
Improved Synthesis of 1-O-Acyl-β-d-Glucopyranose Tetraacetates

Abstract
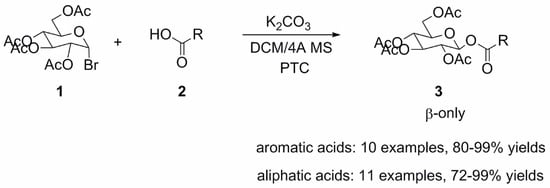
:
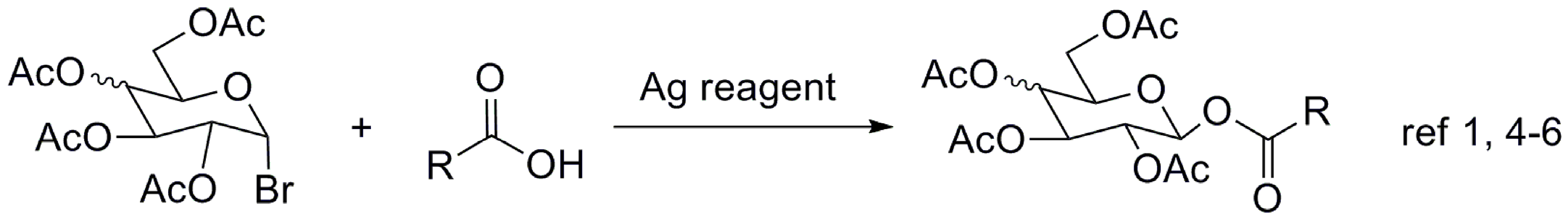
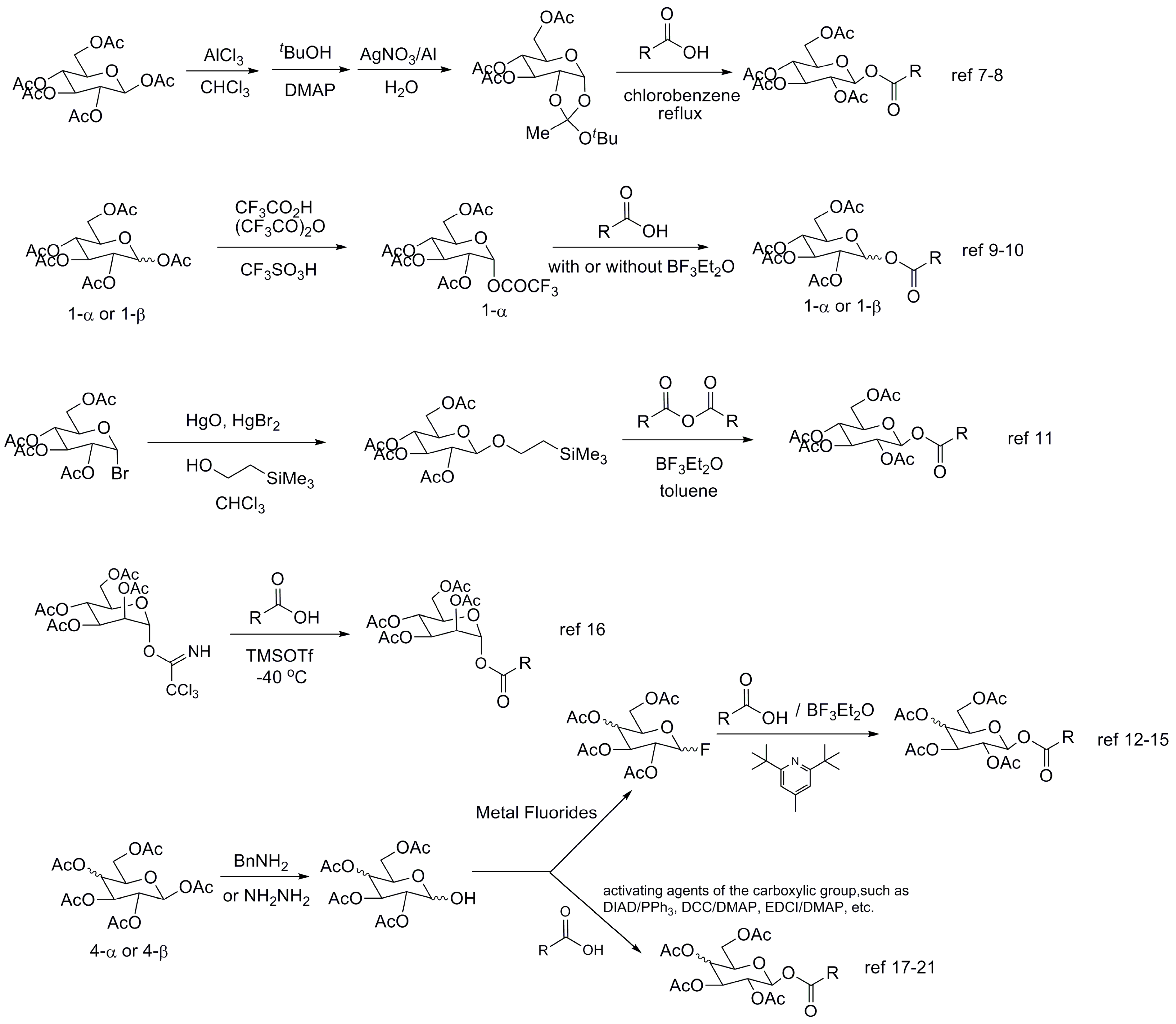
1. Introduction
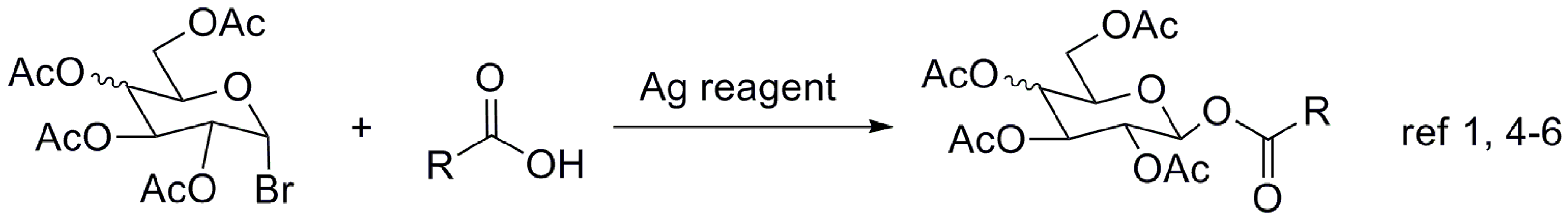
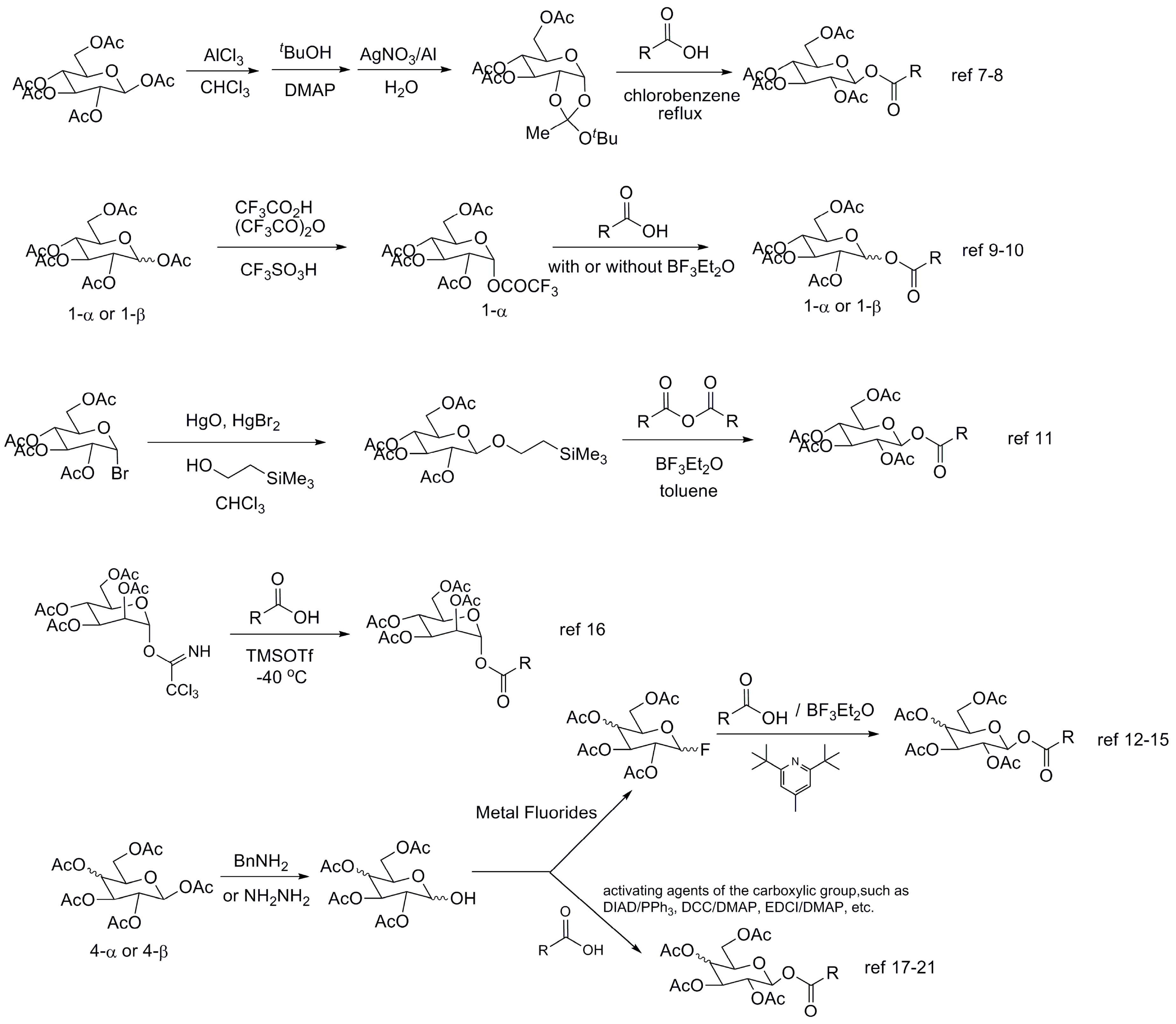
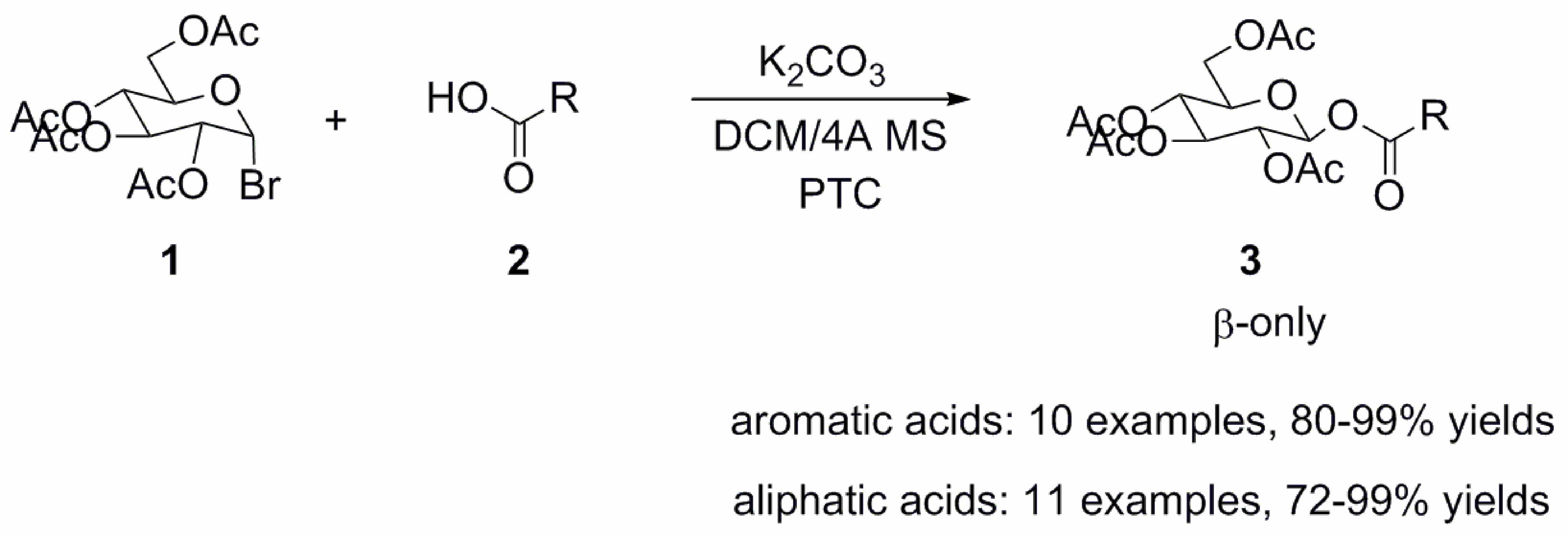
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. General Procedure for the Synthesis of 1-O-Acyl-β-d-Glucopyranose Tetraacetates
3.3. Scaled-Up Synthesis of Compound 3
3.4. Scaled-Up Synthesis of Compound 24
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Nishikawa, Y.; Yoshimoto, K.; Kurono, G.; Michishita, K. Chemical and Biochemical Studies on Carbohydrate Esters. I. Preparation and Properties of 1-O-Acyl-β-d-glucopyranose Tetraacetates. Chem. Pharm. Bull. 1975, 23, 597–603. [Google Scholar] [CrossRef]
- Tschesche, R.; Kammerer, F.J.; Wulff, G. Über Die Antibiotisch Wirksamen Substanzen der Tulpe (Tulipa Gesneriana). Tetrahedron Lett. 1968, 9, 701–706. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Okabe, M.; Yoshimoto, K.; Kurono, G.; Fukuoka, F. Chemical and Biochemical Studies on Carbohydrate Esters. II. Antitumor Activity