
Synthesis of Aminoglycoside-2′-O-Methyl Oligoribonucleotide Fusions

1
Department of Chemistry, University of Turku, Vatselankatu 2, 20014 Turku, Finland
2
Centre of New Technologies, University of Warsaw, Banacha 2c, 02 097 Warsaw, Poland
3
College of Inter-Faculty Individual Studies in Mathematics and Natural Sciences, University of Warsaw, Banacha 2c, 02 097 Warsaw, Poland
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(5), 760; https://doi.org/10.3390/molecules22050760
Submission received: 24 April 2017
/
Revised: 4 May 2017
/
Accepted: 6 May 2017
/
Published: 8 May 2017
(This article belongs to the Special Issue Synthesis and Applications of Oligonucleotide Conjugates)
Abstract
:Phosphoramidite building blocks of ribostamycin (3 and 4), that may be incorporated at any position of the oligonucleotide sequence, were synthesized. The building blocks, together with a previously described neomycin-modified solid support, were applied for the preparation of aminoglycoside-2′-O-methyl oligoribonucleotide fusions. The fusions were used to clamp a single strand DNA sequence (a purine-rich strand of c-Myc promoter 1) to form triple helical 2′-O-methyl RNA/DNA-hybrid constructs. The potential of the aminoglycoside moieties to stabilize the triple helical constructs were studied by UV-melting profile analysis.
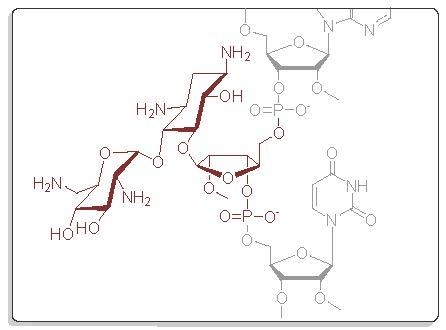
1. Introduction
Aminoglycosides are well-known small molecular ligands for a variety of RNA targets [1] (including bulges and internal loops at ribosomal decoding site [2,3,4,5], several ribozymes [6,7] and important regions of HIV RNAs [8,9,10,11]), and they also show relatively high affinities as groove binders for DNA- and RNA-triple helices and for their hybrids [12,13,14]. Thanks to these binding properties, they may be attractive conjugate groups for oligonucleotide-based probes to provide an extra binding motif in the recognition of the target DNA or RNA [15,16,17,18,19,20]. For example, neomycin has been used to enhance affinity of oligonucleotides to an α-sarcin loop RNA sequence [21] and to HIV-1 trans activation response element (TAR) models [22] via binding to known binding sites for neomycin on these RNA targets. In a favorable case, the cooperative recognition via combined small molecular binding and hybridization may take place [22]. Triple helical recognition of DNA has also been enhanced by appropriately conjugated neomycin ligands [23]. Furthermore, aminoglycoside moieties may improve cellular uptake via lipid-mediated delivery of oligonucleotides [24]. All together, these beneficial properties of the conjugated aminoglycosides may find applications in developing of modern antigene and antisense therapies.
We have previously described aminoglycoside-derived phosphoramidite building blocks (1 and 2) [17] and solid supports (5–7) [22], which may be used for the automated synthesis of 5′- and 3′-aminoglycoside conjugated oligonucleotides, respectively (Figure 1). Herein, the set of the useful building blocks is expanded by appropriately modified ribostamycins (3 and 4) that may be incorporated at any position of the oligonucleotide sequence. The building blocks (3 and 4), together with a previously described neomycin-derived LCAA-CPG-support 7 (long chain alkylamine controlled pore glass), were used for the synthesis of pure fusions of the aminoglycosides and 2′-O-methyl oligoribonucleotides. The aminoglycoside moieties were aimed to stabilize triple helical constructs formed by clamping a purine-rich DNA single strand (a sequence of c-Myc promoter 1, [25]). The effect of the aminoglycoside moieties on the resulting triple helical 2′-O-methyl RNA/DNA-hybrid constructs was evaluated by UV-melting profile analysis.
2. Results and Discussion
2.1. Synthesis of the Phosphoramidite Building Blocks 3 and 4
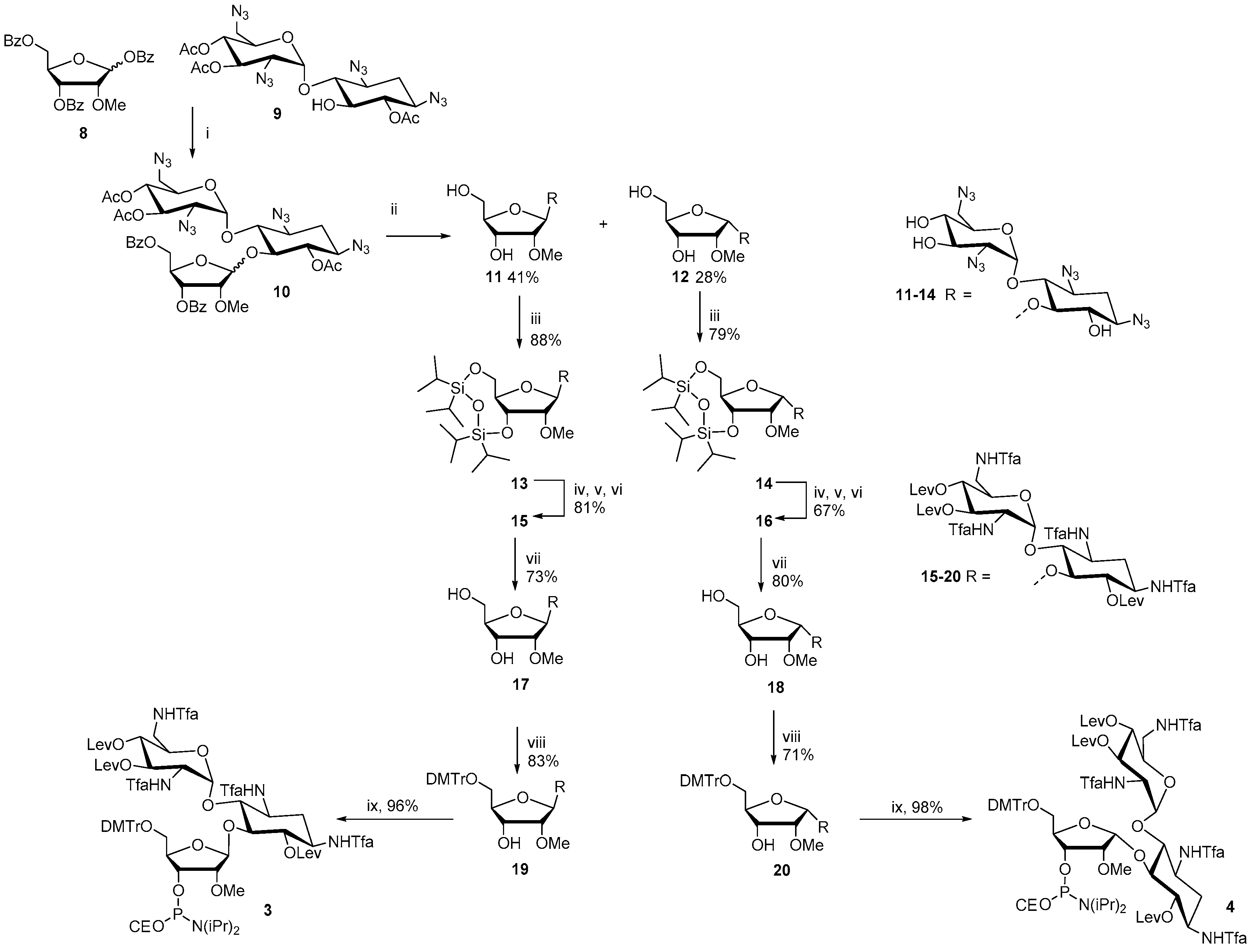
The synthesis of the ribostamycin-derived phosphoramidites 3 and 4 is outlined in Scheme 1. TMSTf-promoted glycosidation between 1,3,5-di-O-benzoyl-2-O-methyl-α/β-d-ribofuranose (8) and 6,3′,4′-tri-O-acetyl-1,3,2′,6′-tetraazido neamine (9) [26] gave an anomeric mixture (β:α = 1:2) of peracylated 2′′-O-methyl ribostamycin (10) in 81% yield. A sodium methoxide-catalyzed transesterification yielded 1,3,2′,6′-tetraazido-2′′-O-methyl ribostamycins that could be isolated as the pure β- (11) and α-anomer (12) by a simple column chromatography. 11 and 12 were then subjected to a treatment with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane to protect the 5′′- and 3′′-hydroxyl groups (13 and 14). The azide masks of 13 and 14 were reduced by a Staudinger reaction, the exposed amino groups were trifluoroacetylated and the hydroxyl groups were levulinoylated to give fully protected 2′′-O-methyl ribostamycins 15 and 16. The 5′′-O, 3′′-O-1,1,3,3-tetraisopropyldisiloxane protection was then removed by a treatment of triethylamine trihydrofluoride and the exposed 5′′-hydroxy group was selectively 4′,4′-dimethoxytritylated to give 19 and 20. The whole protecting group manipulation from 11 to 19 and from 12 to 20 could be carried out in 43% and 30% overall yields, respectively. Phosphitylation of the 3′′-OH group with 2-cyanoethyl N,N-diisopropylphosphoramido chloridite gave 3 and 4 in nearly quantitative yields.
2.2. Synthesis of Aminoglycoside 2′-O-Methyl Oligonucleotide Fusions Using 3 and 4
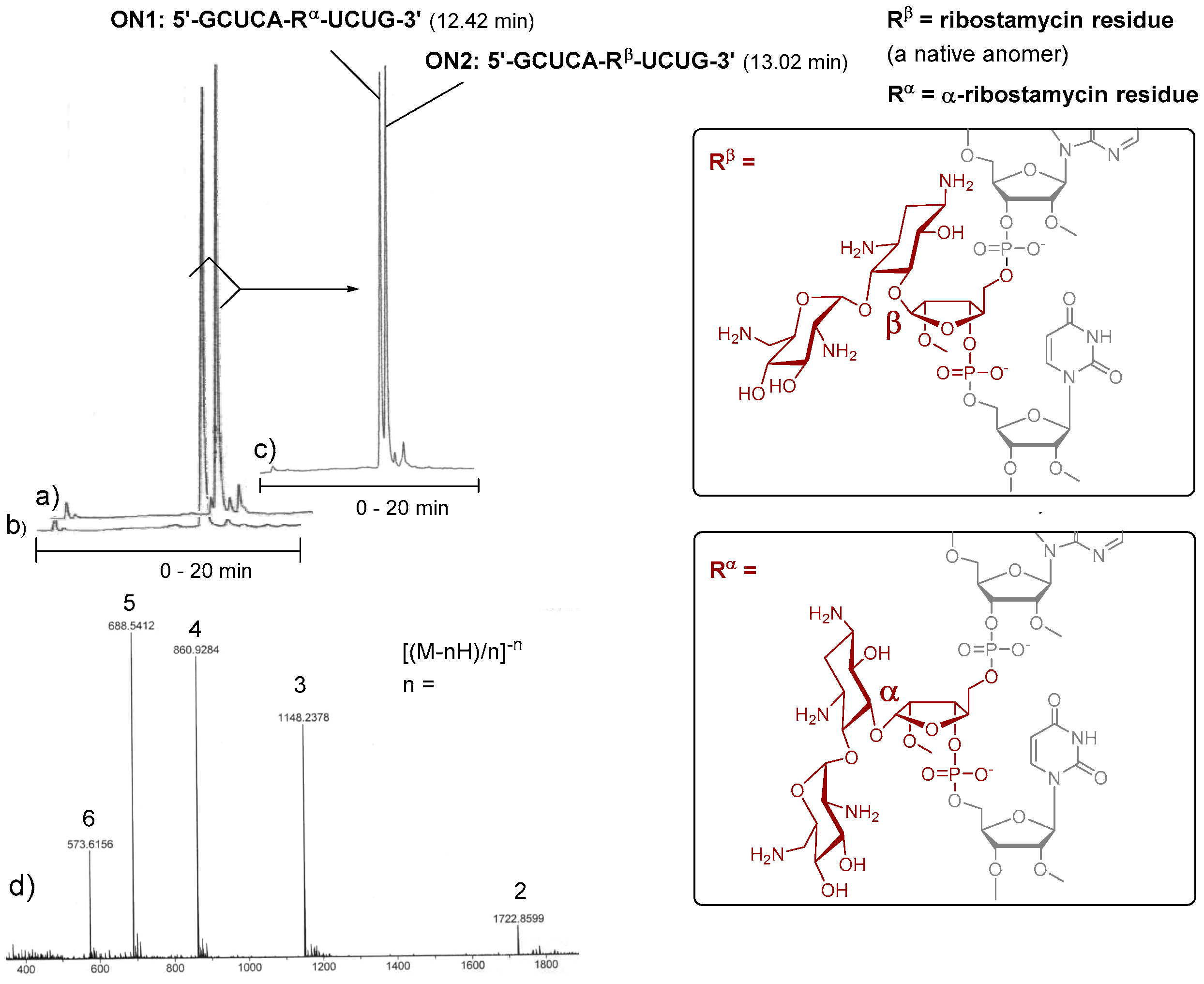
To evaluate the applicability of 3 and 4 in the automated chain assembly, intra-chain fusions of the ribostamycins and a random 2′-O-methyl RNA sequence (5′-GCUCA-R-UCUG-3′, ON1: R = α-ribostamycin residue (Rα) and ON2: R = ribostamycin residue (Rβ)) were first synthesized (Figure 2). The coupling efficiency was evaluated by DMTr-assay. A double phosphoramidite coupling using 0.1 mol L−1 3 and 4 in acetonitrile, benzylthiotetrazol as an activator and a 600 s coupling time (2 × 600 s), followed by the standard oxidation step, gave ca. 95% coupling yield for both building blocks. Otherwise, the oligonucleotides were assembled using the standard RNA coupling cycle (a 300 s coupling time used for the 2′-O-methyl nucleoside building blocks). After the chain assembly, the levulinoyl groups of the ribostamycin moieties were removed on support with a mixture of hydrazinium acetate (NH2NH2·OH2, pyridine, AcOH, 0.124/4/1, v/v/v, 2 × 10 min at 25 °C), and the supports were then subjected to concentrated ammonia (overnight at 55 °C) [17]. The released ON1 and ON2 were analysed by ion exchange HPLC. As seen in the HPLC profiles of the crude products (Figure 2a,b), the automated synthesis could be successfully carried out. Interestingly, the chiral integrity of the ribostamycin moieties also affects the retention times of the conjugates in the HPLC profiles (Figure 2c).
