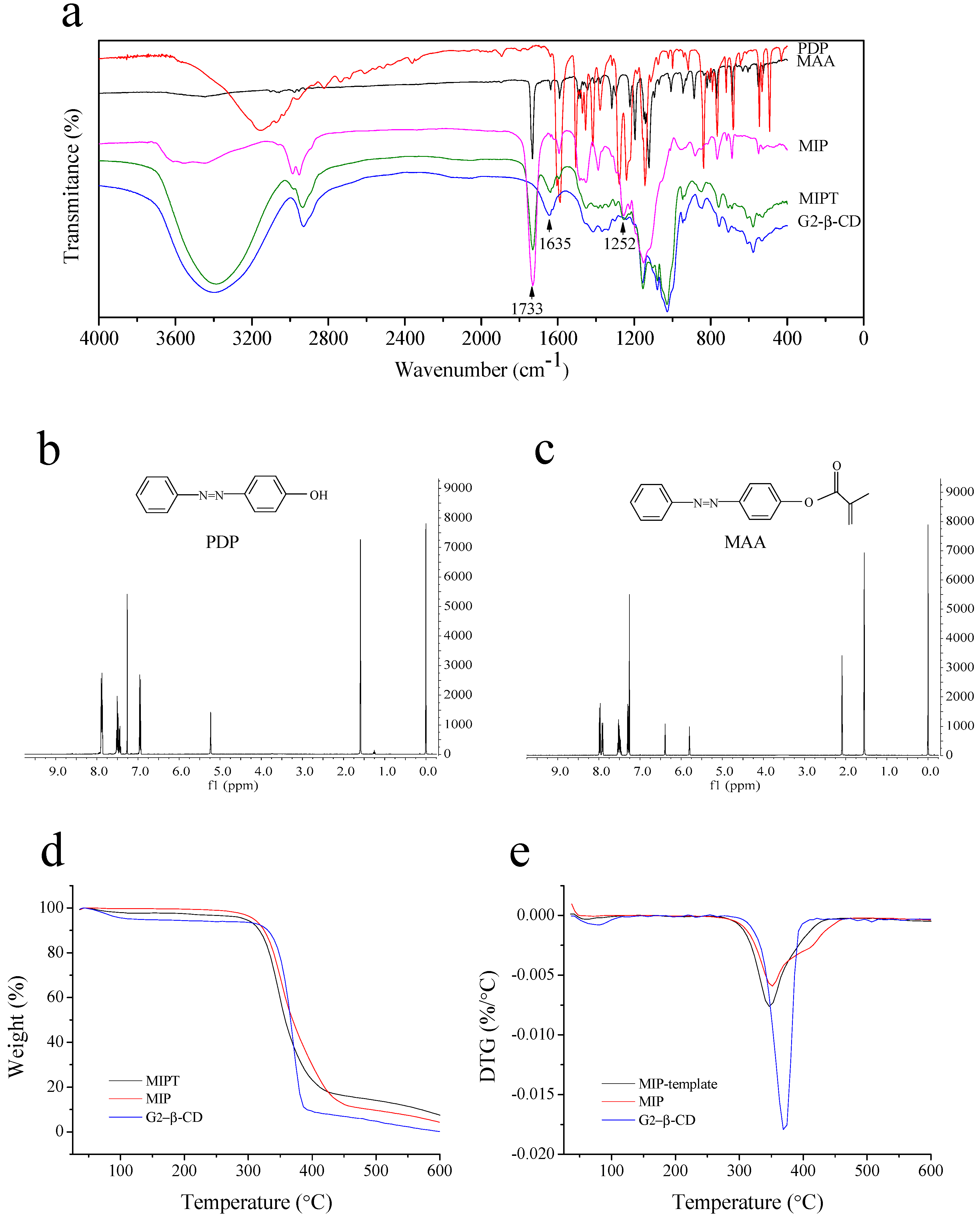
2.1.2. FTIR and 1H-NMR Spectroscopy of PDP and MAA
2.2. Characterization of MIPT and MIP
2.2.1. Morphology of Samples
2.2.2. FTIR Spectroscopy
2.2.3. Thermogravimetric Analysis
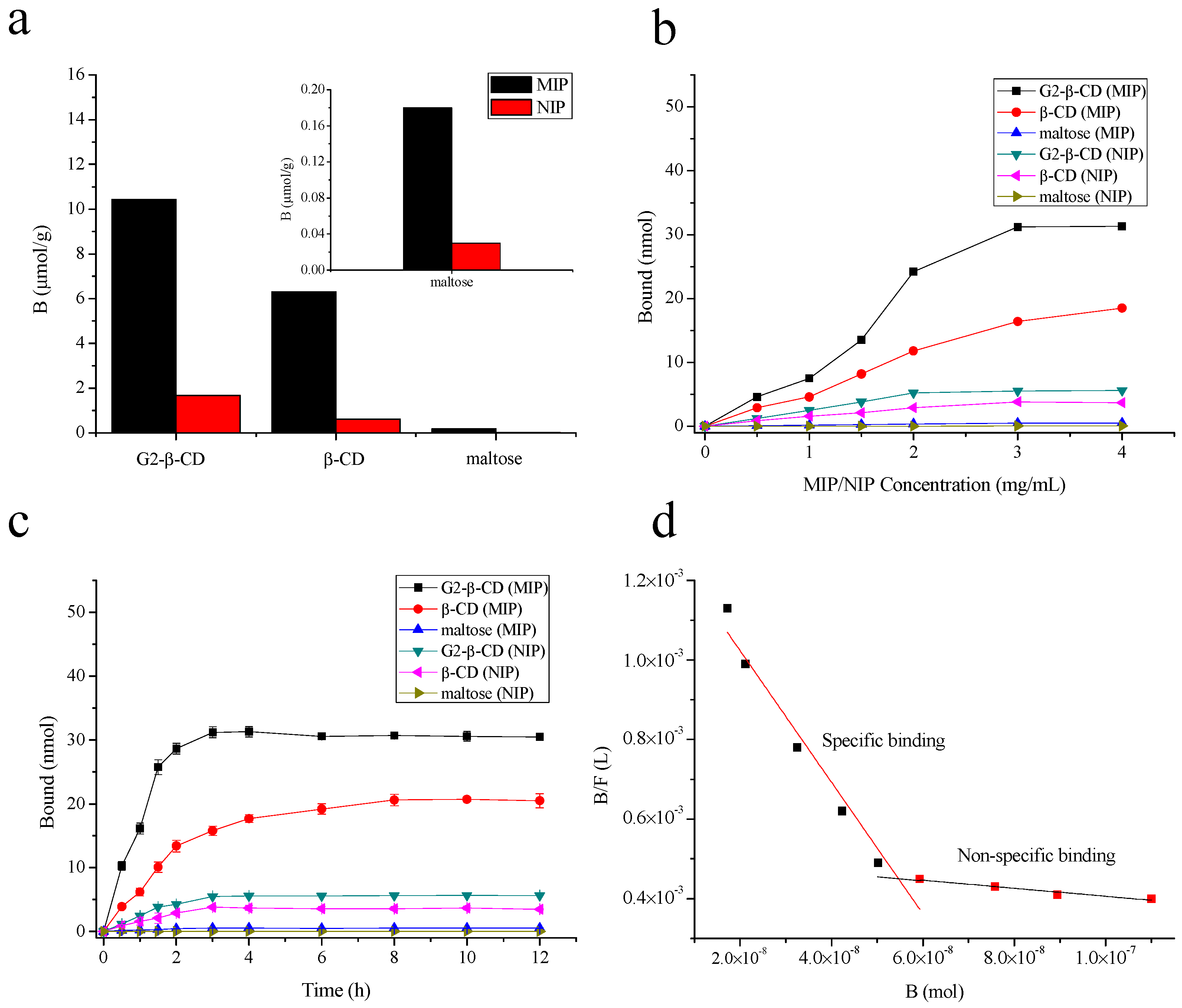
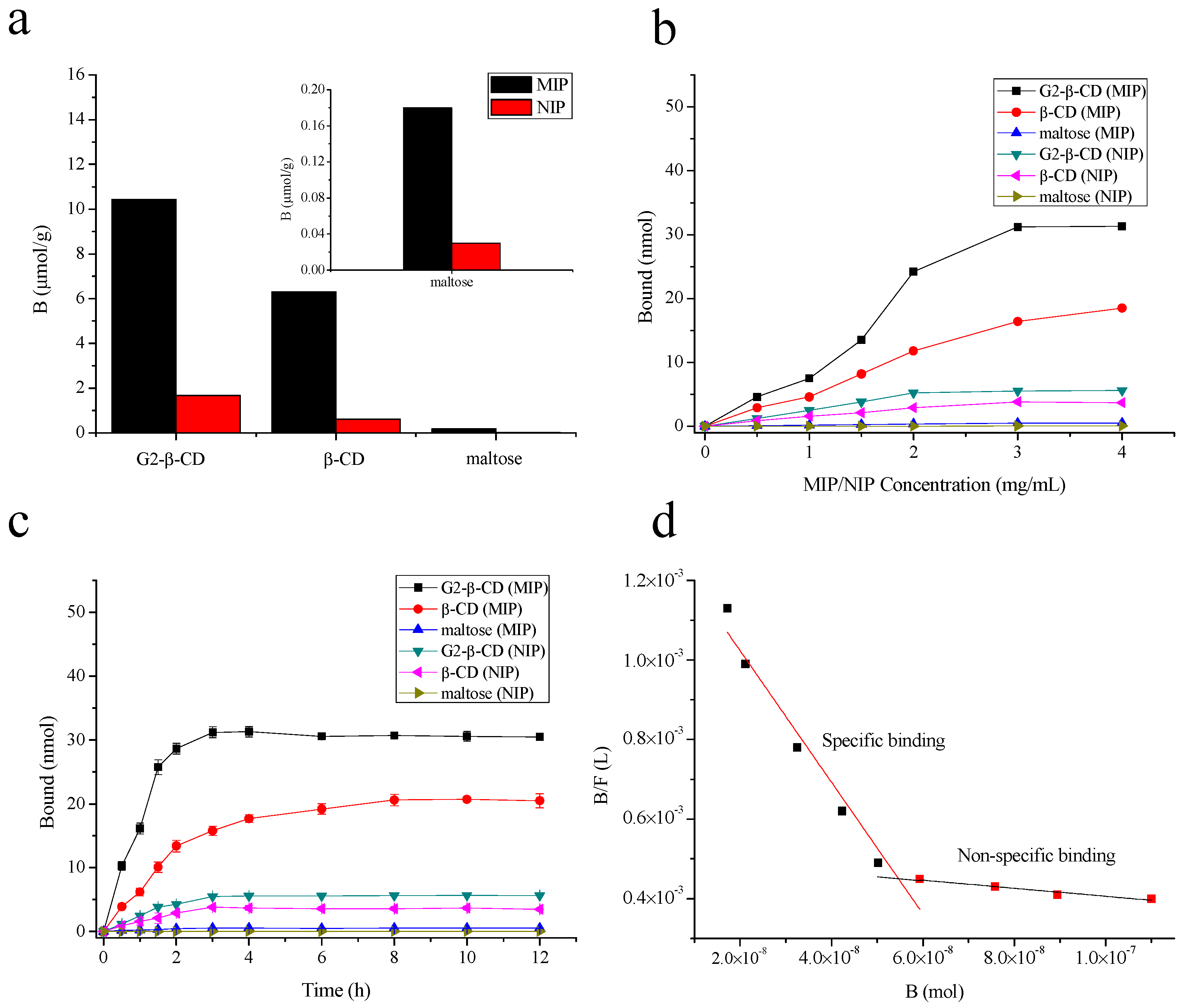
2.3. MIP and NIP Binding Properties
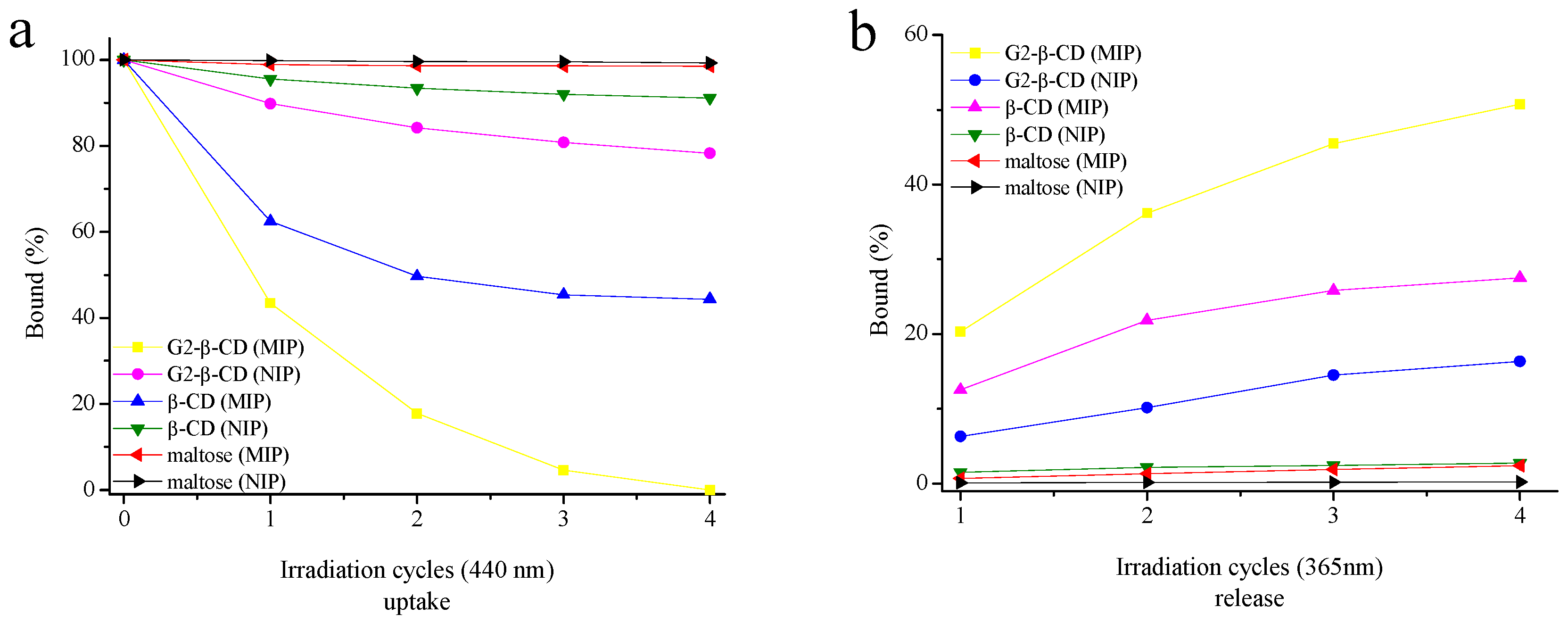
2.4. Photoregulated Release and Uptake of G2-β-CD and Maltose by the MIP
3. Materials and Methods
3.1. Materials
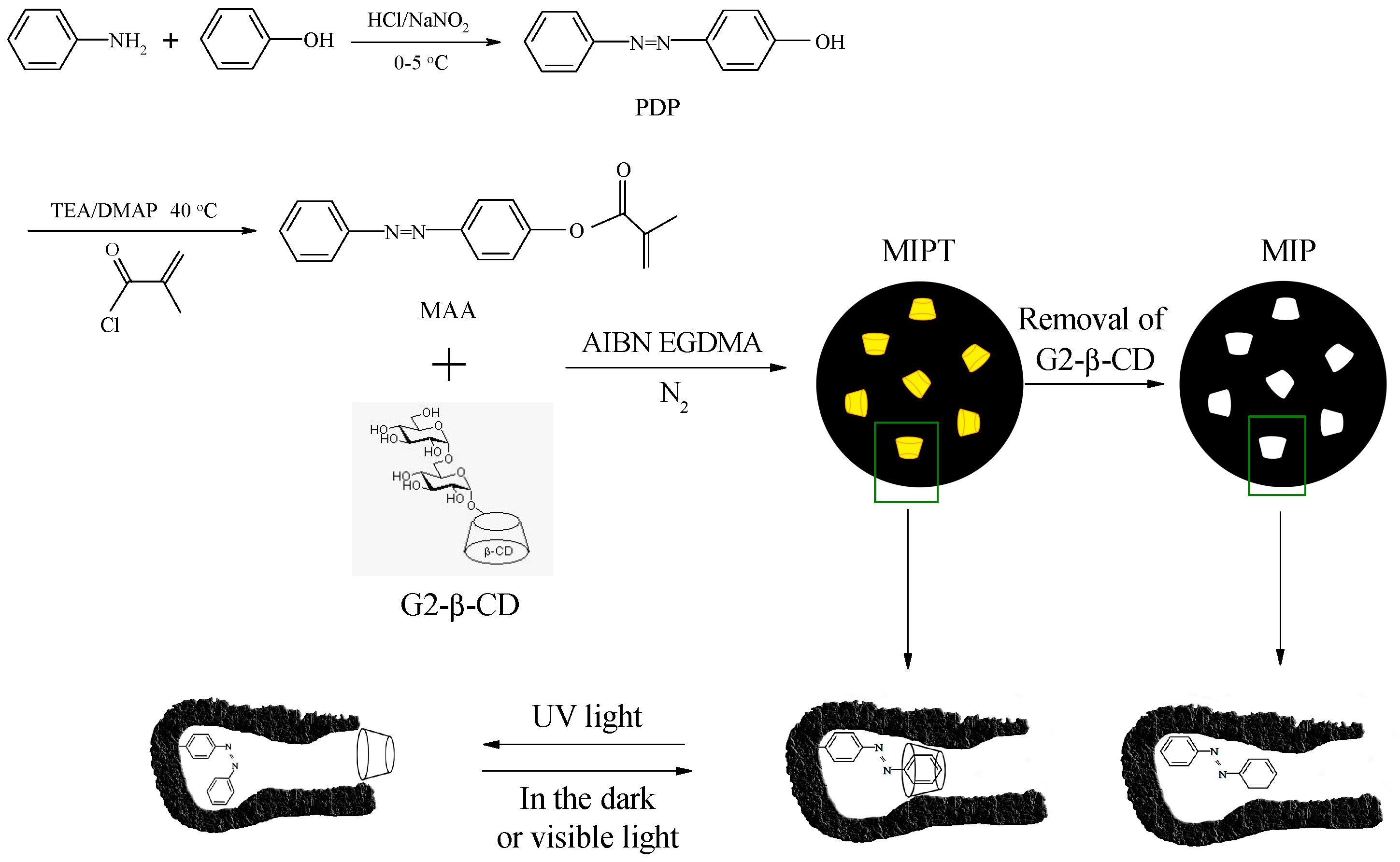
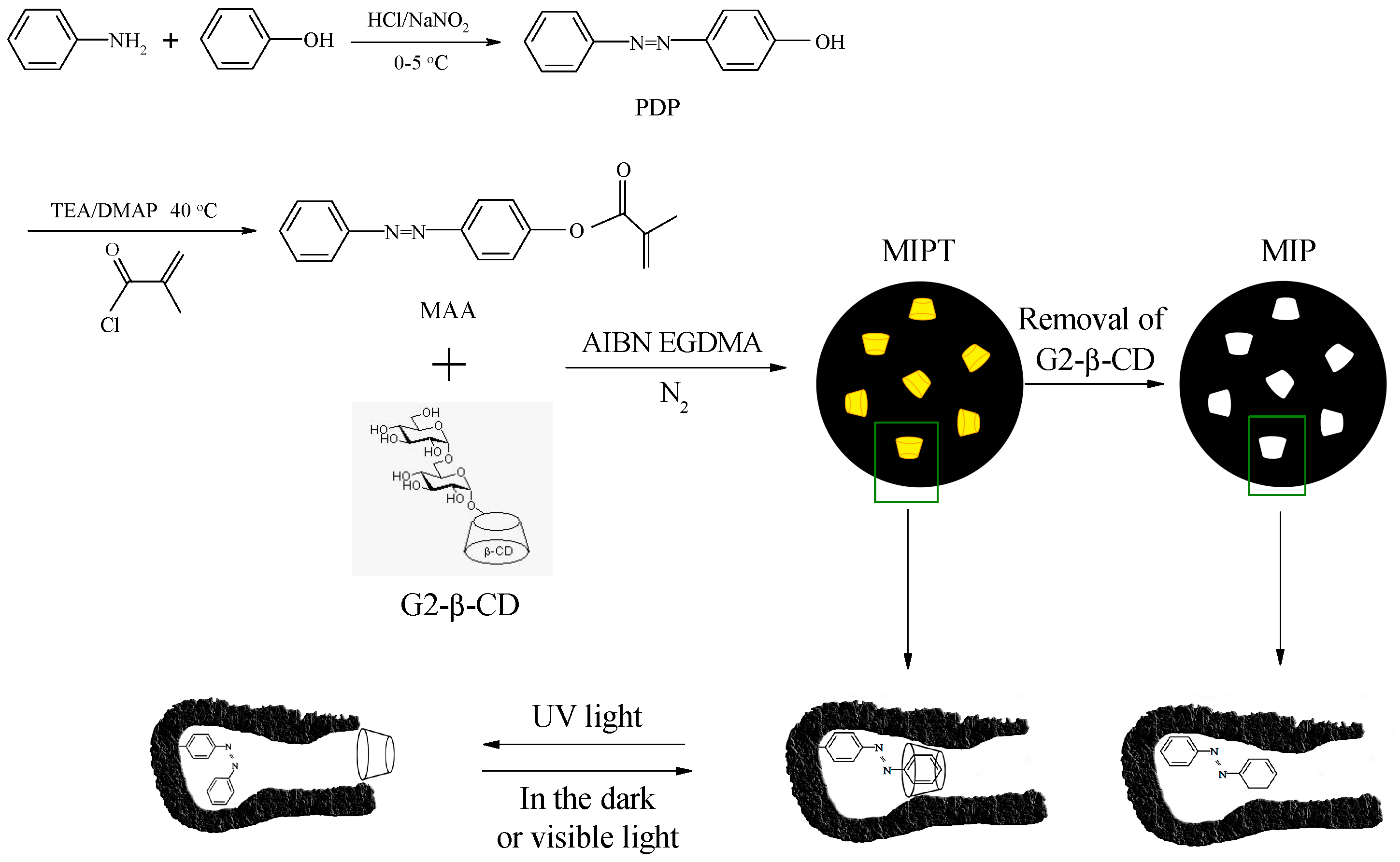
3.2. Synthesis of 4-(Phenyldiazenyl)phenol
3.3. Synthesis of 4-Methacryloyloxy Azobenzene
3.4. Preparation of the MIP
3.5. Characterization of PDP and MAA
3.6. Photoisomerization Studies
3.7. Scanning Electron Microscopy and Transmission Electron Microscopy
3.8. Fourier Transform Infrared Spectroscopy
3.9. Thermogravimetric Analysis
3.10. Binding Selectivity
3.11. Equilibrium Binding Experiments
3.12. Binding Kinetics
3.13. Binding Isotherm
3.14. Photocontrolled Uptake and Release
3.15. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tavornvipas, S.; Arima, H.; Hirayama, F.; Uekama, K.; Ishiguro, T.; Oka, M.; Hamayasu, K.; Hashimoto, H. Some Pharmaceutical Properties of a New Branched Cyclodextrin, 6-O-α-(4-O-α-d-Glucuronyl)-d-glucosylβ-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 391–394. [Google Scholar] [CrossRef]