of Saturated Fatty Acids and Their Ester Derivatives against Ehrlich Ascites Carcinoma. Chem. Pharm. Bull. 1976, 24, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Xu, M.; Yao, W.; Mao, J. Room-temperature Ionic Liquids Enhanced Green Synthesis of β-glycosyl 1-ester. Carbohydr. Res. 2015, 407, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Xiao, G.Q.; Cai, M.S. Studies on Carbohydrates XII. An Improved Koenigs Knorr Method for Highly Stereoselectives Synthesis of 1-O-Acyl-β-d-Galactopyranose Tetraacetates. Chin. Chem. Lett. 1992, 3, 711–712. [Google Scholar]
- Kunz, H.; Wernig, P. New 1-O-Alkenoic Acid Ester(s) of Carbohydrate(s)—Used as Glycosyl Donors in Synthesis of Glycoside(s) and Saccharide(s). Patent D.E. 4009634, 26 March 1990. [Google Scholar]
- Greimel, P.; Lapeyre, M.; Nagatsuka, Y.; Hirabayashi, Y.; Ito, Y. Syntheses of Phosphatidyl-β-D-glucoside Analogues to Probe Antigen Selectivity of Monoclonal Antibody ‘DIM21’. Bioorg. Med. Chem. 2008, 16, 7210–7217. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Hamada, A. Studies on Glycosylation. III. A Novel, Stereospecific Synthesis of 1-O-Acyl- and 1-Aryl-β-d-glucopyranose Tetraacetates via the 1, 2-t-Butyl-orthoacetate. Chem. Pharm. Bull 1976, 24, 1165–1168. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimadate, T. Synthesis of Glycosyl Trifluoroacetates and Their Reactions with Carboxylic Acids. Chem. Pharm. Bull. 1986, 34, 4069–4074. [Google Scholar] [CrossRef]
- Yu, C.; Li, Z.; Cai, M. Studies on Carbohydrates IV. A Novel Highly Stereoselective Synthesis of 1-O-Acyl-β-d-Glucopyranose Tetraacetates via the Glucosyl Trifluoroacetate. Synth. Commun. 1990, 20, 943–948. [Google Scholar] [CrossRef]
- Jansson, K.; Ahlfors, S.; Frejd, T.; Kihlberg, J.; Magnusson, G. 2-(Trimethylsilyl)ethyl Glycosides. Synthesis, Anomeric Deblocking, and Transformation into 1,2-Trans 1-O-Acyl Sugars. J. Org. Chem. 1988, 53, 5629–5647. [Google Scholar] [CrossRef]
- Sim, M.M.; Kondo, H.; Wong, C. Synthesis and Use of Glycosyl Phosphites: An Effective Route to Glycosyl Phosphates, Sugar Nucleotides, and Glycosides. J. Am. Chem. Soc. 1993, 115, 2260–2267. [Google Scholar] [CrossRef]
- Van, T.N.; Claessens, S.; Habonimana, P.; Tehrani, K.A.; Puyvelde, L.V.; Kimpe, N.D. Synthesis of Harounoside, A Naturally Occurring Pentalongin Hydroquinone Bisglucoside. Synlett 2006, 17, 2469–2471. [Google Scholar]