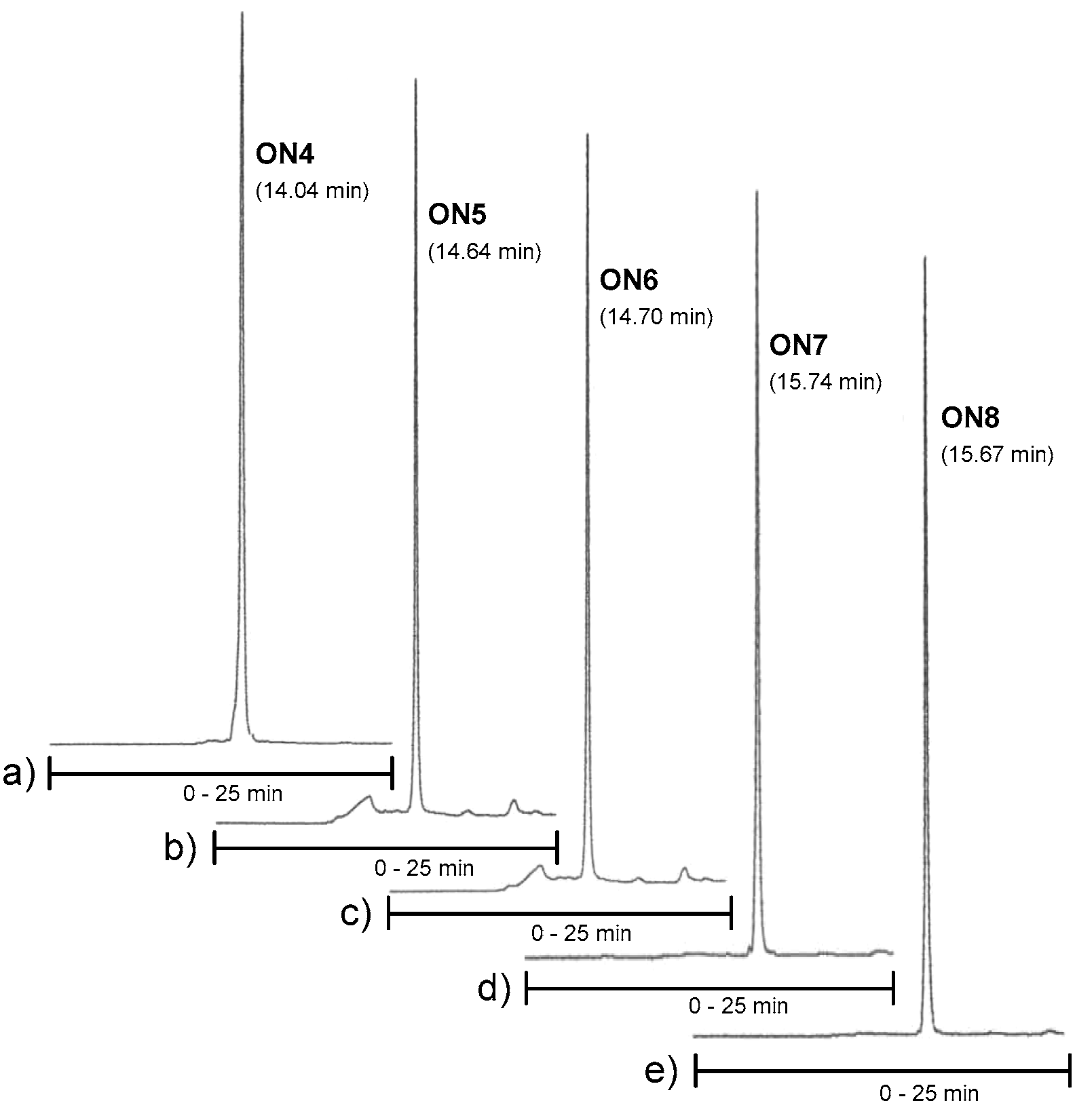
Conjugates ON4–ON6, which aimed to clamp a purine-rich DNA strand (a sequence of C-Myc promoter 1), were then synthesized on our previously described neomycin-derived LCAA-CPG support (7) [22]. A manual coupling of 3 and 4 to support 7 was carried out (see the material and methods) and then the 2′-O-methyl oligoribonucleotide chain, including the 2′-deoxy oligonucleotide turn (TCTCT), was assembled in a standard manner. After the chain assembly, the levulinoyl groups of the ribostamycin moieties were removed on support using the hydrazine acetate treatment as mentioned above. The solid-supported conjugates were then exposed to a mixture of NaOMe in methanol (0.1 mol L−1, for 2 h at 25 °C, i.e., acetyl removal of the neomycin moieties and cleavage of the succinyl linker), and the deprotection was continued by ammonolysis (overnight at 55 °C, i.e., removal of the Tfa-protections and of the Bz-protections of cytosine bases) [22]. Conjugates ON7 and ON8 were synthesized following the procedure described for ON1 and ON2. RP HPLC and MS (ESI-TOF) data of the conjugates ON1, ON2, ON4–ON8 are shown in Figure 2a,b and Figure 3 (and Table S1), respectively. Isolated yields ranged from 10–26%.
2.3. UV-Melting Profile Analysis
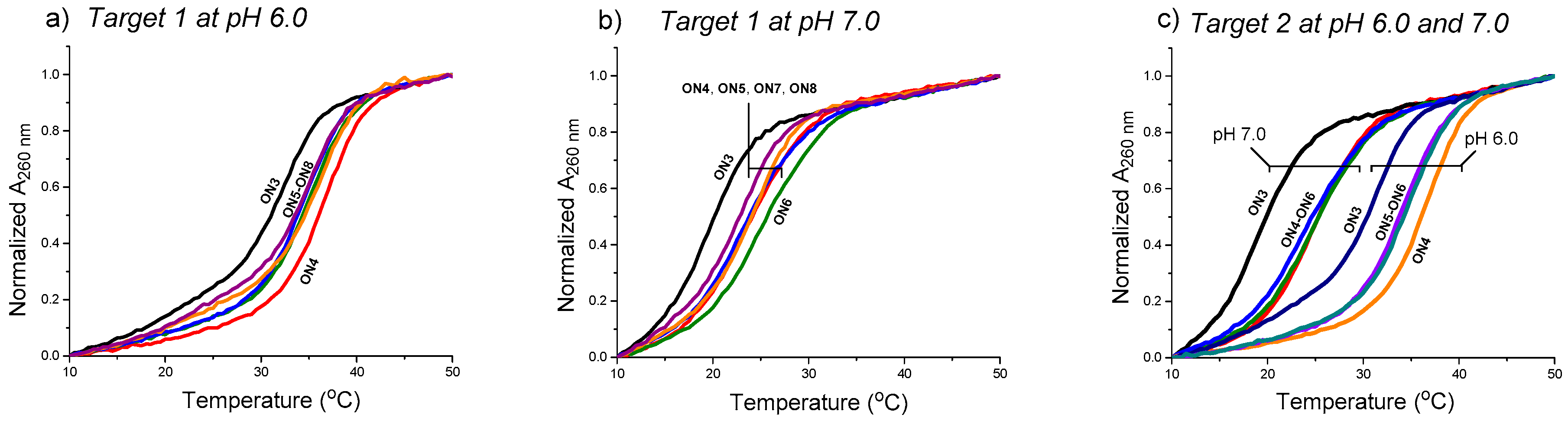
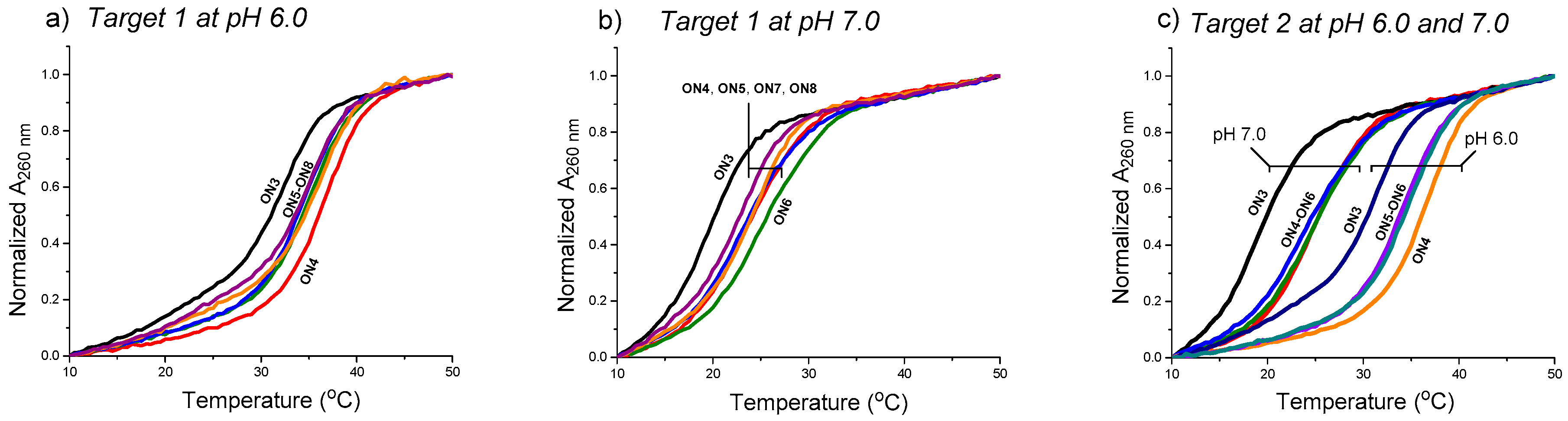
The effect of the aminoglycosides moieties (ribostamycin and neomycin) to stabilize the clamp structures targeted to a purine-rich DNA single strand (a sequence of c-Myc promoter 1) (cf. Scheme 2) has been studied by UV-melting profile experiments (Figure 4 and Table 1). The measurements were carried out using 2 µmol L−1 of each oligonucleotide in a mixture of 10 mmol L−1 sodium cacodylate and 0.1 mol L−1 NaCl at pH 6.0 and 7.0. The temperature was changed at a rate of 0.2 °C min−1. In each case, a biphasic melting curve was observed (the duplex melting range, Tm = 65 °C, excluded in Figure 4) and the Tm3-values were extracted from the first inflection point. The Tm3-values of the triple helical clamps were expectedly higher (ca. 10 °C) at pH 6.0 than at pH 7.0. As shown, the overhanging 3′-aminoglycoside moiety of the conjugates increased the stability of the clamps in each case. The acidic conditions (pH 6.0 vs. 7.0) did not show a marked role in ∆Tm3-values. The 3′-neomycin conjugate (ON4) increased the stability of the clamps (Target 1 and Target 2) by ∆Tm3 = +4.0–+5.8 °C. The incorporation of the ribostamycin unit to the 3′-aminoglycosides overhang (ON5 and ON6) seemed to elicit, however, slightly decreased ∆Tm3-values (e.g., ON4: +5.8 °C vs. ON5: +4.8 °C and ON6: +5.5 °C with Target 2 at pH 7.0). ON6 did an exception with Target 1 at pH 7.0 (ON6: ∆Tm3 = +5.1 vs. ON4: ∆Tm3 = +4.0). A slightly increased stability was also observed, when the ribostamycin was incoporporated into the 2′-deoxy oligoribonucleotide turn (ON7 and ON8: ∆Tm3 = +2.6 °C–+4.3 °C).
