Recent Advances in Asymmetric Organocatalyzed Conjugate Additions to Nitroalkenes

 ,
,  , , and
, , and
Abstract
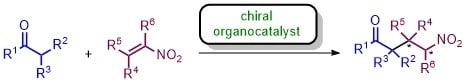
:
{kind=link}
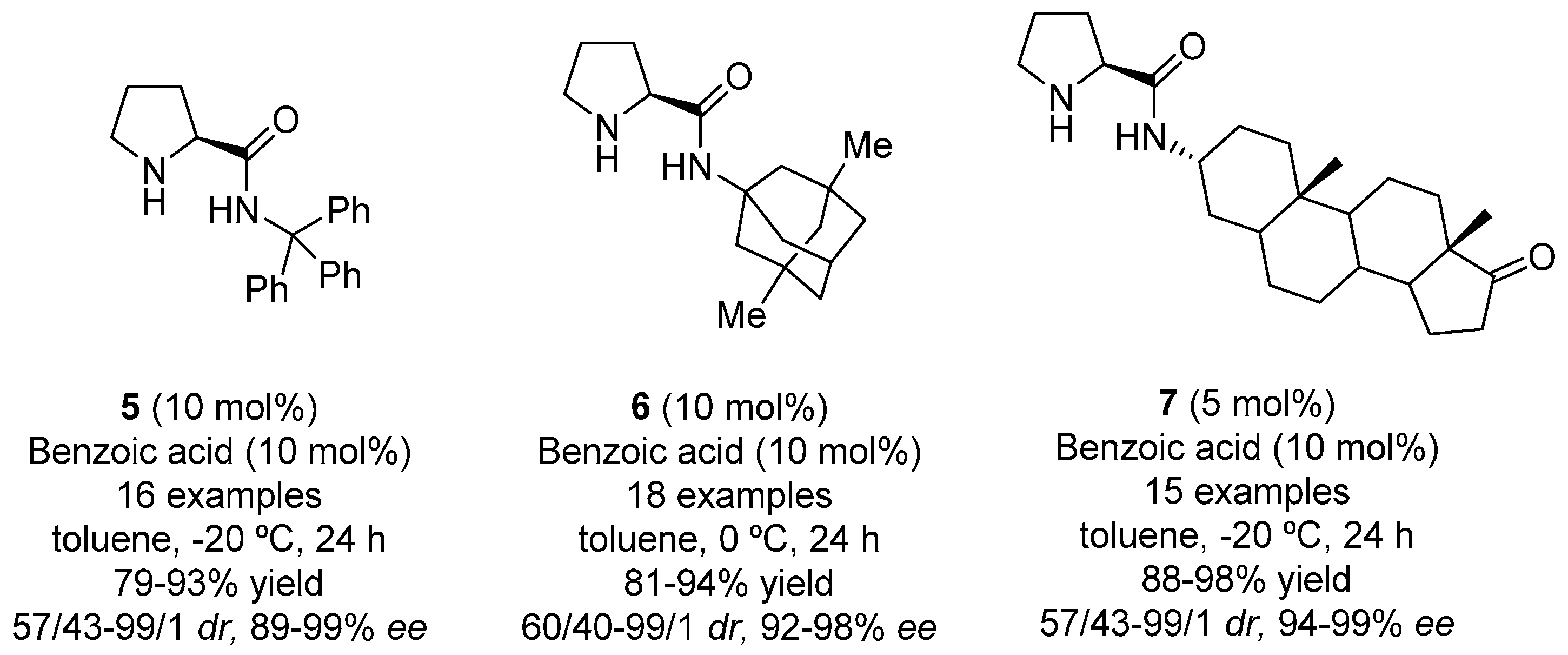{kind=link}
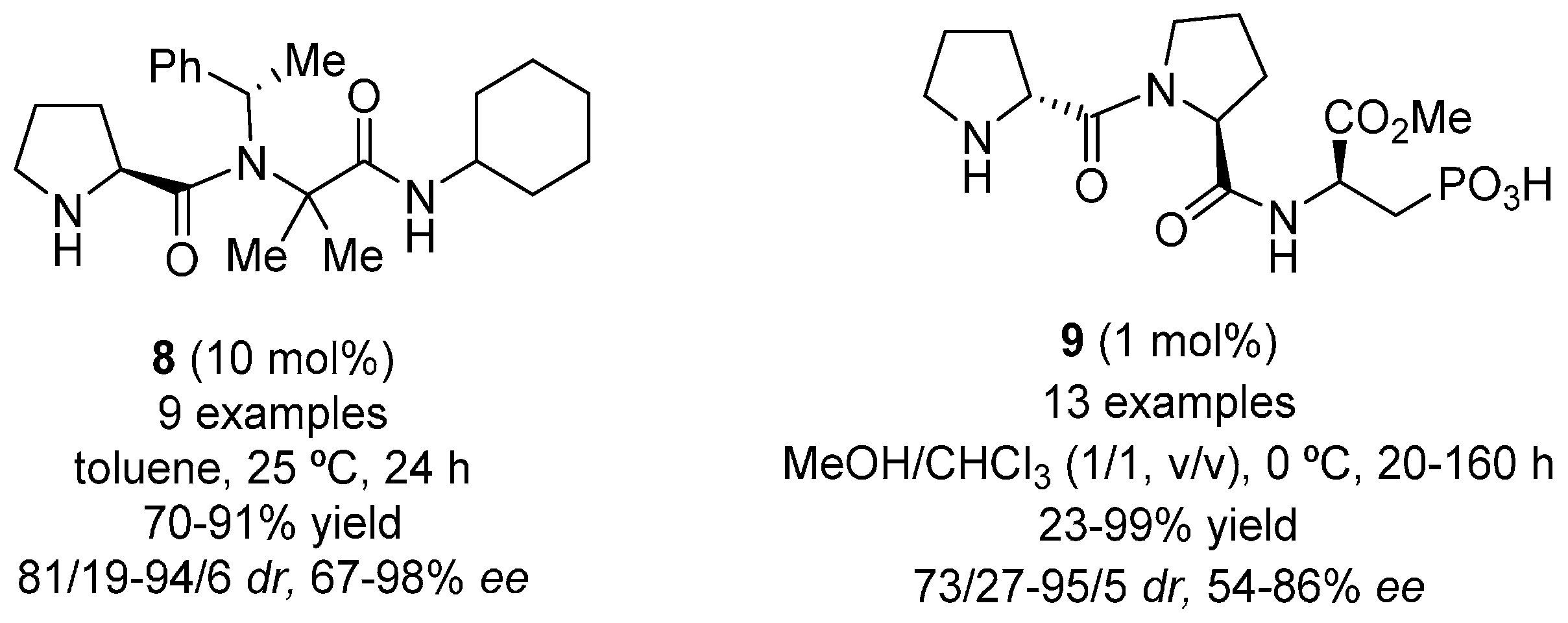{kind=link}
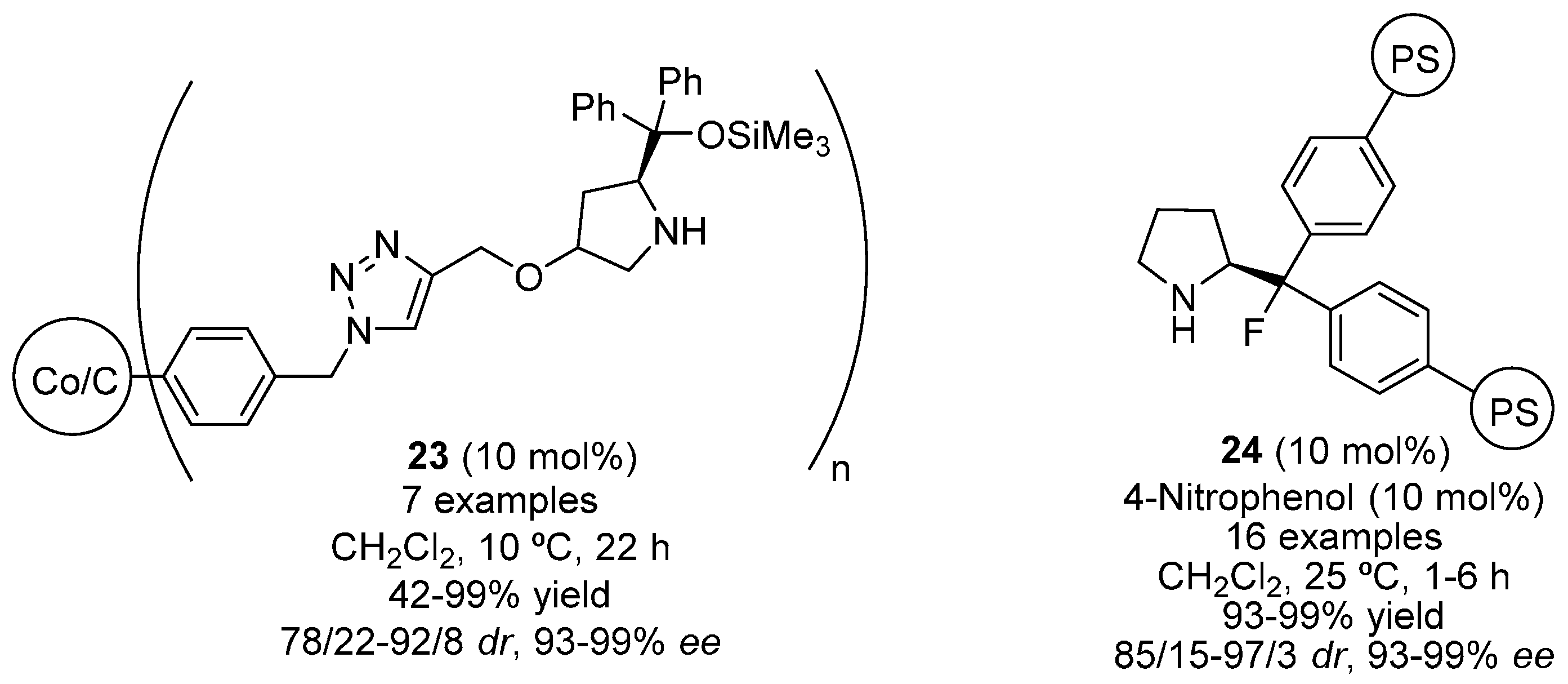{kind=link}
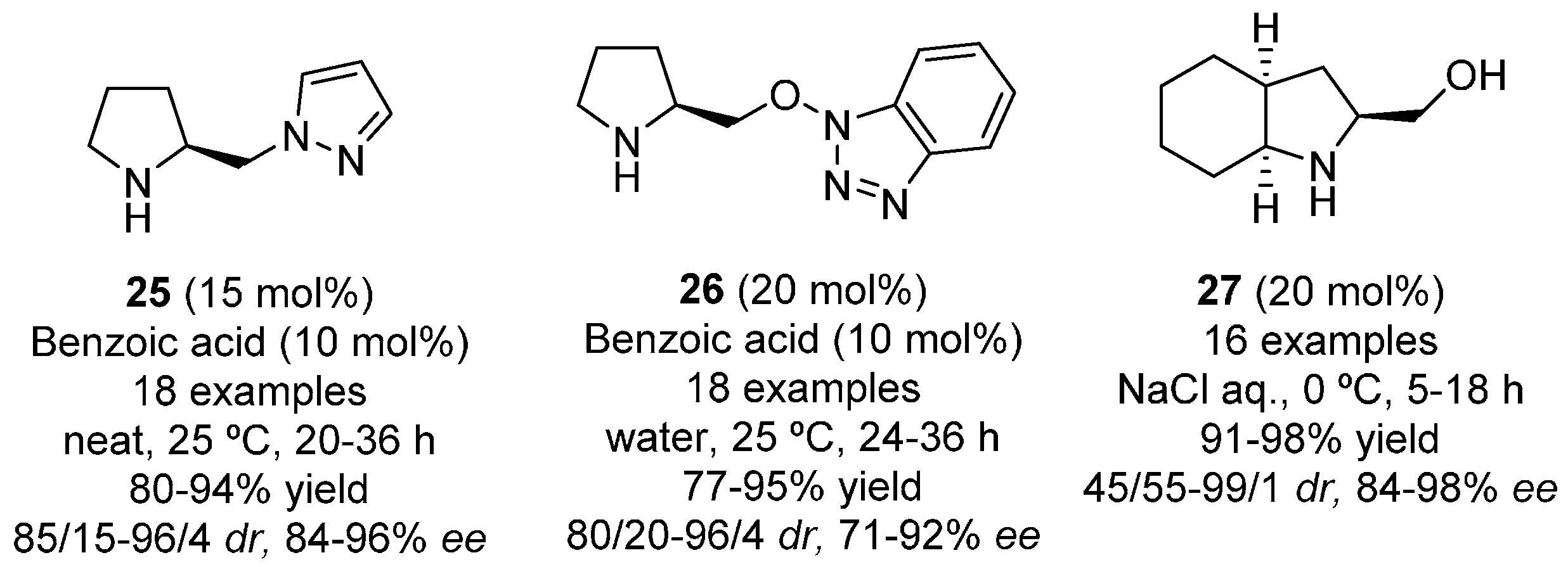{kind=link}
