
Synthesis and Biological Evaluation of Novel 8-Morpholinoimidazo[1,2-a]pyrazine Derivatives Bearing Phenylpyridine/Phenylpyrimidine-Carboxamides


Abstract
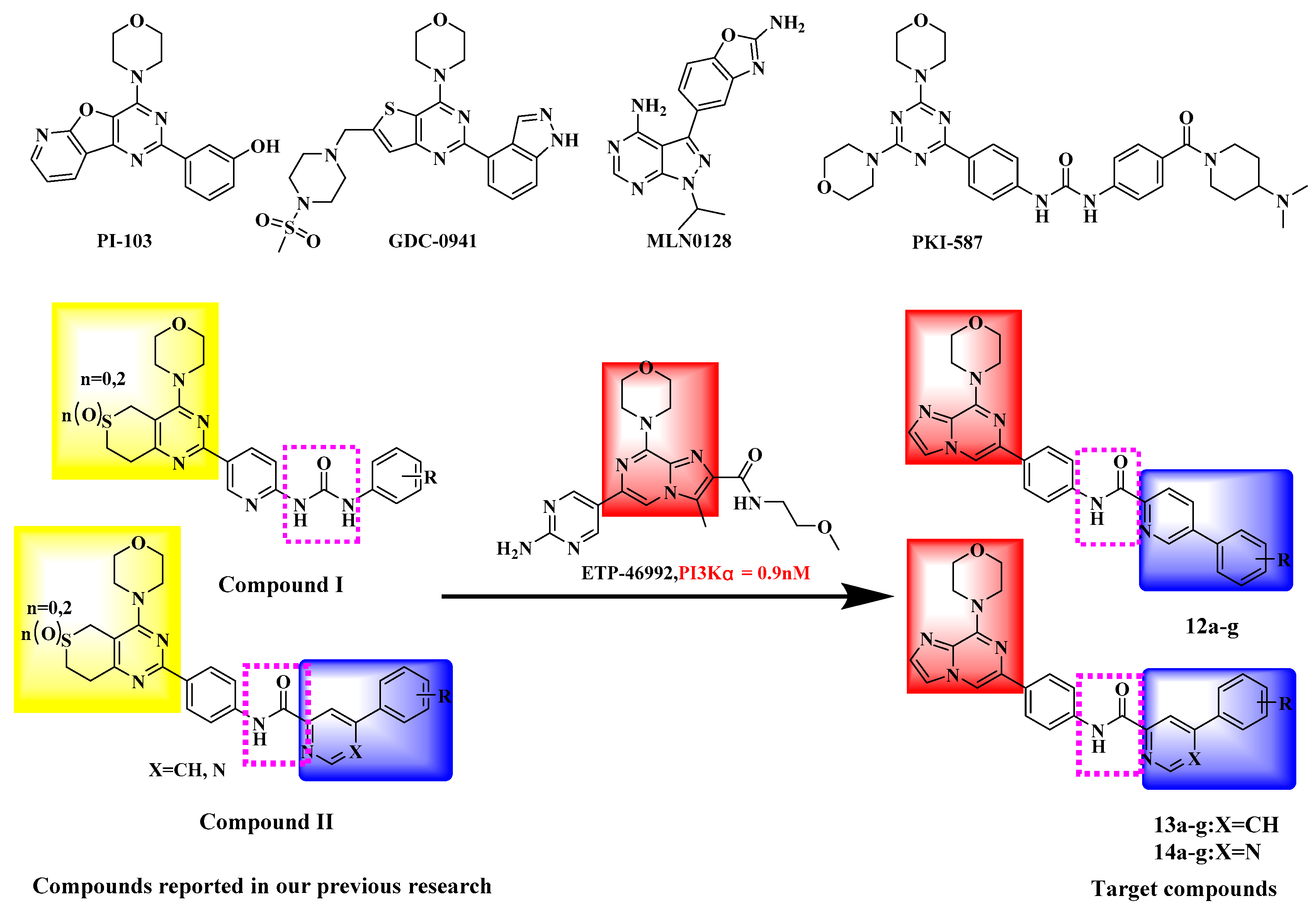
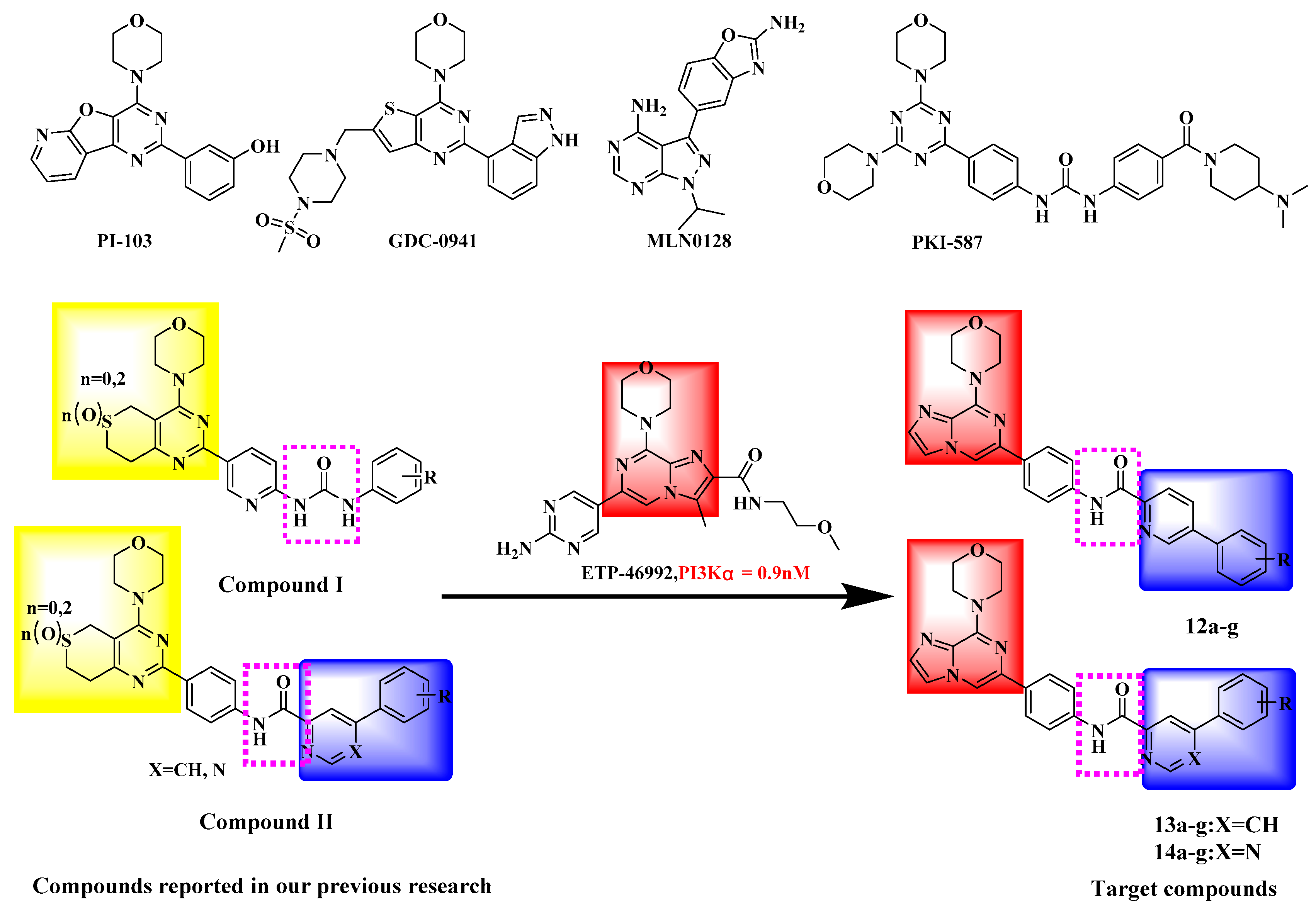
:1. Introduction
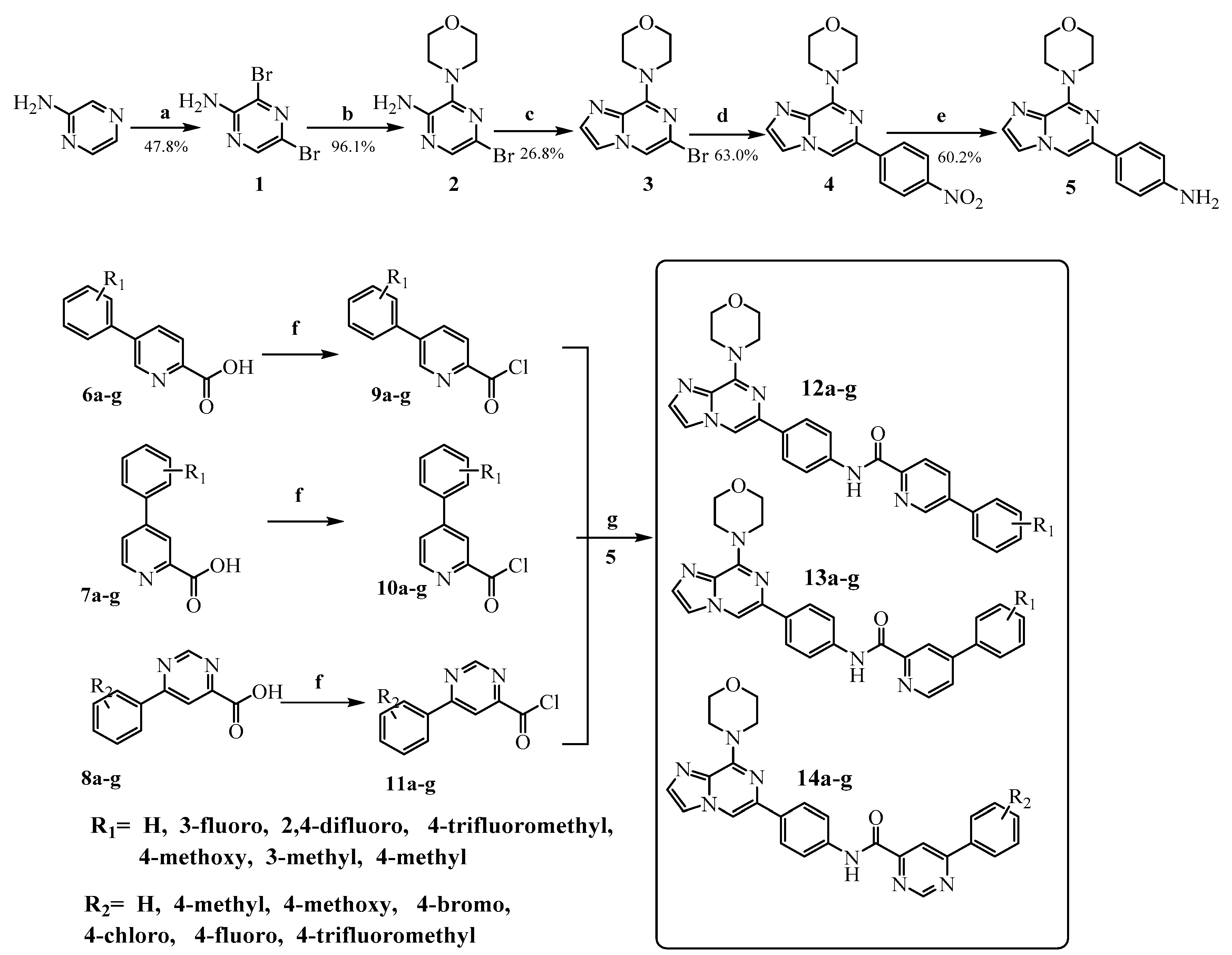
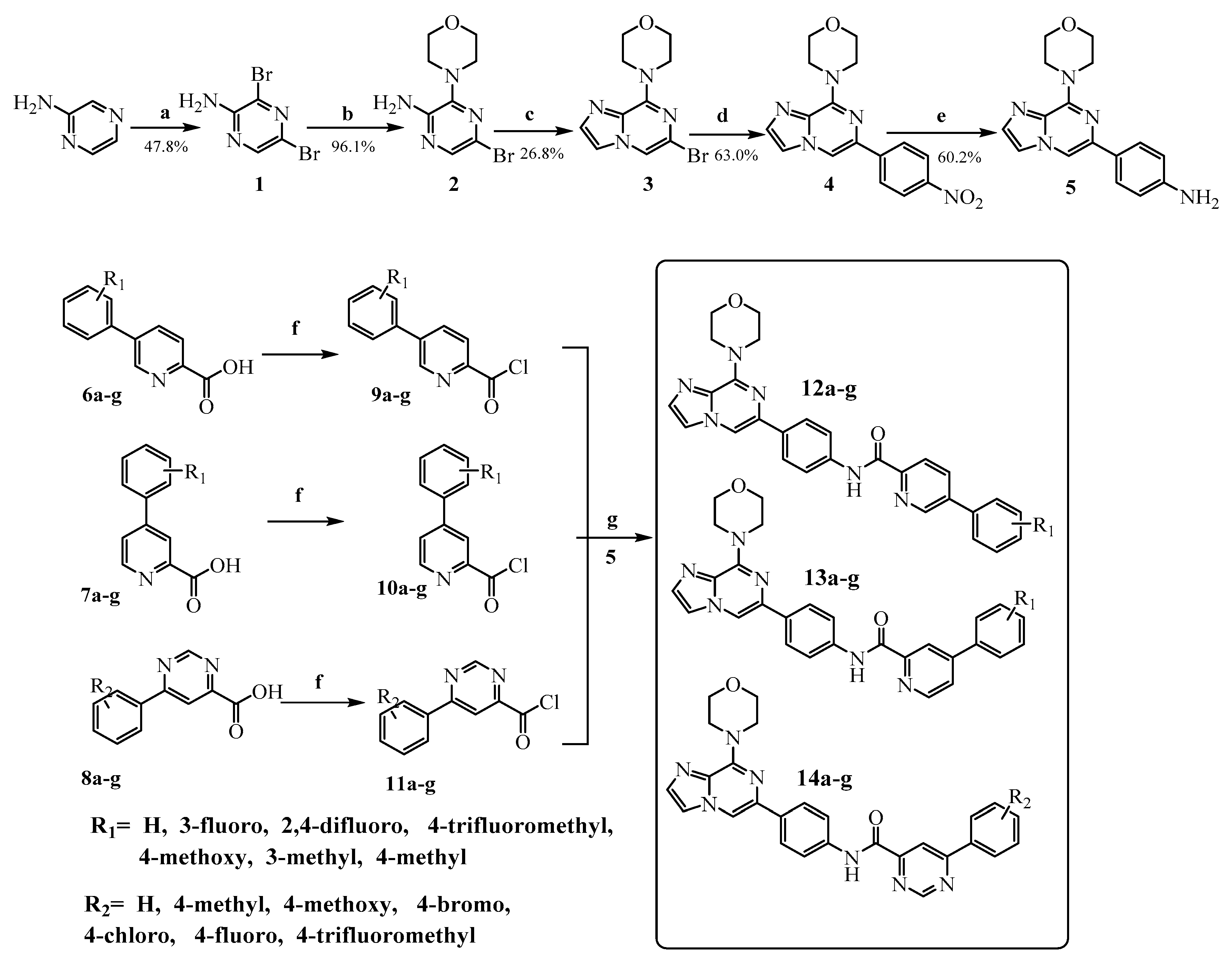
2. Results and Discussion
2.1. Biological Evaluation
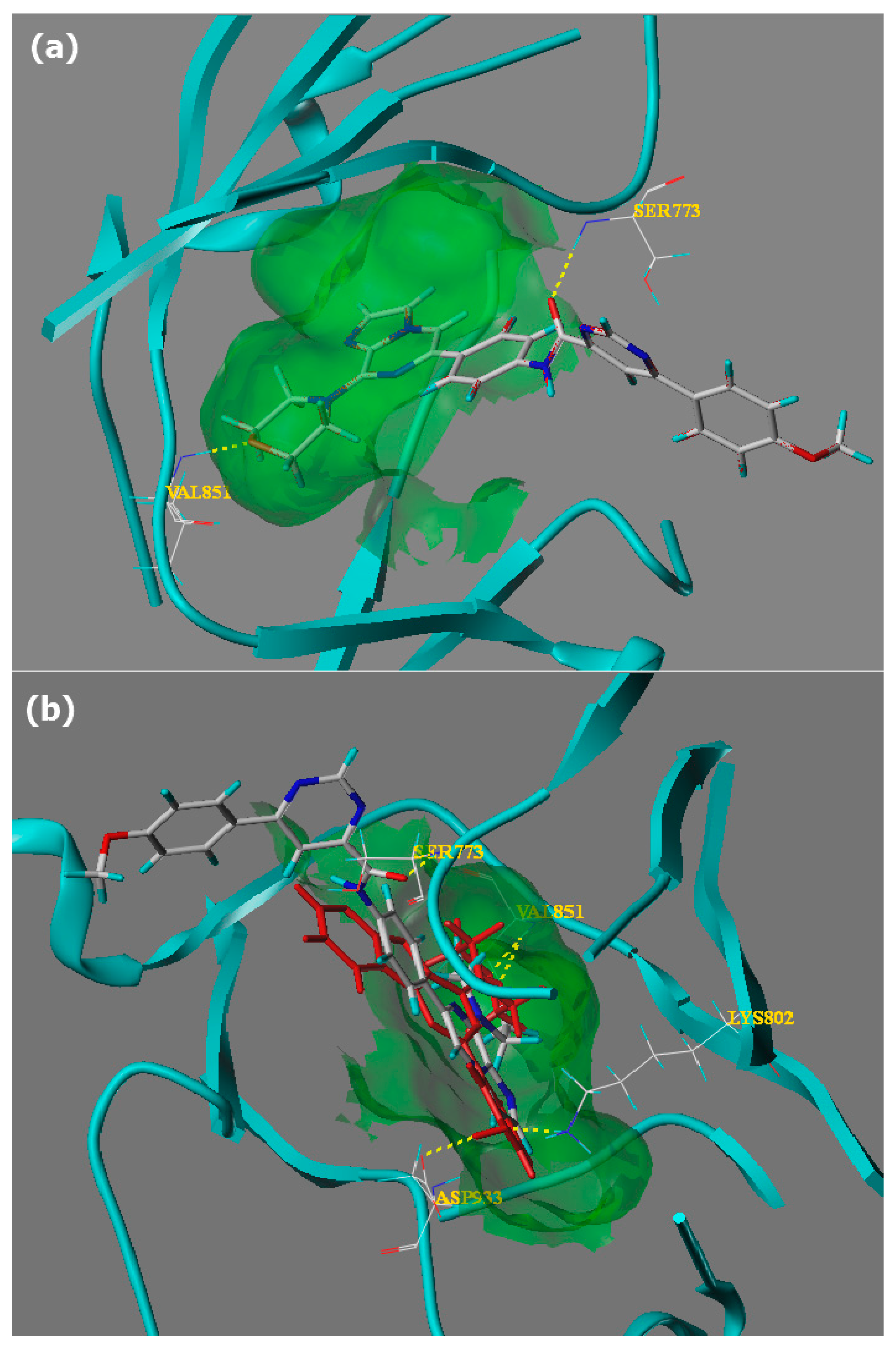
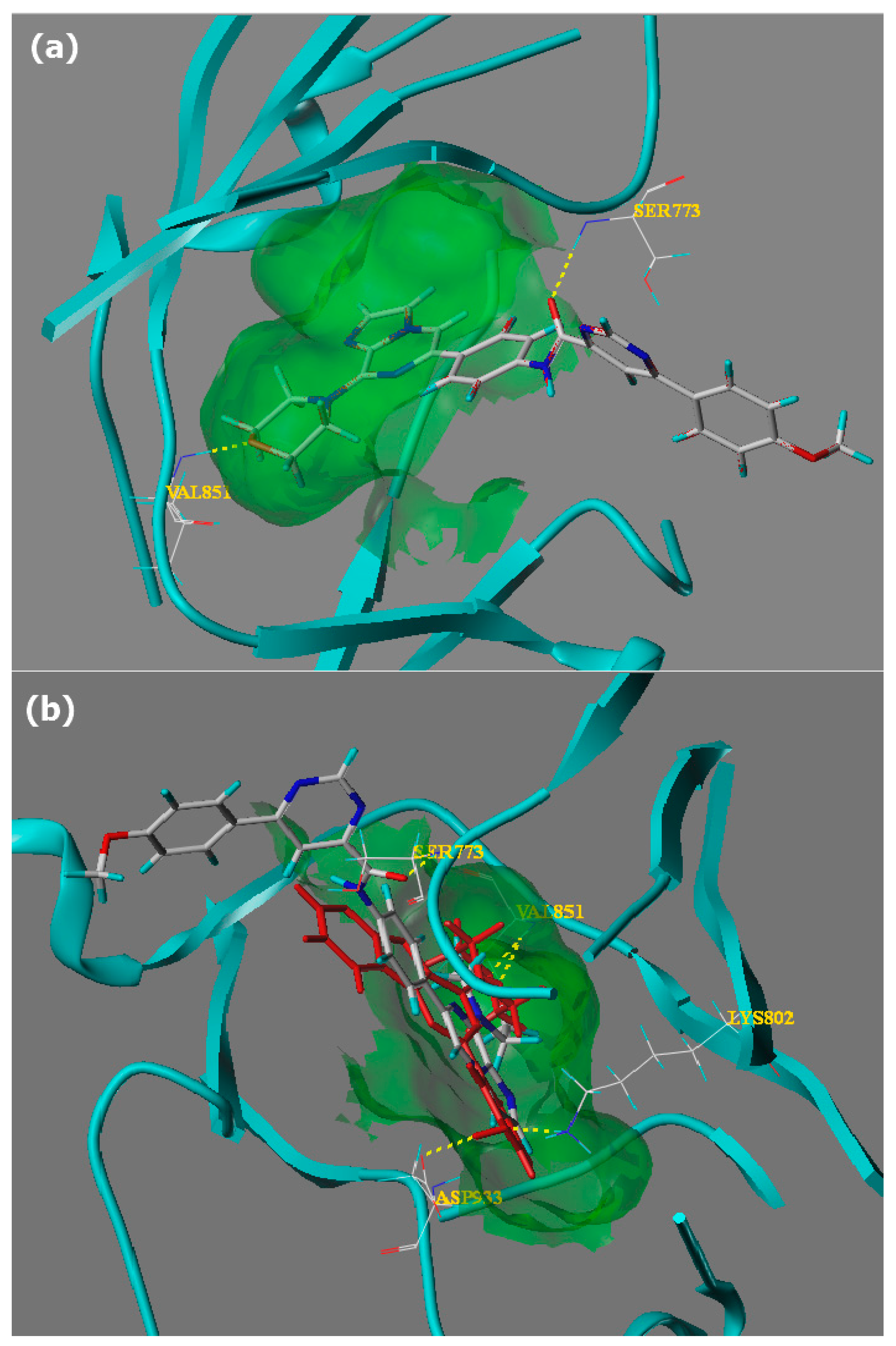
2.2. Molecular Docking Study
3. Experimental Section
3.1. General Information