- Koizumi, K.; Utamura, T.; Sato, M.; Yagi, Y. Isolation and characterization of branched cyclodextrins. Carbohydr. Res. 1986, 153, 55–67. [Google Scholar] [CrossRef]
- Abe, J.-I.; Mizowaki, N.; Hizukuri, S.; Koizumi, K.; Utamura, T. Synthesis of branched cyclomalto-oligosaccharides using Pseudomonas isoamylase. Carbohydr. Res. 1986, 154, 81–92. [Google Scholar] [CrossRef]
- Watanabe, N.; Yamamoto, K.; Tsuzuki, W.; Oya, T.; Kobayashi, S. A novel method to produce branched α-cyclodextrins: Pullulanase-glucoamylase-mixed method. J. Ferment. Bioeng. 1997, 83, 43–47. [Google Scholar] [CrossRef]
- Ammeraal, R.N.; Hedges, A.R.; Gottneid, D.J. Purification and Separation of Branched Beta-Cyclodextrins. U.S. Patent 4840679 A, 8 July 1988. [Google Scholar]
- Lammers, J.N.J.J. Reproducible separation of α- and β-cyclodextrin on charcoal columns. J. Chromatogr. A 1969, 41, 462–466. [Google Scholar] [CrossRef]
- Haupt, K.; Mosbach, K. Molecularly imprinted polymers and their use in biomimetic sensors. Chem. Rev. 2000, 100, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Andersson, H.S.; Andersson, L.I.; Ansell, R.J.; Kirsch, N.; Nicholls, I.A.; O’Mahony, J.; Whitcombe, M.J. Molecular imprinting science and technology: A survey of the literature for the years up to and including 2003. J. Mol. Recognit. 