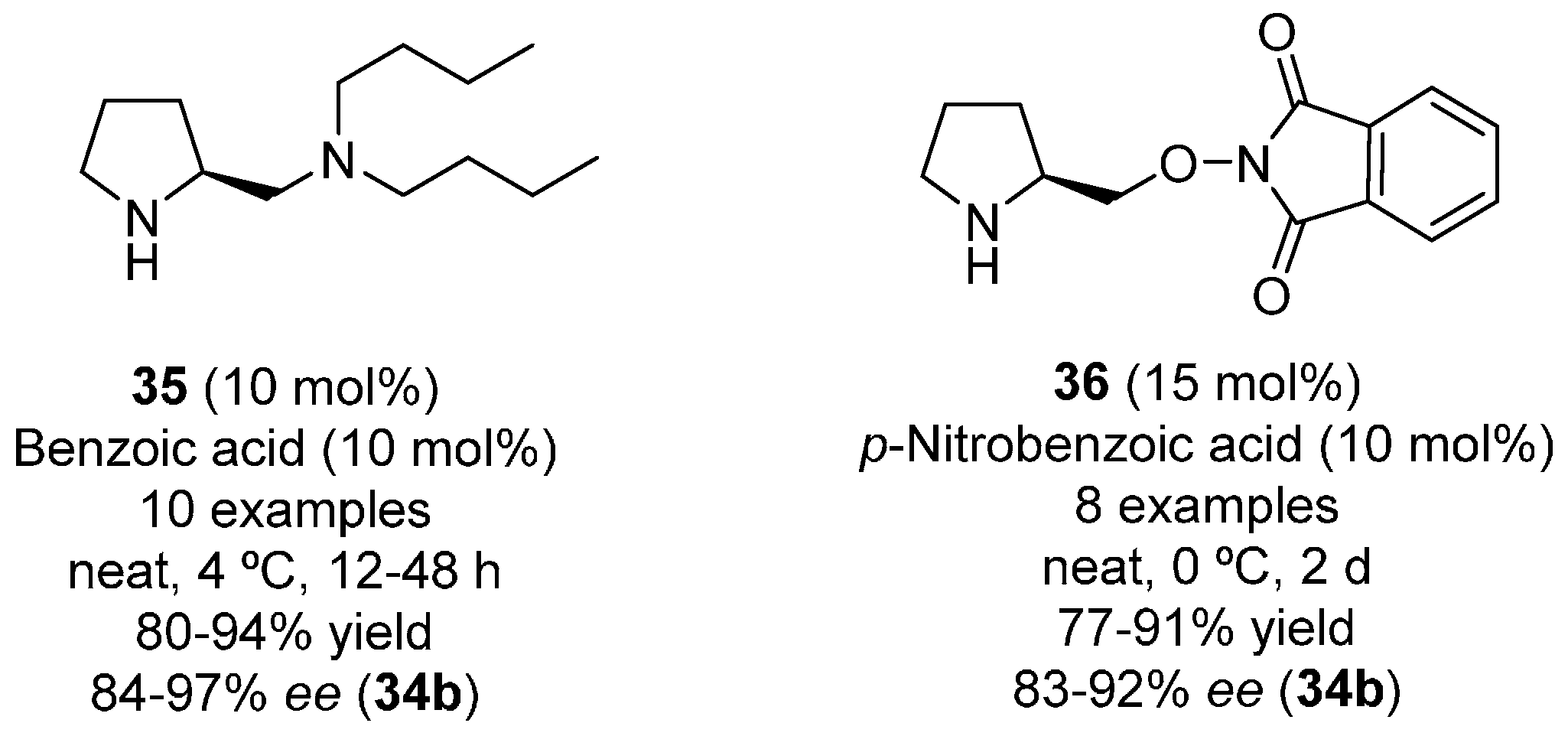{kind=link}
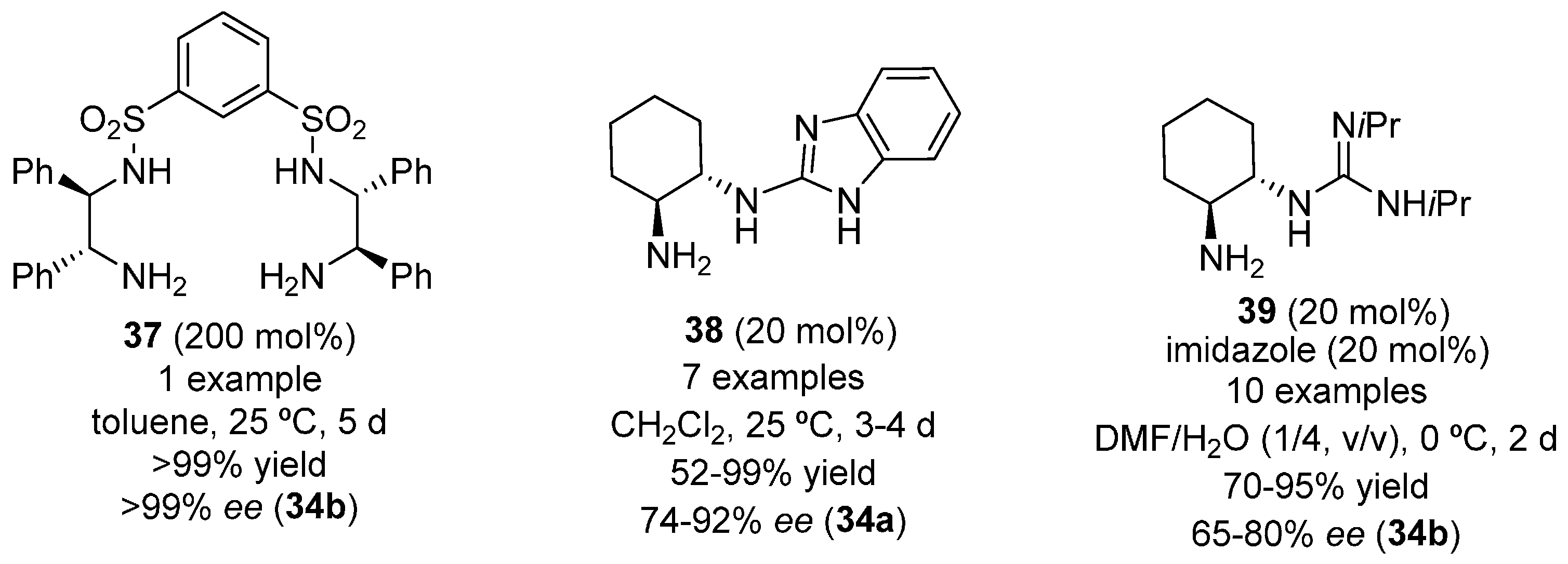{kind=link}
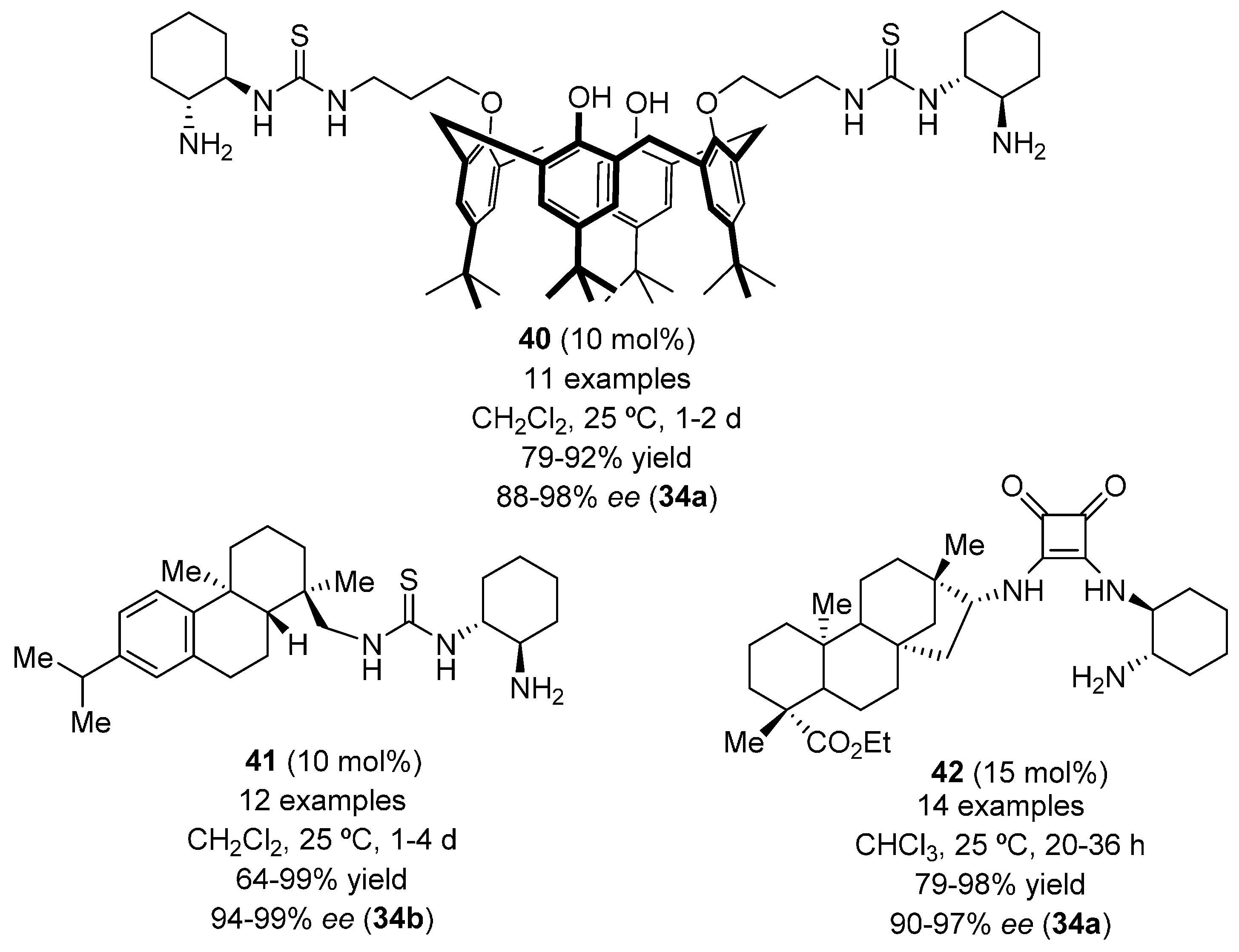{kind=link}
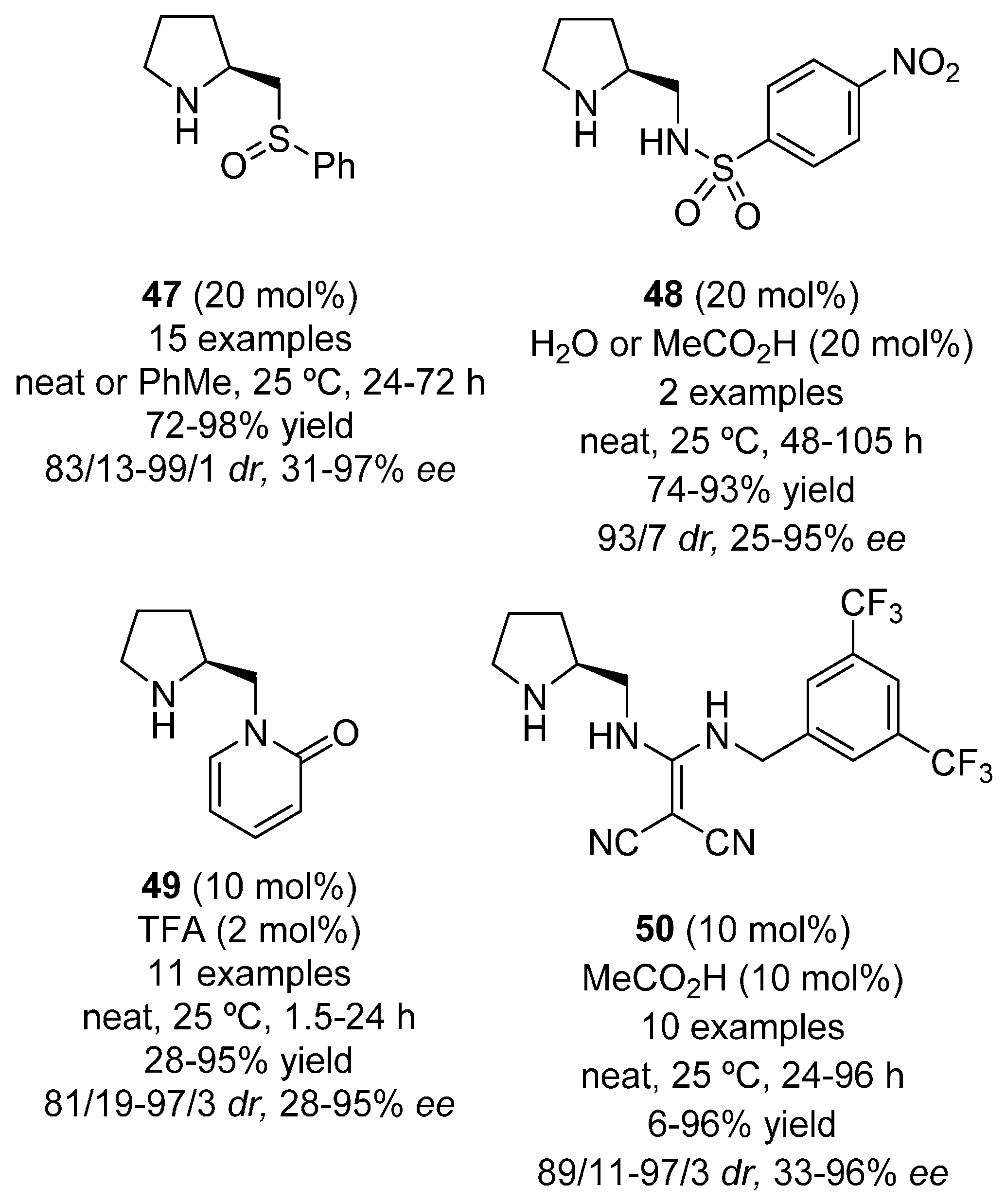{kind=link}
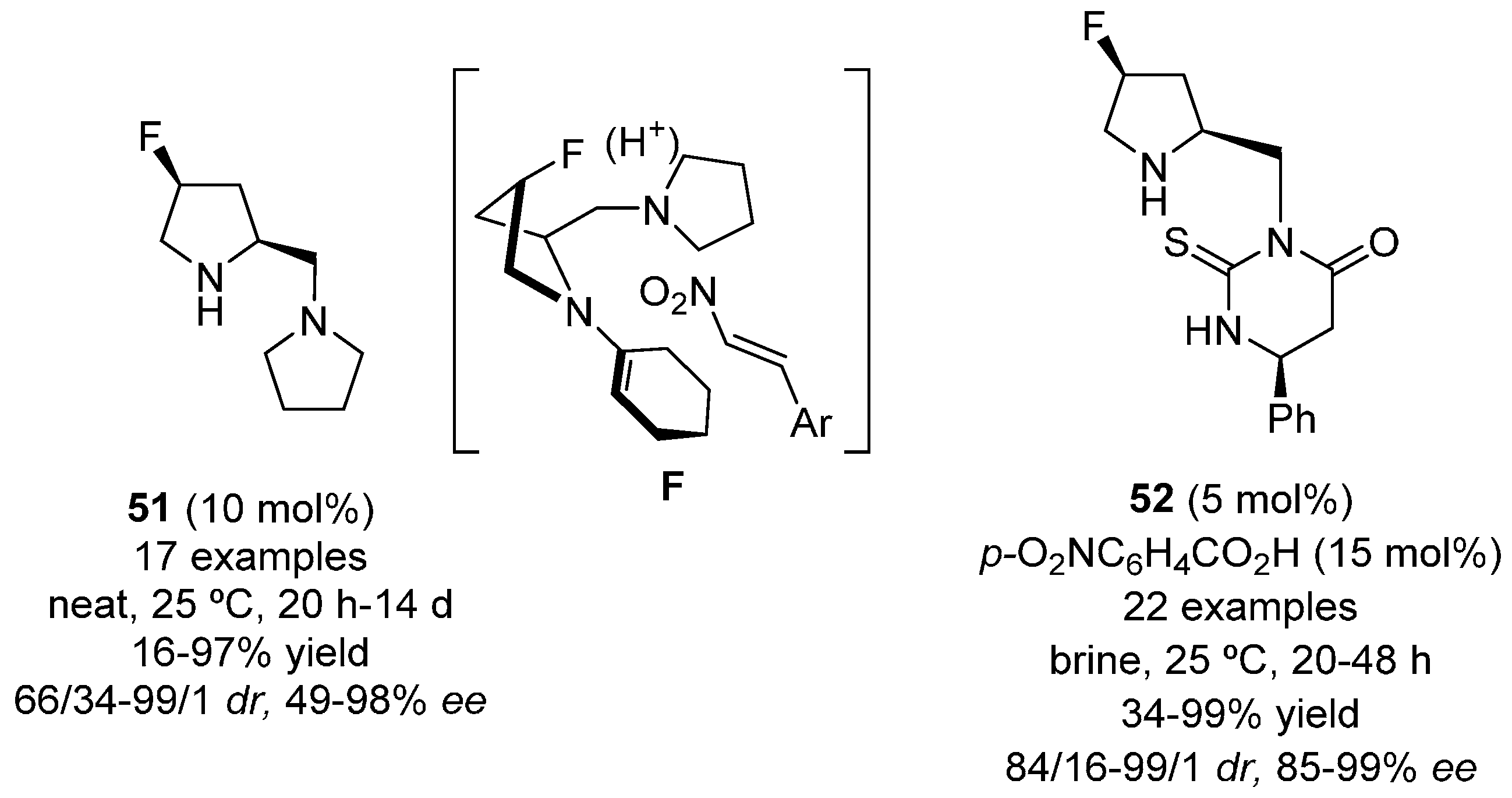{kind=link}
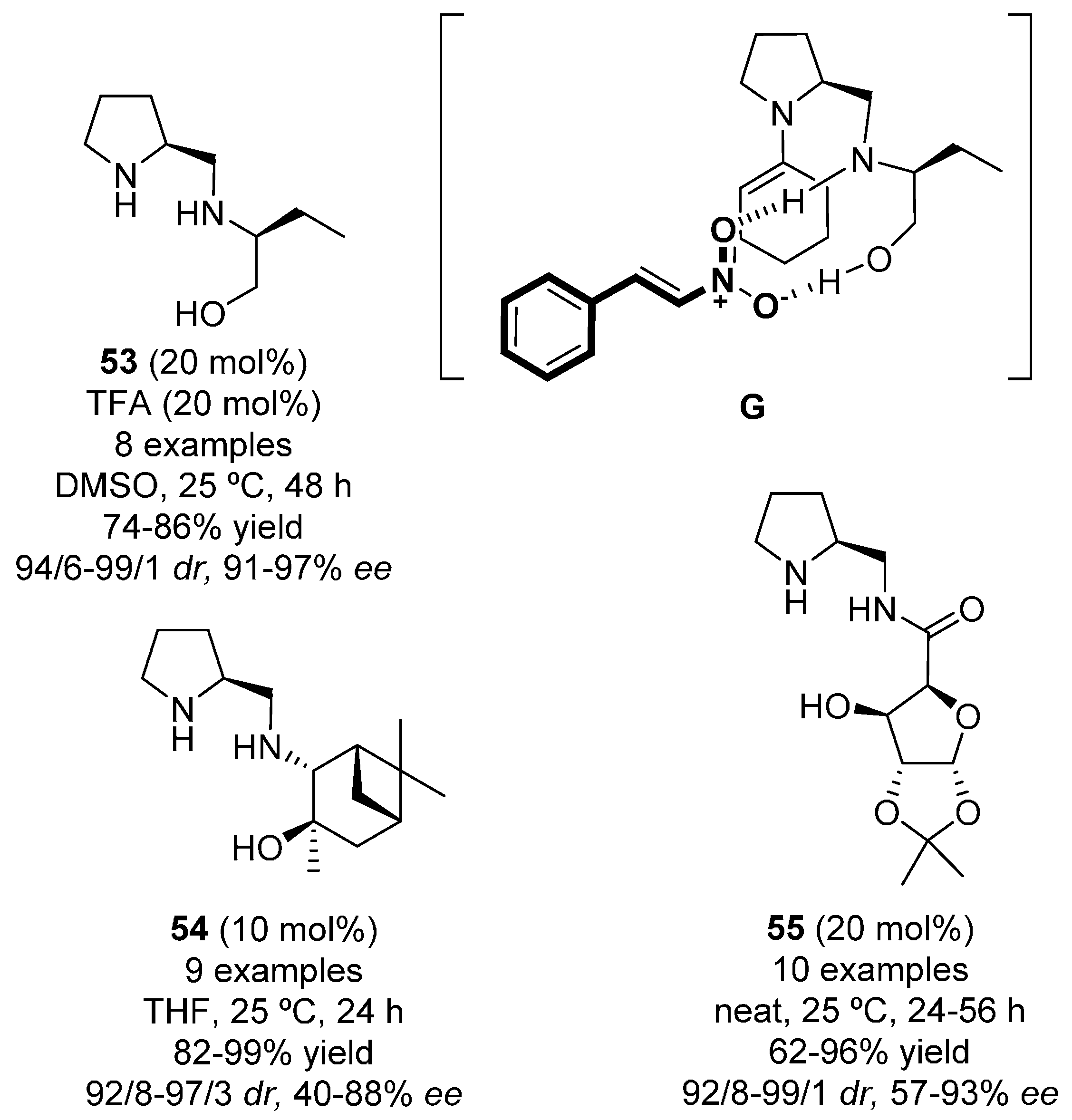{kind=link}
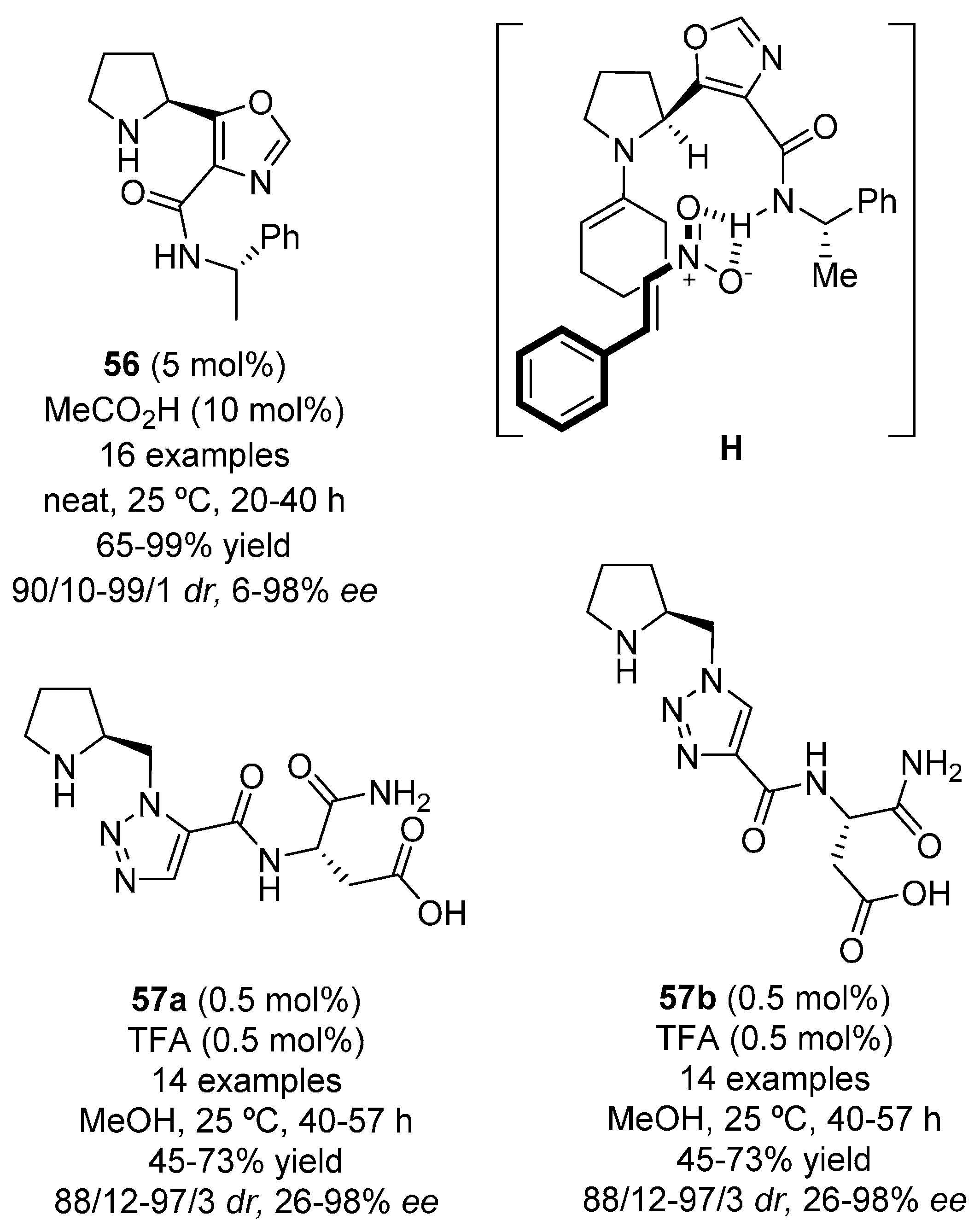{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
