
One-Pot Conversion of Epoxidized Soybean Oil (ESO) into Soy-Based Polyurethanes by MoCl2O2 Catalysis


 ,
,  ,
,  ,
, 
Abstract
:
1. Introduction
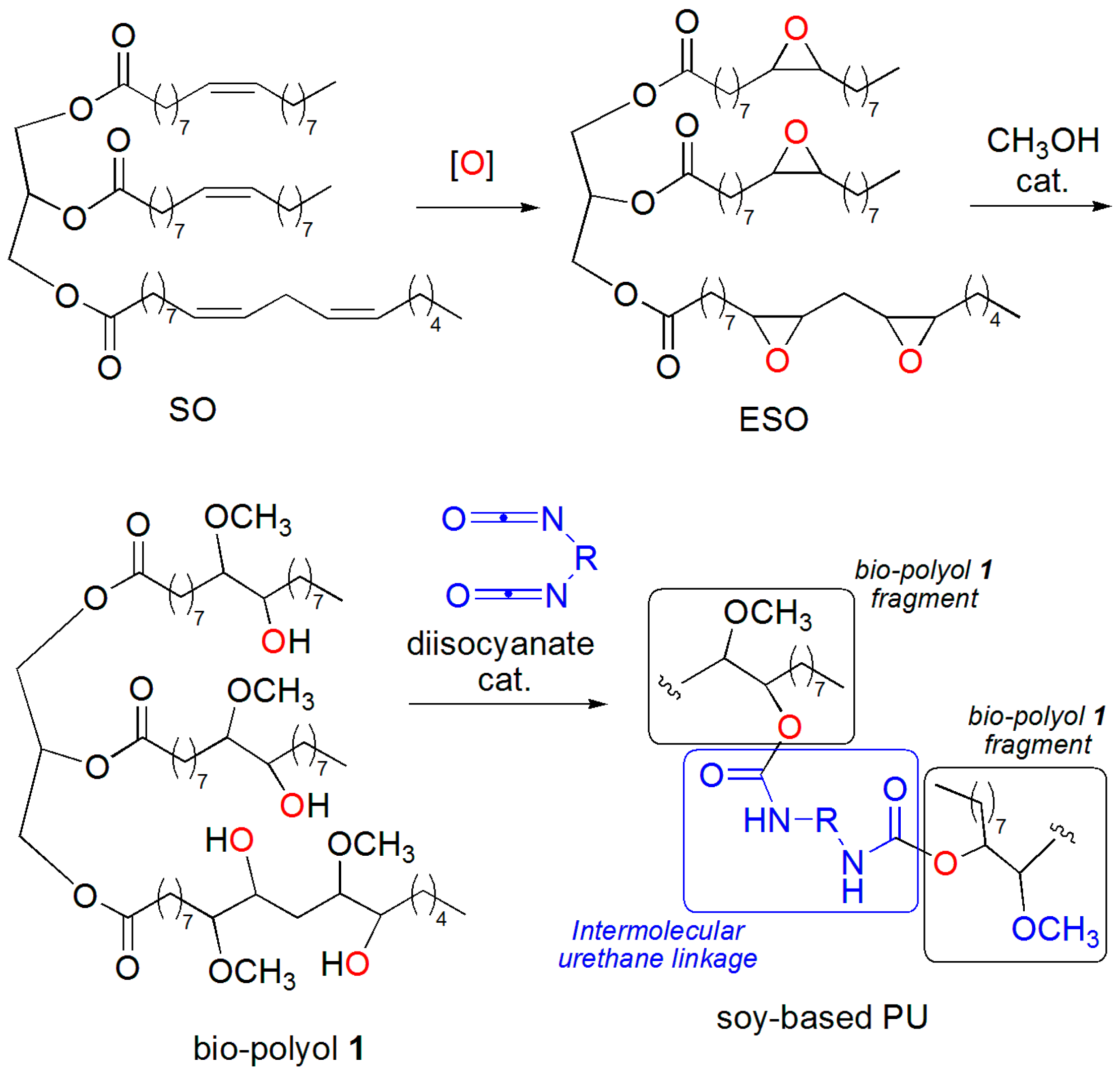
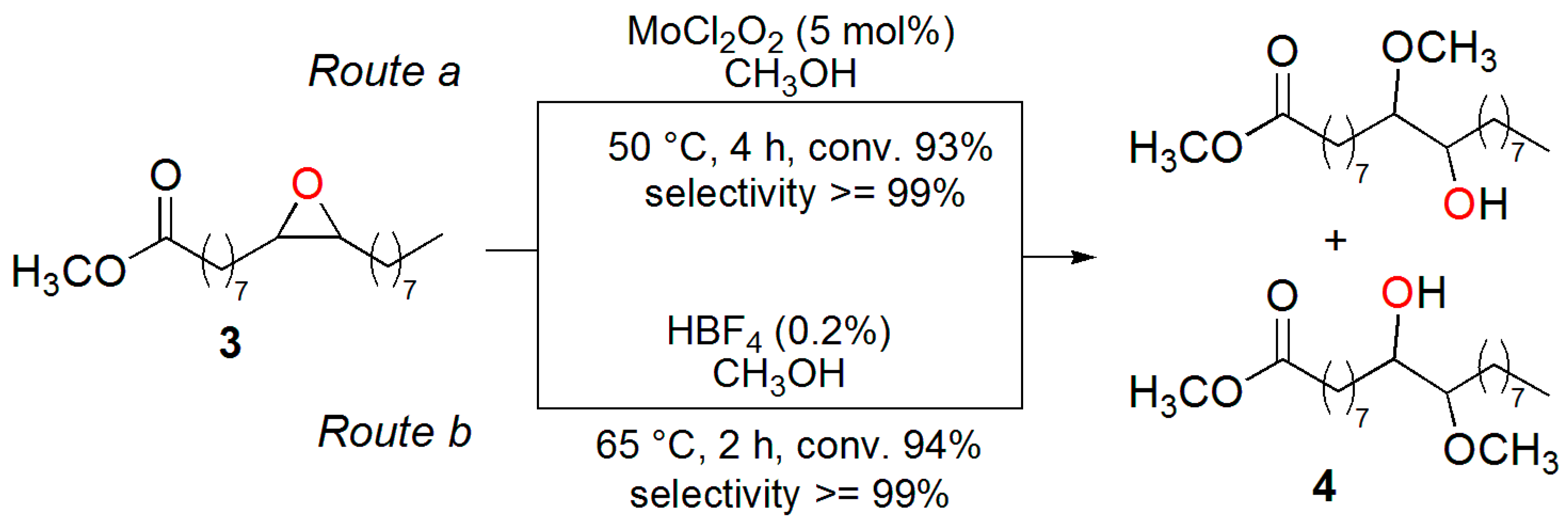
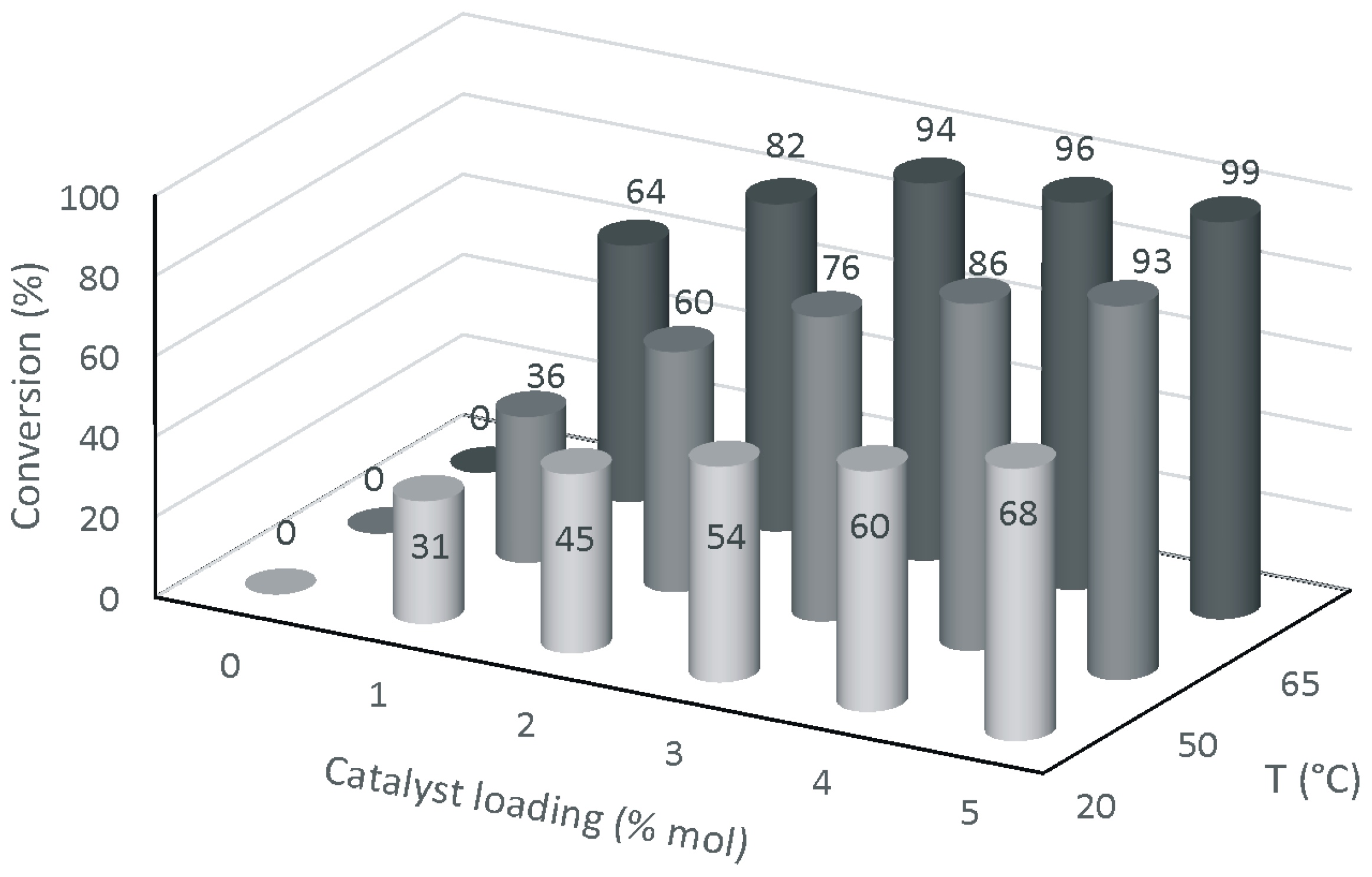
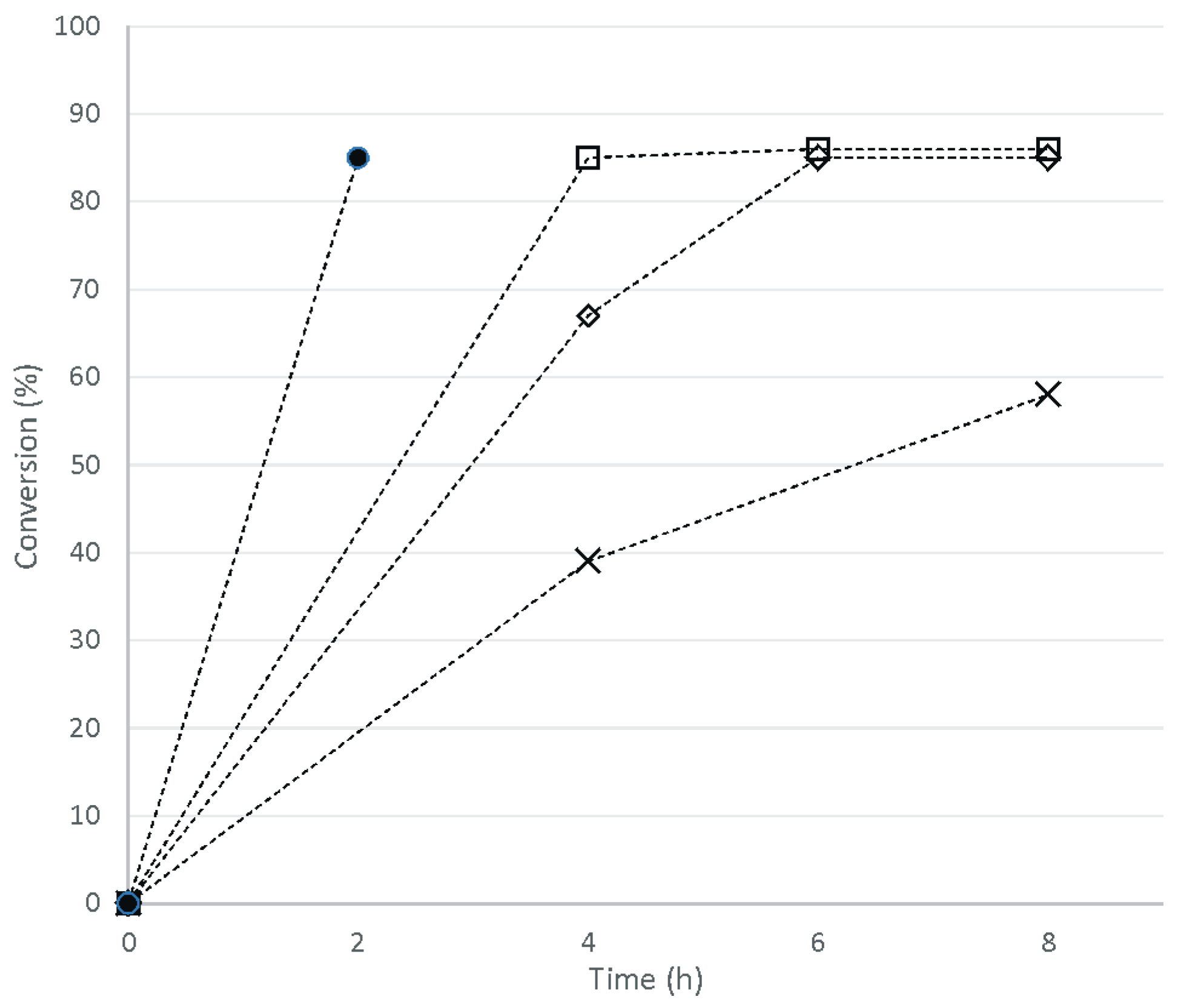
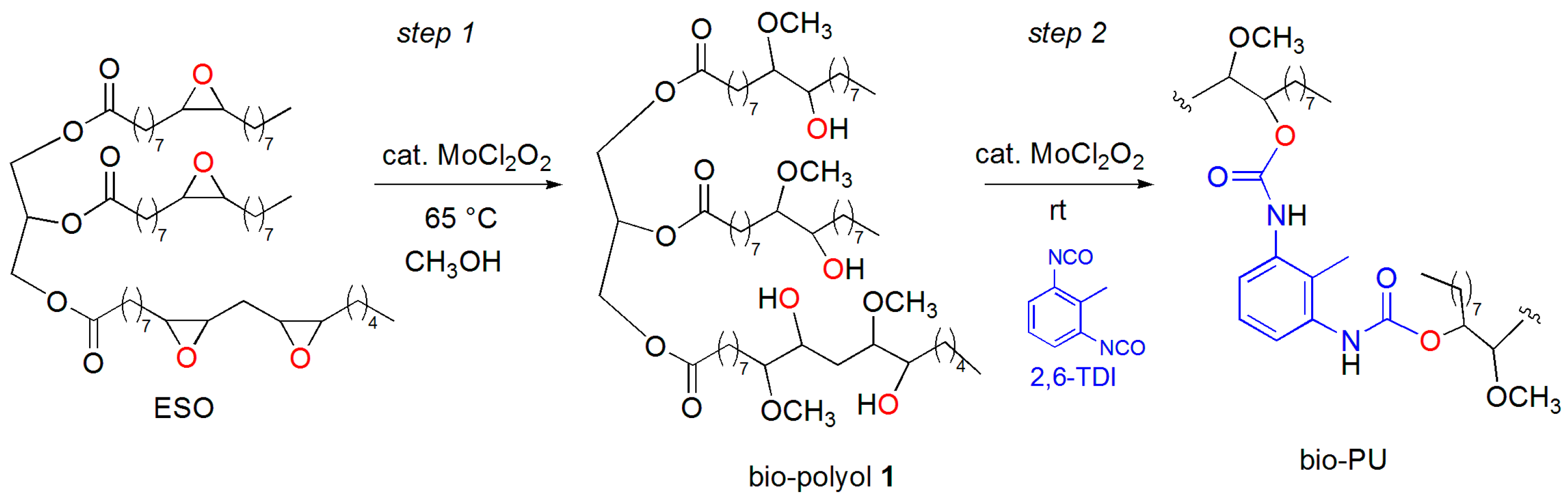