3. Discussion
3.1. Synthesis of the Building Blocks 3 and 4 and of the Aminoglycoside-Oligonucleotide Conjugates ON1, ON2, ON4–ON8
Phosphoramidite building blocks 3 and 4 could be synthesized in relatively high yields (Scheme 1). Despite the multistep synthesis, the overall yields of 3 and 4 (calculated from 6,3′,4′-tri-O-acetyl-1,3,2′,6′-tetraazido neamine 9 [26]) were 19% and 10%, respectively. The chiral integrity of the anomers was confirmed by a NOESY spectrum that showed a correlation between the H1′′ and 2′-O-methoxy groups of the β1′′−5-ribostamycins (e.g., 13). Building blocks 3 and 4 could be efficiently incorporated into oligonucleotide sequences using either an automated double phosphoramidite coupling (2 × 600 s coupling time) or a manual coupling (see details in the Materials and Methods section). In the manual coupling, the concentration of the building blocks could be increased to 0.11 mol L−1 (after the addition of benzylthiotetrazol) that improved the coupling efficiency (Note: In the synthesizer, the initial concentration of the phosphoramidites is 0.1 mol L−1 that is diluted to one half by the solution of benzylthiotetrazol). The O-levulinoyl/N-trifluoroacetyl (Lev/Tfa)-protecting group combination for 3 and 4 was applied. The reason for this protecting group scheme was the selective on-support removal of the Lev protections (by a hydrazinium acetate treatment in the presence of N-Tfa groups) that suppressed plausible O→N acyl migration. The NaOMe-catalyzed methanolysis was sufficient to selectively remove O-acetyl groups from the neomycin moiety (7) and to eliminate N-acylated side products (ON4) [22], but the Lev/Tfa-combination is evidently [17] the superior protecting group scheme, when the number of 1,2-aminoethanol-moieties increases. Thus, two-step- (ON1, ON2, ON4, ON7 and ON8) and three-step-treatments (ON5 and ON6) with hydrazinium acetate, NaOMe/MeOH and ammonolysis were used to release the conjugates.
3.2. The Effect of the Aminoglycoside Moieties of ON1, ON2, ON4–ON8 on the Triple Helical Constructs
According to UV-melting profiles, the aminoglycoside moieties (ribostamycin and neomycin) increased the stability of the clamp structures in each case (ΔTm = +2.2–+5.8 °C), but the effect remained modest. The Watson-Hoogsteen groove (the groove between the pyrimidine strands) of the DNA-triple helix may bind multiple aminoglycosides (neomycin primarily) and the binding has been proposed to be involved in the amino groups of neomycin rings II and IV [27]. The elongated 3′-aminoglycoside overhang of ON5 and ON6 (containing one biosamine and two neamines in the ribose-phosphodiester backbone) probably could not reach the optimal binding contact needed for the groove binding and the stability did not increase compared to 3′-neomycin moiety (ON4: ΔTm3 = +4.0–+5.8 °C). The phosphodiester bond between the neomycin and ribostamycin units may also disturb the binding. The incorporation of the ribostamycin units into the 2′-deoxy oligonucleotide turn increased the stability of the clamps by ΔTm = +2.6–+4.3 °C. The stability of the clamps may hence be further increased by incorporation of aminoglycosides at both terminus of the clamp. Further studies may also be needed to evaluate the influence of the longer spacers between the oligonucleotide and the aminoglycoside overhang.
4. Materials and Methods
4.1. General Remarks
MeCN, pyridine and dichloromethane were dried over 3Å molecular sieves and triethylamine over CaH2. NMR spectra were recorded using a 500 MHz instrument. The chemical shifts for 1H and 13C NMR resonances are given in parts of million from the residual signal of the deuterated solvents (CD3OD and CD3CN). 31P NMR resonance shifts are compared to external H3PO4. Mass spectra were recorded using electrospray ionization (ESI-TOF).
1,3,2′,6′-Tetraazido-2′′-O-methyl ribostamycin (11 and 12). 6,3′,4′-Tri-O-acetyl-1,3,2′,6′-tetraazido neamine (9, 1.6 g, 2.9 mmol) [25] and 1,3,5-di-O-benzoyl-2-O-methyl-α/β-d-ribofuranose (8, 3.4 g, 7.1 mmol) were dissolved in dichloromethane (20 mL) and the mixture was cooled down to 0 °C. Trimethylsilyl trifluoromethanesulfonate (0.58 mL, 0.32 mmol) was slowly added to the mixture and the reaction was stirred at 0 °C for 2 h and then at room temperature for 2 h under a nitrogen atmosphere. Dichloromethane (50 mL) and saturated NaHCO3 (20 mL) were added to the mixture. The organic layer was separated, washed with saturated NaCl, dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (30% EtOAc in petroleum ether) to give 2.13 g (81%) of 10 as colourless oil (anomeric mixture, β:α = 1:2). The peracylated ribostamycine (10) was dissolved in 0.1 mol L−1 methanolic sodium methoxide (5.0 mL). The mixture was stirred at ambient temperature for 1 h, neutralized by addition of strongly acidic cation-exchange resin, and filtered. The filtrate was evaporated to dryness and purified by silica gel chromatography (1. EtOAc, 2. 10% MeOH in CH2Cl2) to give 0.55 g (41%) of 11 (β anomer) and 0.37 g (28%) of 12 (α anomer). 11: 1H NMR (500 MHz, CD3OH): δ 5.89 (d, 1H, J = 3.8 Hz), 5.45 (b, 1H), 4.21–4.17 (m, 2H), 3.92–3.89 (m, 2H), 3.82–3.79 (m, 2H), 3.70–3.63 (m, 3H), 3.58–3.52/m, 2H), 3.54 (s, 3H), 3.48–3.42 (m, 3H), 3.39 (m, 1H), 3.13 (dd, 1H, J = 10.6 Hz & 3.8 Hz), 2.24 (m, 1H), 1.40 (m, 1H); 13C (125 MHz, CD3OH): δ 105.5, 96.7, 84.6, 84.0, 83.1, 76.0, 75.6, 71.8, 71.2, 70.7, 69.8, 63.1, 62.2, 60.6, 59.9, 57.3, 51.2, 31.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C18H28N12NaO10 595.1949, found 595.1930. 12: 1H NMR (500 MHz, CD3OH): δ 5.84 (d, 1H, J = 3.9 Hz), 5.54 (d, 1H, J = 4.6 Hz), 4.27–4.24 (m, 3H), 3.87 (dd, 1H, J = 5.0 Hz, both), 3.81 (dd, 1H, J = 10.0 Hz & 9.1 Hz), 3.70–3.61 (m, 4H), 3.55 (s, 3H), 3.55–3.43 (m, 5H), 3.38 (m, 1H), 3.24 (dd, 1H, J = 10.5 Hz & 4.0 Hz), 2.25 (ddd, 1H, J = 12.9 Hz, 4.4 Hz & 4.4 Hz), 1.44 (ddd, 1H, J = 12.3 Hz, each); 13C (125 MHz, CD3OH): δ 103.0, 97.4, 86.6, 84.6, 81.1, 77.8, 74.9, 71.8, 71.5, 71.1, 68.3, 64.0, 61.8, 60.1, 59.5, 57.6, 51.1, 31.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C18H28N12NaO10 595.1949, found 595.1929.
1,3,2′,6′-Tetraazido-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-2′′-O-methyl ribostamycin (13). 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane (350 μL, 1.1 mmol) was added to a mixture of 11 (0.55 g, 0.96 mmol) in pyridine (3.0 mL). The mixture was stirred overnight at ambient temperature and then dichloromethane and saturated NaHCO3 were added. The organic layer was separated, washed with saturated NaCl, dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (5% MeOH in CH2Cl2) to yield 0.69g (88%) of the product (13) as white foam. 1H NMR (500 MHz, CD3OH): δ 6.10 (d, 1H, J = 3.8 Hz), 5.27 (s, 1H), 4.53 (dd, 1H, J = 9.0 Hz & 4.2 Hz), 4.20 (m, 1H), 4.12 (dd, 1H, J = 12.9 Hz & 1.9 Hz), 3.96 (dd, 1H, J = 12.8 Hz & 2.5 Hz), 3.92–3.86 (m, 3H), 3.67–3.62 (m, 2H), 3.59 (s, 3H), 3.57–3.41 (m, 4H), 3.31–3.24 (m, 2H), 3.07 (dd, 1H, J = 10.5 Hz & 3.8 Hz), 2.24 (m, 1H), 1.36 (ddd, J = 12.5 Hz, each), 1.16–1.06 (m, 28H); 13C (125 MHz, CD3OH): δ 107.8, 96.3, 85.9, 84.8, 80.4, 76.7, 75.2, 71.52, 71.50, 71.1, 70.3, 63.5, 60.9, 60.6, 60.1, 51.5, 31.8, 16.8, 16.62, 16.59, 16.5, 16.40, 16.37, 16.31, 16.29, 16.24, 16.16, 16.1, 13.5, 13.4, 13.1, 12.9, 12.5, 12.4; HRMS (ESI-TOF) m/z: [M + K]+ calcd. for C30H54KN12O11Si2 853.3211, found 853.3148.
1,3,2′,6′-Tetraazido-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-2′′-O-methyl-α1′′−5-ribostamycin (14). The α-anomer (14) was synthesized as described for 13 above. 0.37 g (0.64 mmol) of 12 gave 0.42 g (79%) of 14 as white foam. 1H NMR (500 MHz, CD3OH): δ 5.67 (d, 1H, J = 3.8 Hz), 5.43 (d, 1H, J = 4.1 Hz), 4.40 (dd, 1H, J = 7.7 Hz & 5.1 Hz), 4.28 (m, 1H), 4.20 (m, 1H), 4.04–4.00 (m, 2H), 3.96 (dd, 1H, J = 12.8 Hz & 4.4 Hz), 3.87 (dd, 1H, J = 10.3 Hz & 9.1 Hz), 3.66 (s, 3H), 3.64–3.61 (m, 2H), 3.56–3.43 (m, 5H), 3.40 (dd, 1H, J = 9.6 Hz & 9.2 Hz), 3.24 (dd, 1H, J = 10.5 Hz & 3.9 Hz), 2.24 (m, 1H), 1.46 (ddd, 1H, J = 12.3 Hz, each), 1.16–1.06 (m, 28H); 13C (125 MHz, CD3OH): δ 103.4, 97.9, 87.3, 81.4, 80.0, 77.7, 74.9, 72.0, 71.3, 71.14, 71.08, 63.4, 61.0, 59.9, 59.22, 59.18, 51.1, 31.5, 16.5, 16.42, 16.38, 16.30, 16.29, 16.2, 16.1; HRMS (ESI-TOF) m/z: [M + K]+ calcd. for C30H54KN12O11Si2 853.3211, found 853.3174.