2006, 19, 106–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ye, L.; Mosbach, K. Non-covalent molecular imprinting with emphasis on its application in separation and drug development. J. Mol. Recognit. 2006, 19, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Enzyme-like catalysis by molecularly imprinted polymers. Chem. Rev. 2002, 102, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Hilt, J.Z.; Byrne, M.E. Configurational biomimesis in drug delivery: Molecular imprinting of biologically significant molecules. Adv. Drug Deliv. Rev. 2004, 56, 1599–1620. [Google Scholar] [CrossRef] [PubMed]
- Sellergren, B. Molecularly imprinted polymers: Shaping enzyme inhibitors. Nat. Chem. 2010, 2, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Mosbach, K. Molecular imprinting: Synthetic materials as substitutes for biological antibodies and receptors. Chem. Mater. 2008, 20, 859–868. [Google Scholar] [CrossRef]
- Kanazawa, H. Thermally responsive chromatographic materials using functional polymers. J. Sep. Sci. 2007, 30, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Suksuwan, A.; Lomlim, L.; Rungrotmongkol, T.; Nakpheng, T.; Dickert, F.L.; Suedee, R. The composite nanomaterials containing (R)-thalidomide-molecularly imprinted polymers as a recognition system for enantioselective-controlled release and targeted drug delivery. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Wackerlig, J.; Lieberzeit, P.A. Molecularly imprinted polymer nanoparticles in chemical sensing—Synthesis, characterisation and application. Sens. Actuators B Chem. 2015, 207, 144–157. [Google Scholar] [CrossRef]
- Takeuchi, T.; Akeda, K.; Murakami, S.; Shinmori, H.; Inoue, S.; Lee, W.S.; Hishiya, T. Photoresponsive porphyrin-imprinted polymers prepared using a novel functional monomer having diaminopyridine and azobenzene moieties. Org. Biomol. Chem. 2007, 5, 2368–2374. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Lam, M.H.W.; Yu, H. The fabrication of a photoresponsive molecularly imprinted polymer for the photoregulated uptake and release of caffeine. Adv. Funct. Mater. 2006, 16, 1759–1767. [Google Scholar] [CrossRef]
- Gong, C.; Wong, K.-L.; Lam, M.H.-W. Photoresponsive molecularly imprinted hydrogels for the photoregulated release and uptake of pharmaceuticals in the aqueous media. Chem. Mater. 2008, 20, 1353–1358. [Google Scholar] [CrossRef]