3.2. Chemistry
3.3. In Vitro Cytotoxicity Assays
3.3.1. The Selection of Cancer Cell Lines
3.3.2. MTT Assay In Vitro
3.4. PI3Kα Kinase Assay
3.5. Docking Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I.; et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Kaizawa, H.; Moritomo, H.; Koizumi, T.; Ohishi, T.; Yamano, M.; Okada, M.; Ohta, M.; Tsukamoto, S.; Raynaud, F.I.; et al. Synthesis and biological evaluation of pyrido[3′,2′:4,5] furo[3,2-d]pyrimidine derivatives as novel PI3 kinase p110á inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2438–2442. [Google Scholar] [CrossRef] [PubMed]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The identification of 2-(1H-Indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-yl methyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.M.; Dehnhardt, C.M.; Delos Santos, E.; Chen, Z.; Dos Santos, O.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis (morpholino-1,3,5-triazine) derivatives: Potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: Discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J. Med. Chem. 2010, 53, 2636–2645. [Google Scholar] [CrossRef] [PubMed]
- Martínez González, S.; Hernández, A.I.; Varela, C.; Lorenzo, M.; Ramos-Lima, F.; Cendón, E.; Cebrián, D.; Aguirre, E.; Gomez-Casero, E.; Albarrán, M.I.; et al. Rapid identification of ETP-46992, orally bioavailable PI3K inhibitor, selective versus mTOR. Bioorg. Med. Chem. Lett. 2012, 22, 5208–5214. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Sun, C.; Xu, S.; Wu, C.; Wu, J.; Xu, M.; Zhao, H.; Chen, L.; Zeng, W.; Zheng, P. Design, synthesis, anticancer activity and docking studies of novel 