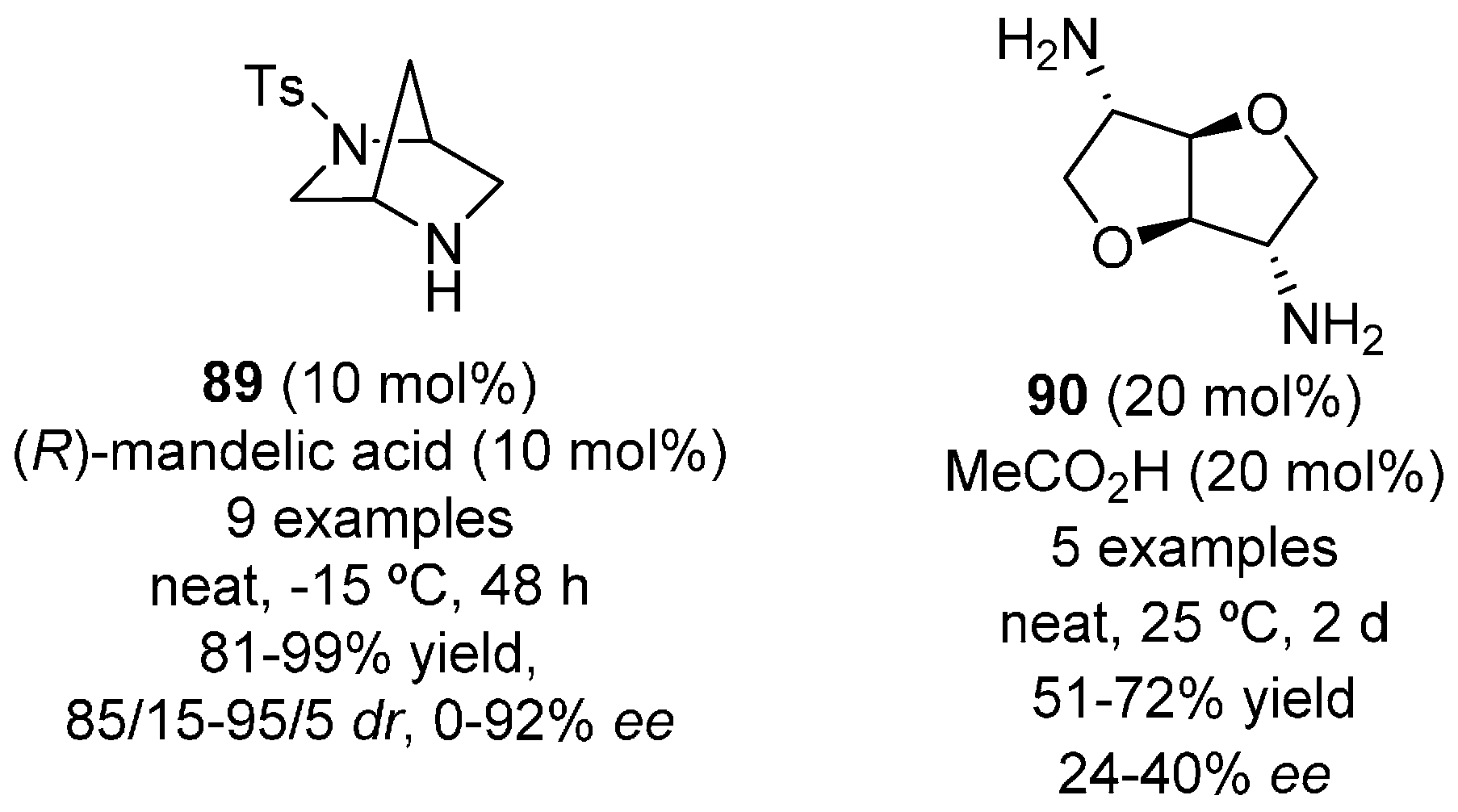{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
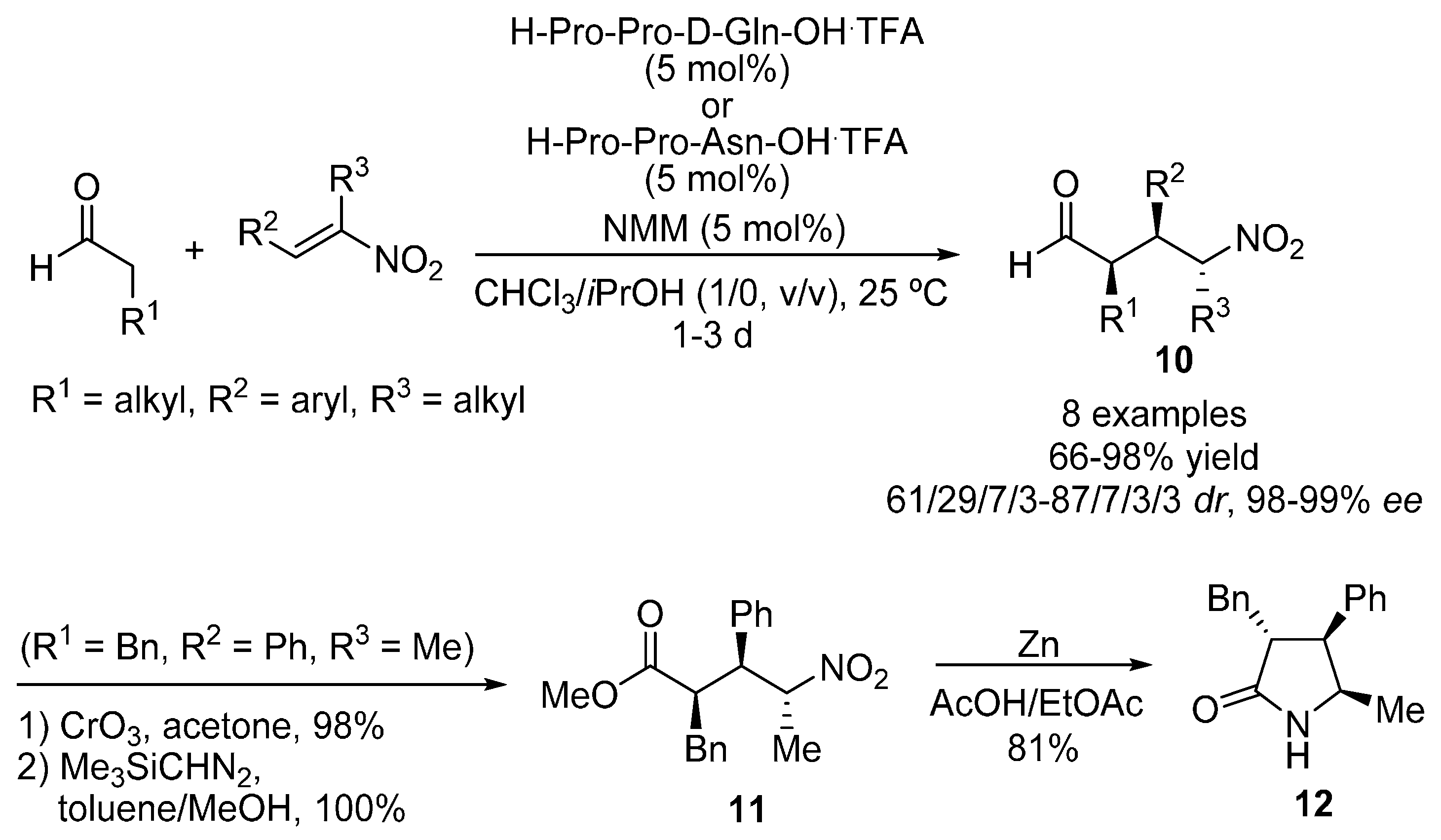{kind=link}
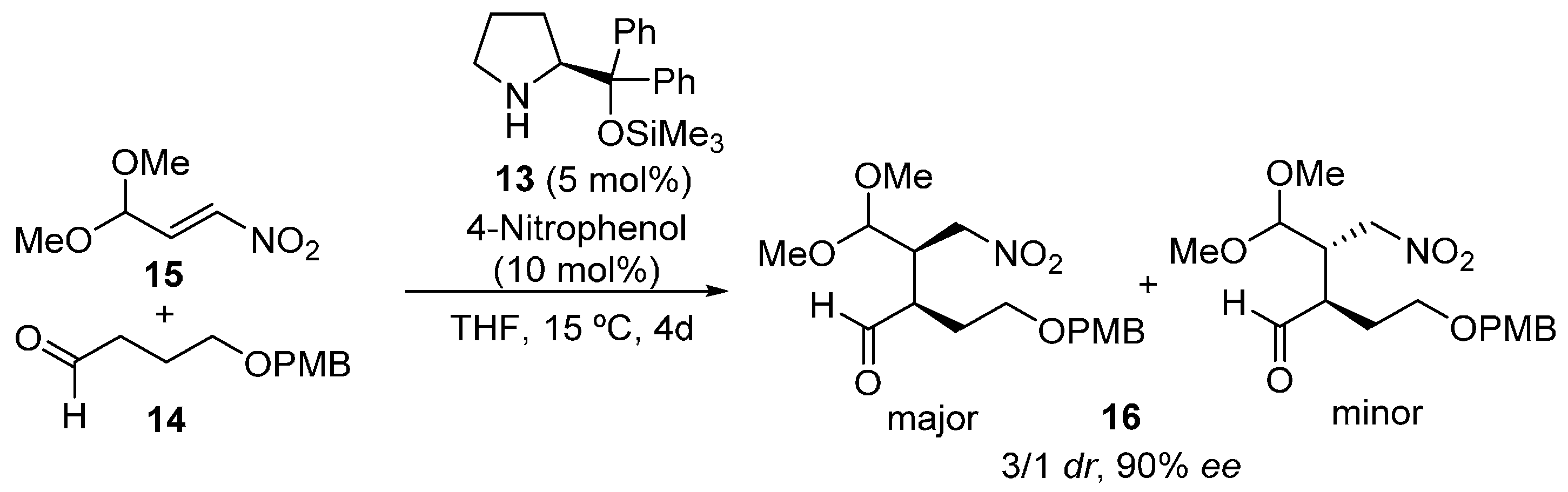{kind=link}
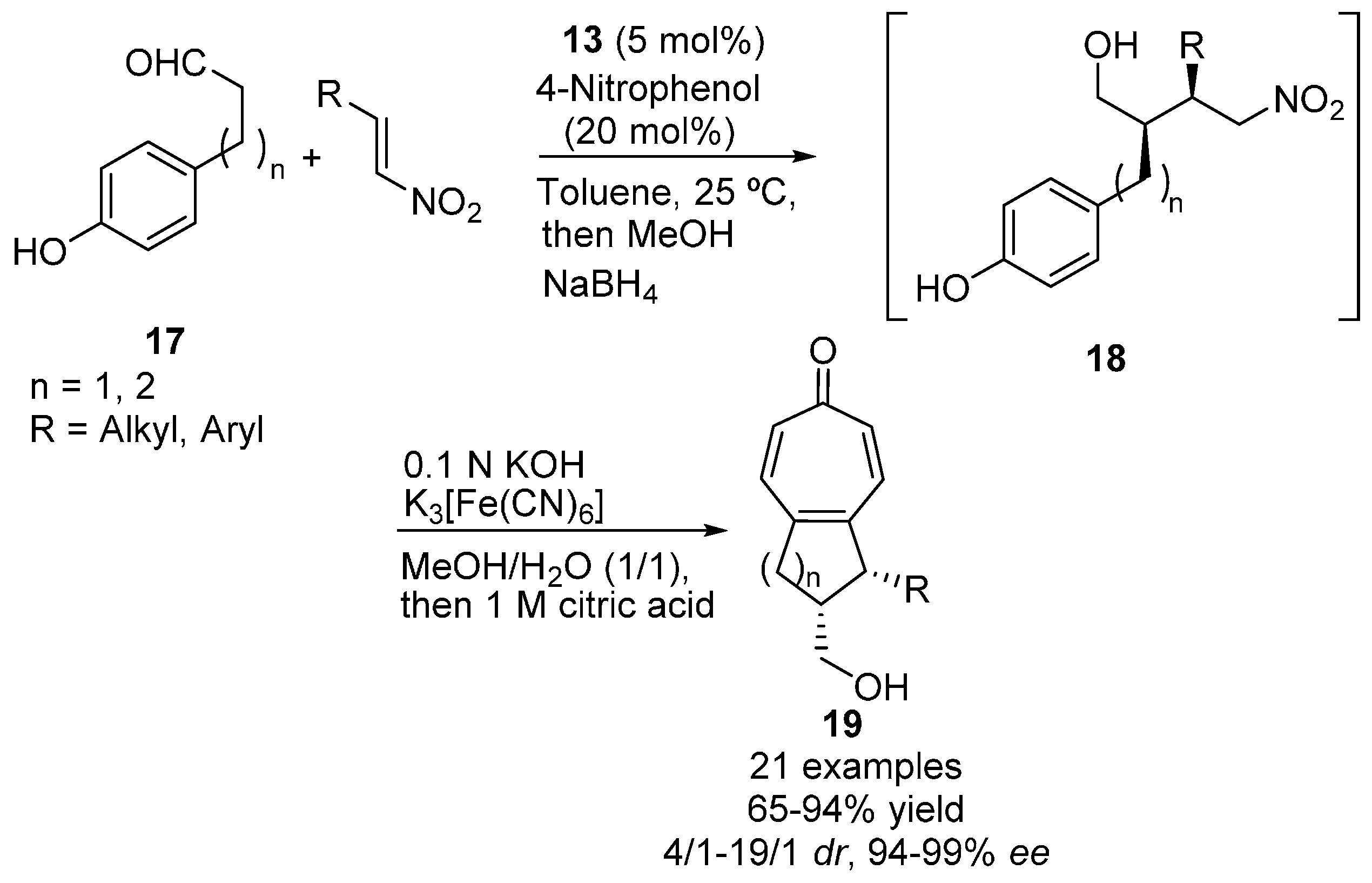{kind=link}
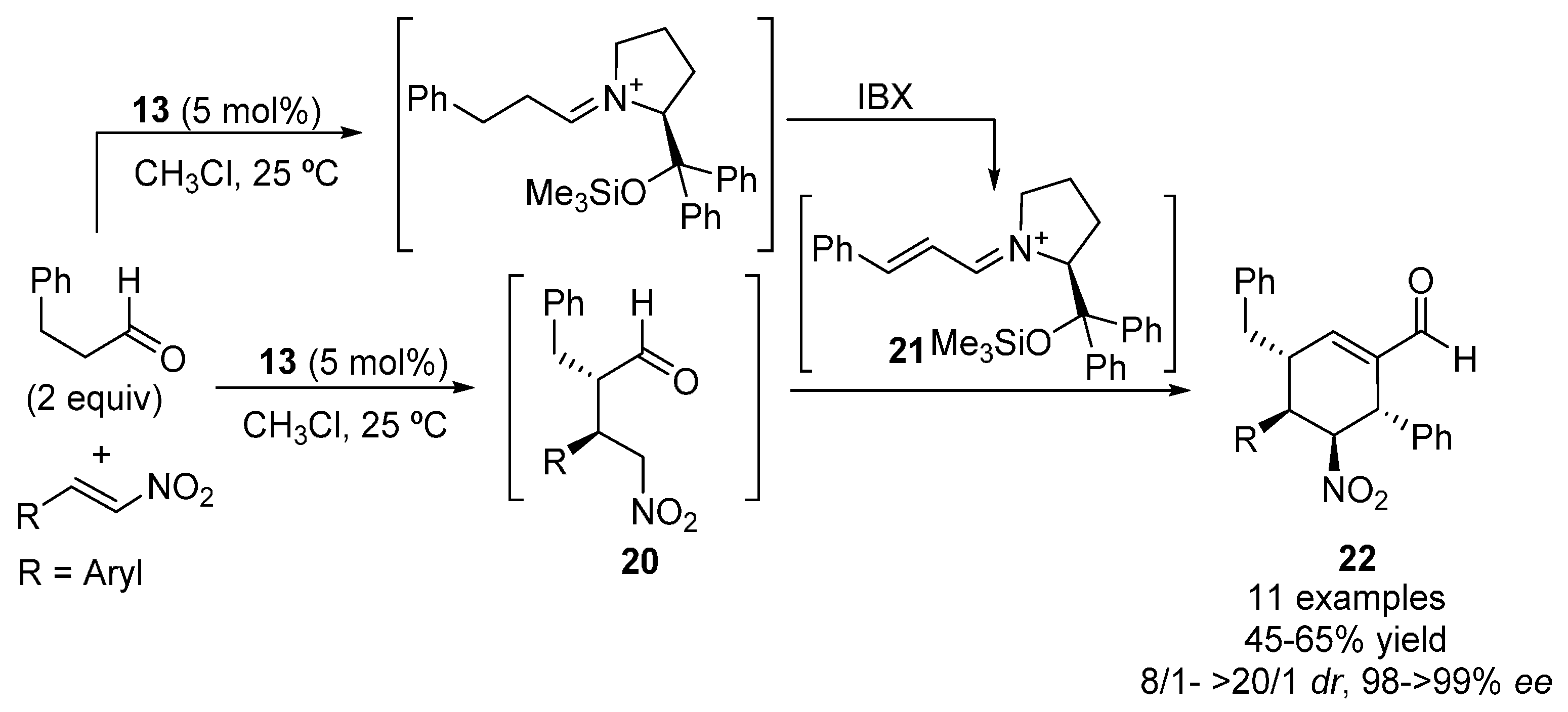{kind=link}
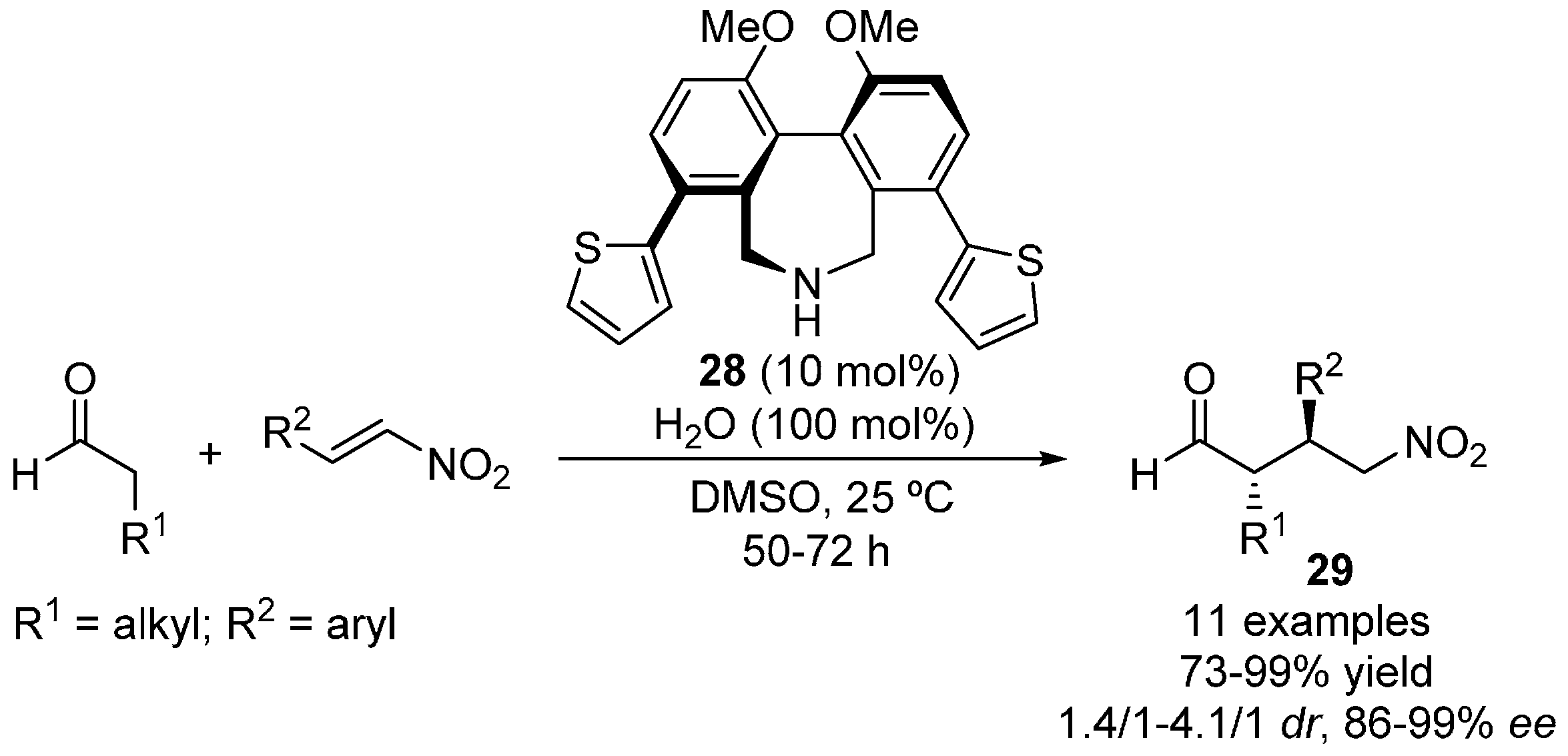{kind=link}