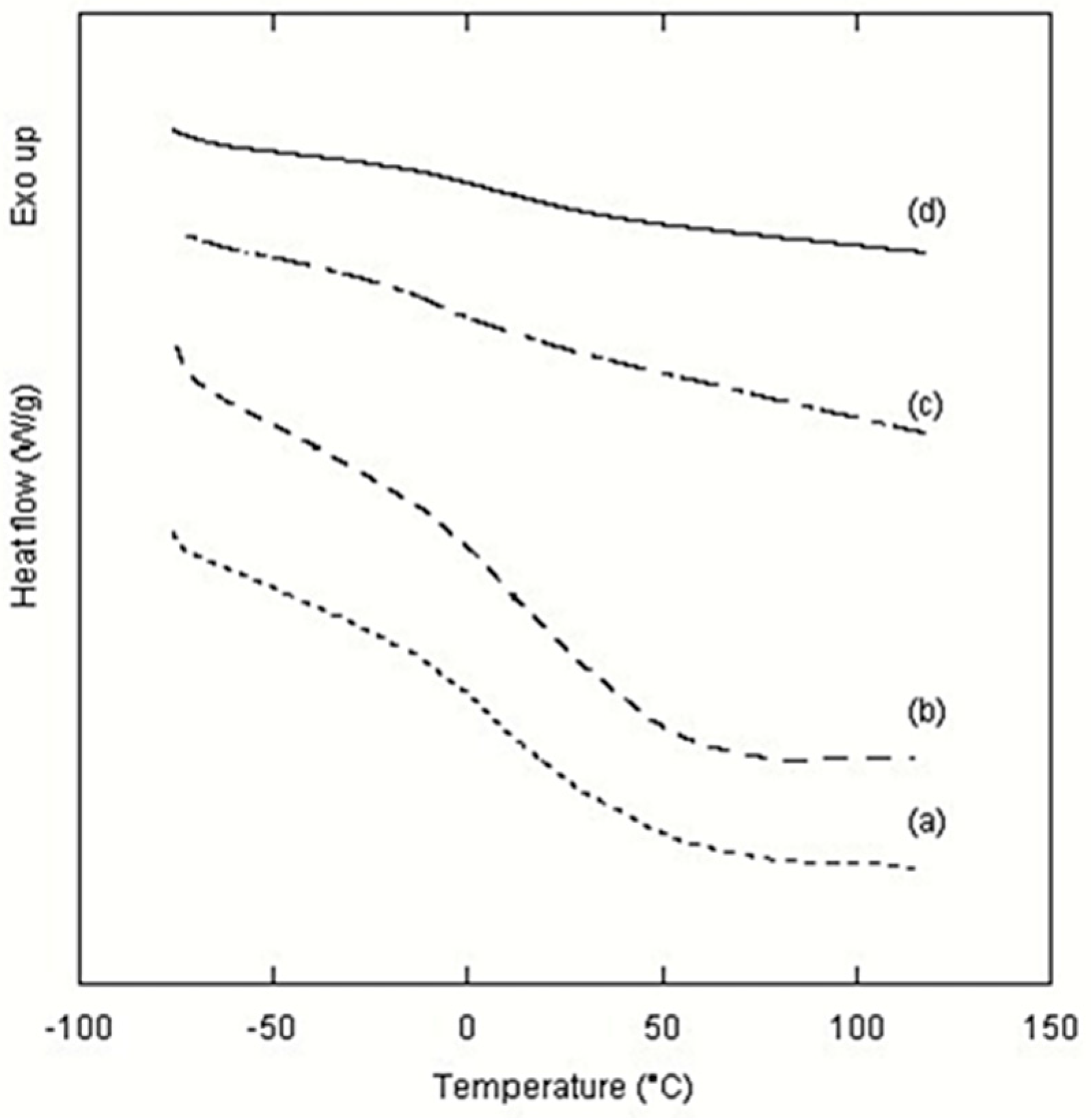
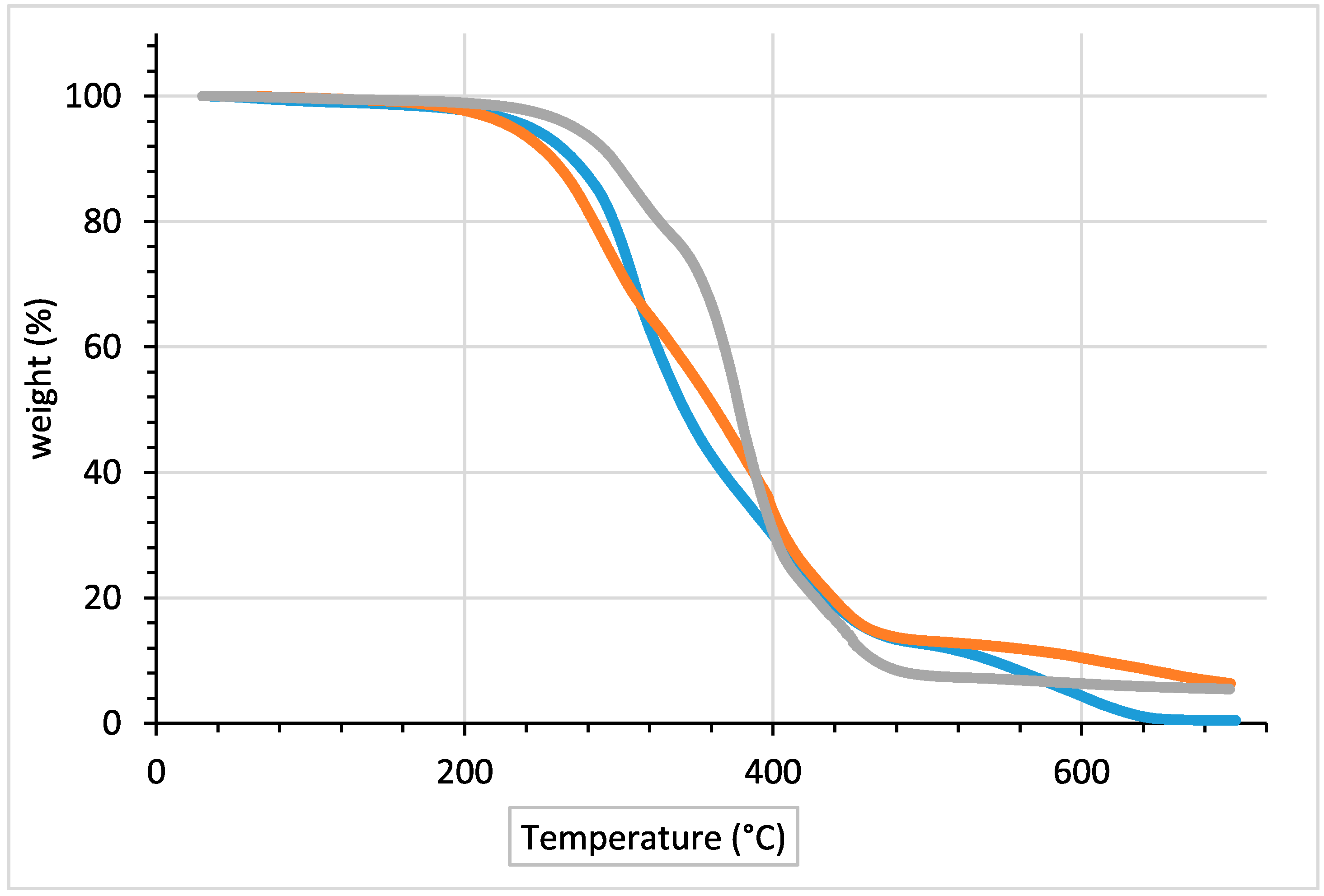
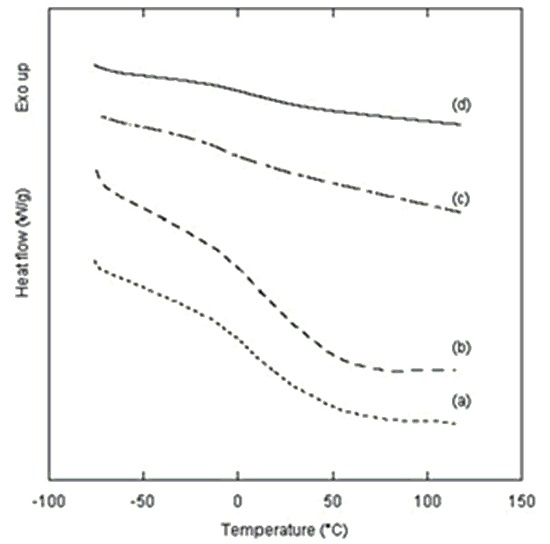
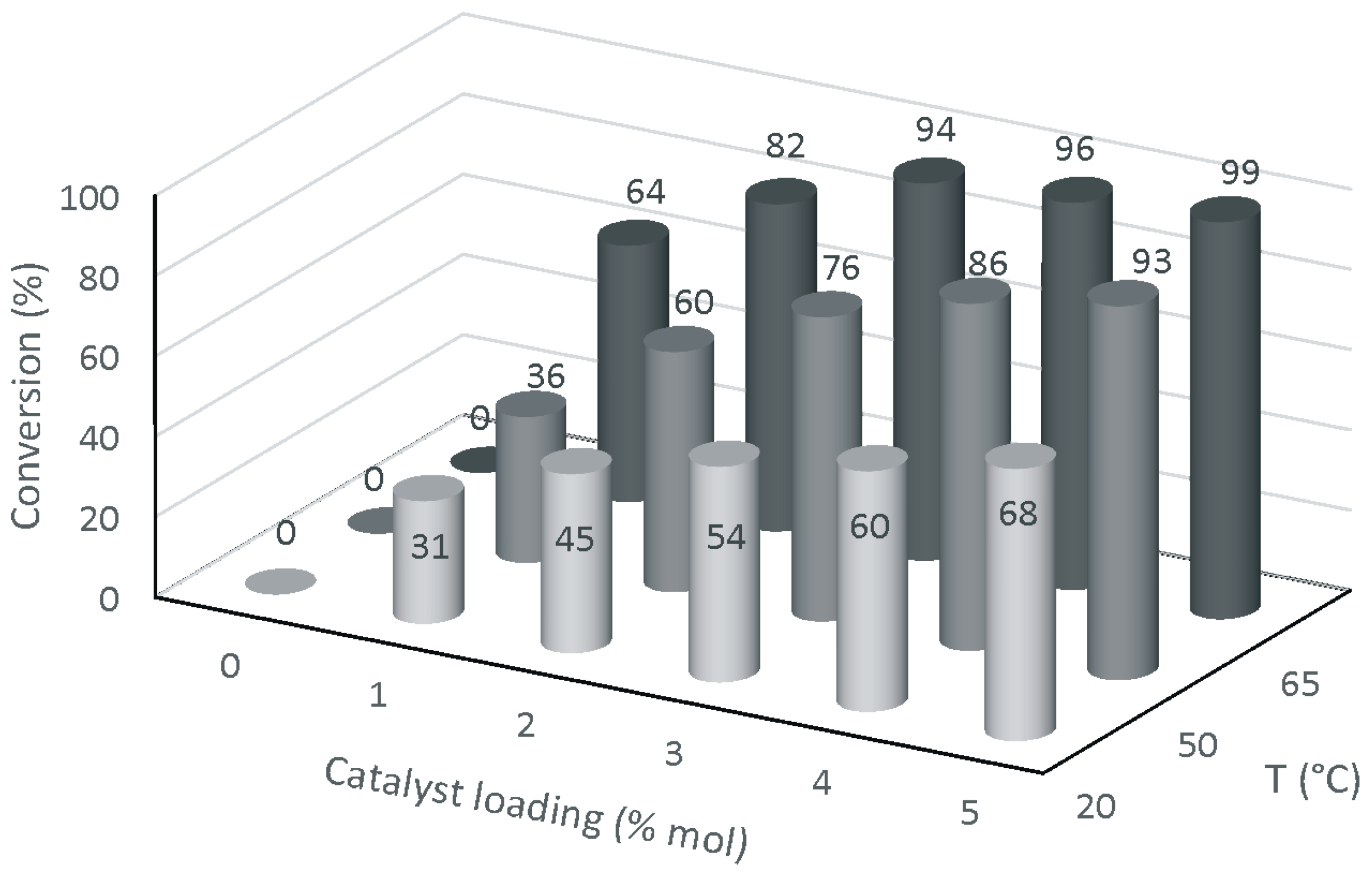
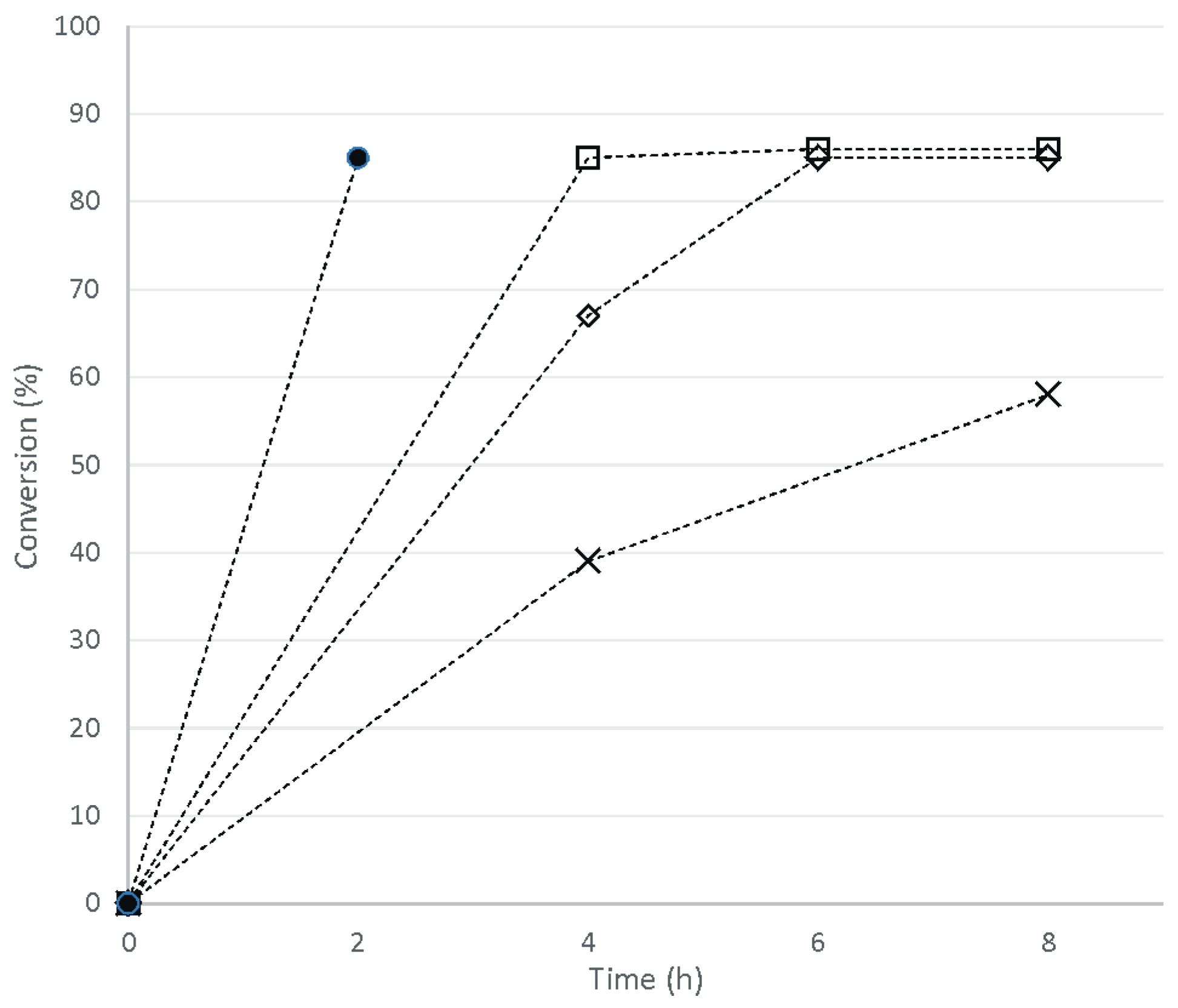
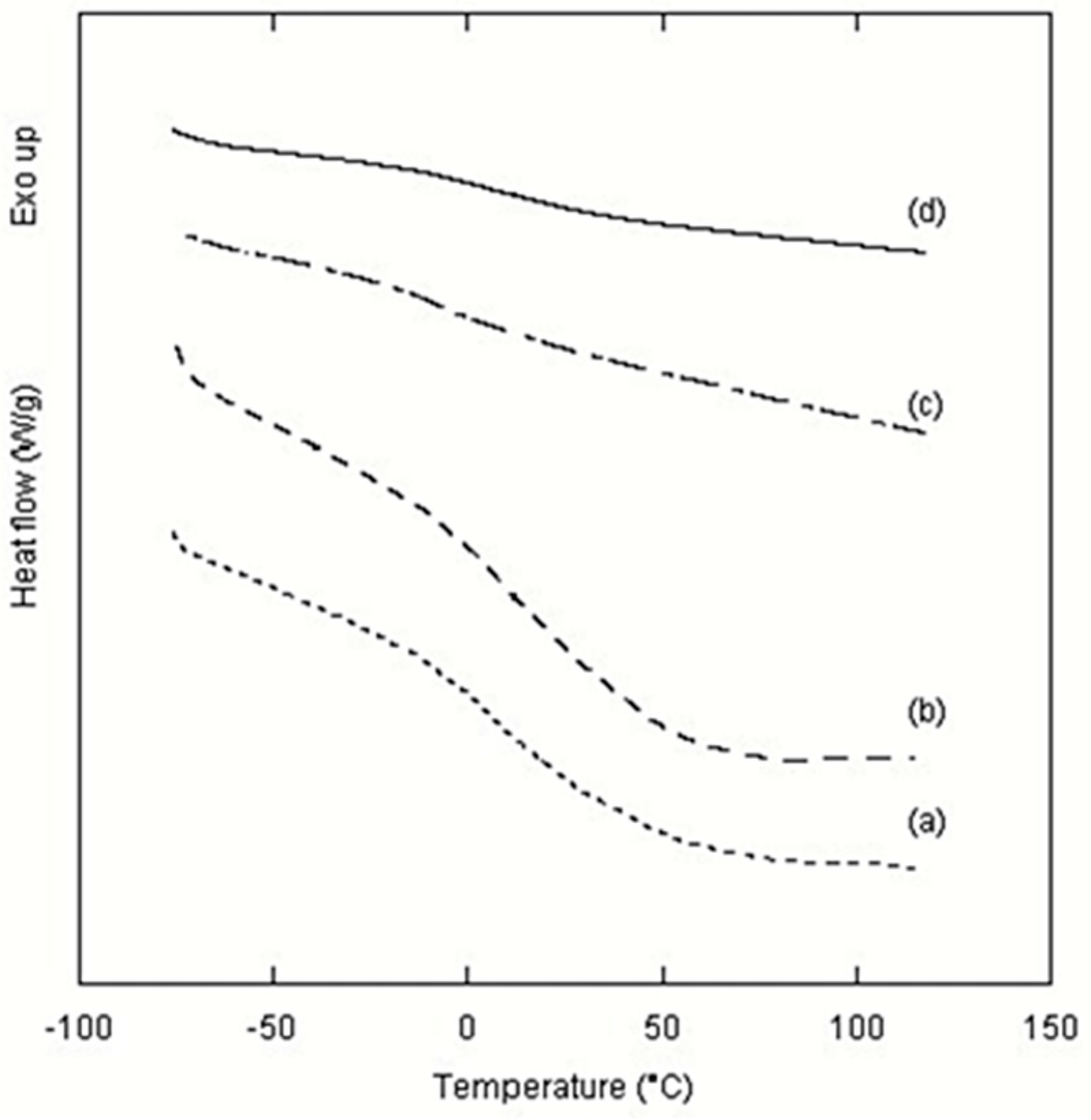
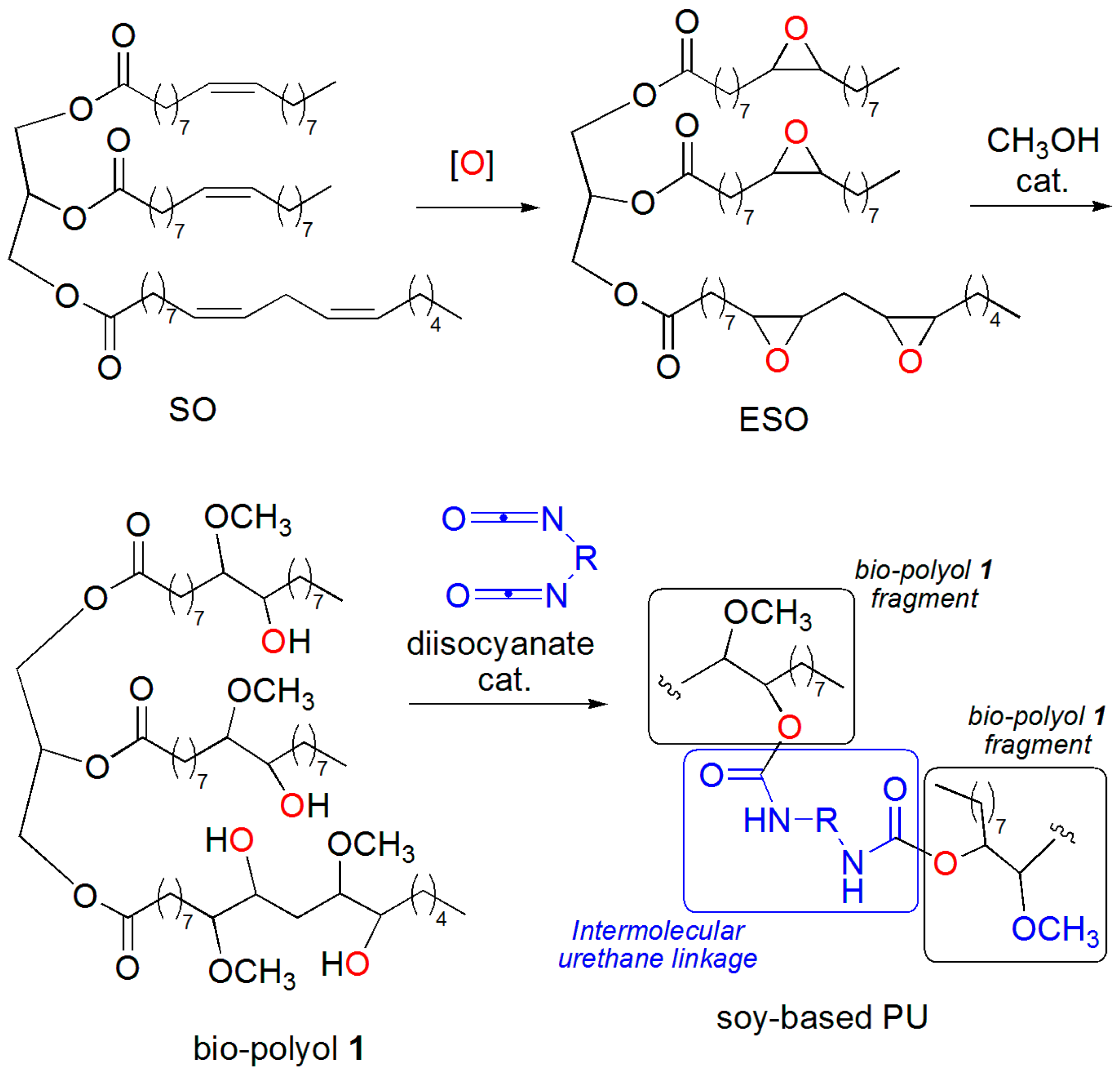
2. Results and Discussion
