
[2 + 2] Photodimerization of Naphthylvinylpyridines through Cation-? Interactions in Acidic Solution

Abstract
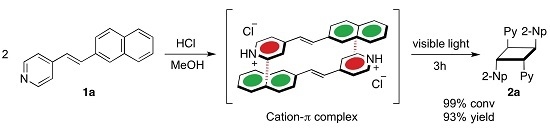
:
1. Introduction
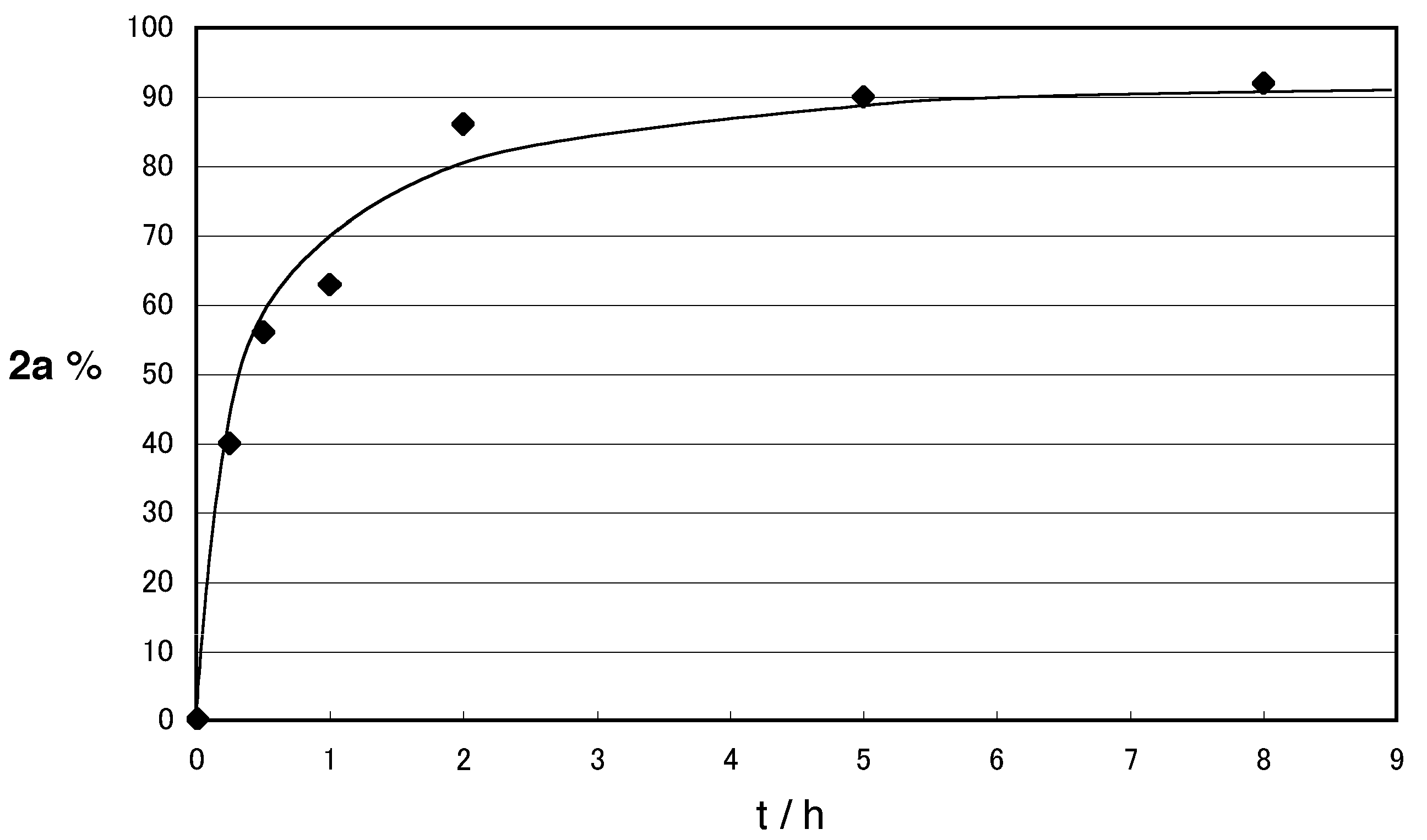
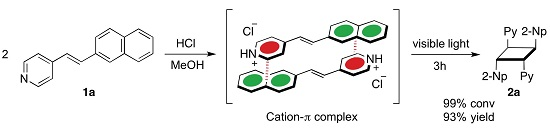
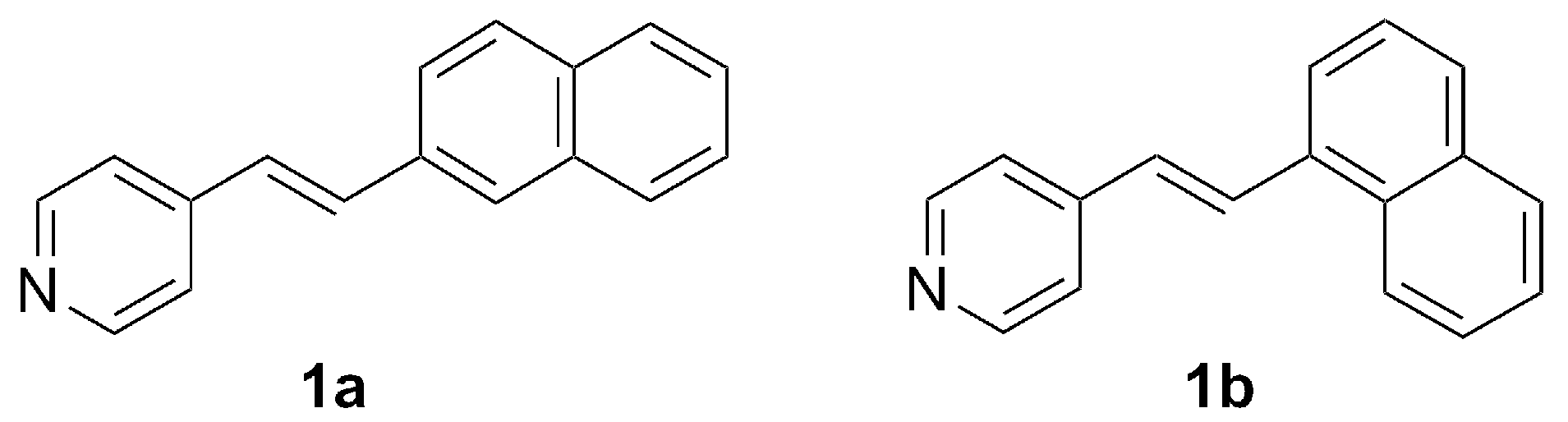
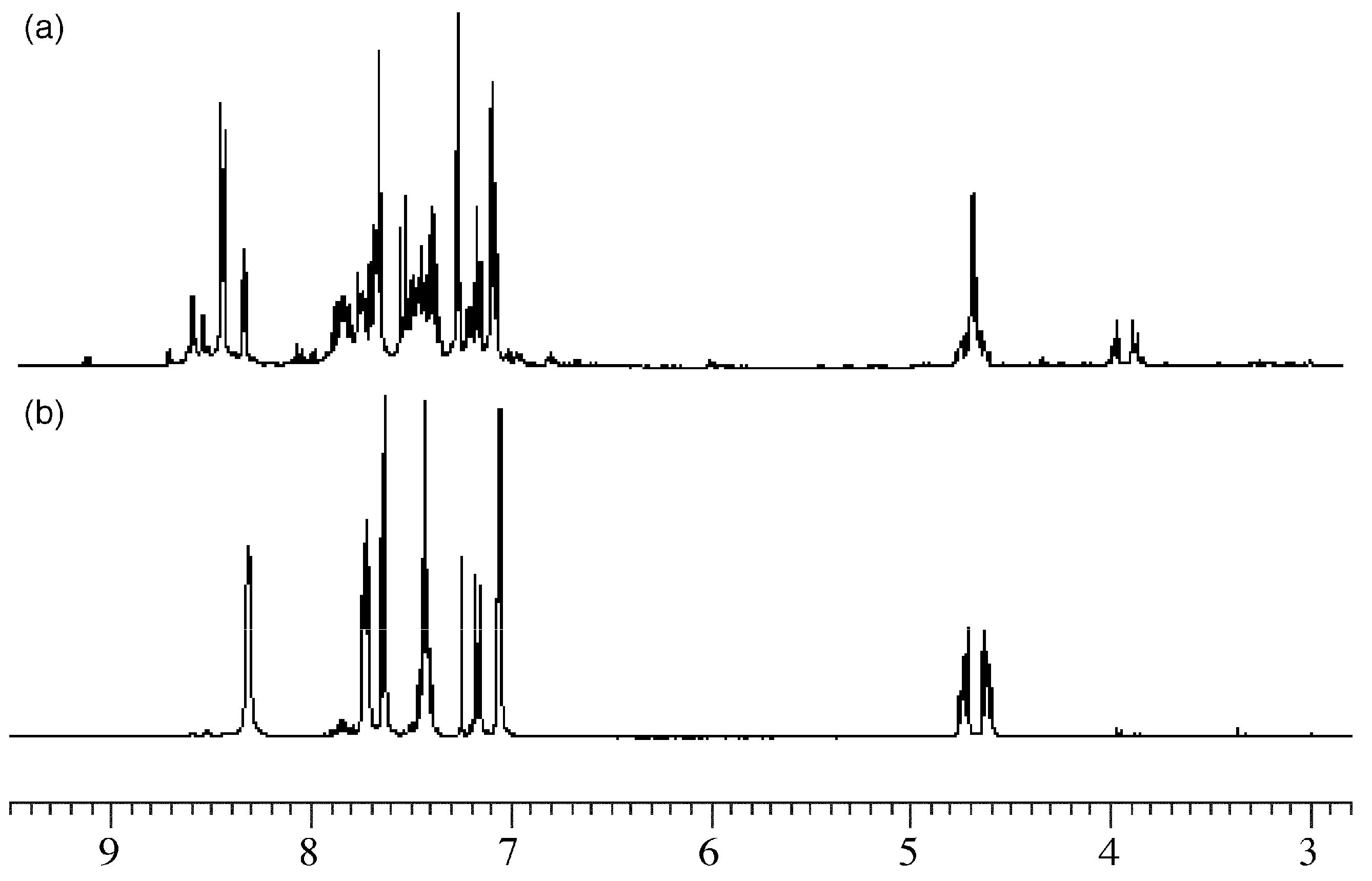
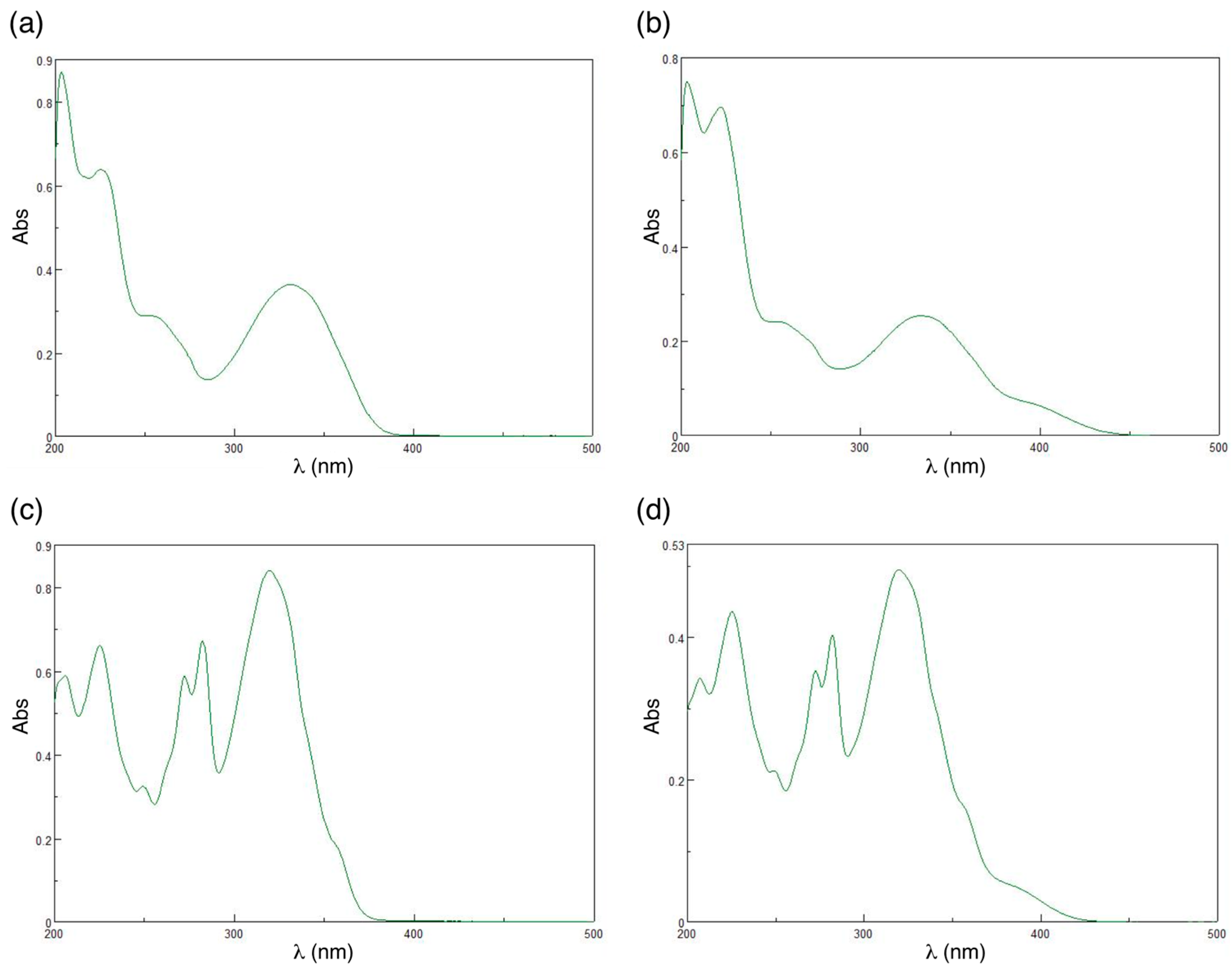
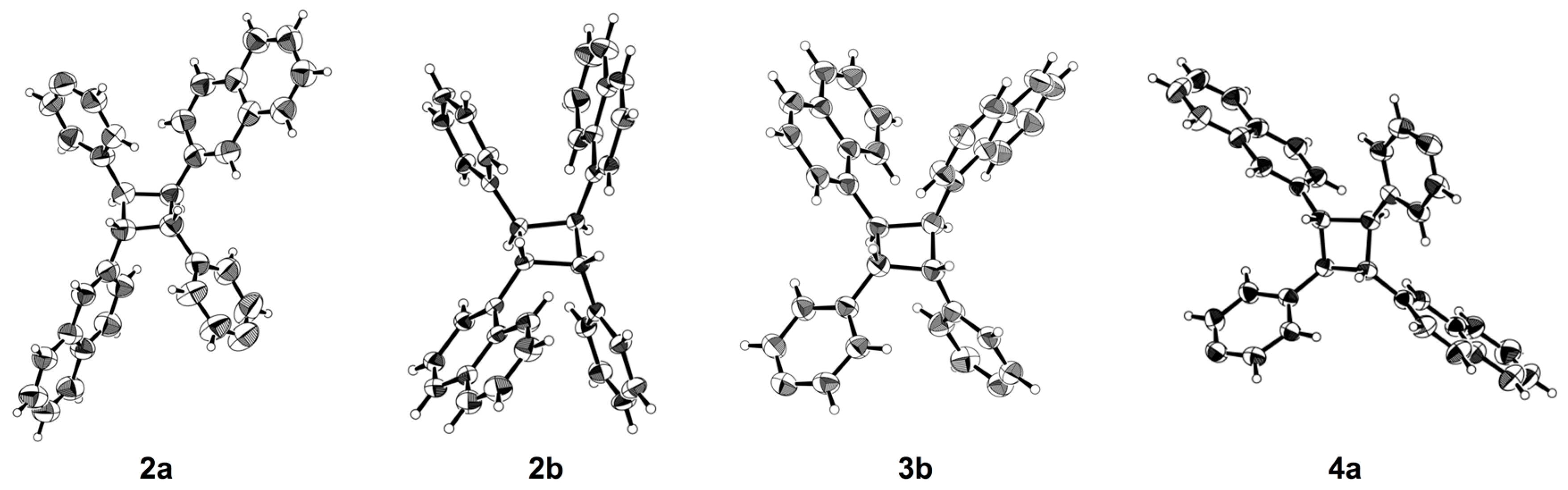
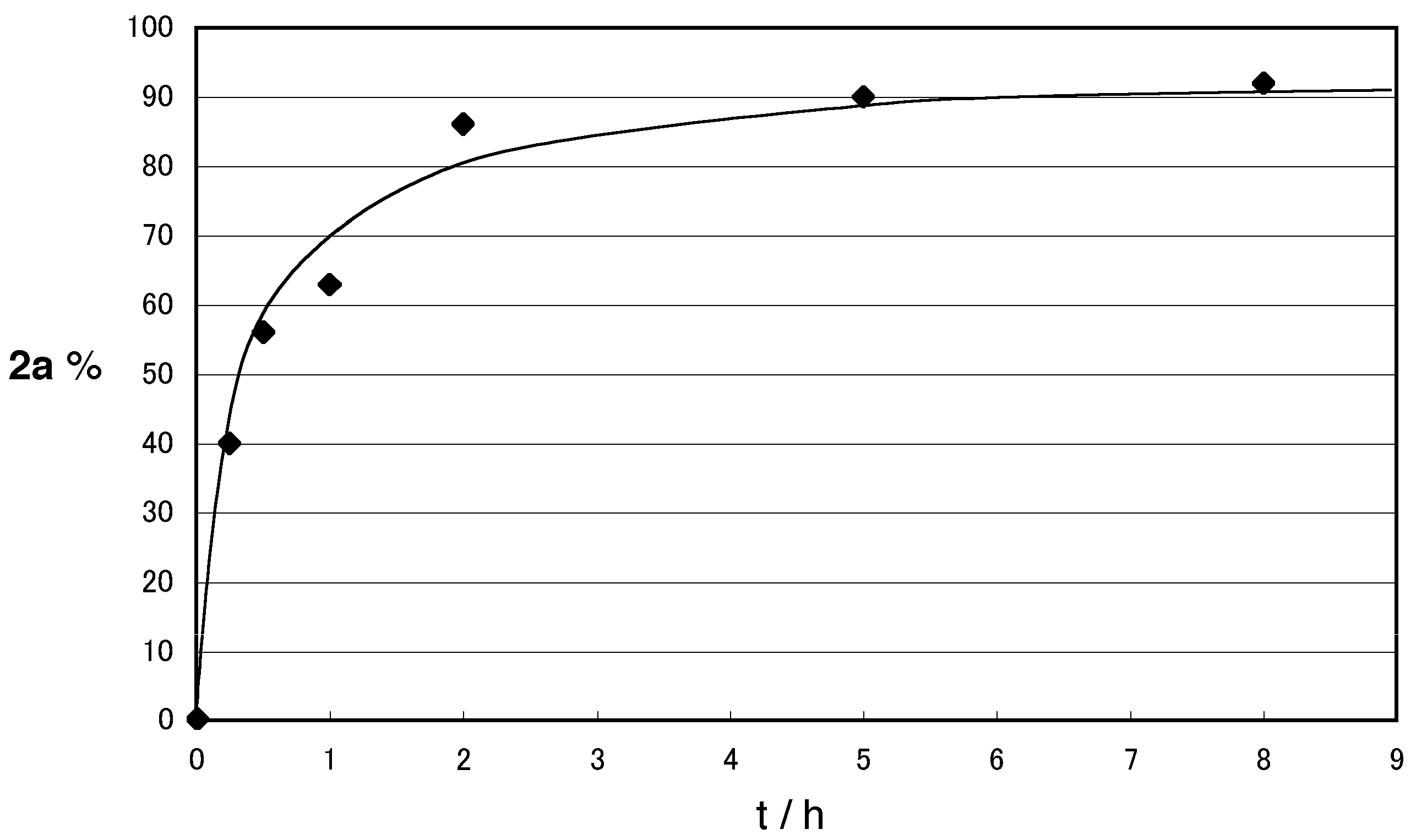
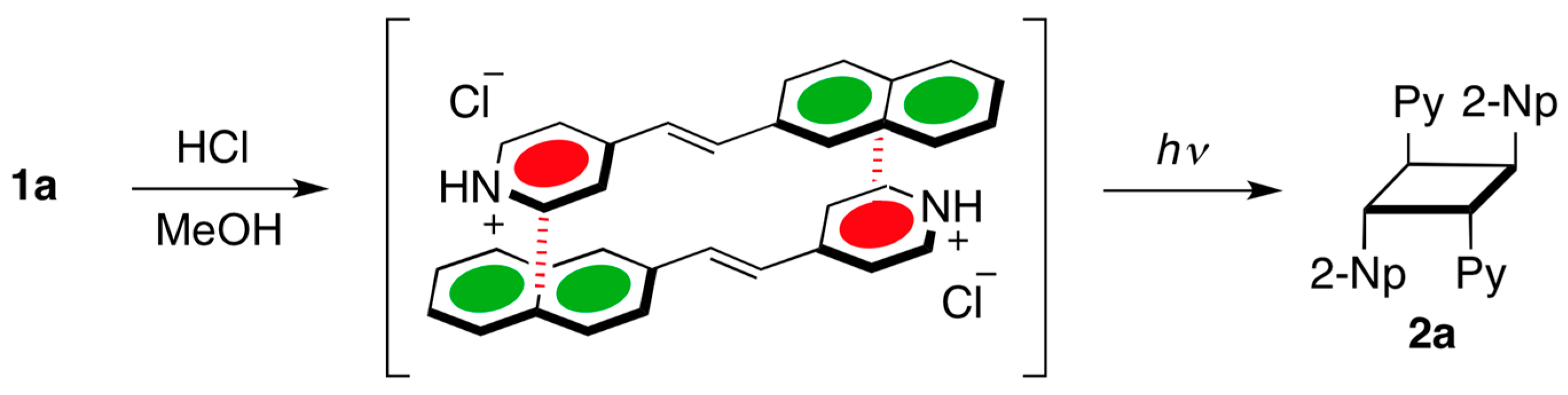
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Representative Procedure for Irradiation of Naphthylvinylpyridines under Acidic Conditions
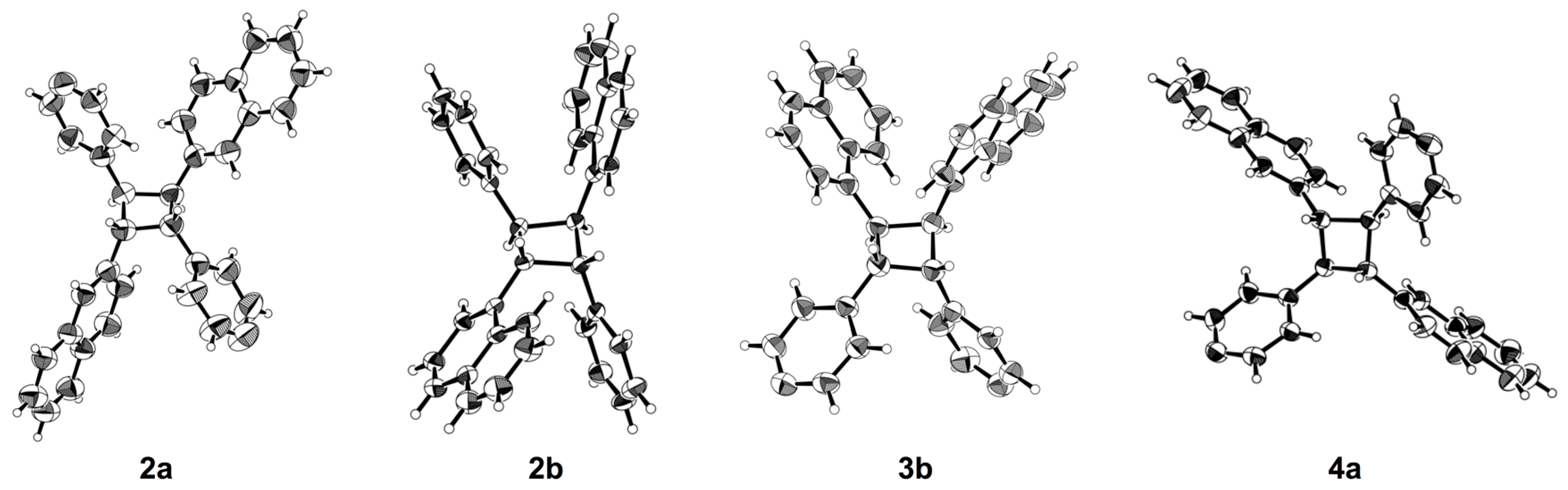
3.3. X-ray Crystallography
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Horspool, W.M. Synthetic Organic Photochemistry; Plenum Press: New York, NY, USA, 1984; pp. 1–143. [Google Scholar]