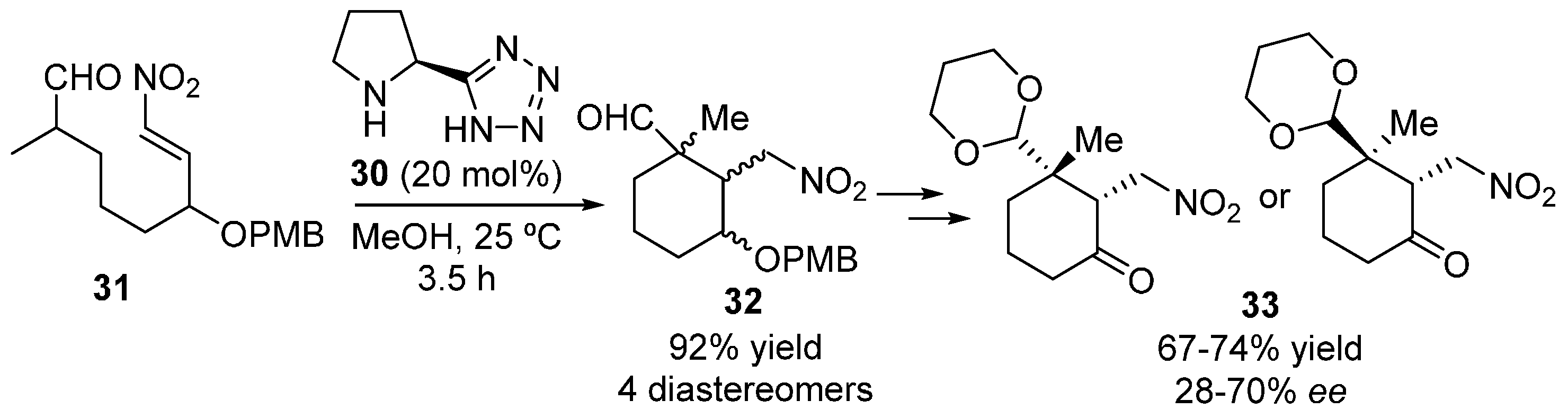{kind=link}
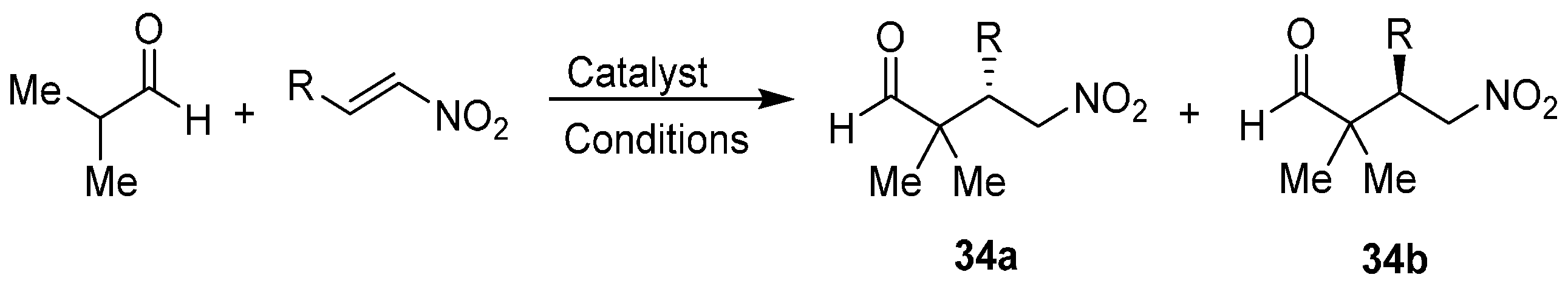{kind=link}
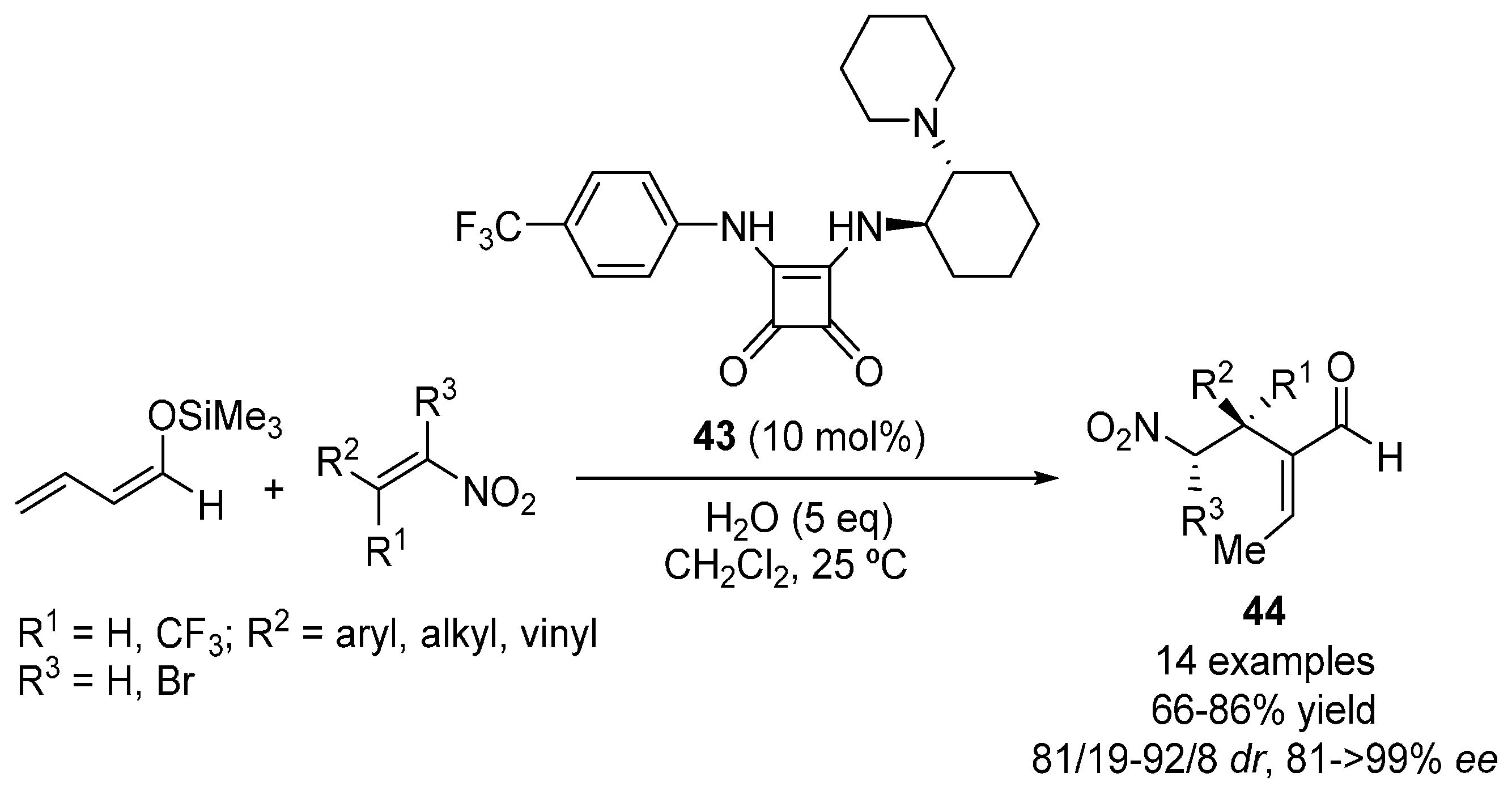{kind=link}
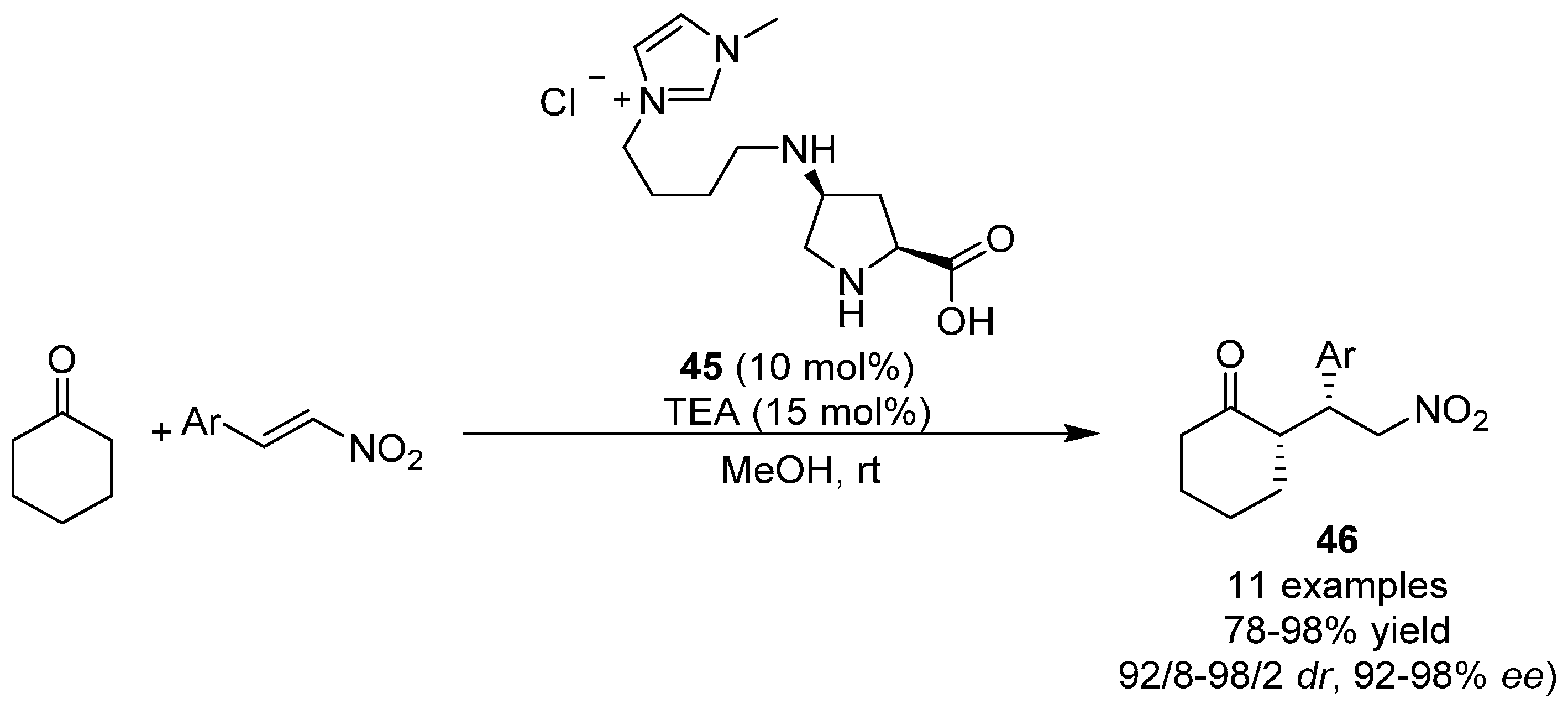{kind=link}
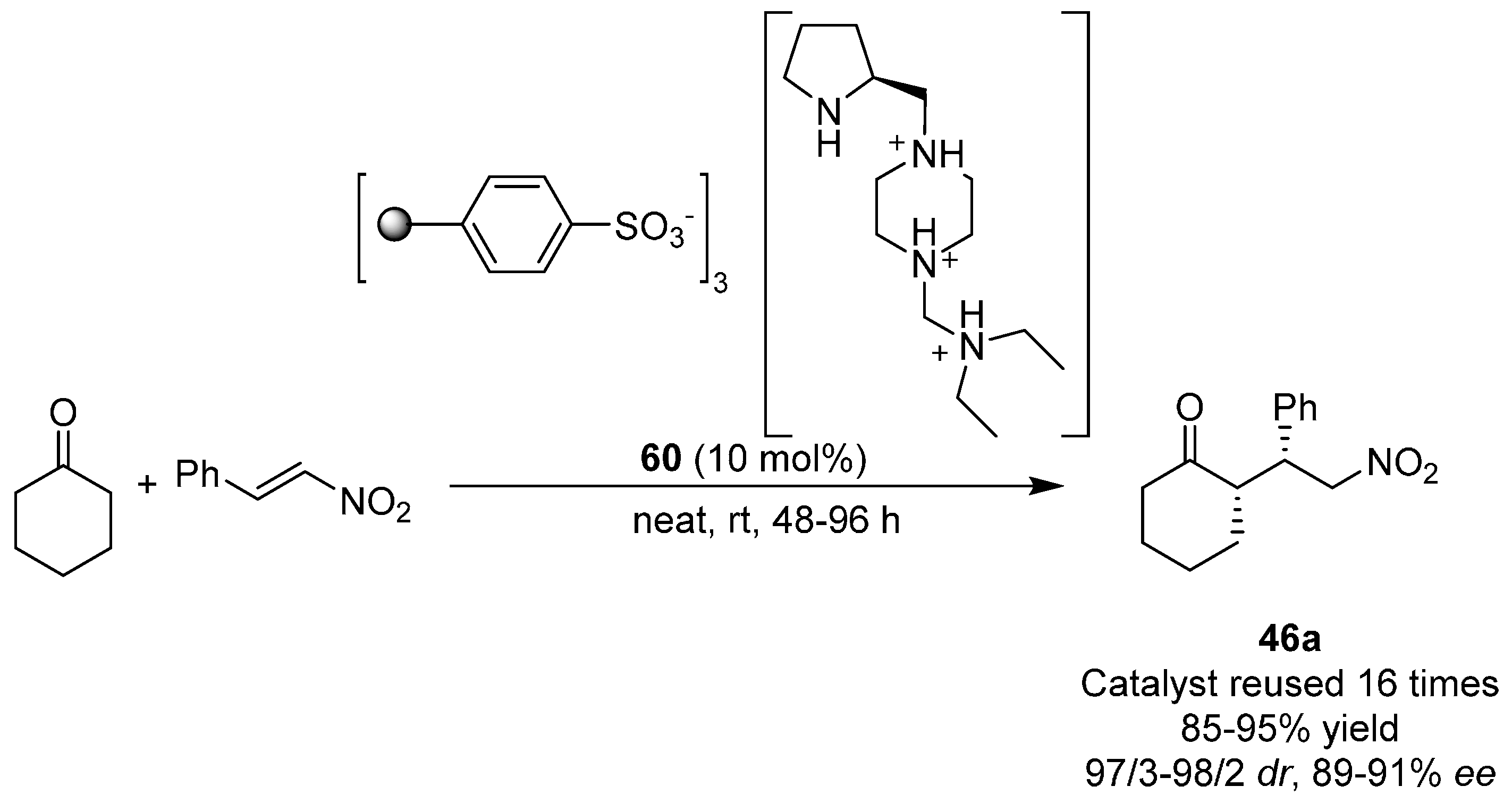{kind=link}
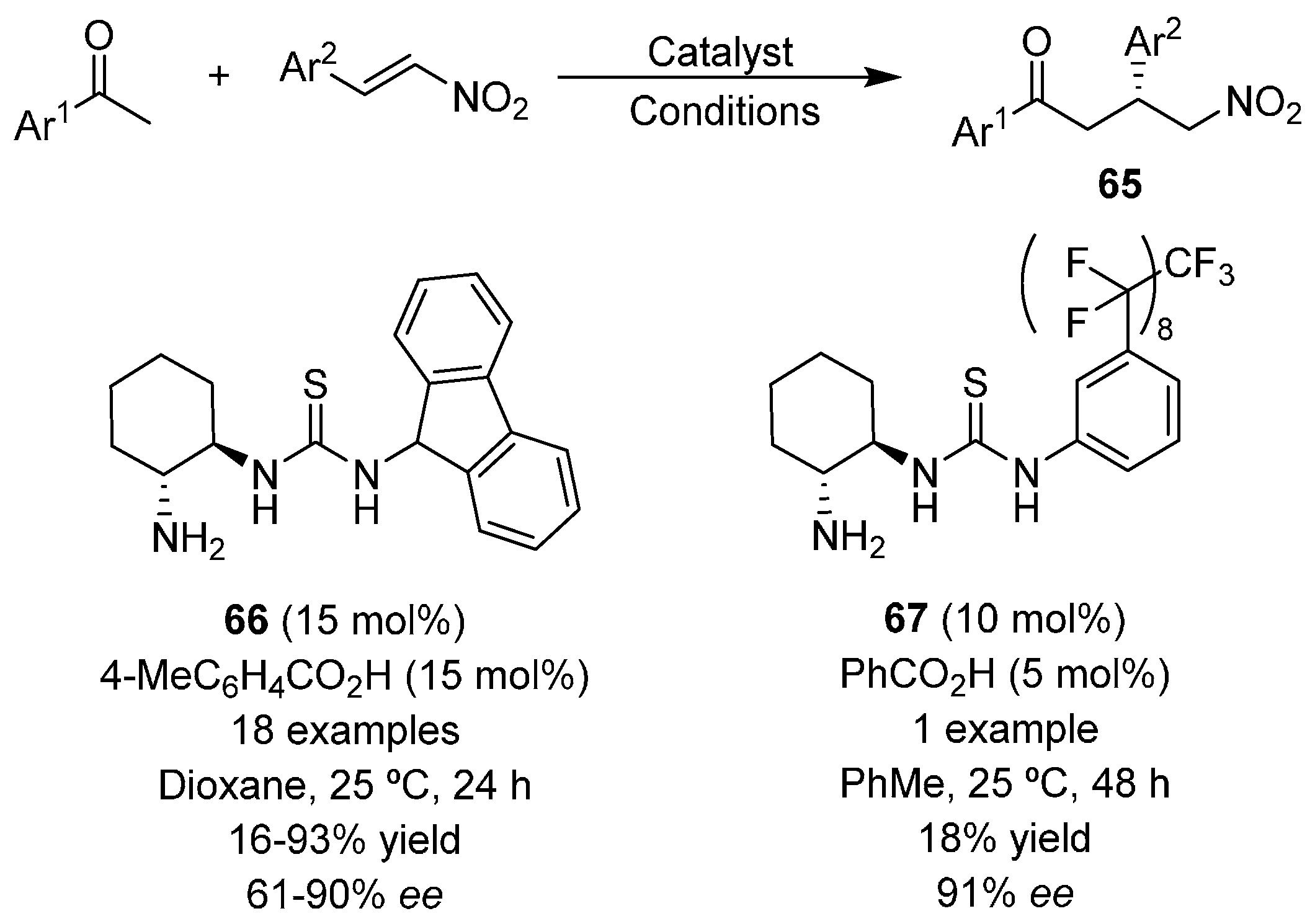{kind=link}
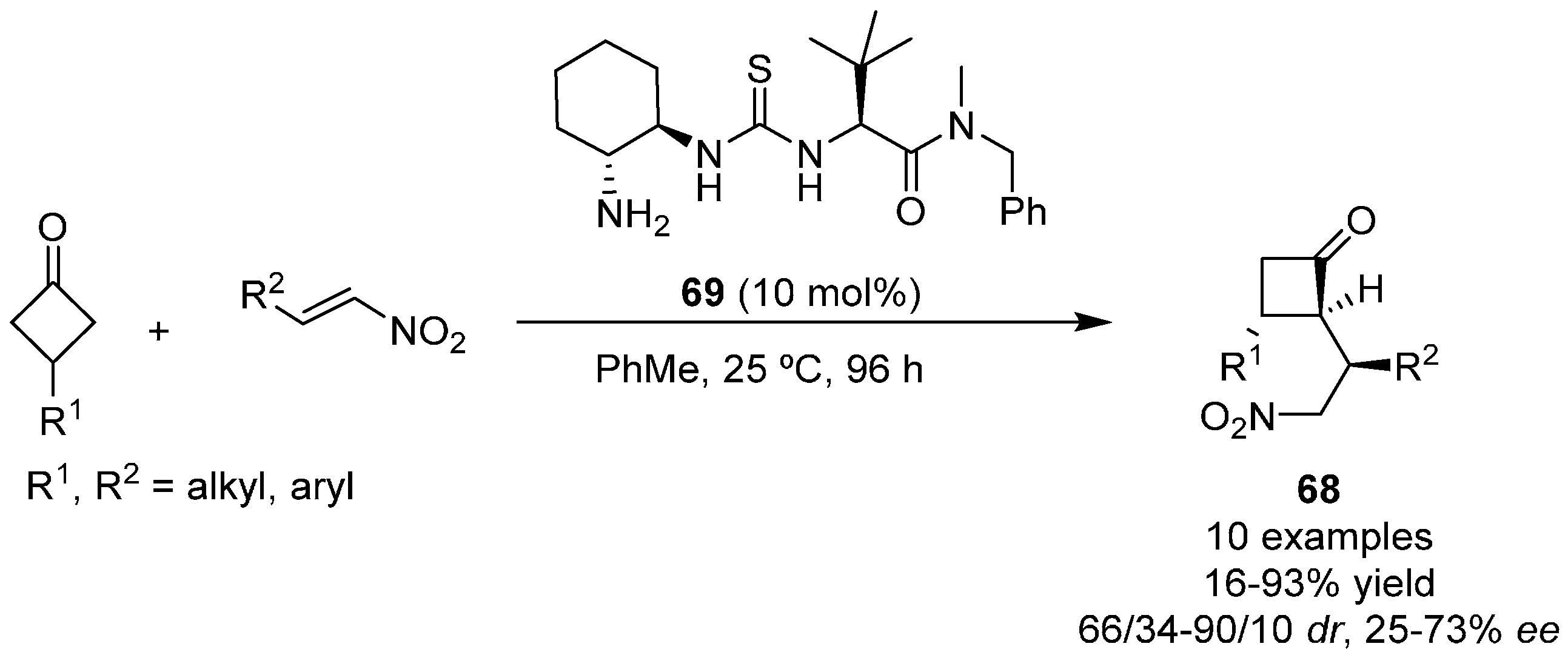{kind=link}