- Gomy, C.; Schmitzer, A.R. Synthesis and photoresponsive properties of a molecularly imprinted polymer. Org. Lett. 2007, 9, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Nakashima, K.; Arahira, M. Production and some properties of branched cyclo-malto-oligosaccharides. Carbohydr. Res. 1989, 192, 223–231. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Rai, A.K. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Sakano, Y.; Sano, M.; Kobayashi, T. Preparation and Enzymatic Hydrolysis of Maltosyl-α-cyclodextrin. Agric. Biol. Chem. 1985, 49, 3391–3398. [Google Scholar] [CrossRef]
- Tang, Q.; Nie, Y.-T.; Gong, C.-B.; Chow, C.-F.; Peng, J.-D.; Hon-Wah Lam, M. Photo-responsive molecularly imprinted hydrogels for the detection of melamine in aqueous media. J. Mater. Chem. 2012, 22, 19812–19820. [Google Scholar] [CrossRef]
- Wang, G.; Tong, X.; Zhao, Y. Preparation of azobenzene-containing amphiphilic diblock copolymers for light-responsive micellar aggregates. Macromolecules 2004, 37, 8911–8917. [Google Scholar] [CrossRef]
- Akiyama, H.; Tamaoki, N. Synthesis and Photoinduced Phase Transitions of Poly(N-isopropylacrylamide) Derivative Functionalized with Terminal Azobenzene Units. Macromolecules 2007, 40, 5129–5132. [Google Scholar] [CrossRef]