4-morpholino-7,8-dihydro-5H-thiopyrano[4,3-d] pyrimidine derivatives as mTOR inhibitors. Bioorg. Med. Chem. 2014, 22, 6746–6754. [Google Scholar] [CrossRef]
- Lei, F.; Sun, C.; Xu, S.; Wang, Q.; OuYang, Y.; Chen, C.; Xia, H.; Wang, L.; Zheng, P.; Zhu, W. Design, synthesis, biological evaluation and docking studies of novel 2-substituted-4-morpholino-7,8-dihydro-5H-thiopyrano[4,3-d]pyrimidine derivatives as dual PI3Kα/mTOR inhibitors. Eur. J. Med. Chem. 2016, 116, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Chen, C.; Xu, S.; Wang, J.; Zhu, Y.; Kong, D.; Tao, H.; Jin, M.; Zheng, P.; Zhu, W. Synthesis and anticancer activity of novel 4-morpholino-7,8-dihydro-5H-thiopyrano[4,3-d]pyrimidine derivatives bearing chromone moiety. Bioorg. Med. Chem. 2016, 24, 3862–3869. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, W.; Sun, C.; Wang, C.; Zhu, W.; Zheng, P. Synthesis and Biological Evaluation of Novel 4-Morpholino-7,8-dihydro-5H-thiopyrano[4,3-d]pyrimidine Derivatives Bearing Phenylpyridine/Phenylpyrimidine-Carboxamides. Molecules 2016, 21, 1447. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, W.; Xu, S.; Wang, J.; Tang, Q.; Wu, C.; Zhao, Y.; Zheng, P. Synthesis, and docking studies of phenylpyrimidine-carboxamide derivatives bearing 1H-pyrrolo[2,3-b]pyridine moiety as c-Met inhibitors. Bioorg. Med. Chem. 2016, 24, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Wang, X.; Wang, Y. The Relationship between the PI3K/Akt/mTOR Signal Transduction Pathway and Non-small Cell Lung Cancer. Chin. J. Lung Cancer 2009, 12, 312–315. [Google Scholar]
- López-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 12a–g, 13a–g and 14a–g are available from the authors.