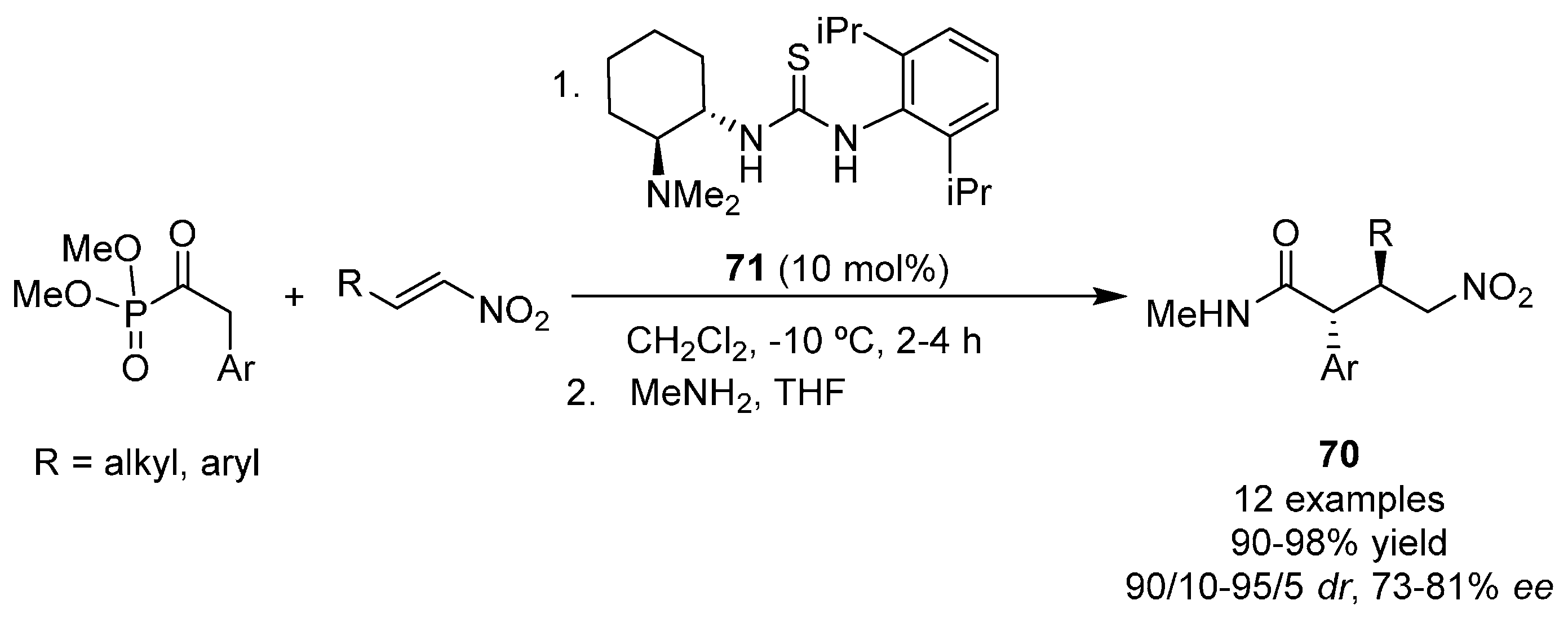{kind=link}
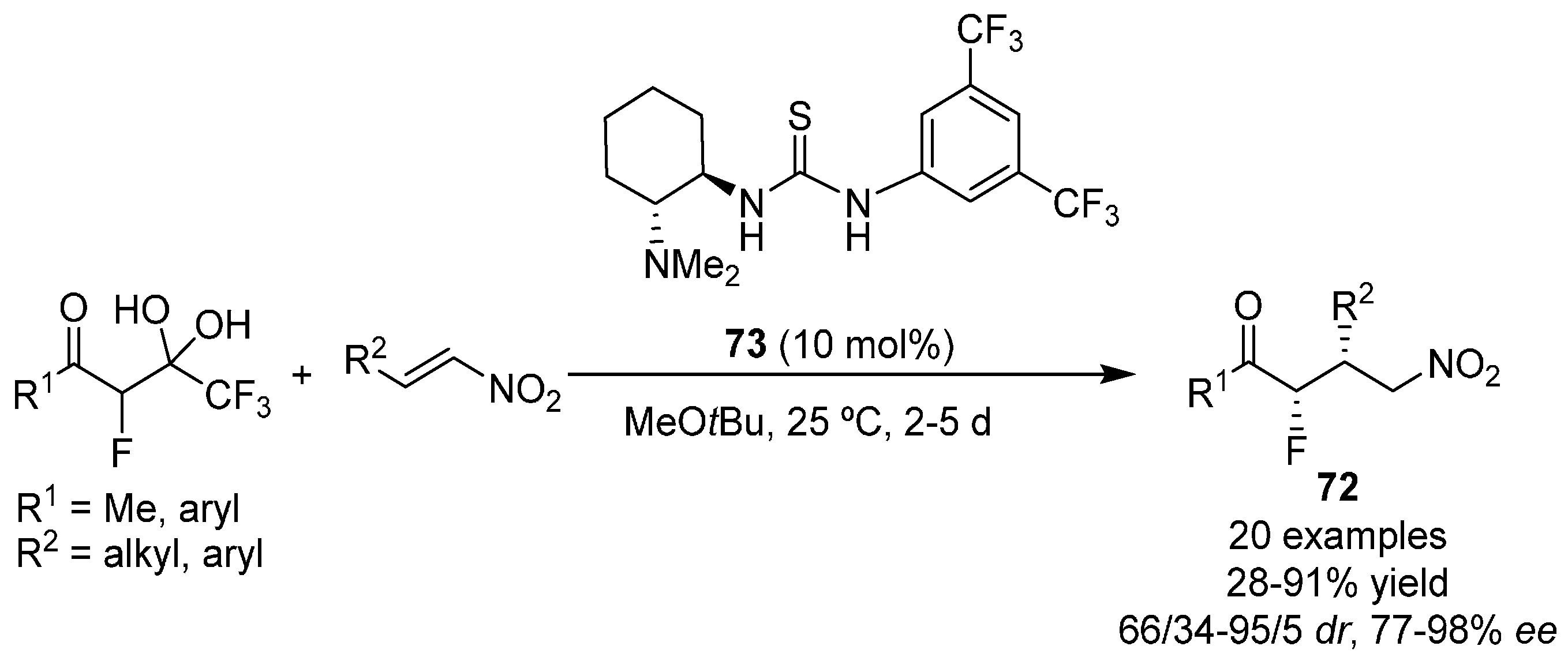{kind=link}
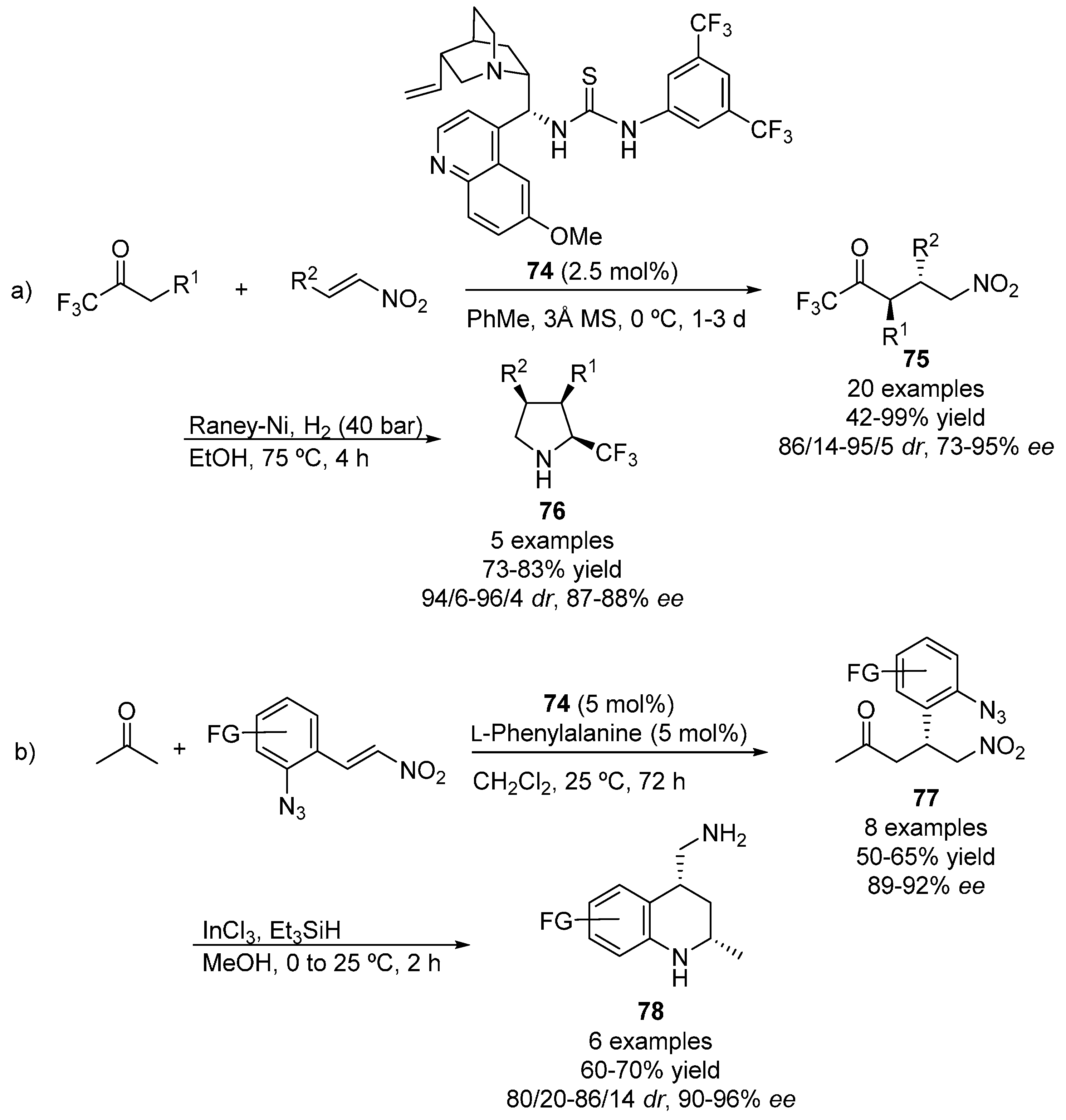{kind=link}
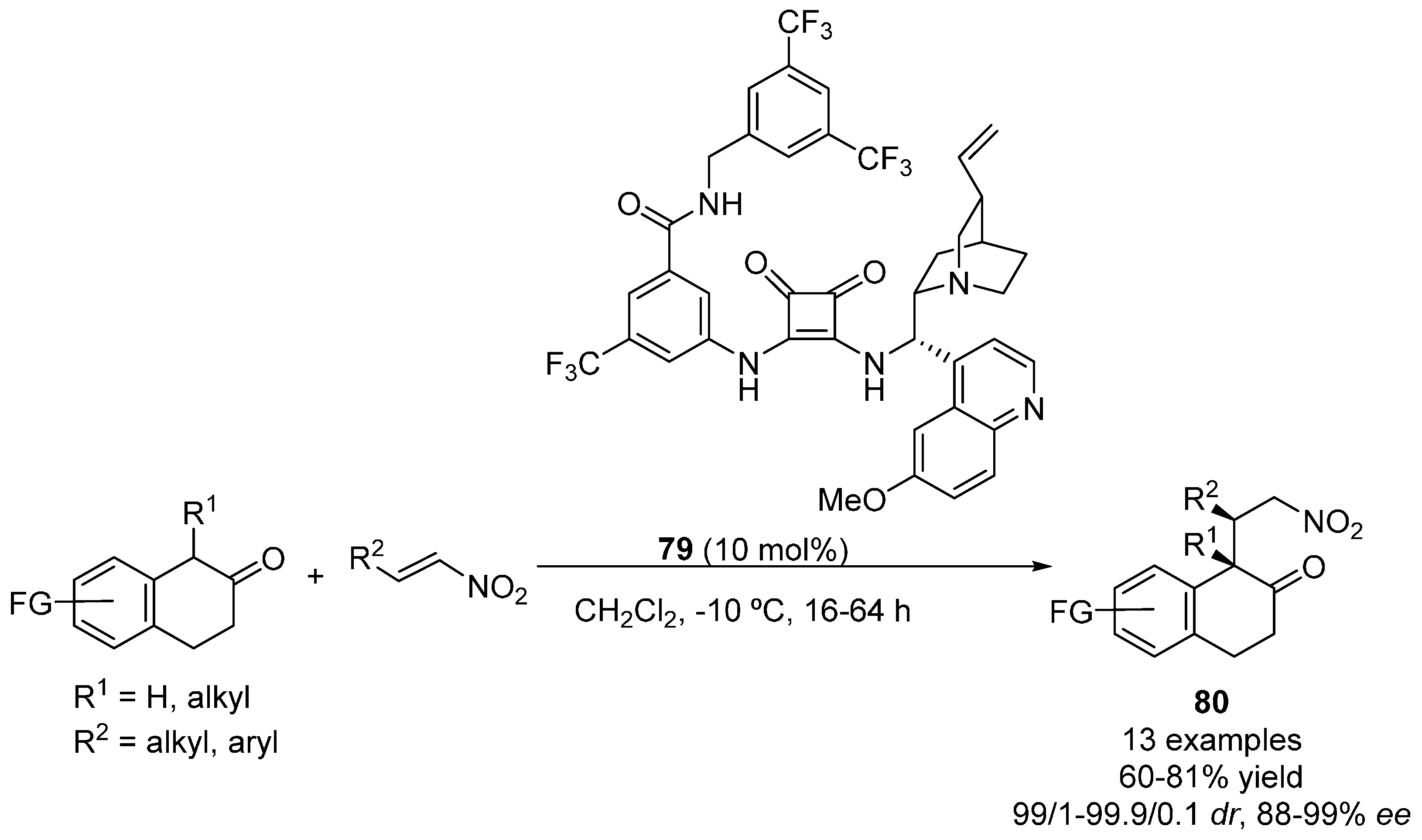{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
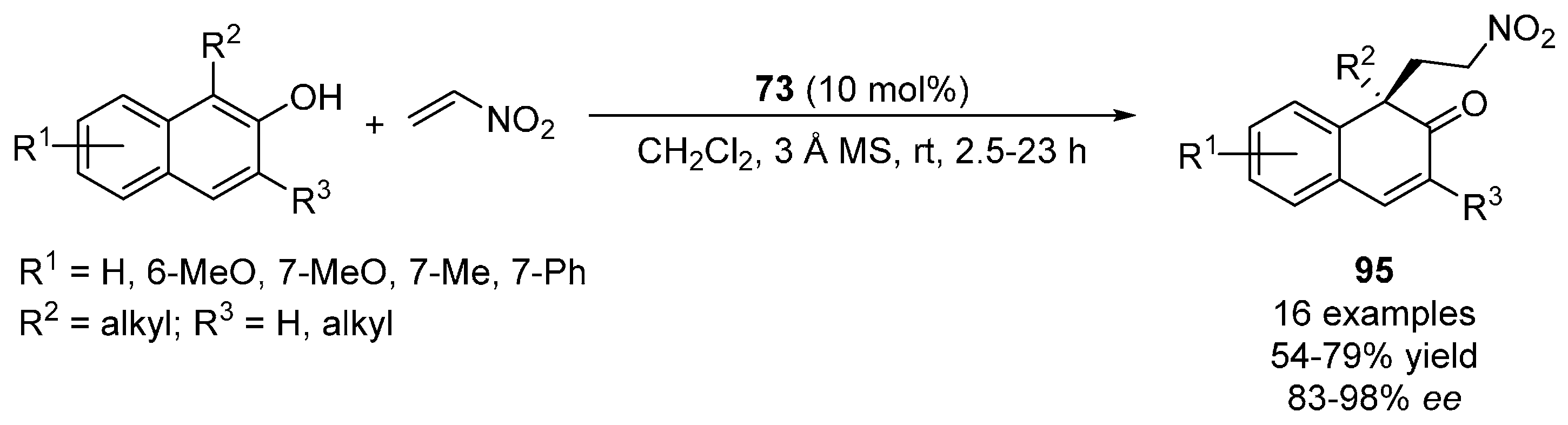{kind=link}
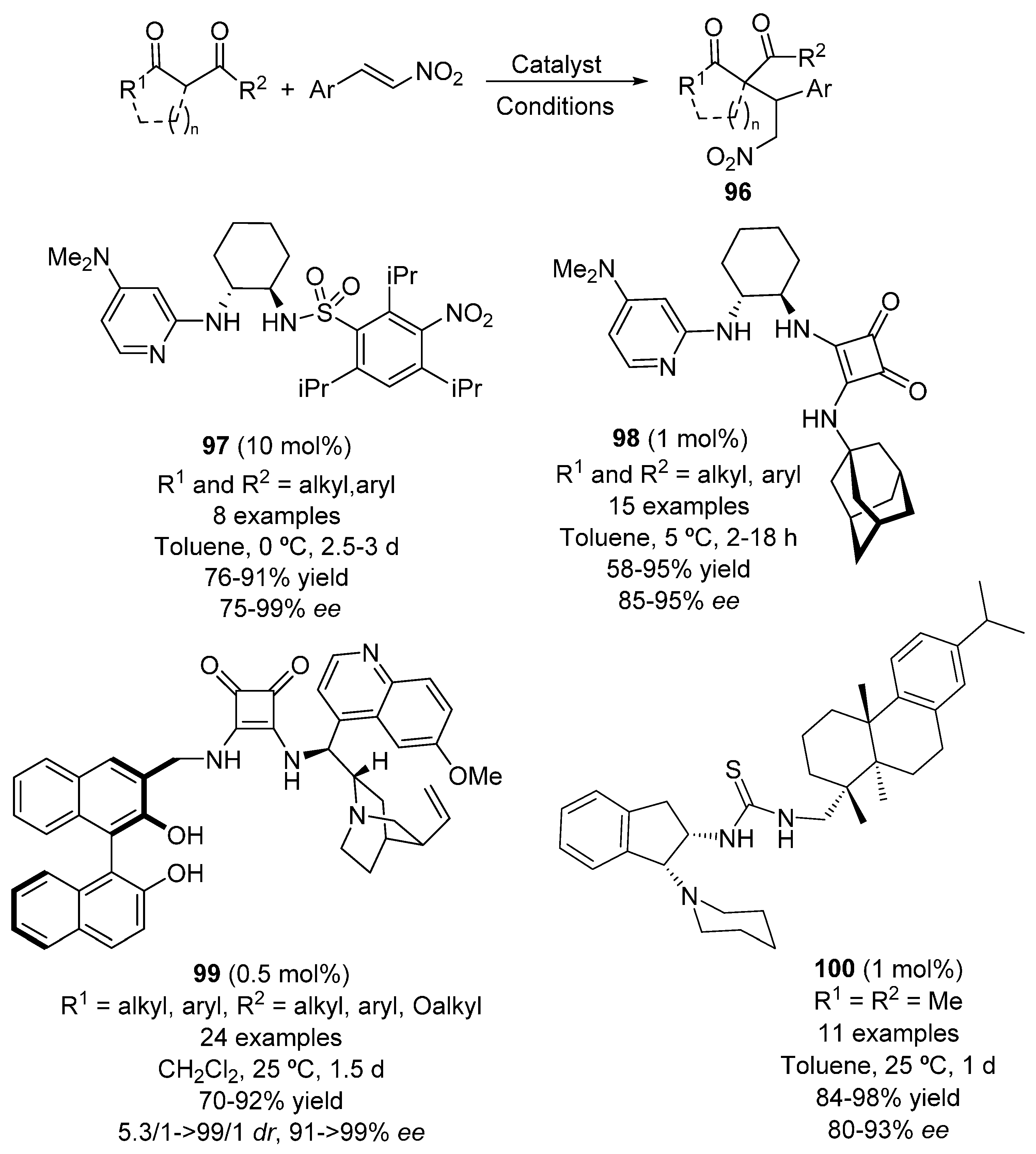{kind=link}