- Fang, L.; Chen, S.; Zhang, Y.; Zhang, H. Azobenzene-containing molecularly imprinted polymer microspheres with photoresponsive template binding properties. J. Mater. Chem. 2011, 21, 2320–2329. [Google Scholar] [CrossRef]
- Gong, C.-B.; Yang, Y.-Z.; Gao, C.; Tang, Q.; Chow, C.-F.; Peng, J.-D.; Lam, M.H.-W. The preparation and characterization of photo-responsive sol–gel materials for 2,4-dichlorophenoxyacetic acid by surface imprinting. J. Sol-Gel Sci. Technol. 2013, 67, 442–450. [Google Scholar] [CrossRef]
- Yang, Y.; Tang, Q.; Gong, C.; Ma, X.; Peng, J.; Lam, M.H. Ultrasensitive detection of bisphenol A in aqueous media using photoresponsive surface molecular imprinting polymer microspheres. New J. Chem. 2014, 38, 1780–1788. [Google Scholar] [CrossRef]
- Liu, W.; Wang, B. Preparation and application of Norfloxacin-MIP/polysulfone blending molecular imprinted polymer membrane. J. Appl. Polym. Sci. 2009, 113, 1125–1132. [Google Scholar] [CrossRef]
- Sheng, Z.; Tian, Y.; Hu, X.; Jin, Z.; Xu, X.; Cheng, A. Identification and releasing characteristics of β-cyclodextrin–phenylethanoid glycosides inclusion complex. J. Incl. Phenom. Macrocycl. Chem. 2014, 79, 437–442. [Google Scholar] [CrossRef]