3. Experimental Section
3.1. Material and Methods
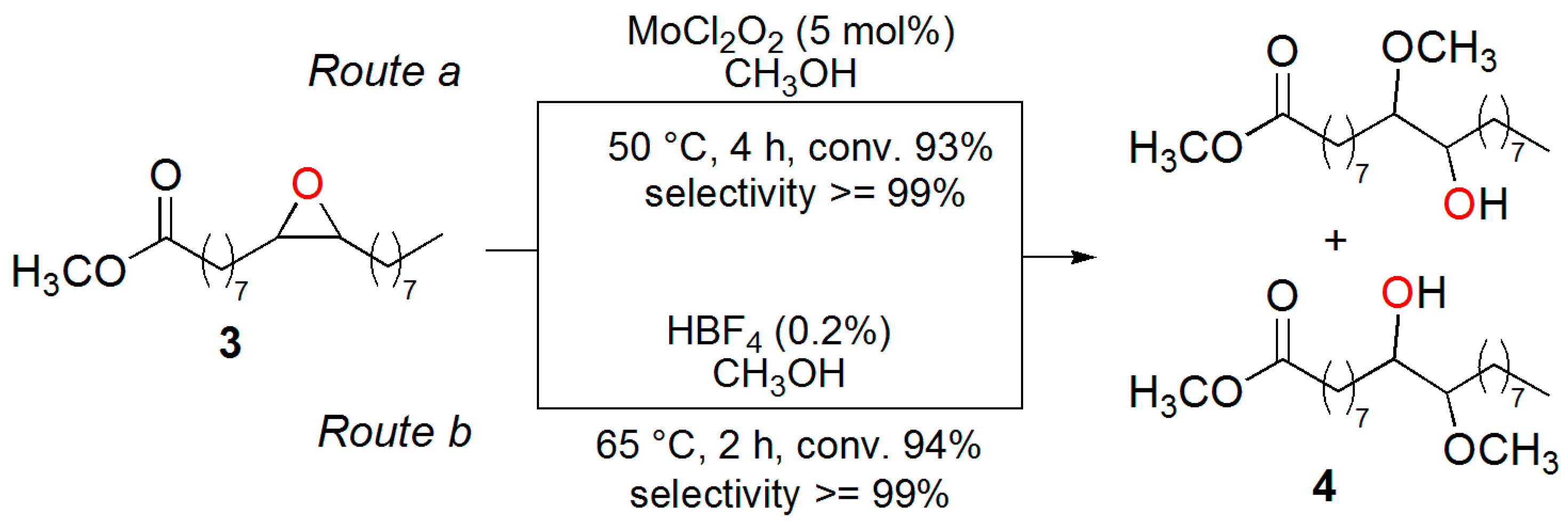
3.2. Synthesis of the Epoxide of Methyl Oleate (3)
3.3. Synthesis of 10(9)-Hydroxy-9(10)-methoxy-octadecanoate (4)
3.4. Synthesis of ESO
3.5. Methanolysis of ESO Using HBF4
Estimation of the E-factor:
3.6. Synthesis of Methyl 10(9)-Hydroxy-9(10)-methoxy-octadecanoate (4) Using MoCl2O2