6,3′,4′-Tri-O-levulinoyl-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin (15). Trimethylphosphine (1 mol L−1 Me3P in toluene, 5.1 mL, 5.1 mmol) was added to a mixture of 13 (0.42 g, 0.51 mmol) in water–dioxane (1:4, v/v, 5.0 mL). The mixture was stirred at ambient temperature for 4 h under nitrogen and then concentrated ammonia (1.0 mL) was added. After overnight reaction, the mixture was evaporated to dryness and the residue was coevaporated with pyridine. The residue was dissolved in methanol (4.0 mL) and triethylamine (2.0 mL), and methyl trifluoroacetate (0.41 mL, 4.1 mmol) was added to the mixture. The reaction was stirred at ambient temperature for 5 h. Saturated NaHCO3 was added and the mixture was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and evaporated to dryness. The residue was dissolved in pyridine (5.0 mL) and then freshly prepared levulinic anhydride (0.66 g, 3.1 mmol) and a catalytic amount of DMAP were added to the mixture. After overnight stirring, methanol and saturated NaHCO3-were added to the mixture and the crude product was extracted with ethyl acetate. The combined organic layers were dried over NaSO4, filtered and evaporated to dryness. The residue was subjected to a silica gel chromatography (30% petroleum ether in ethyl acetate) to yield 0.58 g (81 %) of 15 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.05 (d, 1H, J = 3.7 Hz), 5.51 (dd, 1H, J = 10.1 Hz, both), 4.97–4.90 (m, 3H), 4.37 (d, 1H, J = 3.9 Hz), 4.34 (m, 1H), 4.26–4.11 (m, 2H), 4.08–4.03 (m, 2H), 3.95 (dd, 1H, J = 13.2 Hz & 1.5 Hz), 3.84–3.80 (m, 2H), 3.73 (m, 1H), 3.64 (dd, 1H, J = 14.6 Hz & 3.0 Hz), 3.67 (s, 3H), 3.59–3.43 (m, 2H), 2.83–2.41 (m, 12H), 2.17, 2.16 and 2.15 (3 × s, 9H), 2.04–1.95 (m, 2H), 1.14–1.06 (m, 28H); 13C (125 MHz, CD3OH): δ 206.4, 205, 9, 205.0, 170.9, 170, 8, 170.4, 156.4–155.6 (m), 118.0–111.0 (m), 107.6, 93.8, 82.9, 82.4, 78.7, 73.9, 73.8, 69.0, 68.7, 67.8, 65.4, 58.3, 56.6, 49.9, 49.2, 47.2, 46.7, 37.9, 35.7, 35.6, 35.5, 29.5, 26.7, 26.65, 26.62, 26.57, 26.1, 25.9, 25.8, 25.7, 15.02, 14.93, 14.92, 14.89, 14.88, 14.78, 14.76, 14.7, 14.6, 14.5, 11.9, 11.6, 11.5, 11.0, 10.8; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C53H76F12N4NaO21Si2 1411,4247, found 1411.4283.
6,3′,4′-Tri-O-levulinoyl-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α1′′−5-ribostamycin (16). The α-anomer (16) was synthesized as described for 15 above. 0.41 g (0.50 mmol) of 14 gave 0.42 g (59%) of 16 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.02 (d, 1H, J = 3.9 Hz), 5.24 (dd, 1H, J = 10.7 Hz & 9.6 Hz), 5.06 (dd, 1H, J = 10.1 Hz & 9.1 Hz), 5.02 (d, 1H, J = 3.9 Hz), 4.96 (dd, 1H, J = 9.9 Hz, both), 4.39 (dd, 1H, J = 10.9 Hz & 3.9 Hz), 4.25–4.19 (m, 2H), 4.13 (m, 1H), 4.04–4.00 (m, 2H), 3.97–3.91 (m, 2H), 3.88 (dd, 1H, J = 12.7 Hz & 2.8 Hz), 3.80 (dd, 1H, J = 12.7 Hz & 4.3 Hz), 3.66–3.58 (m, 2H), 3.54 (s, 3H), 3.55–3.50 (m, 1H), 2.85–2.75 (m, 6H), 2.71–2.40 (m, 6H), 2.17 (s, 3H), 2.15 (s, 2 × 3H), 2.04–1.95 (m, 2H), 1.14–1.05 (m, 28H); 13C (125 MHz, CD3OH): δ 207.8, 207.4, 207.2, 172.6, 172.02, 171.98, 158.4–156.8 (m), 119.5–111.5 (m), 102.9, 95.7, 85.3, 81.5, 79.7, 76.1, 74.4, 70.8, 70.2, 68.5, 67.5, 60.5, 58.5, 51.8, 48.8, 48.5, 38.6, 37.2, 37.0, 30.9, 28.3, 28.1, 27.8, 27.5, 27.2, 16.5, 16.4, 16.3, 16.24, 16.21, 16.19, 16.1, 13.3, 13.1, 13.0, 12.6, 12.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C53H76F12N4NaO21Si2 1411,4247, found 1411.4190.
6,3′,4′-Tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin (17). Compound 15 (0.48 g, 0.35 mmol) was dissolved in a mixture of triethylamine trihydrofluoride and acetonitrile (1:10, v/v, 2.0 mL). The reaction was stirred overnight at ambient temperature, 10% aqueous KH2PO4 was added and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 and saturated NaCl, dried with Na2SO4, filtered and evaporated to dryness. The crude product was purified by a silica gel chromatography (10 % MeOH in CH2Cl2) to yield 0.35 g (73 %) of 17 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.11 (d, 1H, J = 3.5 Hz), 5.21 (dd, 1H, J = 10.6 Hz & 9.6 Hz), 5.16 (d, 1H, J = 4.3 Hz), 4.99 (dd, 1H, J = 10.2 Hz & 9.6 Hz), 4.95 (dd, 1H, J = 9.8 Hz, both), 4.38 (dd, 1H, J = 10.8 Hz & 3.9 Hz), 4.24 (m, 1H), 4.15 (m, 1H), 4.12 (dd, 1H, J = 4.1 Hz, both), 4.04–4.01 (m, 2H), 3.85 (dd, 1H, J = 10.0 Hz & 8.8 Hz), 3.78 (m, 1H), 3.65 (dd, 1H, J = 14.4 Hz & 4.0 Hz), 3.56–3.52 (m, 3H), 3.47 (dd, 1H, J = 14.5 Hz & 2.4 Hz), 3.39 (s, 3H), 2.84–2.72 (m, 6H), 2.70–2.41 (m, 6H), 2.19 (s, 3H), 2.17 (s, 3H), 2.14 (s, 3H), 2.01–1.97 (m, 2H); 13C (125 MHz, CD3OH): δ 208.0, 207.7, 207.6, 172.6, 171.98, 171.96, 158.5–156.9 (m), 119.5–112.5 (m), 108.1, 95.1, 84.7, 85.6, 82.9, 75.5, 75.1, 70.1, 69.0, 68.6, 67.5, 61.6, 57.5, 48.7, 48.5, 39.0, 37.3, 37.1, 30.8, 28.3, 28.2, 27.8, 27.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C41H50F12N4NaO20 1169.2724, found 1169.2744.
6,3′,4′-Tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α(1′′−5)-ribostamycin (18). The α-anomer (18) was synthesized as described for 17 above. 0.35 g (0.25 mmol) of 16 gave 0.28 g (80%) of 18 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.09 (d, 1H, J = 2.6 Hz), 5.22 (dd, 1H, J = 10.7 Hz & 9.6 Hz), 5.08–5.04 (m, 2H), 4.94 (dd, 1H, J = 9.9 Hz & 9.8 Hz), 4.36 (dd, 1H, J = 10.9 Hz & 3.9 Hz), 4.22 (m, 1H), 4.17–4.12 (m, 2H), 4.04–3.98 (m, 3H), 3.89 (dd, 1H, J = 10.1 Hz & 8.8 Hz), 3.65 (dd, 1H, J = 14.8 Hz & 3.5 Hz), 3.58–3.54 (m, 3H), 3.49 (dd, 1H, J = 14.4 Hz & 2.0 Hz), 3.35 (s, 3H), 2.86–2.41 (m, 12H), 2.17 (s, 2 × 3H), 2.15 (s, 3H), 2.04–1.94 (m, 2H); 13C (125 MHz, CD3OH): δ 207.9, 207.8, 207.5, 172.9, 172.0, 158.4–156.9 (m), 119.4–112.5 (m), 103.8, 96.1, 86.9, 84.1, 80.9, 77.2, 74.3, 70.2, 68.5, 67.7, 67.6, 61.8, 57.2, 53.5, 48.7, 48.6, 38.6, 37.3, 37.2, 37.1, 30.9, 28.4, 28.3, 28.0, 27.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C41H50F12N4NaO20 1169.2724, found 1169.2716.
5′′-O-(4,4′-Dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin (19). 17 (0.35 g, 0.31 mmol) was dissolved in dry pyridine (2.0 mL) and 4′,4′-dimethoxytritylchloride (0.12 g, 0.34 mmol) was added. The mixture was stirred overnight at ambient temperature, poured to saturated aqueous NaHCO3 and the product was extracted with dichloromethane. The organic layers were combined, dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (10% MeOH in CH2Cl2) to give 0.37 g (83%) of the product (19) as white foam. 1H NMR (500 MHz, CD3OH): δ 7.48 (2H, d, J = 7.5 Hz), 7.35–7.32 (m, 6H), 7.25 (m, 1H), 6.92 (d, 4H, J = 8.8 Hz), 6.16 (d, 1H, J = 4.0 Hz), 5.18 (d, 1H, J = 4.9 Hz), 5.14 (dd, 1H, J = 10.4 Hz & 9.8 Hz), 5.07 (dd, 1H, J = 10.2 Hz & 9.7 Hz), 4.88 (dd, 1H, J = 9.9 Hz, both), 4.23 (m, 1H), 4.16 (m, 1H), 4.05–3.97 (m, 4H), 3.90 (dd, 1H, J = 10.2 Hz & 8.9 Hz), 3.85 (m, 1H), 3.82 (s, 2 × 3H), 3.63 (dd, 1H, J = 4.6 Hz, both), 3.54 (dd, 1H, J = 14.6 Hz & 4.3 Hz), 3.42 (dd, 1H, J = 14.6 Hz & 3.4 Hz), 3.31 (s, 3H), 3.21 (d, 2H, J = 4.0 Hz), 2.84–2.71 (m, 6H), 2.69–2.40 (m, 6H), 2,17 (s, 3H), 2.16 (s, 3H), 2.14 (s, 3H), 2.07 (ddd, 1H, J = 12.8 Hz, each), 2.00 (ddd, 1H, J = 12.8 Hz, 4.6 Hz & 4.6 Hz); 13C (125 MHz, CD3OH): δ 207.9, 207.6, 207.4, 172.6, 172.2, 171.8, 158.84, 158.80, 158.4–156.6 (m), 145.1, 135.7, 135.4, 130.2, 130.1, 128.0, 127.4, 126.4, 119.5–112.5 (m), 112.8, 112.7, 108.3, 94.9, 86.2, 84.8, 83.9, 83.2, 75.1, 75.0, 70.2, 69.2, 69.0, 66.9, 62.5, 57.3, 54.4, 51.2, 48.7, 48.5, 39.1, 37.3, 37.1, 30.8, 28.2, 28.15, 28.12, 27.9, 27.49, 27.46. HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C62H68F12N4NaO22 1471.4031, found 1471.4055.
5′′-O-(4,4′-Dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α1′′−5-ribostamycin (20). The α-anomer (20) was synthesized as described for 19 above. 0.28 g (0.24 mmol) of 18 gave 0.25 g (71%) of 20 as white foam. 1H NMR (500 MHz, CD3OH): δ 7.41 (d, 2H, J = 7.6 Hz), 7.31–7.28 (m, 6H), 7.22 (m, 1H), 6.86 (d, 4H, J = 8.7 Hz), 6.12 (b, 1H), 5.26 (dd, 1H, J = 10.4 Hz & 9.9 Hz), 5.16 (d, 1H, J = 4.3 Hz), 5.06 (dd, 1H, J = 9.9 Hz & 9.4 Hz), 4.95 (dd, 1H, J = 9.9Hz, both), 4.38 (dd, 1H, J = 10.9 Hz & 3.9 Hz), 4.27–4.14 (m, 3H), 4.08–4.04 (m, 3H), 3.91 (dd, 1H, J = 10.2 Hz & 9.1 Hz), 3.81 (m, 1H), 3.79 (s, 2 × 3H), 3.68 (dd, 1H, J = 14.8 Hz & 3.2 Hz), 3.50 (dd, 1H, J = 14.8 Hz & 2.0 Hz), 3.37 (s, 3H), 3.21 (dd, 1H, J = 10.5 Hz & 3.5 Hz), 3.09 (dd, 1H, J = 10.4 Hz & 3.2 Hz), 2.84–2.71 (m, 4H), 2.61–2.41 (m, 8H), 2.14 (s, 3H), 2.12 (s, 3H), 2.02 (s, 3H), 2.00–1.96 (m, 2H); 13C (125 MHz, CD3OH): δ 207.8, 207.4, 207.1, 172.7, 172.9, 171.9, 158.1, 158.4–156.8 (m), 144.9, 135.7, 135.6, 129.9, 127.8, 127.4, 126.5, 119.5–112.5 (m), 112.7, 104.0, 96.2, 86.1, 85.8, 81.1, 77.4, 74.3, 70.2, 68.6, 67.8, 67.7, 63.6, 57.2, 54.3, 51.7, 48.73, 48.70, 38.6, 37.13, 37.05, 31.0, 28.4, 28.17, 28.15, 28.0, 27.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C62H68F12N4NaO22 1471.4031, found 1471.3997.