- Batt, D.K.; Garala, K.C. Preparation and evaluation of inclusion complexes of diacerein with β-cyclodextrin and hydroxypropyl β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2013, 77, 471–481. [Google Scholar] [CrossRef]
- Egyed, O.; Weiszfeiler, V. Structure determination of copper(II)-β-cyclodextrin complex by Fourier transform infrared spectroscopy. Vib. Spectrosc. 1994, 7, 73–77. [Google Scholar] [CrossRef]
- Li, C.E.; Zhong, S.A.; Li, X.J.; Guo, M. Silica particles coated with azobenzene-containing photoresponsive molecule-imprinted skin layer. Colloid Polym. Sci. 2013, 291, 2049–2059. [Google Scholar] [CrossRef]
- Yang, K.; Liu, Z.; Mao, M.; Zhang, X.; Zhao, C.; Nishi, N. Molecularly imprinted polyethersulfone microspheres for the binding and recognition of bisphenol A. Anal. Chim. Acta 2005, 546, 30–36. [Google Scholar] [CrossRef]
- Tang, Q.; Meng, X.; Jiang, H.; Zhou, T.; Gong, C.; Fu, X.; Shi, S. Synthesis and characterization of photo- and pH-responsive nanoparticles containing amino-substituted azobenzene. J. Mater. Chem. 2010, 20, 9133–9139. [Google Scholar] [CrossRef]
- Kiskan, B.; Dogan, F.; Durmaz, Y.Y.; Yagci, Y. Synthesis, characterization and thermally-activated curing of azobenzene-containing benzoxazines. Des. Monomers Polym. 2008, 11, 473–482. [Google Scholar] [CrossRef]