- Shimizu, M.; Togo, H.; Yokoyama, M. Chemistry of Glycosyl Fluorides. Synthesis 1998, 30, 799–822. [Google Scholar] [CrossRef]
- Oyama, K.; Kondo, T. Highly Efficient β-Glucosylation of the Acidic Hydroxyl Groups, Phenol and Carboxylic Acid, with an Peracetylated Glucosyl Fluoride Using a Combination of BF3·Et2O and DTBMP as a Promoter. Synlett 1999, 10, 1627–1629. [Google Scholar] [CrossRef]
- Pakulski, Z.A.; Cmoch, P.; Oklestkova, L.; Strnad, M. Saccharide Lupane Derivatives, Their Use and Pharmaceutical Compositions Containing These Derivatives. Patent W.O. 2009094958, 6 August 2009. [Google Scholar]
- Smith, A.B.; Halc, K.J.; Rivero, R.A. An Efficient Synthesis of Glycosyl Esters Exploiting the Mitsunobu Reaction. Tetrahedron Lett. 1986, 27, 5813–5816. [Google Scholar] [CrossRef]
- Watanabe, Y.; Ishimaru, M.; Ozaki, S. Proximately Assisted and Chemoselectively Cleavable Protecting Groups for Alcohols, 2-[2-(Arylmethyloxy)ethyl]benzoic Esters. Chem. Lett. 1994, 23, 2163–2166. [Google Scholar] [CrossRef]
- Sangmam, C.; Winum, J.; Lucas, M.; Montero, J.; Chavis, C. A Simple, General and Efficient Method for O and N-retinoylation. Application to the Synthesis of 2-Retinoyl-lecithin. Synth. Commun. 1998, 28, 2945–2958. [Google Scholar] [CrossRef]