3.7. Methanolysis of ESO Using MoCl2O2
Estimation of the E-Factor:
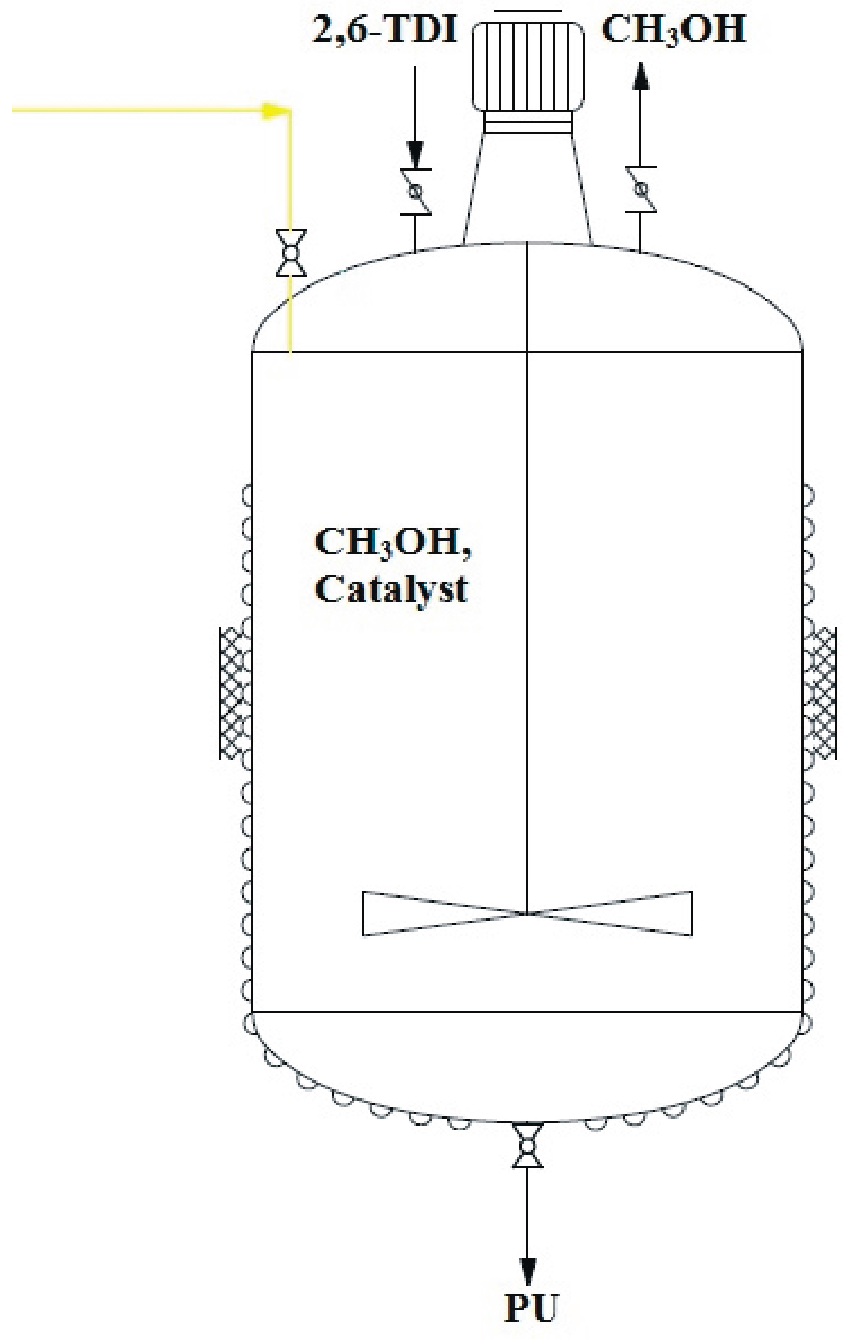
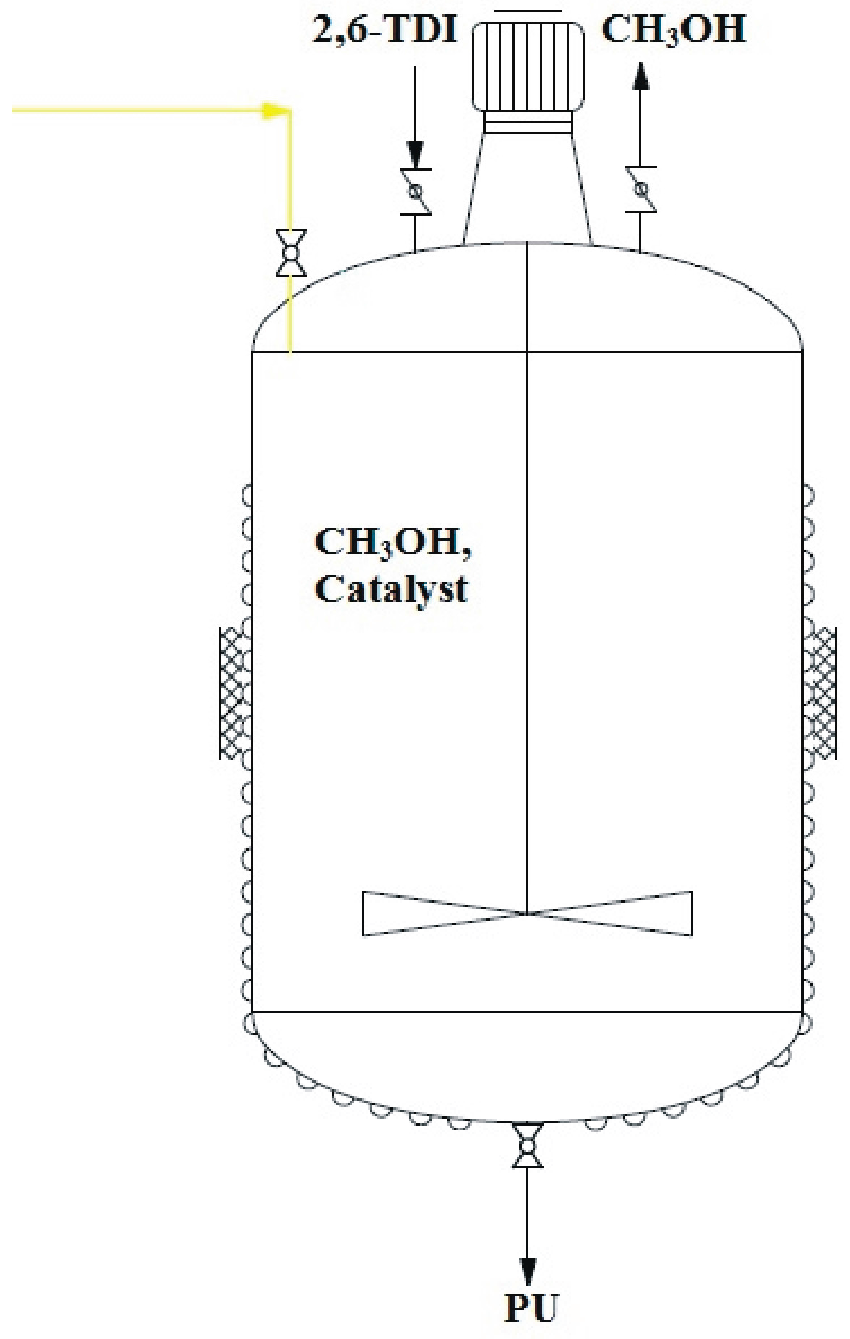
3.8. One-Pot Synthesis of Bio-Based PU
3.9. Synthesis Bio-Based PU without Catalyst
4. Conclusions
- The synthesis of PU is carried out in a short time and at room temperature, a great advantage when compared with reactions carried out without catalysts;
- The efficiency and selectivity of the methanolysis step provides a pure polyol intermediate, which could be used directly in subsequent reactions, avoiding the costly and time-consuming purification procedures that are necessary when using the classical method based on HBF4; and
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Petrovic, Z.S. Polyurethanes from Vegetable Oils. Polym. Rev. 2008, 48, 109–155. [Google Scholar] [CrossRef]
- Imhof, P.; van der Waal, J.C. (Eds.) Catalytic Process Development for Renewable Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013.