- Coyle, J.C. Photochemistry in Organic Synthesis; The Royal Society of Chemistry: London, UK, 1986; pp. 163–188. [Google Scholar]
- Griesbeck, A.G.; Mattay, J. Synthetic Organic Photochemistry; Marcel Dekker: New York, NY, USA, 2005; pp. 141–160. [Google Scholar]
- Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev. 2016, 116, 9748–9815. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V. Organic photochemistry in organized media. Tetrahedron 1986, 42, 5753–5839. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Venkatesan, K. Photochemical reactions of organic crystals. Chem. Rev. 1987, 87, 433–481. [Google Scholar] [CrossRef]
- Toda, F. Solid State Organic Chemistry: Efficient Reactions, Remarkable Yields, and Stereoselectivity. Acc. Chem. Res. 1995, 28, 480–486. [Google Scholar] [CrossRef]
- Gamlin, J.N.; Jones, R.; Leibovich, M.; Patrick, B.; Scheffer, J.R.; Trotter, J. The ionic auxiliary concept in solid state organic photochemistry. Acc. Chem. Res. 1996, 29, 203–209. [Google Scholar] [CrossRef]
- Ito, Y. Solid-state photoreactions in two-component crystals. Synthesis 1998, 1–32. [Google Scholar] [CrossRef]
- Nagarathinam, M.; Peedikakkal, A.M.P.; Vittal, J.J. Stacking of double bonds for photochemical [2 + 2] cycloaddition reactions in the solid state. Chem. Commun. 2008, 5277–5288. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, I.G.; MacGillivray, L.R. Metal-mediated reactivity in the organic solid state: From self-assembledcomplexes to metal–organic frameworks. Chem. Soc. Rev. 2007, 36, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, L.R.; Papaefstathiou, G.S.; Friščić, T.; Hamilton, T.D.; Bucar, D.-K.; Chu, Q.; Varshney, D.B.; Georgiev, I.G. Supramolecular control of reactivity in the solid state: from templates to ladderanes to metal-organic frameworks. Acc. Chem. Res. 2008, 41, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Skiredj, A.; Beniddir, M.A.; Joseph, D.; Leblanc, K.; Bernadat, G.; Evanno, L.; Poupon, E. Spontaneous