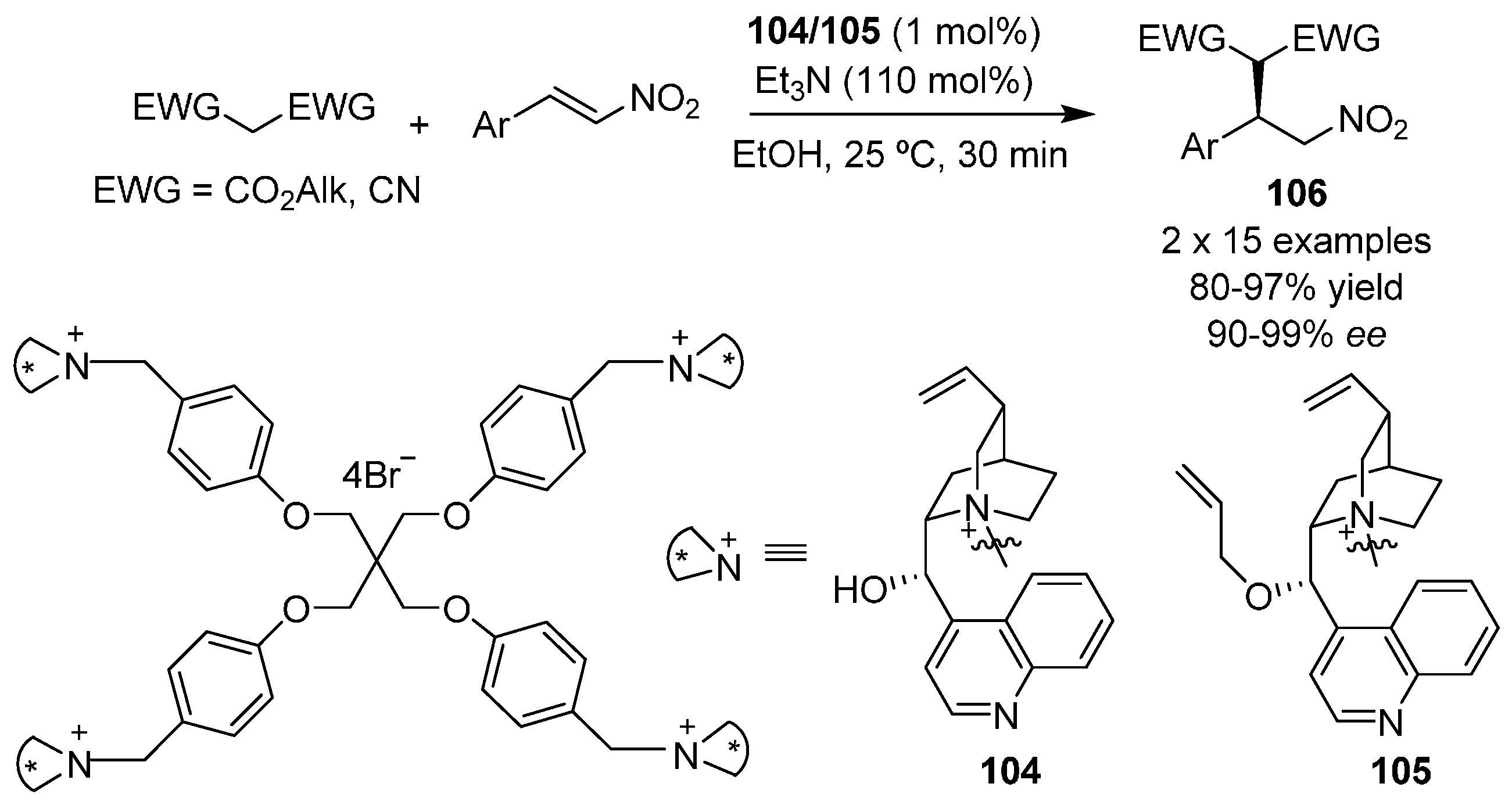{kind=link}
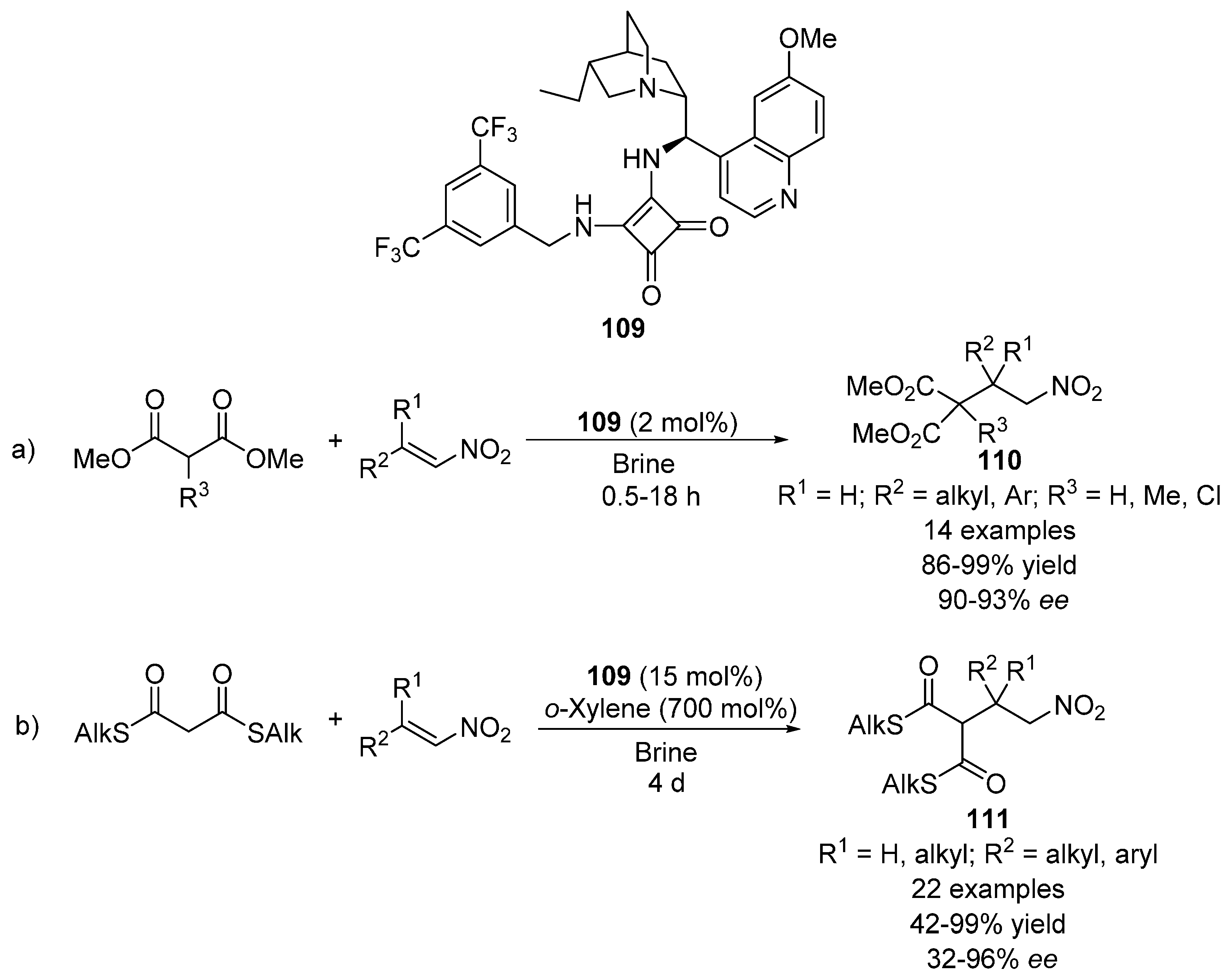{kind=link}
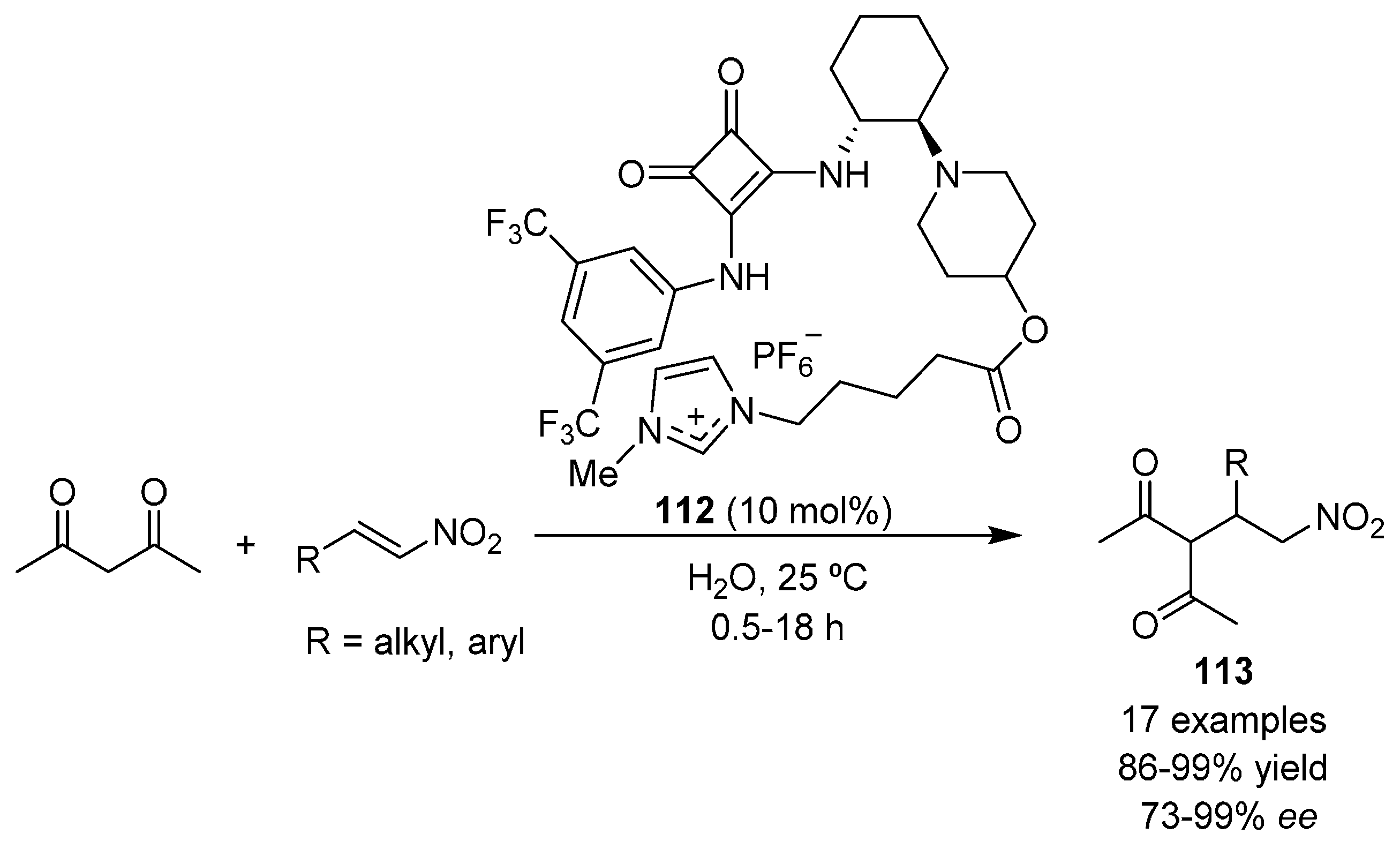{kind=link}
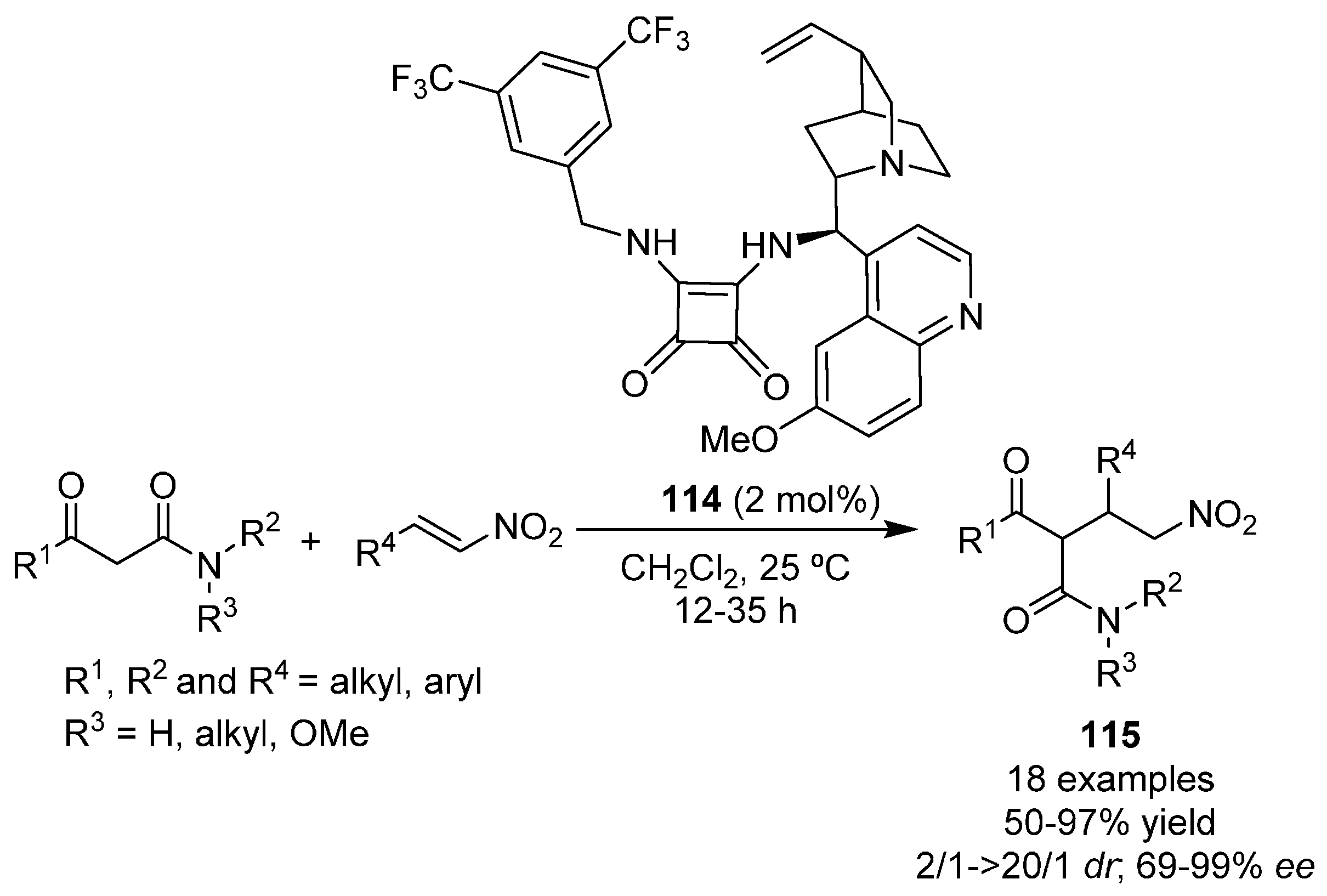{kind=link}
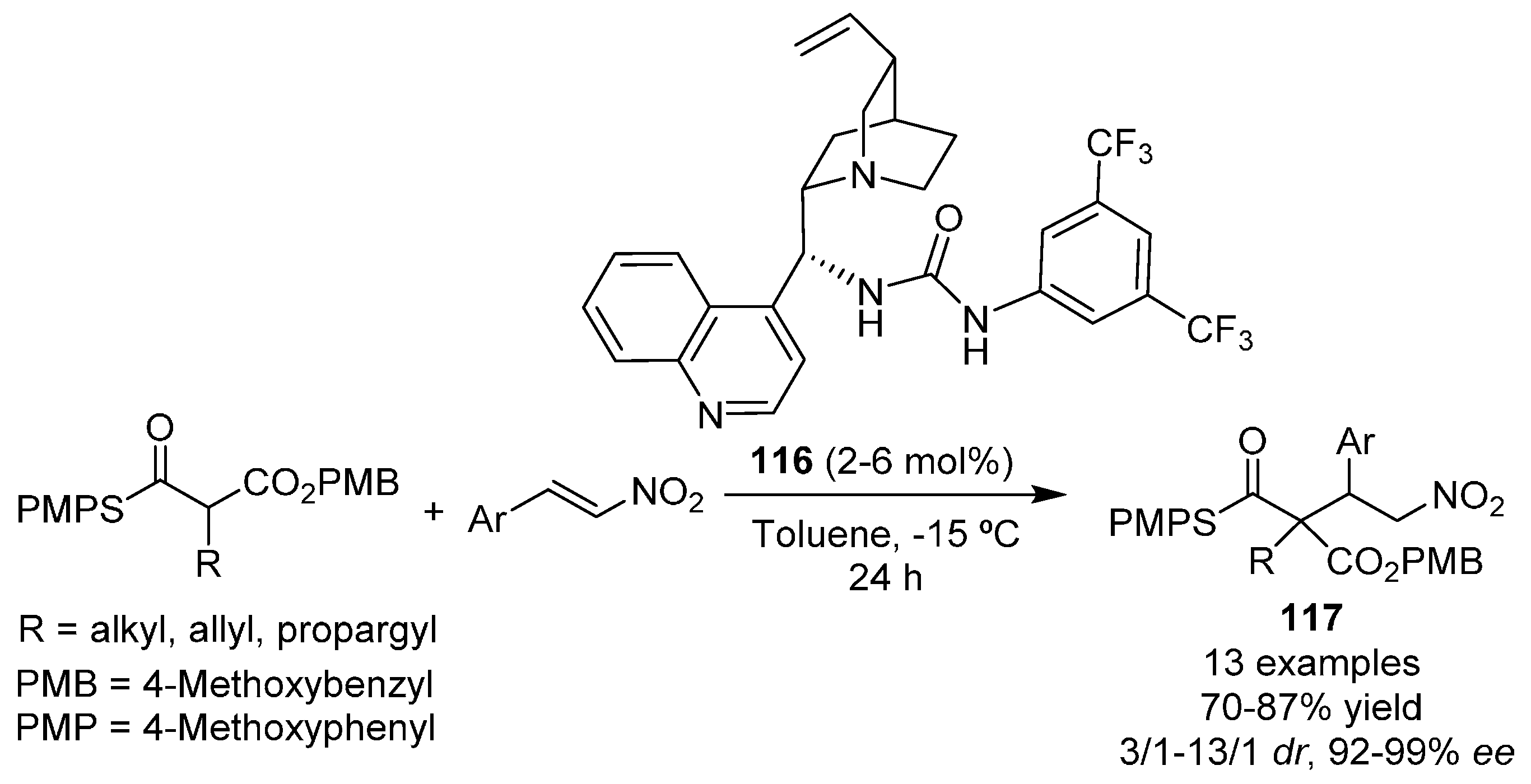{kind=link}
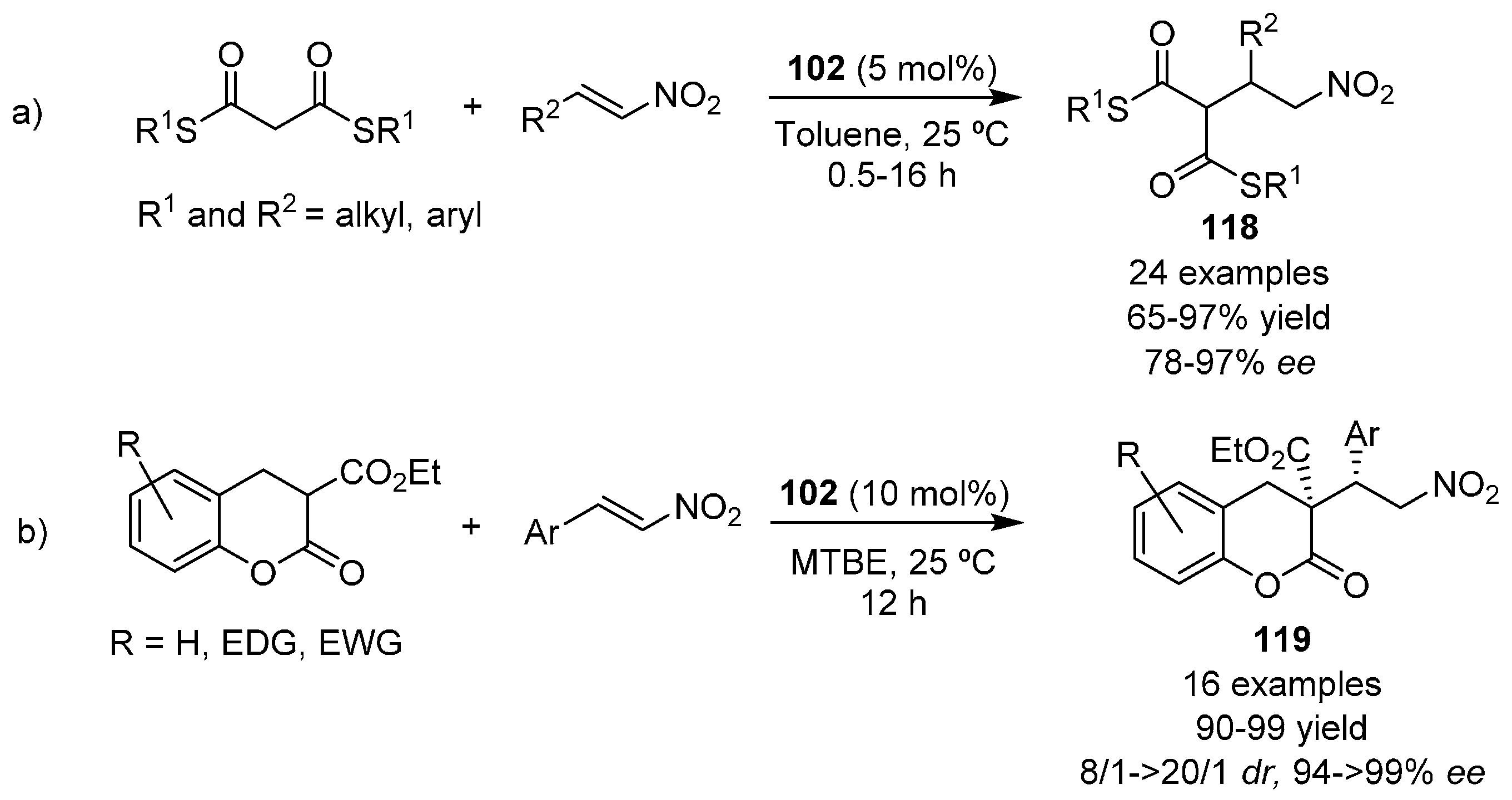{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
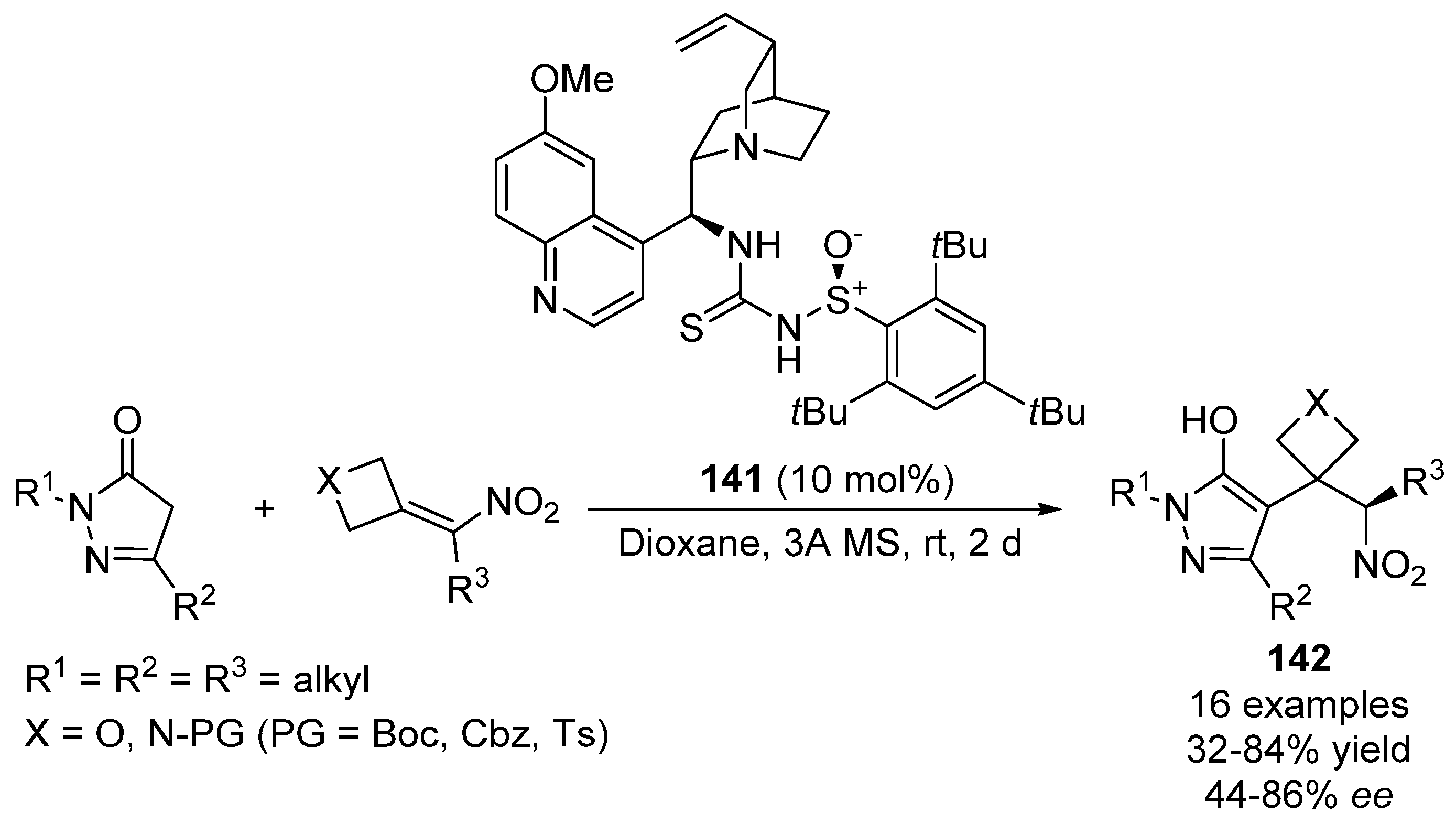{kind=link}