- Noreen, A.; Zia, K.M.; Zuber, M.; Tabasum, S.; Zahoor, A.F. Bio-based polyurethane: An efficient and environment friendly coating systems: A review. Prog. Org. Coat. 2016, 91, 25–32. [Google Scholar] [CrossRef]
- Miao, S.; Wang, P.; Su, Z.; Zhang, S. Vegetable-oil-based polymers as future polymeric biomaterials. Acta Biomater. 2014, 10, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Liu, G.; Qi, H.; Curtis, J.M. Preparation and characterization of high-solid polyurethane coating systems based on vegetable oil derived polyols. Prog. Org. Coat. 2013, 76, 1151–1160. [Google Scholar] [CrossRef]
- Nohra, B.; Candy, L.; Blanco, J.-F.; Guerin, C.; Raoul, Y.; Mouloungui, Z. From Petrochemical Polyurethanes to Biobased Polyhydroxyurethanes. Macromolecules 2013, 46, 3771–3792. [Google Scholar] [CrossRef] [Green Version]
- Desroches, M.; Escouvois, M.; Auvergne, R.; Caillol, S.; Boutevin, B. From Vegetable Oils to Polyurethanes: Synthetic Routes to Polyols and Main Industrial Products. Polym. Rev. 2012, 52, 38–79. [Google Scholar] [CrossRef]
- Pfister, D.P.; Xia, Y.; Larock, R.C. Recent Advances in Vegetable Oil-Based Polyurethanes. ChemSusChem 2011, 4, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; Levin, E.; Rodriguez, C.; Williard, P.G.; Stanton, A.; Socha, A.M. Equilibrium studies of canola oil transesterification using a sodium glyceroxide catalyst prepared from a biodiesel waste stream. Fuel Process. Technol. 2016, 146, 70–75. [Google Scholar] [CrossRef]
- Fowler, P.; Hughes, J.M.; Elias, R.M. Biocomposite: Technology, enviromental credentials and marked forces. J. Sci. Food Agric. 2006, 86, 1781–1789. [Google Scholar] [CrossRef]
- Liu, Z.; Bai, G.; Huang, Y.; Ma, Y.; Du, F.; Li, F. Reflection and absorption contributions to the electromagnetic interference shielding of single-walled carbon nanotube/polyurethane composites. Carbon 2007, 45, 821–827. [Google Scholar] [CrossRef]
- Annese, C.; D’Accolti, L.; Giambastiani, G.; Mangone, A.; Milella, A.; Tuci, G.; Fusco, C. Tunable Epoxidation of Single-Walled Carbon Nanotubes by Isolated Methyl (trifluoromethyl) dioxirane. Eur. J. Org. Chem. 2014, 1666–1671. [Google Scholar] [CrossRef]
- Bloise, E.; Becerra-Herrera, M.; Mele, G.; Sayago, A.; Carbone, L.; D’Accolti, L.; Mazzetto, S.E.; Vasapollo, G. Sustainable preparation of cardanol-based nanocarriers with embedded natural phenolic compounds. ACS Sustain. Chem. Eng. 2014, 2, 1299–1304. [Google Scholar] [CrossRef]
- D’Accolti, L.; Annese, C.; De Riccardis, A.; De Giglio, E.; Cafagna, D.; Fanelli, F.; Fusco, C. Dioxirane-Mediated Heterogeneous Epoxidations with Potassium Caroate: A Solid Catalyst Bearing Anchored Ketone Moieties. Eur. J. Org. Chem. 2012, 4616–4621. [Google Scholar]
- Limnios, D.; Kokotos, C.G. 2,2,2-Trifluoroacetophenone: An organocatalyst for an environmentally friendly epoxidation of alkenes. J. Org. Chem. 2014, 79, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Fang, Z.; Ji, D.; Chen, K.; Wan, Z.; Li, X.; Gan, H.; Tang, S.; Zhang, K.; Guo, K. Epoxidation of Soybean Oil by Continuous Micro-Flow System with Continuous Separation. Org. Process Res. Dev. 2013, 17, 1137–1141. [Google Scholar] [CrossRef]
- Khot, S.N.; Lascala, J.J.; Can, E.; Morye, S.S.; Williams, G.I.; Palmese, G.R.; Kusefoglu, S.H.; Wool, R.P. Development and application of triglyceride-based polymers and composites. J. Appl. Polym. Sci. 2001, 82, 703–723. [Google Scholar] [CrossRef]
- Neves, P.; Amarante, T.R.; Valente, A.A.; Pillinger, M.; Gonçalves, I.S. Catalytic Application of an Octamolybdate Salt (H3biim)4[β-Mo8O26] in Olefin Epoxidation (H2biim = 2,2′-biimidazole). Catal. Lett. 2016, 146, 841–850. [Google Scholar] [CrossRef]
- Amarante, T.R.; Neves, P.; Tomé, C.; Abrantes, M.; Valente, A.A.; Almeida Paz, F.A.; Pillinger, M.; Gonçalves, I.S. Novel Heterotetranuclear V2Mo2 or V2W2 Complexes with 4,4′-Di-tert-butyl-2,2′-bipyridine: Syntheses, Crystal Structures, and Catalytic Activities. Inorg. Chem. 2012, 51, 3666–3676. [Google Scholar] [CrossRef]
- Farias, M.; Martinelli, M.; Pagliocchi Bottega, D. Epoxidation of soybean oil using a homogeneous catalytic system based on a molybdenum (VI). Appl. Catal. A Gen. 2010, 384, 213–219. [Google Scholar] [CrossRef]
- Vanoye, L.; Hamami, Z.E.; Wang, J.; de Bellefon, C.; Fongarland, P.; Favre-Réguillon, A. Epoxidation of methyl oleate with molecular oxygen: Implementation of Mukaiyama reaction in flow. Eur. J. Lipid Sci. Technol. 2016, 118. [Google Scholar] [CrossRef]
- Silva, A.L.; Bordado, J.C. Recent Developments in Polyurethane Catalysis: Catalytic Mechanisms Review. Catal. Rev. 2004, 46, 31–51. [Google Scholar] [CrossRef]
- Blank, W.J.; He, Z.A.; Hessell, E.T. Catalysis of the isocyanate-hydroxyl reaction by non-tin catalysts. Prog. Org. Coat. 1999, 35, 19–29. [Google Scholar] [CrossRef]
- Dworakowska, S.; Bogdał, D.; Zaccheria, F.; Ravasio, N. The role of catalysis in the synthesis of polyurethane foams based on renewable raw materials. Catal. Today 2014, 223, 148–156. [Google Scholar] [CrossRef]
- Cotugno, P.; Monopoli, A.; Ciminale, F.; Milella, A.; Nacci, A. Palladium-Catalyzed Cross-Coupling of Styrenes with Aryl Methyl Ketones in Ionic Liquids: Direct Access to Cyclopropanes. Angew. Chem. 2014, 53, 13563–13567. [Google Scholar] [CrossRef] [PubMed]
- Annese, C.; D’Accolti, L.; Fusco, C.; Curci, R. Selective Hydroxylation of Methane by Dioxiranes under Mild Conditions. Org. Lett. 2011, 13, 2142–2144. [Google Scholar] [CrossRef] [PubMed]
- Mele, G.; Annese, C.; De Riccardis, A.; Fusco, C.; Palmisano, L.; Vasapollo, G.; D’Accolti, L. Turning lipophilic phthalocyanines/TiO2 composites into efficient photocatalysts for the conversion of CO2 into formic acid under UV–Vis light irradiation. Appl. Catal. A 2014, 481, 169–172. [Google Scholar] [CrossRef]
- Monopoli, A.; Cotugno, P.; Palazzo, G.; Ditaranto, N.; Mariano, B.; Cioffi, N.; Ciminale, F.; Nacci, A. Ullmann homocoupling catalysed by gold nanoparticles in water and ionic liquid. Adv. Synth. Catal. 2012, 354, 2777–2788. [Google Scholar] [CrossRef]
- Jeyakumar, K.; Chand, D.K. Application of molybdenum (VI) dichloride dioxide (MoO2Cl2) in organic transformations. J. Chem. Sci. 2009, 121, 111–123. [Google Scholar] [CrossRef]
- Stock, C.; Brückner, B. Mild and High-Yielding Molybdenum (VI) Dichloride Dioxide-Catalyzed Formation of Mono-, Di-, Tri-, and Tetracarbamates from Alcohols and Aromatic or Aliphatic. Adv. Synth. Catal. 2012, 354, 2309–2330. [Google Scholar] [CrossRef]
- D’Accolti, L.; Denora, N.; La Piana, G.; Marzulli, D.; Siwy, Z.S.; Fusco, C.; Annese, C. Synthesis and Biological Evaluation of a Valinomycin Analog Bearing a Pentafluorophenyl Active Ester Moiety. J. Org. Chem. 2015, 80, 12646–12650. [Google Scholar] [CrossRef] [PubMed]
- Annese, C.; Fanizza, I.; Calvano, C.D.; D’Accolti, L.; Fusco, C.; Curci, R.; Williard, P.G. Selective synthesis of hydroxy analogues of valinomycin using dioxiranes. Org. Lett. 2011, 13, 5096–5099. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, K.; Chand, D.K. Ring-opening reactions of epoxides catalyzed by molybdenum (VI) dichloride dioxide. Synthesis 2008, 807–819. [Google Scholar]
- Bunker, S.P.; Wool, R.P. Synthesis and characterization of monomers and polymers for adhesives from methyl oleate. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 451–458. [Google Scholar] [CrossRef]
- Palaskar, D.V.; Boyer, A.; Cloutet, E.; Alfos, C.; Cramail, H. Synthesis of Biobased Polyurethane from Oleic and Ricinoleic Acids as the Renewable Resources via the AB-Type Self-Condensation Approach. Biomacromolecules 2010, 11, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Yang, L.; Lin, B.; Wang, C.; Shi, G. Synthesis and characterization of the different soy-based polyols by ring opening of epoxidized soybean oil with methanol, 1,2-ethanediol and 1,2-propanediol. J. Am. Oil Chem. Soc. 2009, 86, 261–267. [Google Scholar] [CrossRef]
- Wang, C.S.; Yang, L.T.; Ni, B.L.; Shi, G. Polyurethane networks from different soy-based polyols by the ring opening of epoxidized soybean oil with methanol, glycol, and 1,2-propanediol. J. Appl. Polym. Sci. 2009, 114, 125–131. [Google Scholar] [CrossRef]
- Chen, C.T.; Kuo, J.H.; Pawar, V.D.; Munot, Y.S.; Weng, S.S.; Ku, C.H.; Liu, C.Y. Nucleophilic acyl substitutions of anhydrides with protic nucleophiles catalyzed by amphoteric, oxomolybdenum species. J. Org. Chem. 2005, 70, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Helping Our World Work Better. Available online: https://www.astm.org/ (accessed on 20 February 2017).