2-Cyanoethyl [5′′-O-(4,4′-dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin-3′′-O-yl] N,N-diisopropylphosphoramidite (3). Triethylamine (0.16 mL, 1.3 mmol) and 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (0.12 g, 0.52 mmol) were added to a mixture of 19 (0.37 g, 0.26 mmol) in dry dichloromethane (1.0 mL). The mixture was stirred at ambient temperature for 1 h under nitrogen and filtered through a short silica gel column (5% Et3N in EtOAc). The product fractions were evaporated to dryness to give 0.40 g (96%) of the product (3) as white foam. 1H NMR (500 MHz, CD3OH): δ 8.00 (d, 1H, J = 8.7 Hz), 7.48–7.44 (2H), 7.38–7.27 (m, 6H), 7.20–7.25 (m, 2H), 6.95–6.92 (m, 4H), 6.13 and 6.03 (2 × d, 1H, J = 3.8 Hz and 3.9 Hz), 5.15 (d, 1H, J = 6.2 Hz), 5.12–5.08 (m, 1H), 5.01–4.97 (m, 1H), 4.92–4.83 (m, 1H), 4.35–3.05 (m, 13H), 3.82 (s, 6H), 3.26 and 3.23 (2 × s, 3H), 2.81–2.31 (m, 14H), 2.13, 2.11 and 2.10 (3 × s, 9H), 2.15–1.96 (m, 2H), 1.32–1.08 (m, 12H); 13C (125 MHz, CD3OH): δ 207.01, 206.99, 206.45, 206.42, 206.1, 172.9, 172.8, 172.3, 172.2, 171.91, 171.88, 158.8, 157.4–156.6 (m), 145.2, 145.0, 135.61, 135.59, 135.43, 135.37, 130.31, 130.28, 128.1, 127.9, 126.94, 126.90, 118.6, 118.2, 119.5–112.5 (m), 113.1, 113.0, 108.4, 108.3, 94.5, 94.4, 86.5, 86.3, 84.3, 84.2, 83.8, 83.7, 83.3, 83.2, 75.0, 74.8, 74.7, 71.9, 71.8, 70.9, 70.8, 70.1, 70.0, 68.9, 68.8, 67.2, 67.0, 63.1, 62.9, 58.9, 58.8, 58.2, 58.1, 58.0, 57.9, 57.8, 57.6, 57.5, 55.0, 51.6, 51.5, 48.9, 48.8, 48.7, 48.6, 43.2, 43.1, 42.9, 42.8, 39.5, 39.4, 37.6, 37.5, 37.3, 37.2, 31.0, 28.9, 28.8, 28.8, 28.7, 28.0, 27.6, 24.1, 24.0, 23.9, 23.9, 23.8, 23,7, 20.0, 19.9, 19.9, 19.8; 31P (200 MHz, CD3CN): δ 150.4, 148.9; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C71H86F12N6O23P 1649.5290, found 1649.5232.
2-Cyanoethyl [5′′-O-(4,4′-dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α1′′−5-ribostamycin-3′′-O-yl] N,N-diisopropylphosphoramidite (4). The α-anomer (4) was synthesized as described for 3 above. 0.25 g (0.17 mmol) of 20 gave 0.28 g (98%) of 4 as white foam. 1H NMR (500 MHz, CD3OH): δ 8.00–7.90 (m, 1H), 7.55–7.37 (m, 4H), 7.37–7.16 (m, 7H), 6.91–6.87 (m, 4H), 6.18 and 6.87 (2 × b, 1H), 5.30–4.94 (m, 4H), 4.45–3.00 (m, 13H), 3.80 and 3.79 (2 × s, 6H), 3.38 and 3.28 (2 × s, 3H), 2.80–2.28 (m, 14H), 2.08–1.88 (m, 2H), 2.11, 2.10, 2.09, 2.02, 2.00 and 1.99 (6 × s, 9H), 1.33–1.14 (m, 12H); 13C (125 MHz, CD3OH): δ 207.0, 206.9, 206.5, 206.2, 206.0, 172.8, 172.6, 172.5, 172.4, 172.1, 172.0, 158.7, 157.5–156.4 (m), 145.2, 145.0, 136.0, 135.7, 135.7, 135.5, 130.1, 130.0, 129.9, 129.9, 127.9, 127.9, 127.8, 127.8, 126.9, 119.4, 118.4, 116.9–110.4 (m), 113.1, 113.1, 113.0, 104.3, 104.0, 96.2, 95.8, 86.1, 85.9, 84.7, 81.8, 80.9, 74.2, 73.8, 70.0, 69.7, 68.9, 68.3, 68.3, 67.8, 63.6, 63.1, 60.0, 59.1, 58.6, 57.7, 57.5, 57.5, 57.4, 54.9, 54.9, 52.0, 51.7, 49.6, 49.2, 48.7, 48.7, 43.2, 42.8, 39.2, 38.7, 37.4, 37.3, 37.3, 37.2, 31.1, 31.0, 28.8, 28.7, 28.2, 28.0, 27.6, 24.0, 23.9, 23.8, 23.7, 20.2, 20.1, 19.8, 19.7; 31P (200 MHz, CD3CN): δ 150.95, 149.60; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C71H86F12N6O23P 1649.5290, found 1649.5221.
4.2. Synthesis of Oligonucleotides ON1, ON2, ON4–ON8
Oligonucleotides ON1–ON8 were synthesized on a 0.5 or 1.0 µmol scale using an automatic DNA/RNA synthesizer (Applied Biosystems 3400 DNA synthesizer, Waltham, MA, USA). Benzylthiotetrazol was used as an activator. The chain assembly with commercially available nucleoside phosphoramidites (Glen Research, Manchester, CA, USA) was carried out as usual (with a 300 s coupling time). For the coupling of 3 and 4, a double coupling procedure with an extended 600 s coupling time was used. According to DMTr-cation assay, 3 and 4 could be incorporated into oligonucleotide chains (ON1, ON2, ON7 and ON8) with ca. 95% coupling yield. For the synthesis of ON5 and ON6, a manual coupling of 3 and 4 to the neomycin-derived LCAA-CPG-support (7) was, however, applied: A solution of 3 and 4 (0.20 mol L−1 in acetonitrile, 100 µL, 20 µmol) was mixed with a solution of benzylthiotetrazol (BnSTet, 0.25 mol L−1, in dry acetonitrile, 80 µL, 20 µmol). The mixture was suspended with support 7 (L = 10 µmol g−1, 50 mg, 0.5 µmol) and the suspension was mixed for 10 min under nitrogen at ambient temperature. The suspension was loaded to a DNA-synthesis column, washed with acetonitrile and then a mixture of acetic anhydride, lutidine and N-methyl imidazol in THF (5:5:8:82, v/v/v/v) was introduced to the column (i.e., the capping step). After 5 min incubation time, the capping solution was washed away and then a mixture of 0.02 mol L−1 I2 in pyridine and H2O in THF (1:21:213, v/v/v) was introduced to the column (i.e., the oxidation step). After 1 min incubation, the iodine solution was removed, the support was washed with acetonitrile, dried, and the column was then coupled to the DNA/RNA synthesizer. According to DMTr-cation assay ca. 90% coupling yield for 3 and 4 was obtained. The RNA chain assembly with these supports were then carried out as usual. After the chain assembly, the solid supported oligonucleotides (ON1, ON2, ON5–ON8) bearing 3 and 4 were treated with a mixture of hydrazine acetate to selectively remove the levulinoyl protections: The synthesis column was removed from the synthesizer, a solution of hydrazinium acetate (NH2NH2·OH2, pyridine, AcOH, 0.124/4/1, v/v/v, 2 × 10 min at 25 °C) was manually introduced to the column, and then the support was washed with pyridine, acetonitrile and dichloromethane and dried. NaOMe-catalyzed transesterification was also used to selectively remove the acetyl groups of the neomycin moieties of ON4–ON6: The dried supports were removed from the synthesis columns, placed into micro centrifuge tubes and suspended in a mixture of 0.1 mol L−1 NaOMe in methanol (1.0 mL). The mixtures were mixed for 2 h at ambient temperature and then 1.0 mol L−1 aqueous NH4Cl (0.11 mL) was added. The supports were removed by filtration and the filtrates were evaporated to dryness. The partially deprotected residues of ON4–ON6 and the solid-supported ON1, ON2, ON7 and ON8 were finally subjected to concentrated ammonia (overnight at 55°) to completely deprotect the conjugates. The conjugates were purified by HPLC (Figure 2a,b and Figure 3) and their authenticity was verified by MS (ESI-TOF) spectroscopy (Table S1). The isolated yield of the conjugates ranged from 10% to 26%.
4.3. UV-Melting Temperature Studies
The melting curves (absorbance vs. temperature) were measured at 260 nm on a PerkinElmer Lambda 35 UV-Vis spectrometer equipped with a multiple cell holder and a Peltier temperature controller. An internal thermometer was also used. The temperature was changed at a rate of 0.2 °C min−1. Each Tm3-value was determined to be the maximum of the first derivative of the melting curve.
5. Conclusions
In this primarily synthetic study, synthesis of phosphoramidite building blocks of 2′′-O-methyl ribostamycins 3 and 4 that may be incorporated at any position of the oligonucleotide sequence, have been described. According to DMTr-assay and HPLC analysis of the released conjugates, the building blocks (3 and 4) could be efficiently incorporated into oligonucleotide sequences using a double phosphoramidite coupling (2 × 600 s) and using benzylthiotetrazol as an activator. 3 and 4 and a neomycin-derived solid support (7) were used for the preparation of aminoglycoside conjugates of 2′-O-methyl and 2′-deoxy oligoribonucleotide hybrids that were aimed to clamp a purine-rich DNA single strand (a sequence of c-Myc promoter 1). The potential of the intrachain ribostamycin and 3′-multiaminoglycoside overhangs to act as groove binders to stabilize triple helical region of the DNA-2′-O-methyl RNA clamps was demonstrated. According to UV-melting profile analysis, slightly increased clamp stability was observed.
Supplementary Materials
The supplementary materials are available online.
Acknowledgments
The financial support from the Academy of Finland (251539 and 256214) is gratefully acknowledged.