| Compd. | R | Yield (%) | IC50 (μM) a | ||
|---|---|---|---|---|---|
| A549 | PC-3 | MCF-7 | |||
| 12a | H | 96.1 | 58.73 ± 1.99 | 50.28 ± 2.04 | ND |
| 12b | 3-fluoro | 79.4 | NA | NA | NA |
| 12c | 2,4-difluoro | 90.2 | NA | NA | NA |
| 12d | 4-trifluoromethyl | 83.5 | 15.27 ± 0.88 | 69.70 ± 1.13 | ND |
| 12e | 4-methoxy | 93.3 | NA | NA | NA |
| 12f | 3-methyl | 72.6 | 19.11 ± 1.10 | 26.78 ± 1.47 | ND |
| 12g | 4-methyl | 89.1 | 68.12 ± 0.89 | 42.32 ± 0.86 | ND |
| 13a | H | 97.0 | NA | NA | NA |
| 13b | 3-fluoro | 88.0 | NA | NA | NA |
| 13c | 2,4-difluoro | 91.2 | NA | NA | NA |
| 13d | 4-trifluoromethyl | 81.8 | 41.99 ± 1.49 | 55.64 ± 1.32 | 62.48 ± 1.99 |
| 13e | 4-methoxy | 93.1 | 23.95 ± 0.81 | 59.68 ± 2.22 | 74.92 ± 1.14 |
| 13f | 3-methyl | 91.5 | 10.53 ± 0.82 | 65.61 ± 0.52 | 74.19 ± 1.86 |
| 13g | 4-methyl | 98.0 | 11.23 ± 1.08 | NA | 7.89 ± 0.81 |
| 14a | H | 82.7 | 10.75 ± 1.62 | NA | NA |
| 14b | 4-methyl | 79.5 | 8.88 ± 0.97 | 25.04 ± 1.19 | 6.69 ± 2.04 |
| 14c | 4-methoxy | 77.8 | 6.39 ± 1.04 | 12.65 ± 0.95 | 10.23 ± 1.62 |
| 14d | 4-bromo | 90.9 | 14.36 ± 1.01 | 37.14 ± 0.97 | NA |
| 14e | 4-chloro | 93.2 | 17.38 ± 1.01 | NA | NA |
| 14f | 4-fluoro | 80.8 | 58.17 ± 4.67 | NA | 47.06 |
| 14g | 4-trifluoromethyl | 77.6 | NA | 96.63 ± 2.58 | ND |
| Compound II b,c | - | - | 8.37 ± 0.10 | 11.34 ± 0.11 | 9.26 ± 0.82 |
| GDC-0941 b | - | - | 6.99 ± 0.21 | 0.20 ± 0.08 | 0.07 ± 0.03 |

{kind=link}
{kind=link}
{kind=link}
| Compound No. | PI3Kα |
|---|---|
| IC50 (μM) a or 10 μM Inhibitory % a | |
| 14b | 58.0 ± 4.4 ≅10 μM |
| 14c | 1.25 ± 0.13 |
| Compound II b,c | 7.39 ± 0.19 [11] |
| GDC-0941 b | 0.003 [4] |
| PI-103 b | 0.075 ± 0.018 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Sun, C.; Chen, C.; Zheng, P.; Zhou, Y.; Zhou, H.; Zhu, W. Synthesis and Biological Evaluation of Novel 8-Morpholinoimidazo[1,2-a]pyrazine Derivatives Bearing Phenylpyridine/Phenylpyrimidine-Carboxamides. Molecules 2017, 22, 310. https://doi.org/10.3390/molecules22020310
Xu S, Sun C, Chen C, Zheng P, Zhou Y, Zhou H, Zhu W. Synthesis and Biological Evaluation of Novel 8-Morpholinoimidazo[1,2-a]pyrazine Derivatives Bearing Phenylpyridine/Phenylpyrimidine-Carboxamides. Molecules. 2017; 22(2):310. https://doi.org/10.3390/molecules22020310
Chicago/Turabian StyleXu, Shan, Chengyu Sun, Chen Chen, Pengwu Zheng, Yong Zhou, Hongying Zhou, and Wufu Zhu. 2017. "Synthesis and Biological Evaluation of Novel 8-Morpholinoimidazo[1,2-a]pyrazine Derivatives Bearing Phenylpyridine/Phenylpyrimidine-Carboxamides" Molecules 22, no. 2: 310. https://doi.org/10.3390/molecules22020310
APA StyleXu, S., Sun, C., Chen, C., Zheng, P., Zhou, Y., Zhou, H., & Zhu, W. (2017). Synthesis and Biological Evaluation of Novel 8-Morpholinoimidazo[1,2-a]pyrazine Derivatives Bearing Phenylpyridine/Phenylpyrimidine-Carboxamides. Molecules, 22(2), 310. https://doi.org/10.3390/molecules22020310