- Sattar, R.; Kausar, A.; Siidiq, M. Thermal, mechanical and electrical studies of novel shape memory polyurethane/polyaniline blends. Chin. J. Polym. Sci. 2015, 33, 1313–1324. [Google Scholar] [CrossRef]
- Sakulsaknirmitr, W.; Wirasate, S.; Pipatpanyanugoon, K.; Atorngitjawat, P. Structure and thermal properties of polyurethanes synthesized from cardanol diol. J. Polym. Environ. 2015, 23, 216–226. [Google Scholar] [CrossRef]
- Jiao, L.; Xiao, H.; Wang, Q.; Sun, J. Thermal Degradation Characteristics of Rigid Polyurethane Foam and the Volatile Products Analysis with TG-FTIR-MS. Polym. Degrad. Stab. 2013, 98, 2687–2696. [Google Scholar] [CrossRef]
- He, J.-J.; Jiang, L.; Sun, J.-H.; Lo, S. Thermal degradation study of pure rigid polyurethane in oxidative and non-oxidative atmospheres. J. Anal. Appl. Pyrolysis 2016, 120, 269–283. [Google Scholar] [CrossRef]
- Zlatanic´, A.; Lava, C.; Zhang, W.; Petrovic´, Z.S. Effect of Structure on Properties of Polyols and Polyurethanes Based on Different Vegetable Oils. J. Polym. Sci. B Polym. Phys. 2004, 42, 809–819. [Google Scholar] [CrossRef]
- Javni, I.; Petrovic´, Z.S.; Guo, A.; Fuller, R. Thermal Stability of Polyurethanes Based on Vegetable Oils. J. Appl. Polym. Sci. 2000, 77, 1723–1734. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E Factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Maity, P.; Gopinath, C.S.; Bhaduri, S.; Lahiri, G.K. Applications of a high performance platinum nanocatalyst for the oxidation of alcohols in water. Green Chem. 2009, 11, 554–561. [Google Scholar]
- For HBF4. Available online: https://echa.europa.eu/it/substance-information/-/substanceinfo/100.037.165 (accessed on 20 February 2017). For MoO2Cl2. Available online: https://echa.europa.eu/it/substance-information/-/substanceinfo/100.157.480 (accessed on 20 February 2017).
- Pilot apparatus designed by Eng. Luciano Vassallo—Piazza Fontana 14/H, 23868 Valmadrera (LC).
- Sample Availability: Samples of the compounds bio-polyol 1 and Bio-PU are available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Density (Kg/dm3) | Viscosity (cP) | n. OH a (mg of KOH/g) | n. OH/Molecule | n. Epoxy/Molecule | E-Factor b |
|---|---|---|---|---|---|---|
| 1 from HBF4 | 1.001761 | 4551 | 191 | 3.45 | ---- | 1.45 |
| 1 from MoCl2O2 | 1.003678 | 4567 | 188 | 3.40 | ---- | 0.24 |
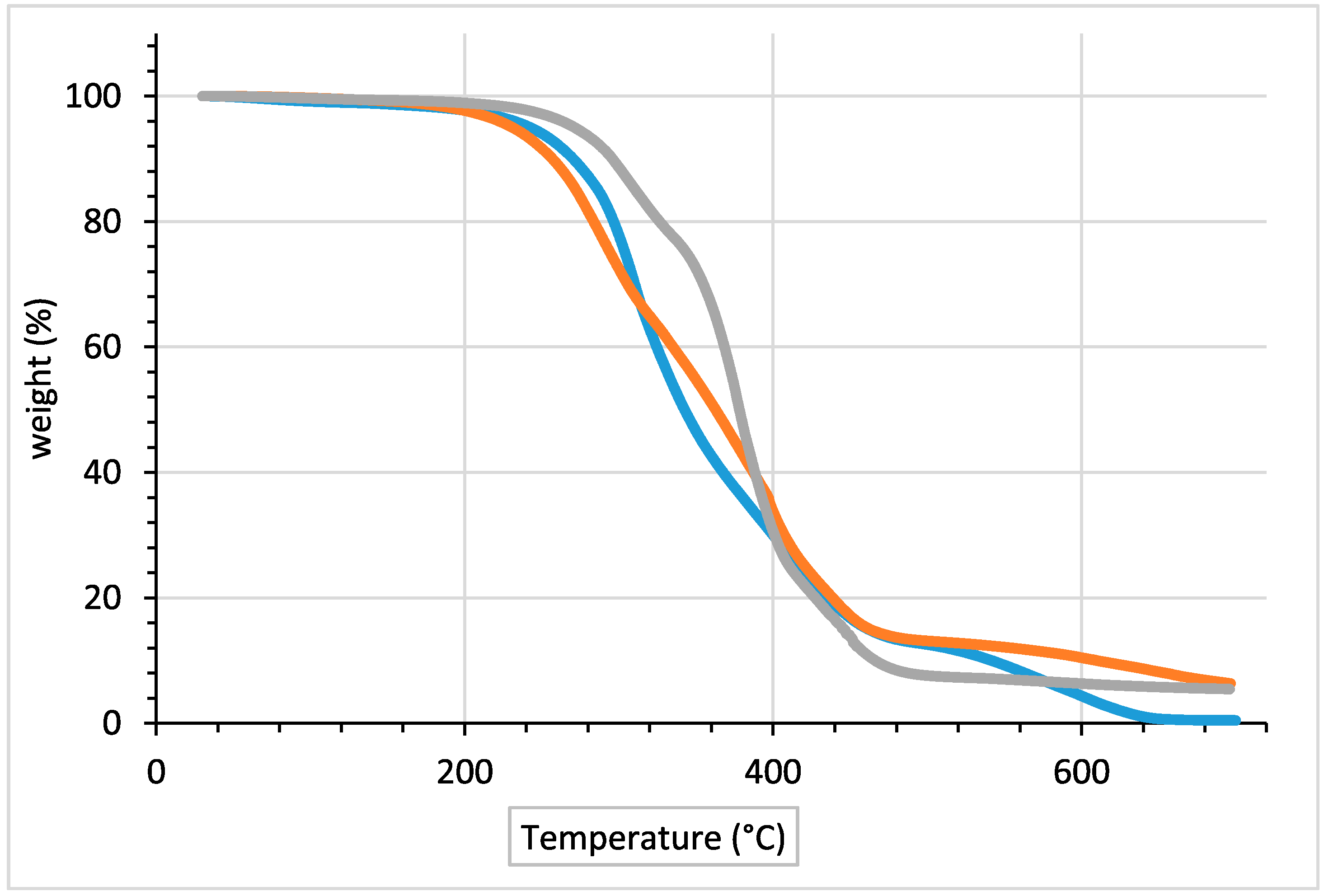
| Material | Td5% (°C) | Tmax (°C) | Td70% (°C) | Td90% (°C) | Mresidue (%) |
|---|---|---|---|---|---|
| Literature procedure | 272.1 | 378.3 | 401.02 | 465.98 | 5.4 |
| BIO PUR with 1 mol % MoCl2O2 catalyst | 242.0 | 312.4 | 401.02 | 540.0 | 0.5 |
| BIO PUR with 2 mol % MoCl2O2 catalyst | 231.5 | 291.7 | 408.0 | 609.0 | 6.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantone, V.; Annese, C.; Fusco, C.; Fini, P.; Nacci, A.; Russo, A.; D’Accolti, L. One-Pot Conversion of Epoxidized Soybean Oil (ESO) into Soy-Based Polyurethanes by MoCl2O2 Catalysis. Molecules 2017, 22, 333. https://doi.org/10.3390/molecules22020333
Pantone V, Annese C, Fusco C, Fini P, Nacci A, Russo A, D’Accolti L. One-Pot Conversion of Epoxidized Soybean Oil (ESO) into Soy-Based Polyurethanes by MoCl2O2 Catalysis. Molecules. 2017; 22(2):333. https://doi.org/10.3390/molecules22020333
Chicago/Turabian StylePantone, Vincenzo, Cosimo Annese, Caterina Fusco, Paola Fini, Angelo Nacci, Antonella Russo, and Lucia D’Accolti. 2017. "One-Pot Conversion of Epoxidized Soybean Oil (ESO) into Soy-Based Polyurethanes by MoCl2O2 Catalysis" Molecules 22, no. 2: 333. https://doi.org/10.3390/molecules22020333
APA StylePantone, V., Annese, C., Fusco, C., Fini, P., Nacci, A., Russo, A., & D’Accolti, L. (2017). One-Pot Conversion of Epoxidized Soybean Oil (ESO) into Soy-Based Polyurethanes by MoCl2O2 Catalysis. Molecules, 22(2), 333. https://doi.org/10.3390/molecules22020333