Author Contributions
Lotta Granqvist performed the oligonucleotide synthesis and UV-melting profile analysis, Andrzej Kraszewski performed part of the oligonucleotide synthesis, Ville Tähtinen carried out NMR measurements to verify the anomeric integrity of the ribostamycins, Pasi Virta (the corresponding author) supervised the project and performed part of the building block synthesis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thomas, J.R.; Hergenrother, P.J. Targeting of RNA with Small Molecules. Chem. Rev. 2008, 108, 1171–1224. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Gorini, L.; Davis, B.D. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol. Pharmacol. 1965, 1, 93–106. [Google Scholar] [PubMed]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, U.; Noller, H.F. Footprinting the sites of interaction of antibiotics with catalytic group I intron RNA. Science 1993, 260, 1500–1503. [Google Scholar] [CrossRef] [PubMed]
- Francois, B.; Russell, R.J.M.; Murray, J.B.; Aboul-ela, F.; Masquida, B.; Vicens, Q.; Westhof, E. Crystal structures of complexes between aminoglycosides and decoding site oligonucleotides: Role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acid Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, U.; Davies, J.; Schroeder, R. Antibiotic inhibition of group I ribozyme function. Nature 1991, 353, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Herman, T. Drugs targeting the ribosome. Curr. Opin. Struct. Biol. 2005, 15, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.-Y.; Galan, A.A.; Halim, N.S.; Mack, D.P.; Morelan, D.W.; Sanders, K.B.; Truong, H.N.; Czarnik, A.W. Inhibition of an HIV-1 Tat-derived peptide binding to TAR RNA by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 1995, 5, 2755–2760. [Google Scholar] [CrossRef]
- Zapp, M.L.; Stern, S.; Green, M.R. Small molecules that selectively block RNA bindingo f HIV-1 rev protein inhibit rev function and viral production. Cell 1993, 74, 969–978. [Google Scholar] [CrossRef]
- Tam, V.K.; Kwong, D.; Tor, Y. Fluorescent HIV-1 dimerization initiation site: Design, properties, and use for ligand discovery. J. Am. Chem. Soc. 2007, 129, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Ennifar, E.; Paillart, J.-C.; Bodlenner, A.; Walter, P.; Weibel, J.-M.; Aubertin, A.-M.; Pale, P.; Dumas, P.; Marquet, R. Targeting the dimerization initiation site of HIV-1 RNA with aminoglycosides: From crystal to cell. Nucleic Acid Res. 2006, 34, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Coffee, R.L., Jr. DNA triple helix stabilization by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 2000, 10, 1897–1899. [Google Scholar] [CrossRef]
- Arya, D.P.; Xue, L.; Tennant, P. Cobining the best in triplex recognition: synthesis and nucleic acid binding of a BQQ-neomycin conjugate. J. Am. Chem. Soc. 2003, 125, 8070–8071. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Coffee, R.L., Jr.; Charles, I. Neomycin induced hybrid triplex formation. J. Am. Chem. Soc. 2001, 123, 11093–11094. [Google Scholar] [CrossRef] [PubMed]
- Riguet, E.; Tripathi, S.; Désire, J.; Pandey, V.N.; Décout, J.-L. A peptide nucleic acid-nemaine conjugate that targets and cleaves HIV-1 TAR RNA inhibits viral replication. J. Med. Chem. 2004, 47, 4806–4809. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, B.; Tripathi, S.; Désire, J.; Baussanne, I.; Décout, J.-L.; Pandey, V.N. Mechanism of RNA cleavage catalyzed by sequence specific polyamide nucleic acid-neamine conjugate. Oligonucleotides 2007, 17, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Ketomäki, K.; Virta, P. Synthesis of aminoglycoside conjugates of 2′-O-methyl oligoribonucleotides. Bioconj. Chem. 2008, 19, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Kiviniemi, A.; Virta, P.; Lönnberg, H. Solid-supported synthesis and c lick conjugation of 4′-C-alkyne functionalized oligodeoxyribonucleotides. Bioconj. Chem. 2010, 21, 1890–1901. [Google Scholar] [CrossRef] [PubMed]
- Kiviniemi, A.; Virta, P.; Lönnberg, H. Utilization of intrachain 4′-C-azidomethylthymidine for preparation of oligodeoxyribonucleotide conjugates by click chemistry in solution and on a solid support. Bioconj. Chem. 2008, 19, 1726–1734. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Xing, L.; Cai, L.; Jin, H.W.; Zhao, P.; Yang, Z.-J.; Zhang, L.-R.; Zhang, L.-H. Studies on the synthesis of neamine-dinucleosides and neamine-PNA conjugates and their interaction with RNA. Bioorg. Med. Chem. Lett. 2008, 18, 5355–5358. [Google Scholar] [CrossRef] [PubMed]
- Irudayasamy, C.; Xi, H.; Arya, D.P. Sequence-specific targeting of RNA with an oligonucleotide-neomycin conjugate. Bioconj. Chem. 2007, 18, 160–169. [Google Scholar]
- Kiviniemi, A.; Virta, P. Synthesis of aminoglycoside-3′-conjugates of 2′-O-methyl oligoribonucleotides and their invasion to a 19F labeled HIV-1 TAR model. Bioconj. Chem. 2011, 22, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Tähtinen, V.; Granqvist, L.; Virta, P. Synthesis of C-5, C-2′ and C-4′-neomycin-conjugated triplex forming oligonucleotides and their affinity to DNA-duplexes. Bioorg. Med. Chem. 2015, 23, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Napoli, S.; Carbone, G.M.; Catapano, C.; Shaw, N.; Arya, D.P. Neomycin improves cationic lipid-mediated transfection of DNA in human cells. Bioorg. Med. Chem. Lett. 2005, 15, 3467–3469. [Google Scholar] [CrossRef] [PubMed]
- Geny, S.; Moreno, P.M.D.; Krzywkowski, T.; Gissberg, O.; Andersen, N.K.; Isse, A.J.; El-Madani, A.M.; Lou, C.; Pabon, Y.V.; Anderson, B.A.; et al. Next-generation bis-locked nucleic acids with stacking linker and 2′-glycylamino-LNA show enhanced DNA invasion into supercoiled duplexes. Nucleic Acids Res. 2016, 44, 2007–2019. [Google Scholar] [CrossRef] [PubMed]
- Alper, P.B.; Hung, S.-C.; Wong, C.-H. Metal catalyzed diazo transfer. Tetrahedron Lett. 1996, 37, 6029–6032. [Google Scholar] [CrossRef]
- Arya, D.P. New Approaches towards Recognition of Nucleic Acid Triple Helices. Acc. Chem. Res. 2011, 44, 134–146. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3 and 4 are available from the authors. |
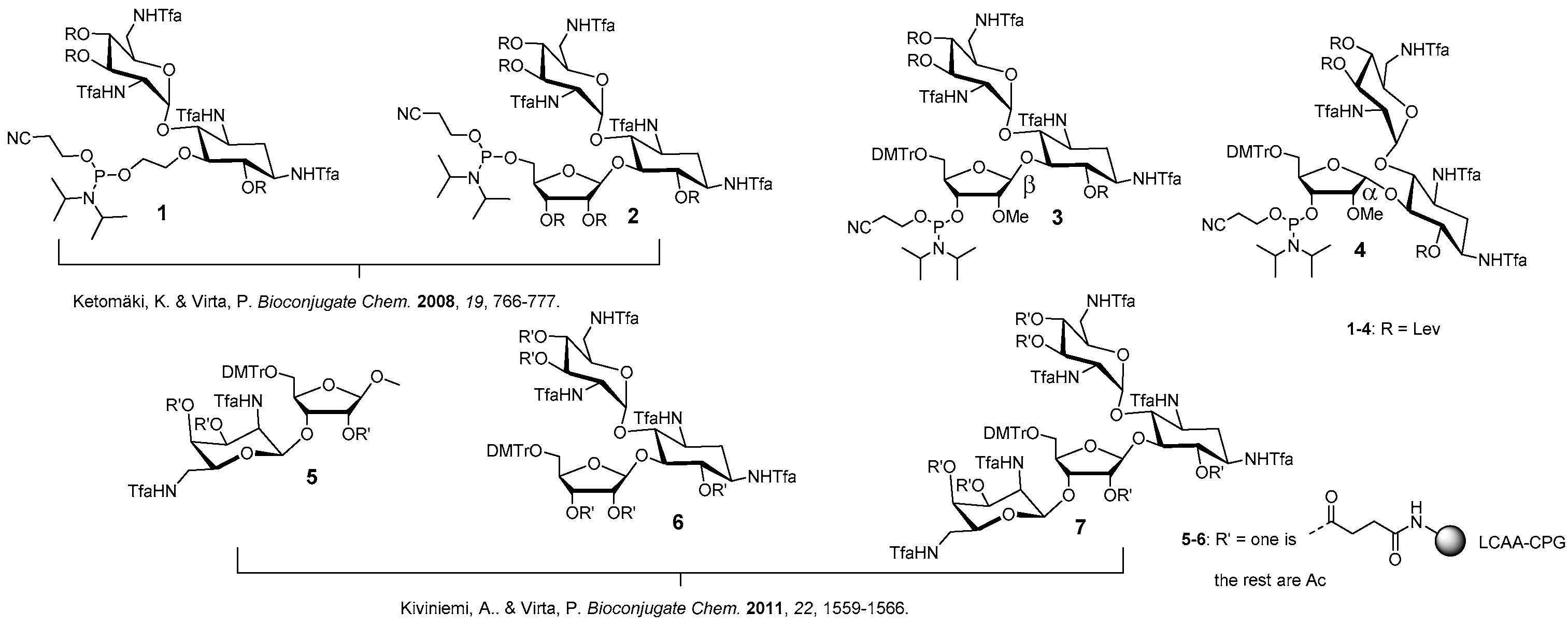
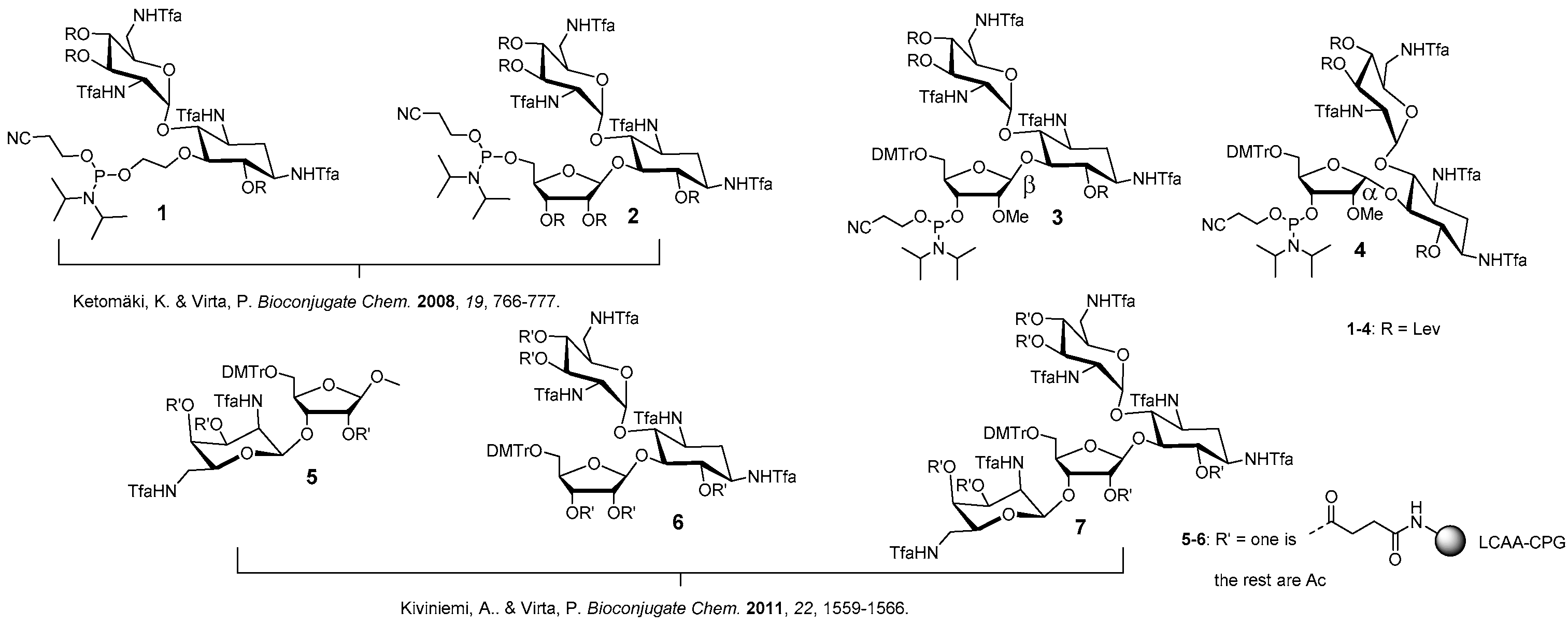
Figure 1.
Phosphoramidite building blocks 1–4 and solid supports 5–7 useful for the automated synthesis of aminoglycoside-oligonucleotide conjugates. 3 and 4 introduced in the present study.
Figure 1.
Phosphoramidite building blocks 1–4 and solid supports 5–7 useful for the automated synthesis of aminoglycoside-oligonucleotide conjugates. 3 and 4 introduced in the present study.
Scheme 1.
(i) TMSTf, DCM, (ii) NaOMe, MeOH, (iii) ClSi(iPr)2OSi(iPr)2Cl, Py, (iv) PMe3, toluene, aq NH3, dioxane, (v) TfaOMe, TEA, MeOH, (vi) Lev2O, DMAP, Py, (viii) DMTrCl, Py, (ix) ClP(NiPr2)OCE, TEA, DCM.
Scheme 1.
(i) TMSTf, DCM, (ii) NaOMe, MeOH, (iii) ClSi(iPr)2OSi(iPr)2Cl, Py, (iv) PMe3, toluene, aq NH3, dioxane, (v) TfaOMe, TEA, MeOH, (vi) Lev2O, DMAP, Py, (viii) DMTrCl, Py, (ix) ClP(NiPr2)OCE, TEA, DCM.