- Umpleby Ii, R.J.; Baxter, S.C.; Rampey, A.M.; Rushton, G.T.; Chen, Y.; Shimizu, K.D. Characterization of the heterogeneous binding site affinity distributions in molecularly imprinted polymers. J. Chromatogr. B 2004, 804, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hisamatsu, M.; Yamada, T. Production of Maltosyl β-Cyclodextrin by a Bioreactor System with Pullulanase Immobilized on Partially Deacetylated Chitin. Starch Stärke 1989, 41, 239–242. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, H.; Wang, J.; Meng, Q.; Xu, X.; Fan, T.; Jin, Z. Preparation of Photoirradiation Molecular Imprinting Polymer for Selective Separation of Branched Cyclodextrins. Molecules 2017, 22, 288. https://doi.org/10.3390/molecules22020288
Fan H, Wang J, Meng Q, Xu X, Fan T, Jin Z. Preparation of Photoirradiation Molecular Imprinting Polymer for Selective Separation of Branched Cyclodextrins. Molecules. 2017; 22(2):288. https://doi.org/10.3390/molecules22020288
Chicago/Turabian StyleFan, Haoran, Jinpeng Wang, Qingran Meng, Xueming Xu, Tianming Fan, and Zhengyu Jin. 2017. "Preparation of Photoirradiation Molecular Imprinting Polymer for Selective Separation of Branched Cyclodextrins" Molecules 22, no. 2: 288. https://doi.org/10.3390/molecules22020288