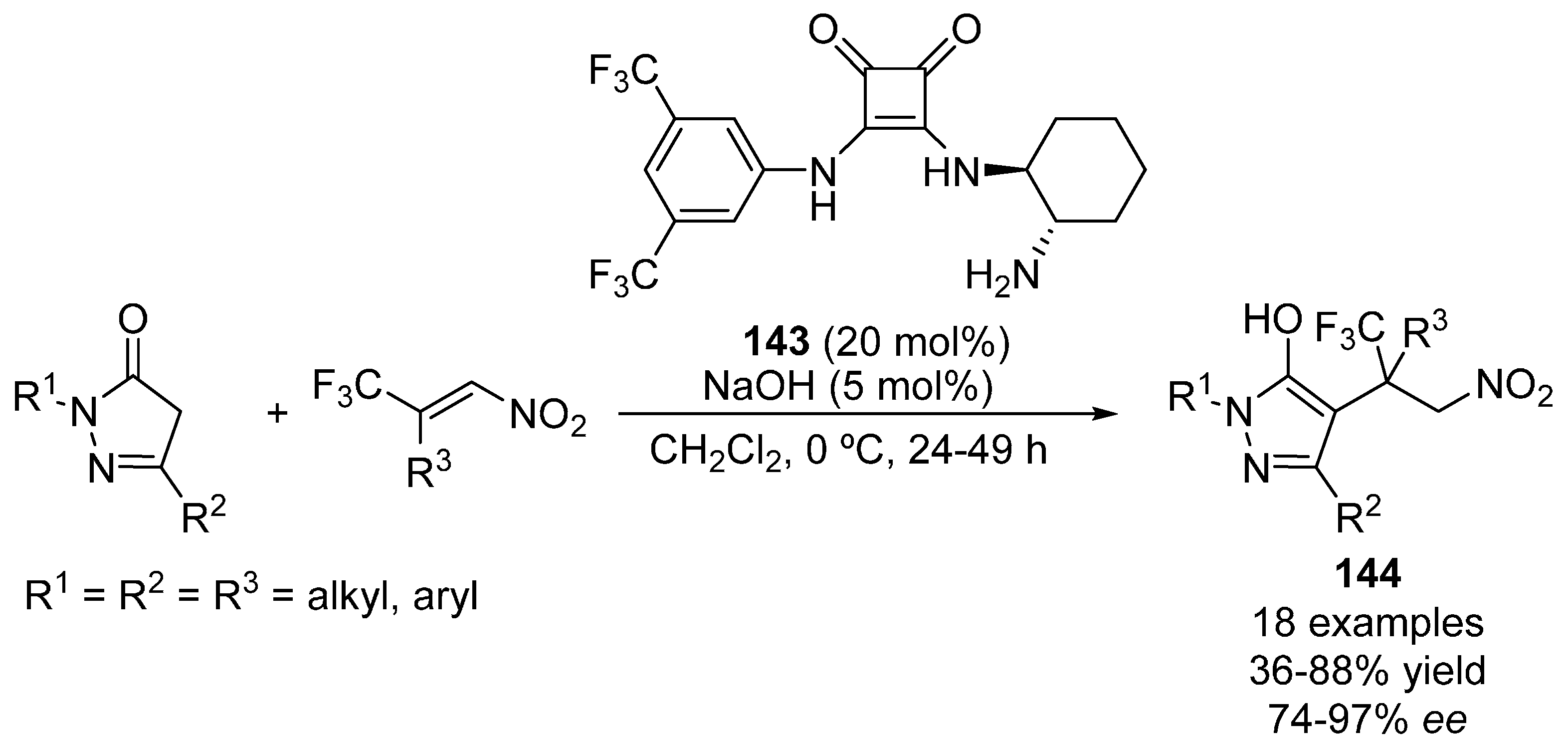{kind=link}
{kind=link}
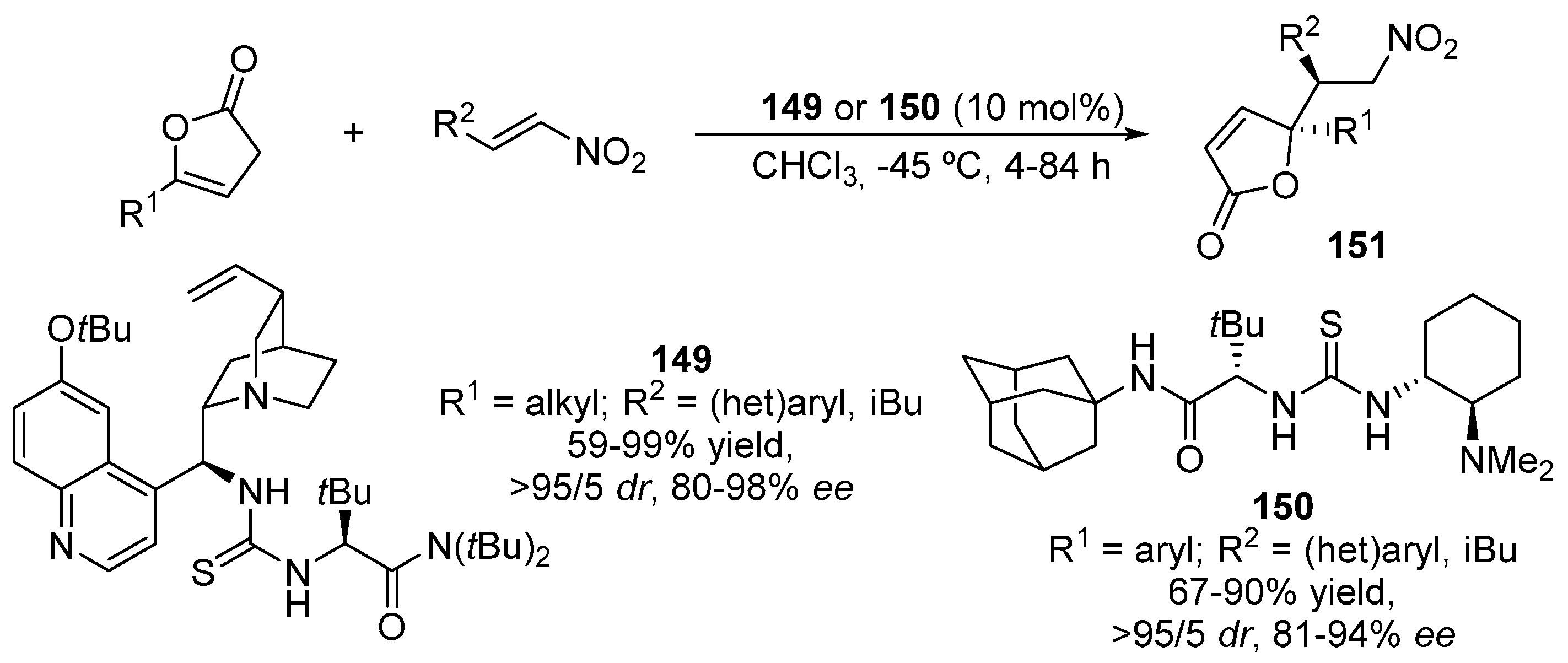{kind=link}
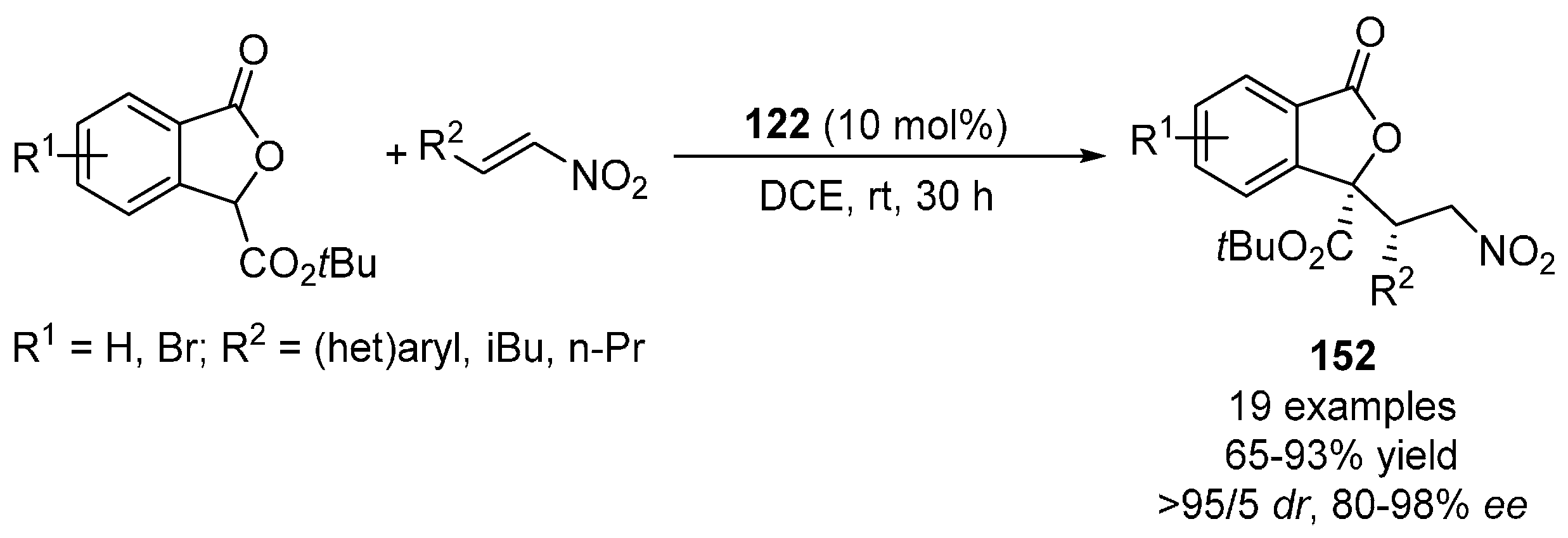{kind=link}
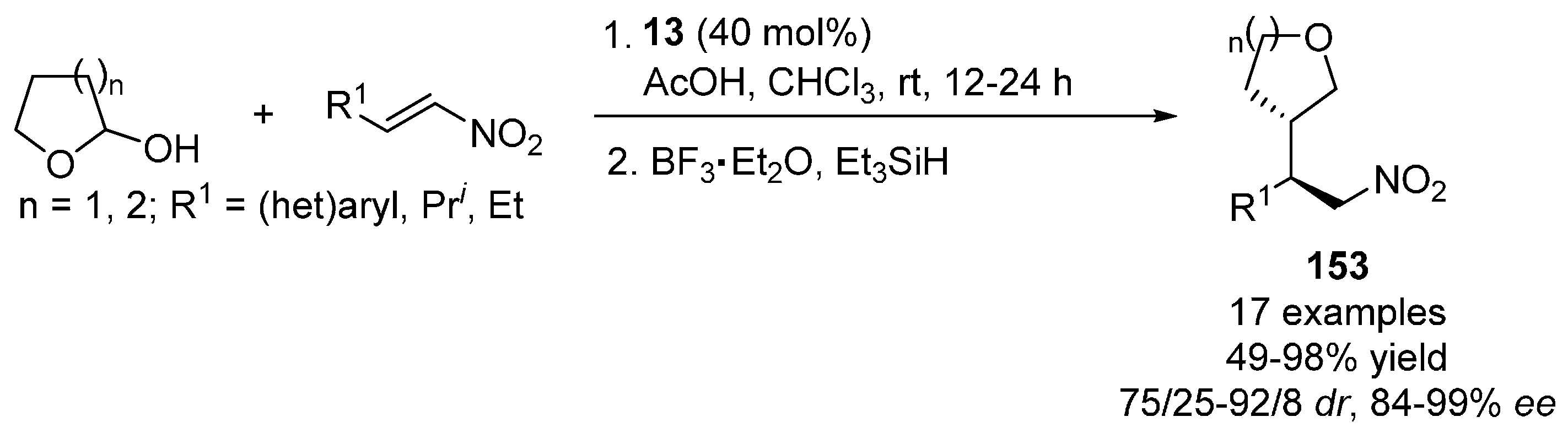{kind=link}
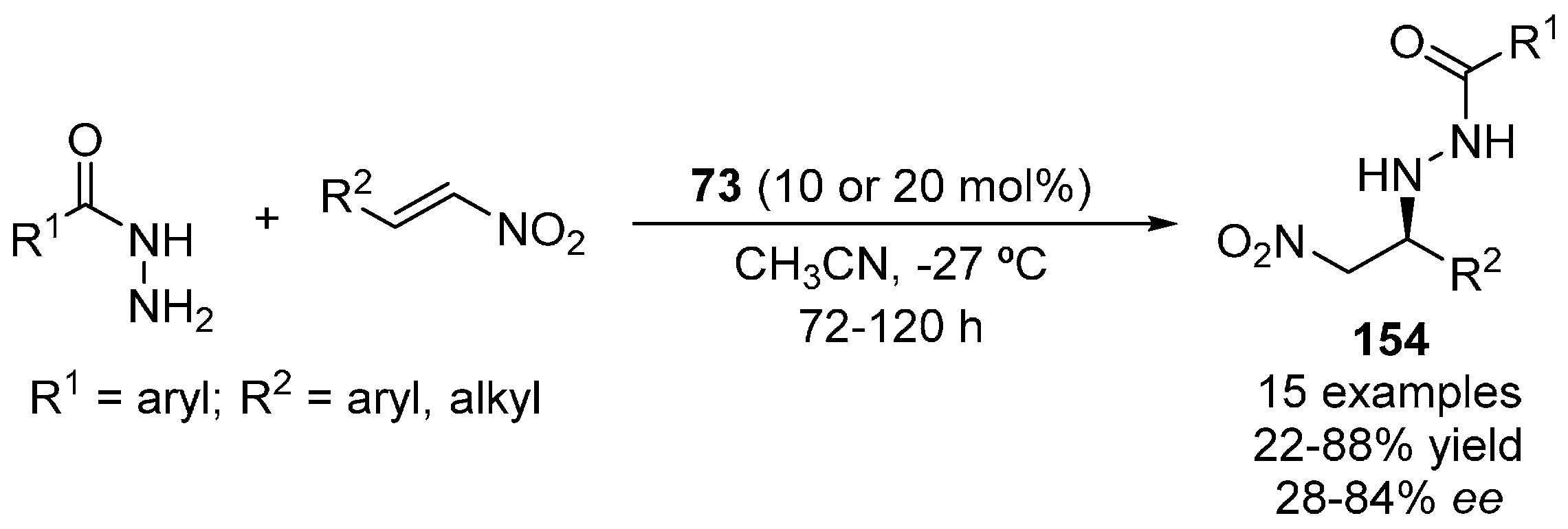{kind=link}
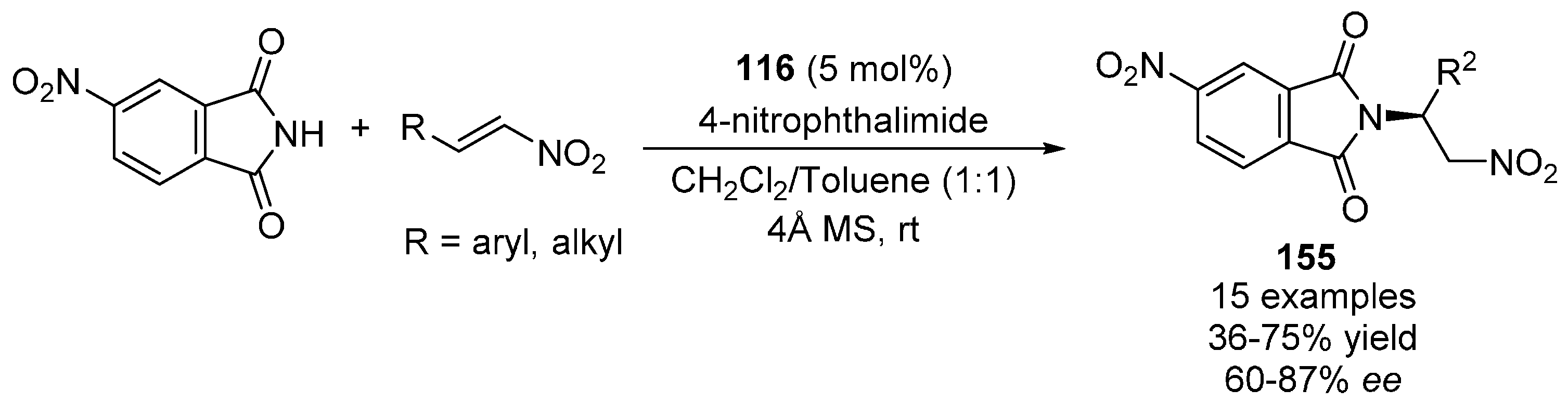{kind=link}