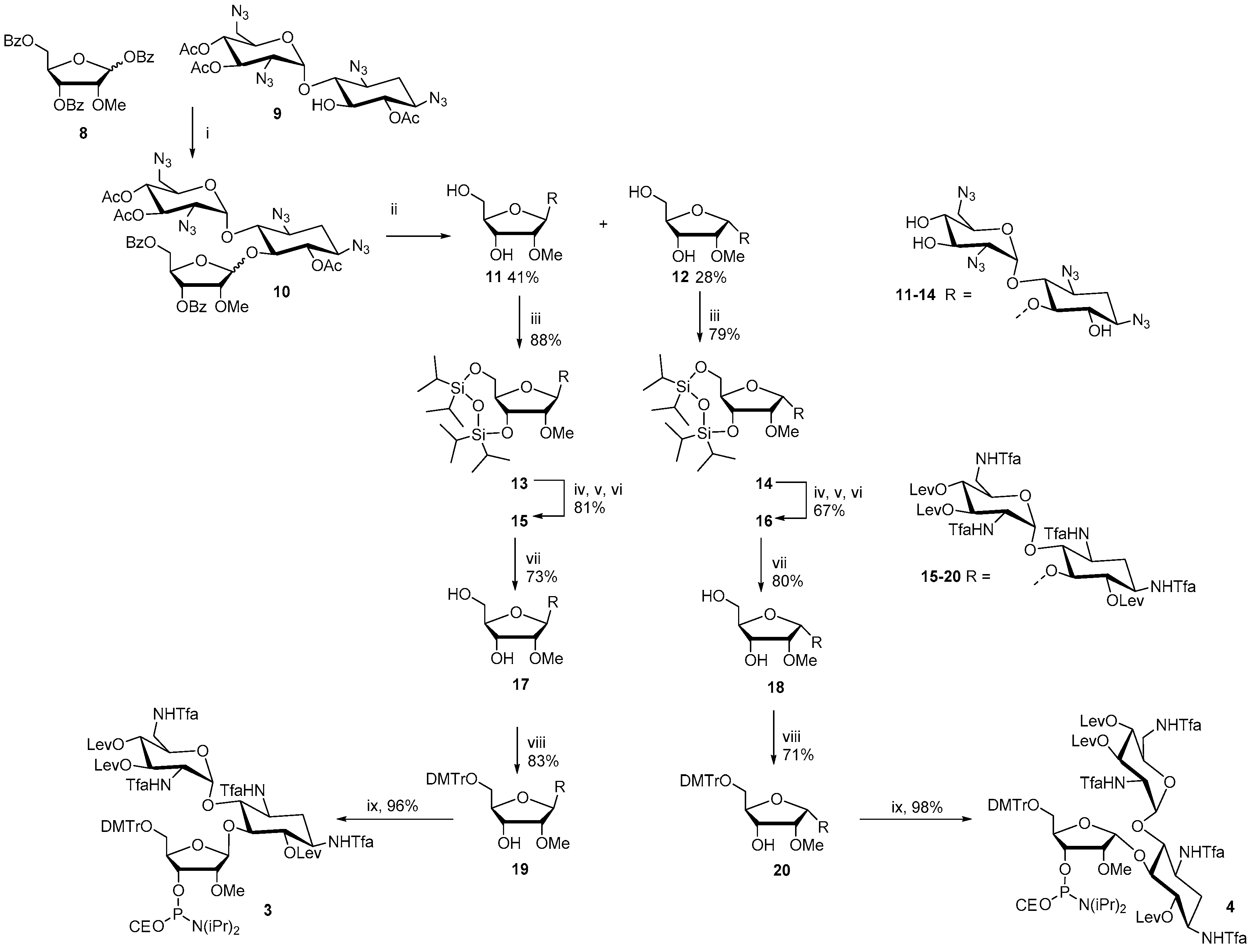
Figure 2.
Intra-chain fusions of ribostamycins and a 2′-O-methyl oligoribonucleotide (the test syntheses for the evaluation of the coupling efficiency of 3 and 4). Ion exchange HPLC profiles of crude ON2 (a), of crude ON1 (b) and of a mixture of ON1 and ON2 (1:1) (c); MS (ESI-TOF) spectrum of ON2 (d); HPLC conditions: An analytical (150 × 5 mm, monolithic) ion exchange column, flow rate 0.5 mL min−1, detection at 260 nm, a gradient elution (0–20 min) from 17 mmol L−1 to 100 mmol L−1 NaClO4 in aqueous 20 mmol L−1 Tris.
Figure 2.
Intra-chain fusions of ribostamycins and a 2′-O-methyl oligoribonucleotide (the test syntheses for the evaluation of the coupling efficiency of 3 and 4). Ion exchange HPLC profiles of crude ON2 (a), of crude ON1 (b) and of a mixture of ON1 and ON2 (1:1) (c); MS (ESI-TOF) spectrum of ON2 (d); HPLC conditions: An analytical (150 × 5 mm, monolithic) ion exchange column, flow rate 0.5 mL min−1, detection at 260 nm, a gradient elution (0–20 min) from 17 mmol L−1 to 100 mmol L−1 NaClO4 in aqueous 20 mmol L−1 Tris.
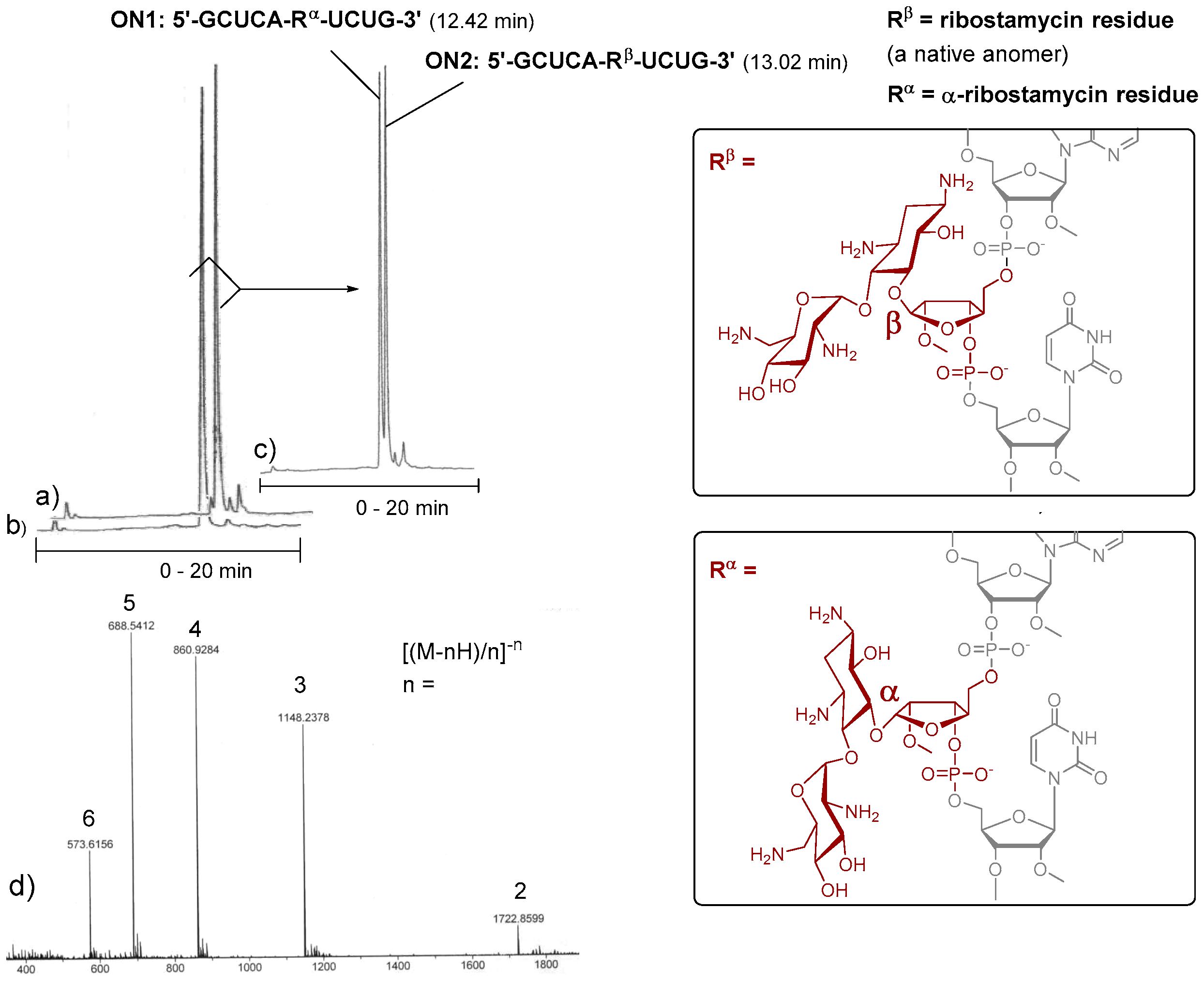
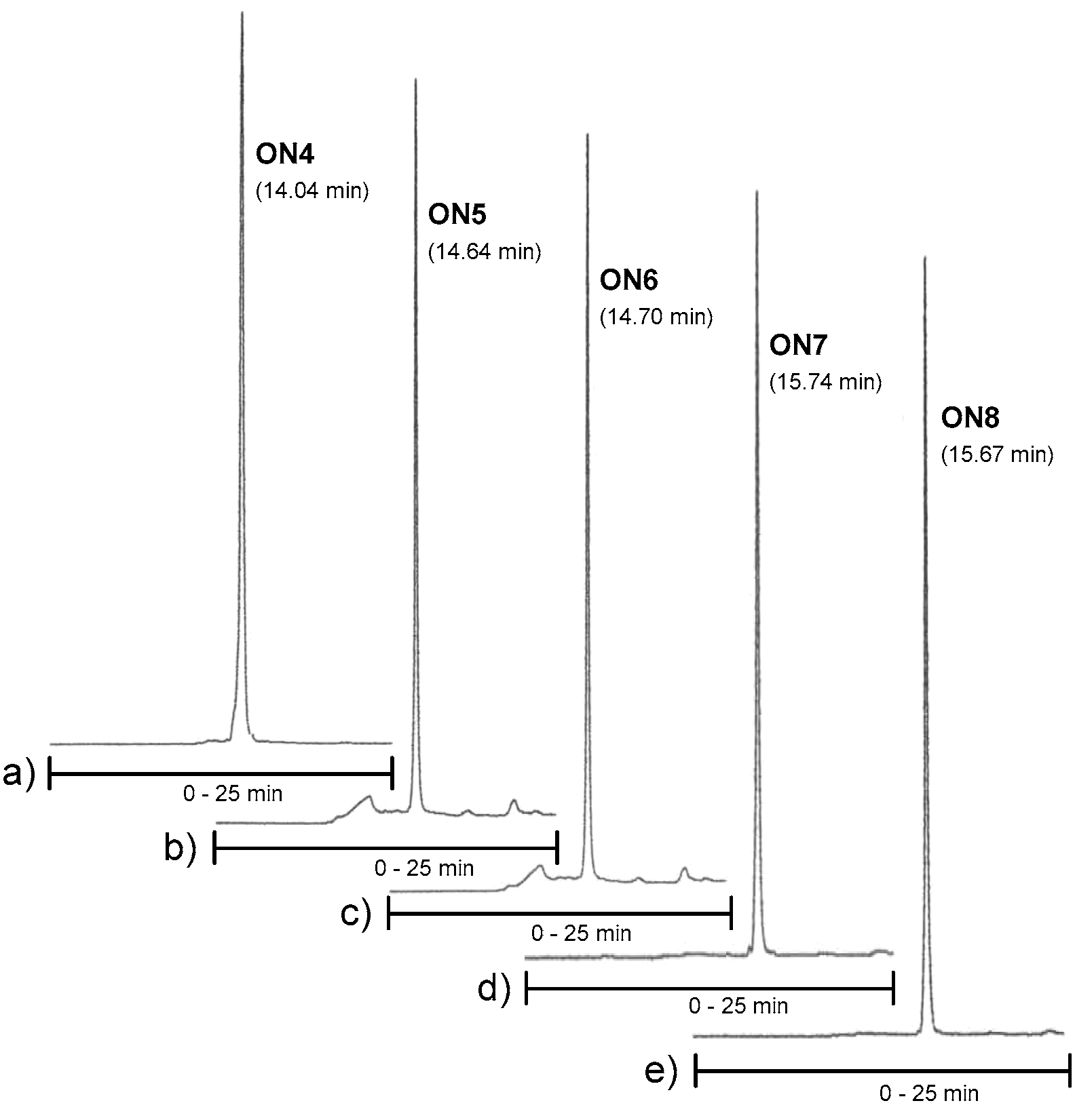
Figure 3.
RP HPLC profiles of ON4–ON6. Conditions: An analytical RP HPLC column (C18, 250 × 5 mm, 5 µm), flow rate: 1.0 mL min−1, detection at 260 nm, a gradient elution (0–25 min) from 0 to 50% acetonitrile in 0.1 mol L−1 aqueous triethylammonium acetate.
Figure 3.
RP HPLC profiles of ON4–ON6. Conditions: An analytical RP HPLC column (C18, 250 × 5 mm, 5 µm), flow rate: 1.0 mL min−1, detection at 260 nm, a gradient elution (0–25 min) from 0 to 50% acetonitrile in 0.1 mol L−1 aqueous triethylammonium acetate.