- Binkowski, C.; Lequart, V.; Hapiot, F.; Tilloy, S.; Cecchelli, R.; Monflier, E.; Martin, P. Adamantoylated Monosaccharides: New Compounds for Modification of the Properties of Cyclodextrin-containing Materials. Carbohydr. Res. 2005, 340, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, J.; Zhu, Y.; Zhang, F.; Yu, B. An Efficient Approach to the Synthesis of Nucleosides: Gold(I)-Catalyzed N-Glycosylation of Pyrimidines and Purines with Glycosyl ortho-Alkynyl Benzoates. Angew. Chem. Int. Ed. 2011, 50, 4933–4936. [Google Scholar] [CrossRef] [PubMed]
- Bliard, C.; Massiot, G.; Nazabadioko, S. Glycosylation of Acids under Phase Transfer Conditions. Partial Synthesis of Saponins. Tetrahedron Lett. 1994, 35, 6107–6108. [Google Scholar] [CrossRef]
- Krishnamurty, H.G.; Dabholkar, K.; Maheshwari, N. Polymer Supported Synthesis of 2,3,4,6-Tetra-O-Acetyl-β-d-Glucopyranosyl Esters of Aromatic Carboxylic Acids. Synth. Commun. 1987, 17, 1323–1329. [Google Scholar] [CrossRef]
- Please see NMR data of compound 3 in Supplementary Materials.
- Please see 2D-NMR data of compound 8 in Supplementary Materials.