biomimetic formation of (±)-dictazole B under irradiation with artificial sunlight. Angew. Chem. Int. Ed. 2014, 53, 6419–6424. [Google Scholar] [CrossRef] [PubMed]
- Serrano, E.; Juan, A.; Garca-Montero, A.; Soler, T.; Jimenez-Marquez, F.; Cativiela, C.; Gomez, M.V.; Urriolabeitia, E.P. Stereoselective Synthesis of 1,3-Diaminotruxillic acid derivatives: an advantageous combination of C-H-ortho-Palladation and On-Flow [2 + 2]-Photocycloaddition in microreactors. Chem. Eur. J. 2016, 22, 144–152. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, L.R. Organic Synthesis in the Solid State via Hydrogen-Bond-Driven Self-Assembly. J. Org. Chem. 2008, 73, 3311–3317. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.; Sivaguru, J. Supramolecular photochemistry as a potential synthetic tool: photocycloaddition. Chem. Rev. 2016, 116, 9914–9993. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.C.; Dougherty, D.A. The cation-π interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Uematsu, N.; Yamashita, K. Role of cation-π interactions in the photodimerization of trans-4-styrylpyridines. J. Am. Chem. Soc. 2007, 129, 12100–12101. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nojiri, Y.; Sugawara, M. [2 + 2] Photodimerization of (Z)-4-styrylpyridine through a cation–π interaction: Formation of cis-cis-trans dimers. Tetrahedron Lett. 2010, 51, 2533–2535. [Google Scholar] [CrossRef]
- Yamada, S.; Kusafuka, M.; Sugawara, M. [2 + 2] Photodimerization of bispyridylethylenes by a controlled shift of the protonation equilibrium. Tetrahedron Lett. 2013, 54, 3997–4000. [Google Scholar] [CrossRef]
- Yamada, S.; Kawamura, C. [4 + 4] Photodimerization of azaanthracenes in both solution and solid phase controlled by cation-π interactions. Org. Lett. 2012, 14, 1572–1575. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Tokugawa, Y. Cation-π controlled solid-state photodimerization of 4-Azachalcones. J. Am. Chem. Soc. 2009, 131, 2098–2099. [Google Scholar] [CrossRef] [PubMed]
- Amunugama, R.; Rodgers, M.T. Cation-π interactions with a model for an extended π network: Absolute binding energies of alkali metal cation–naphthalene complexes determined by threshold collision-induced dissociation and theoretical studies. Int. J. Mass Spectrom. 2003, 227, 1–20. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ikuta, S. A theoretical study on the conformations, energetics, and solvation effects on the cation–π interaction between monovalent ions Li+, Na+, and K+ and naphthalene molecules. J. Mol. Struct. 1999, 468, 85–94. [Google Scholar] [CrossRef]
- Hewlins, M.J.E.; Salter, R. The photochemical cyclodehydrogenation route to polycyclic azaarenes. Synthesis 2007, 2164–2174. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97; Computer Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sample Availability: Not available.




| Entry | Compd. | HCl (eq.) | Solvent b | Conv. (%) c | Products (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | (Z)-1 | |||||
| 1 | 1a d | 0 | THF | 99 | 21 | 46 | 14 | 17 | 0 |
| 2 | 1a | 0 | MeOH e | 82 | 22 | 50 | 12 | 4 | 11 |
| 3 | 1a | 1 | MeOH | 98 | 90 | 6 | 4 | 0 | 0 |
| 4 | 1a | 3 | MeOH | 99 | 93 | 2 | 3 | 2 | 0 |
| 5 | 1b | 0 | MeOH | 91 | 45 | 35 | 6 | 0 | 14 |
| 6 | 1b | 1 | MeOH f | 91 | 75 | 0 | 0 | 0 | 25 |
| 7 | 1b | 1 | MeOH/H2O | 93 | 94 | 0 | 0 | 0 | 6 |
| 8 | 1b | 3 | MeOH/H2O | 93 | 94 | 0 | 0 | 0 | 6 |

| Entry | Compd. | HCl (eq.) | Solvent | Conv. (%) | Products (%) b,c | ||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | (Z)-1 | |||||
| 1 | 1a | 0 | THF | 7 | 0 | 0 | 0 | 0 | 100 |
| 2 | 1a | 1 | MeOH | 99 | 93 | 3 | 2 | 2 | 0 |
| 3 | 1b | 0 | MeOH | 48 | 46 | 39 | 0 | 0 | 15 |
| 4 | 1b | 1 | MeOH/H2O | 88 | 90 | 0 | 0 | 0 | 10 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, S.; Nojiri, Y. [2 + 2] Photodimerization of Naphthylvinylpyridines through Cation-? Interactions in Acidic Solution. Molecules 2017, 22, 491. https://doi.org/10.3390/molecules22030491
Yamada S, Nojiri Y. [2 + 2] Photodimerization of Naphthylvinylpyridines through Cation-? Interactions in Acidic Solution. Molecules. 2017; 22(3):491. https://doi.org/10.3390/molecules22030491
Chicago/Turabian StyleYamada, Shinji, and Yuka Nojiri. 2017. "[2 + 2] Photodimerization of Naphthylvinylpyridines through Cation-? Interactions in Acidic Solution" Molecules 22, no. 3: 491. https://doi.org/10.3390/molecules22030491
APA StyleYamada, S., & Nojiri, Y. (2017). [2 + 2] Photodimerization of Naphthylvinylpyridines through Cation-? Interactions in Acidic Solution. Molecules, 22(3), 491. https://doi.org/10.3390/molecules22030491