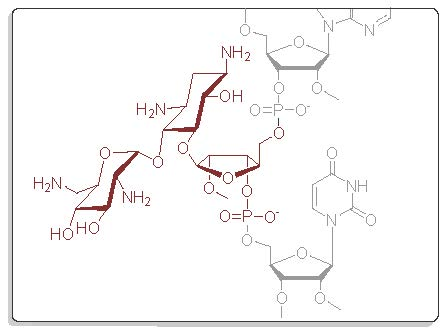
Scheme 2.
Synthesis of fusions of aminoglycosides and 2′-O-methyl 2′-deoxy oligoribonucleotide hybrids (ON4–ON6). The proposed clamp structures between ON4–ON8 and the purine-rich sequence of C-Myc promoter 1 (Target 1 and Target 2) are described. (i) phosphoramidite coupling cycle using benzylthiotetrazol as an activator; (ii) hydrazinium acetate (NH2NH2·OH2, pyridine, AcOH, 0.124/4/1, v/v/v, 2 × 10 min at 25 °C) (ON5–ON8), 0.1 mol L−1 NaOMe in methanol (2 h at 25 °C) (ON4–ON6), concentrated ammonia (overnight at 55 °C) (ON3–ON8).
Scheme 2.
Synthesis of fusions of aminoglycosides and 2′-O-methyl 2′-deoxy oligoribonucleotide hybrids (ON4–ON6). The proposed clamp structures between ON4–ON8 and the purine-rich sequence of C-Myc promoter 1 (Target 1 and Target 2) are described. (i) phosphoramidite coupling cycle using benzylthiotetrazol as an activator; (ii) hydrazinium acetate (NH2NH2·OH2, pyridine, AcOH, 0.124/4/1, v/v/v, 2 × 10 min at 25 °C) (ON5–ON8), 0.1 mol L−1 NaOMe in methanol (2 h at 25 °C) (ON4–ON6), concentrated ammonia (overnight at 55 °C) (ON3–ON8).
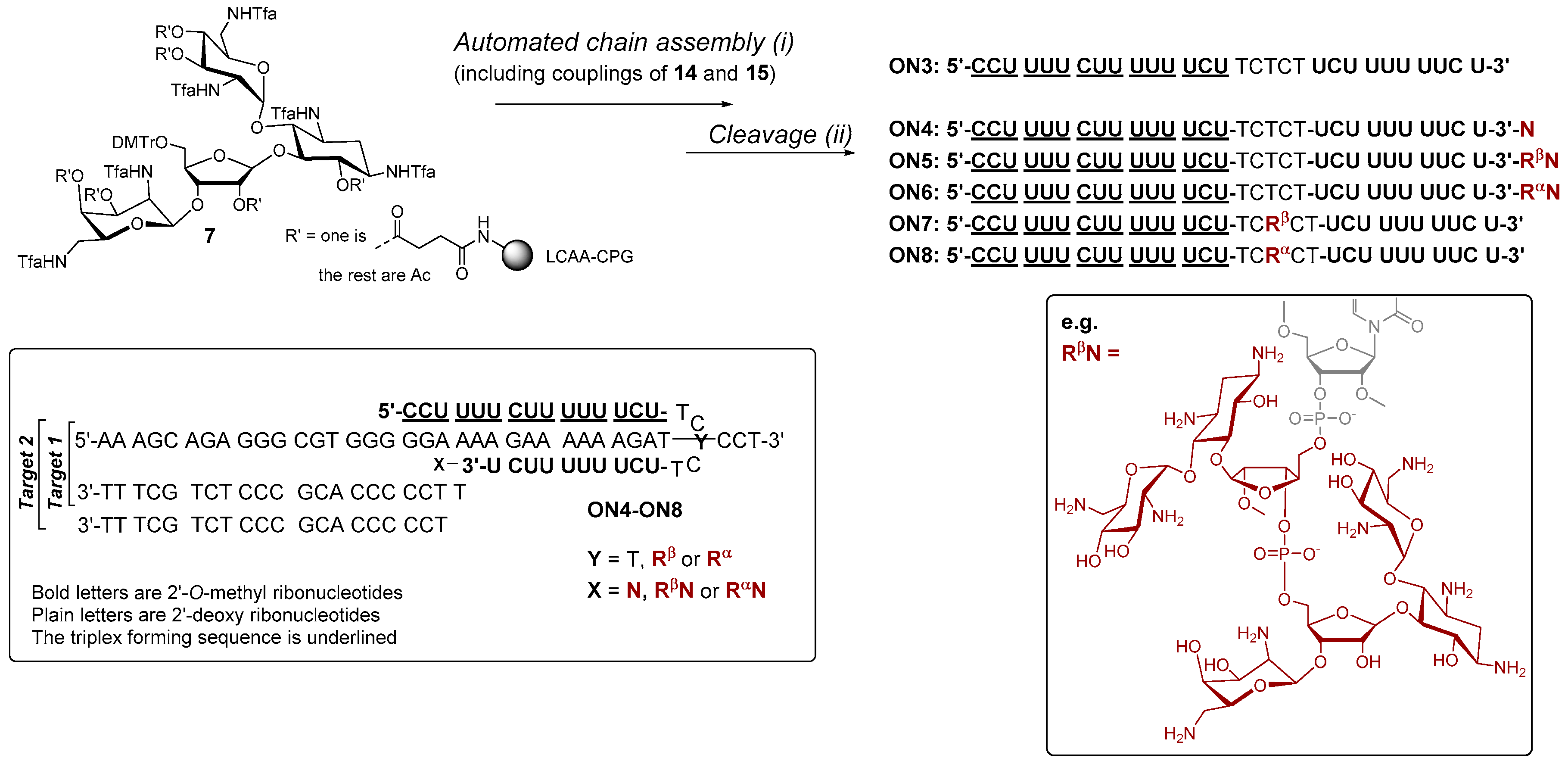
Figure 4.
UV-melting profiles of the triple helical constructs (the duplex melting range, Tm = 65 °C excluded). Conditions: 2.0 each oligonucleotide (Target 1 or 2 + ON3–ON8, cf. Figure 3), 10 mmol L−1 sodium cacodylate, pH 6.0 (a,c) and pH 7.0 (b,c), 0.1 mol L−1 NaCl, 0.2 °C/min, detection at 260 nm.
Figure 4.
UV-melting profiles of the triple helical constructs (the duplex melting range, Tm = 65 °C excluded). Conditions: 2.0 each oligonucleotide (Target 1 or 2 + ON3–ON8, cf. Figure 3), 10 mmol L−1 sodium cacodylate, pH 6.0 (a,c) and pH 7.0 (b,c), 0.1 mol L−1 NaCl, 0.2 °C/min, detection at 260 nm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
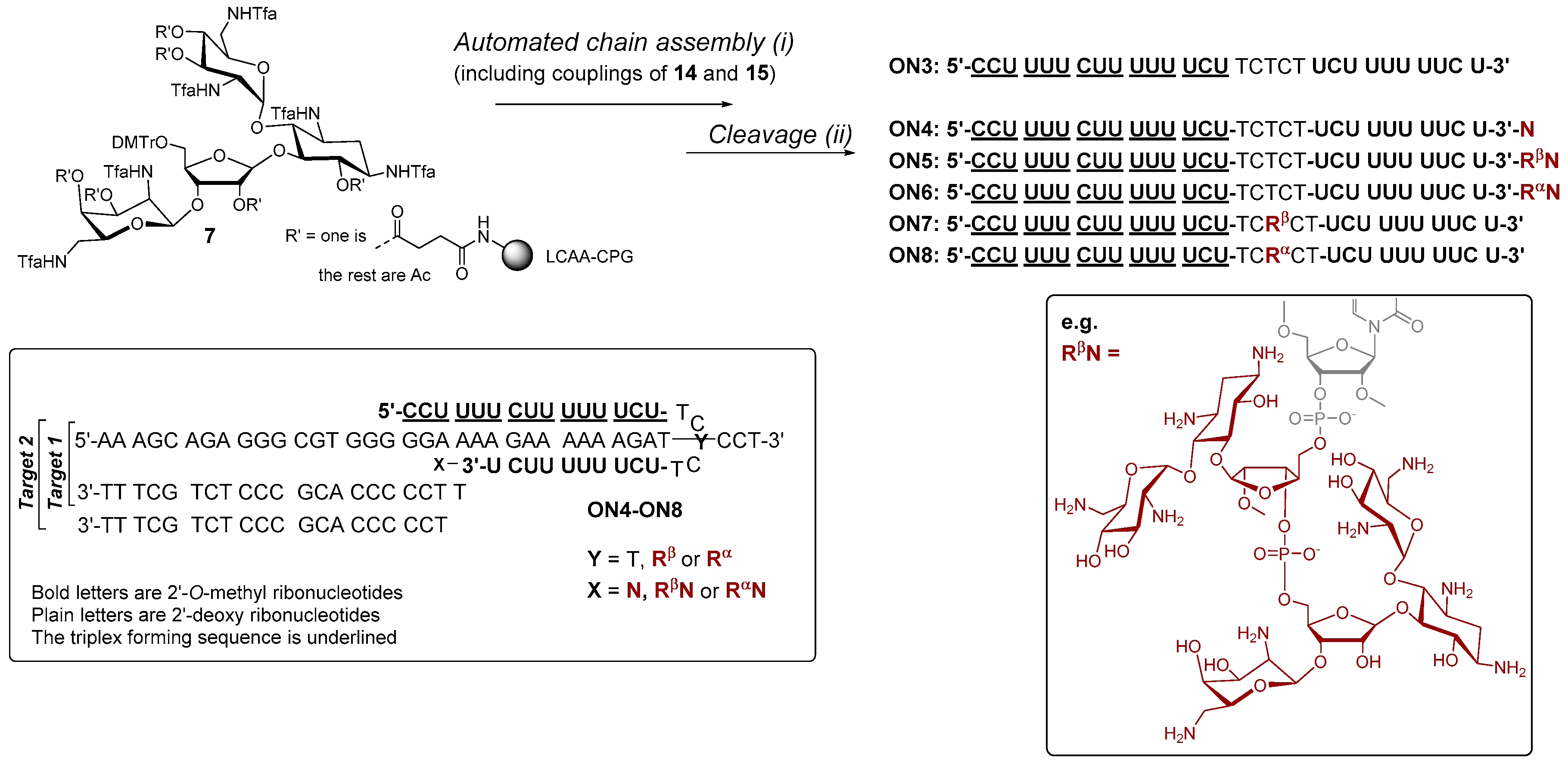{kind=link}
Table 1.
UV thermal melting temperatures (Tm3-values) of the triple helices.
| Duplex | ON3 | ON4 | ON5 | ON6 | ON7 | ON8 |
|---|---|---|---|---|---|---|
| Target 1 (pH 6.0) | 32.3 | 36.8 (+4.5) | 34.5 (+2.2) | 35.2 (+2.9) | 34.9 (+2.6) | 36.1 (+3.8) |
| Target 1 (pH 7.0) | 20.2 | 24.2 (+4.0) | 23.2 (+3.0) | 25.3 (+5.1) | 23.0 (+2.8) | 24.5 (+4.3) |
| Target 2 (pH 6.0) | 31.1 | 36.9 (+5.8) | 34.8 (+3.7) | 34.3 (+3.2) | n.a. | n.a. |
| Target 2 (pH 7.0) | 19.4 | 25.2 (+5.8) | 24.2 (+4.8) | 24.9 (+5.5) | n.a. | n.a. |
Notes: ∆Tm3-values are shown in parentheses in comparison to the Tm3-values of Target 1 or Target 2 in the presence of ON3. Tm3-values are average values extracted from three 0.2 °C min−1 heating and cooling ramps. Error limit in each measurement is in maximum ±0.3 °C.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Granqvist, L.; Kraszewski, A.; Tähtinen, V.; Virta, P. Synthesis of Aminoglycoside-2′-O-Methyl Oligoribonucleotide Fusions. Molecules 2017, 22, 760. https://doi.org/10.3390/molecules22050760
AMA Style
Granqvist L, Kraszewski A, Tähtinen V, Virta P. Synthesis of Aminoglycoside-2′-O-Methyl Oligoribonucleotide Fusions. Molecules. 2017; 22(5):760. https://doi.org/10.3390/molecules22050760
Chicago/Turabian StyleGranqvist, Lotta, Andrzej Kraszewski, Ville Tähtinen, and Pasi Virta. 2017. "Synthesis of Aminoglycoside-2′-O-Methyl Oligoribonucleotide Fusions" Molecules 22, no. 5: 760. https://doi.org/10.3390/molecules22050760