- Drillaud, N.; Banaszak-Léonard, E.; Pezron, I.; Len, C. Synthesis and Evaluation of a Photochromic Surfactant for Organic Reactions in Aqueous Media. J. Org. Chem. 2012, 77, 9553–9561. [Google Scholar] [CrossRef] [PubMed]
- Anomeric H shift for β-configuration is located at δ = 5.93 and anomeric H shift for α-configuration is located at δ = 6.57 according to known data.
- Anomeric C shift for β-configuration is located at δ = 92.3 according to known data.
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
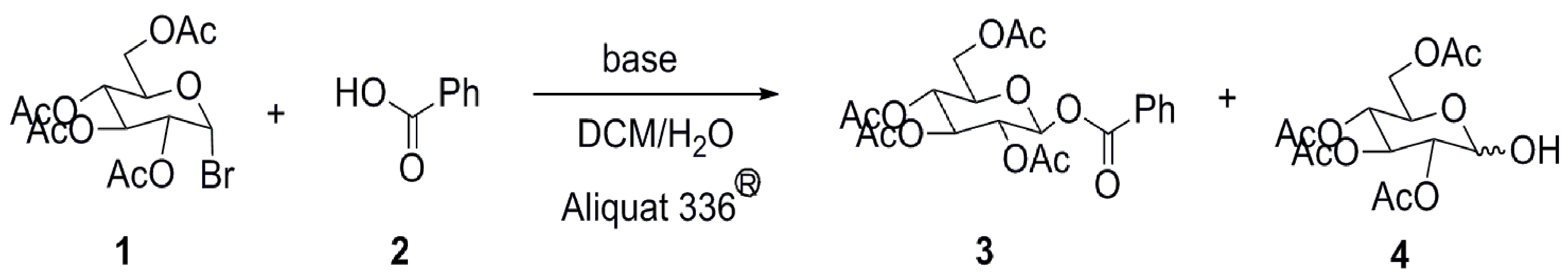
| Entry | Base | H2O | 3 b | 4 b |
|---|---|---|---|---|
| 1 | K2CO3 | 5 mL (278 mmol) c | 35% | 55% |
| 2 | K2CO3 | 2.5 mL (139 mmol) | 54% | 35% |
| 3 | K2CO3 | 0.5 mL (27.8 mmol) | 78% | 12% |
| 4 | K2CO3 | - | 88% | 4% |
| 5 | K2CO3 | - d | 94% | trace |
| 6 | Na2CO3 | - d | 80% | trace |
| 7 | NaHCO3 | - d | 69% | trace |
| 8 | Cs2CO3 | - d | 90% | trace |
| 9 | NaOH | - d | NR | trace |
| 10 | Et3N | - d | NR | trace |
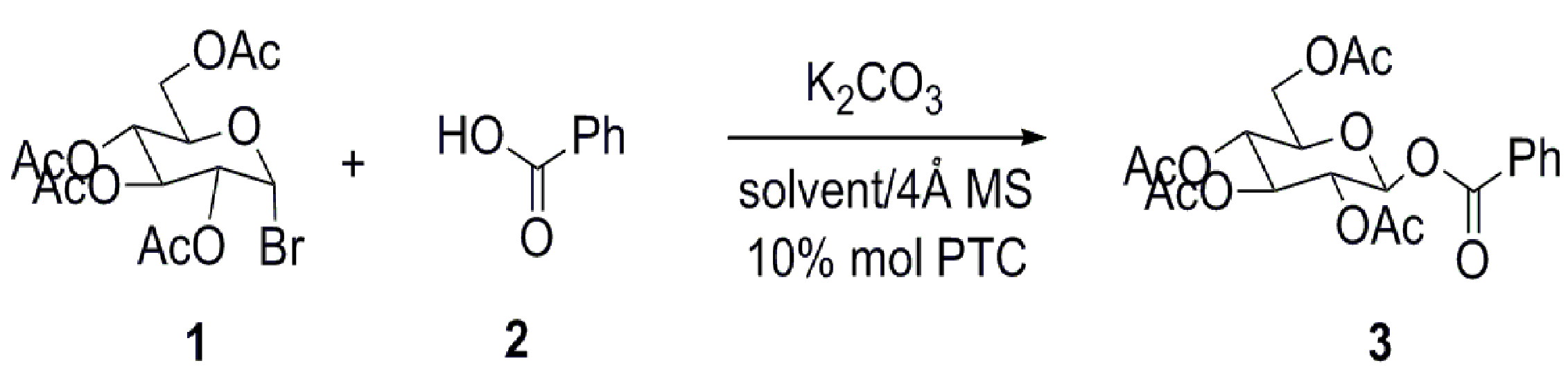
| Entry | PTC | Solvent | 3 b |
|---|---|---|---|
| 1 | TBAB | DCM | 99% |
| 2 | TEAB | DCM | 99% |
| 3 | BEAC | DCM | 96% |
| 4 | CMAB | DCM | 97% |
| 5 | - | DCM | NR |
| 6 | TEAB | THF | <10% |
| 7 | TEAB | CH3CN | 78% |
| 8 | TEAB | DMF | <10% |
| Entry | Aromatic Acids | Product | Yield b (%) |
|---|---|---|---|
| 1 | 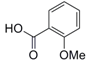 5 |  6 | 95 c |
| 2 | 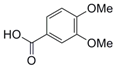 7 | 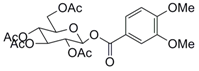 8 | 98 |
| 3 | 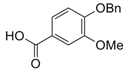 9 | 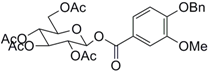 10 | 91 c |
| 4 | 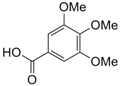 11 | 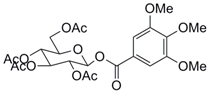 12 | 80 |
| 5 | 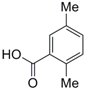 13 |  14 | 96 |
| 6 | 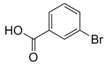 15 |  16 | 99 |
| 7 | 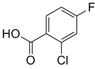 17 | 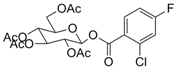 18 | 85 c |
| 8 | 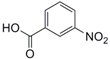 19 |  20 | 90 |
| 9 |  21 |  22 | 99 c |
| Entry | Aliphatic Acids | Product | Yield b (%) |
|---|---|---|---|
| 1 |  23 |  24 | 95 c |
| 2 | 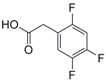 25 | 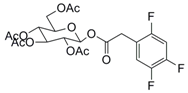 26 | 97 |
| 3 | 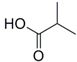 27 |  28 | 91 |
| 4 |  29 |  30 | 99 c |
| 5 | 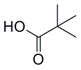 31 |  32 | 72 |
| 6 | 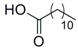 33 | 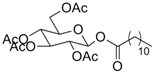 34 | 92 c |
| 7 |  35 | 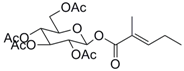 36 | 96 |
| 8 |  37 | 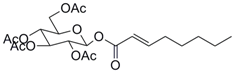 38 | 79 |
| 9 | 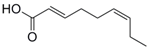 39 | 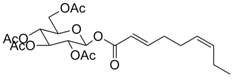 40 | 94 |
| 10 | 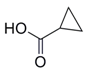 41 | 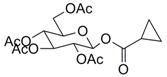 42 | 99 c |
| 11 |  43 |  44 | 78 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Lu, H.; Chen, Y.; Yu, W.; Dai, H.; Pan, X. Improved Synthesis of 1-O-Acyl-β-d-Glucopyranose Tetraacetates. Molecules 2017, 22, 662. https://doi.org/10.3390/molecules22040662
Chen Y, Lu H, Chen Y, Yu W, Dai H, Pan X. Improved Synthesis of 1-O-Acyl-β-d-Glucopyranose Tetraacetates. Molecules. 2017; 22(4):662. https://doi.org/10.3390/molecules22040662
Chicago/Turabian StyleChen, Yu, Huan Lu, Yanyu Chen, Wansheng Yu, Hui Dai, and Xianhua Pan. 2017. "Improved Synthesis of 1-O-Acyl-β-d-Glucopyranose Tetraacetates" Molecules 22, no. 4: 662. https://doi.org/10.3390/molecules22040662
APA StyleChen, Y., Lu, H., Chen, Y., Yu, W., Dai, H., & Pan, X. (2017). Improved Synthesis of 1-O-Acyl-β-d-Glucopyranose Tetraacetates. Molecules, 22(4), 662. https://doi.org/10.3390/molecules22040662