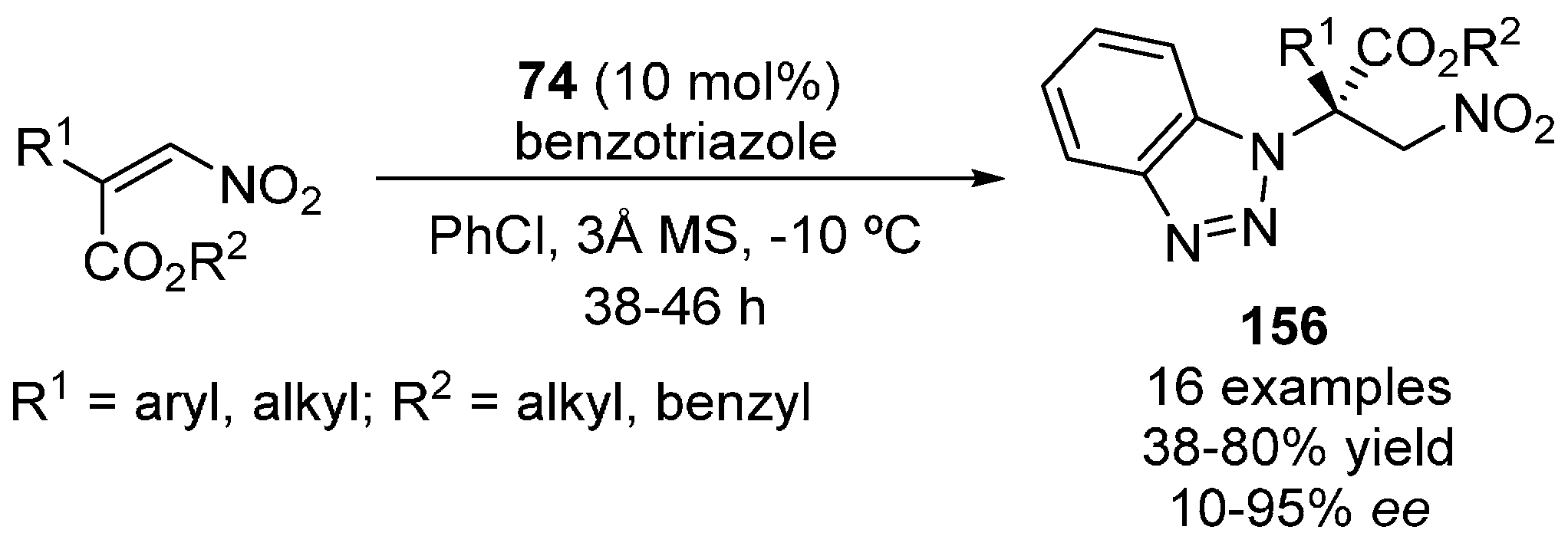{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
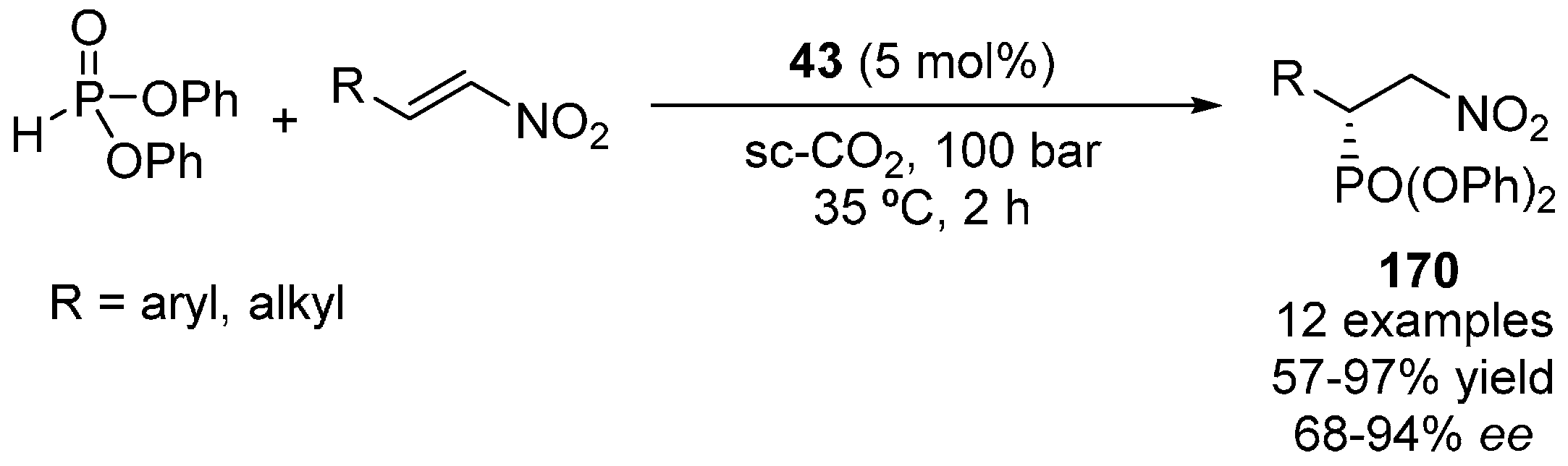{kind=link}
1. Introduction
2. Carbon Nucleophiles
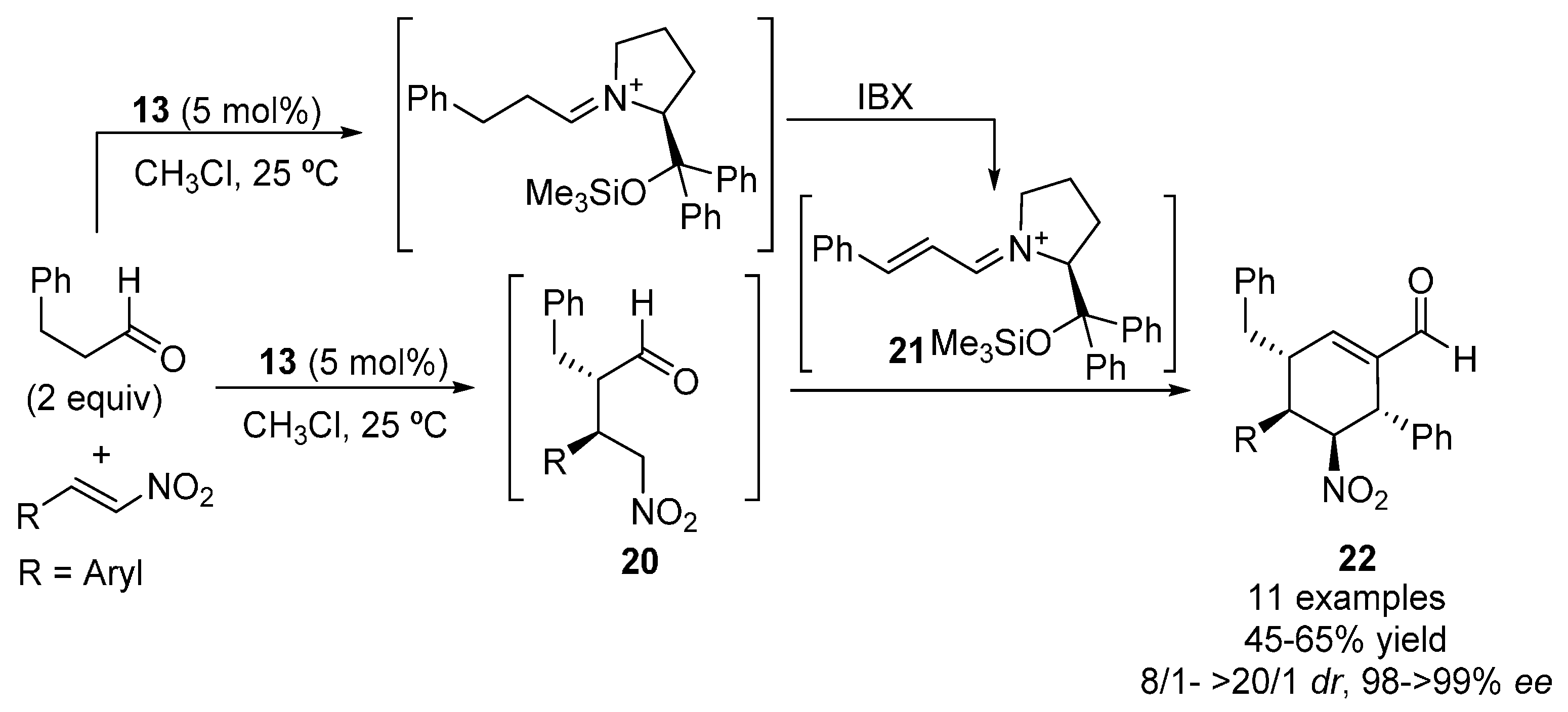
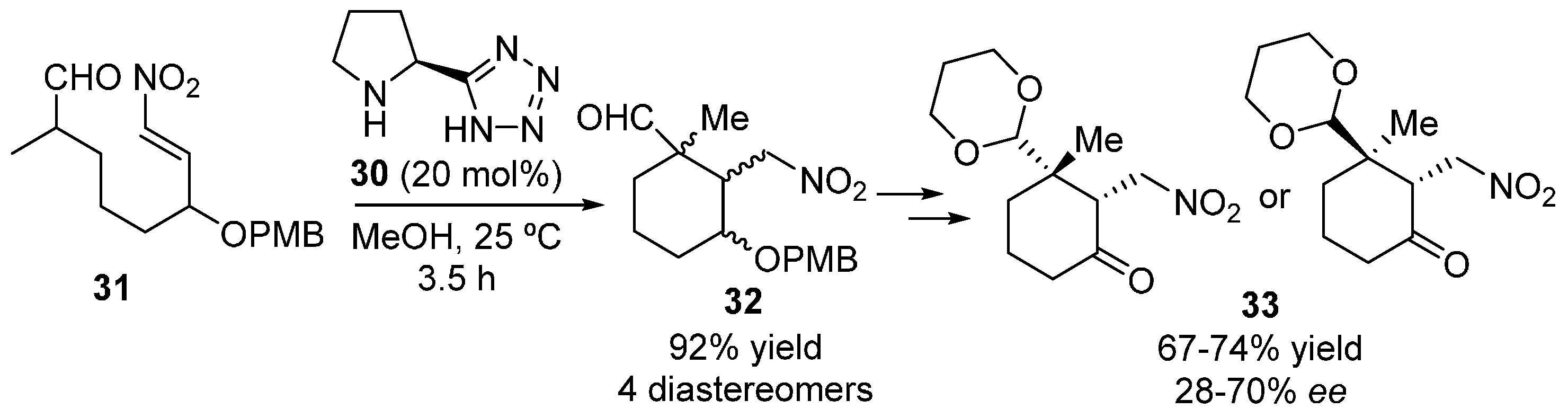
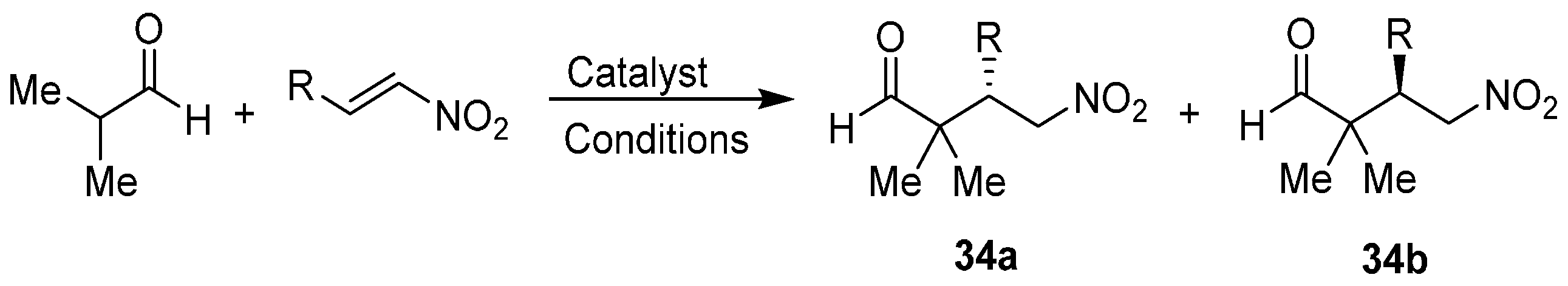
2.1. Aldehydes
2.2. Ketones
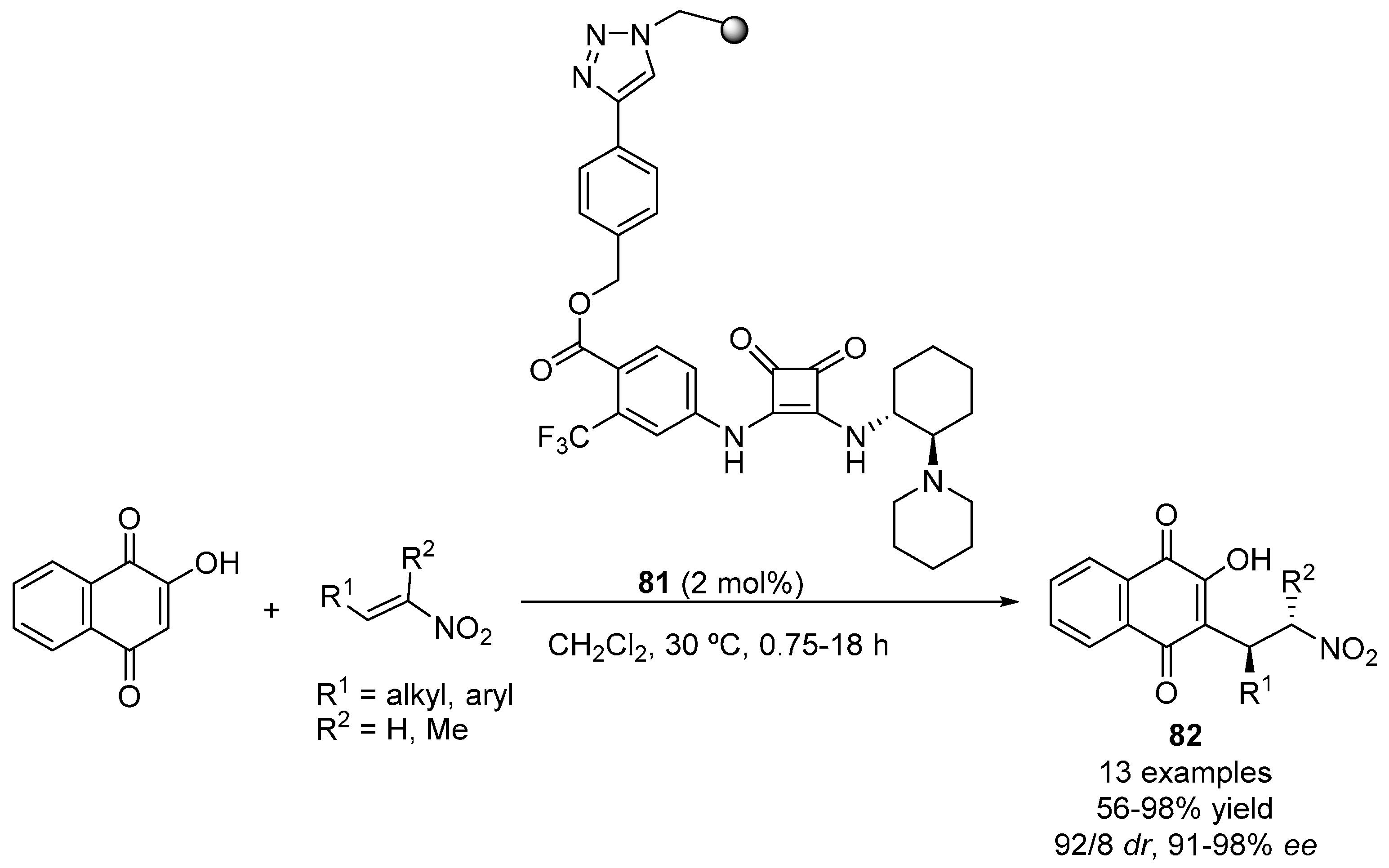
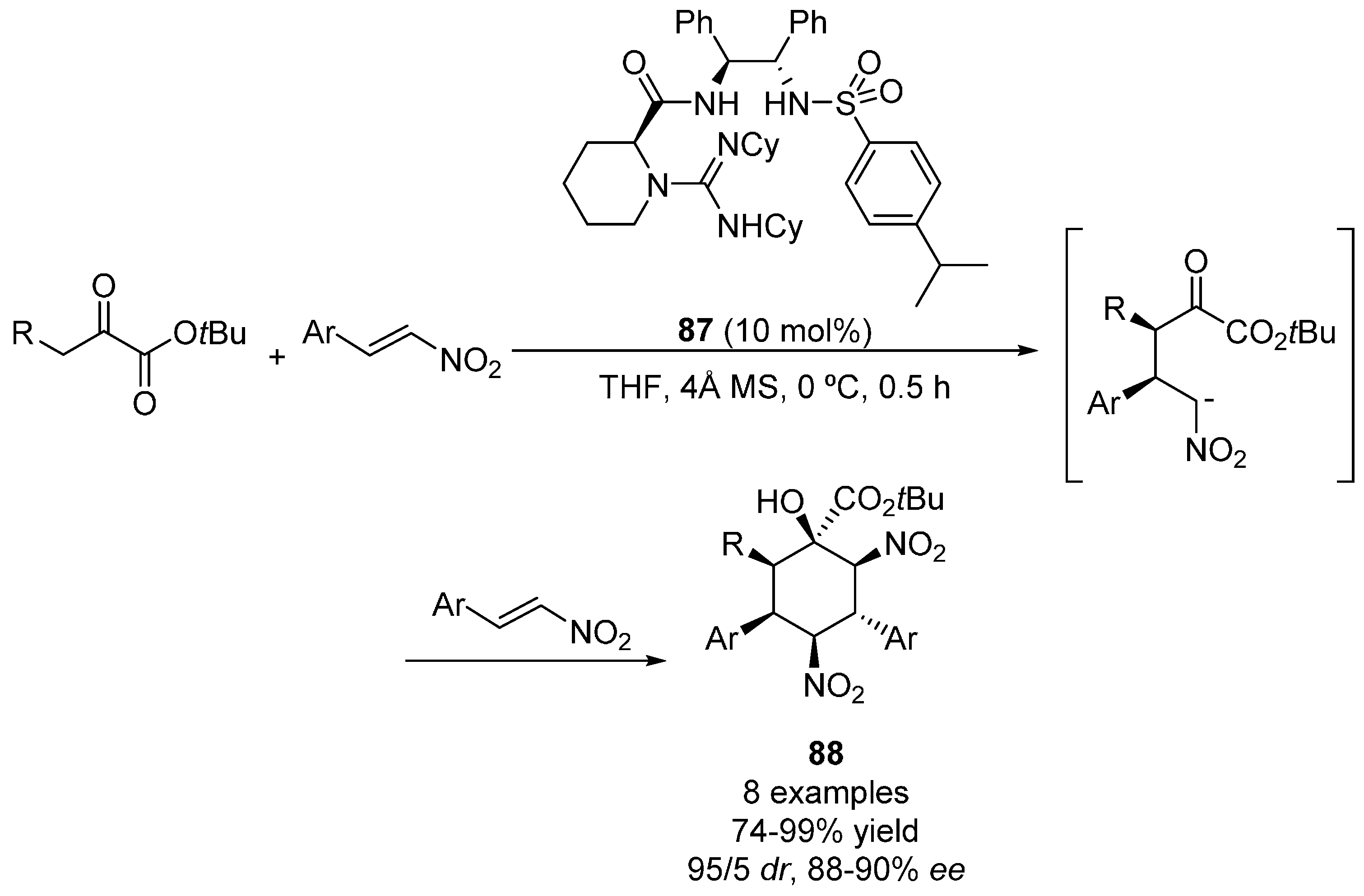
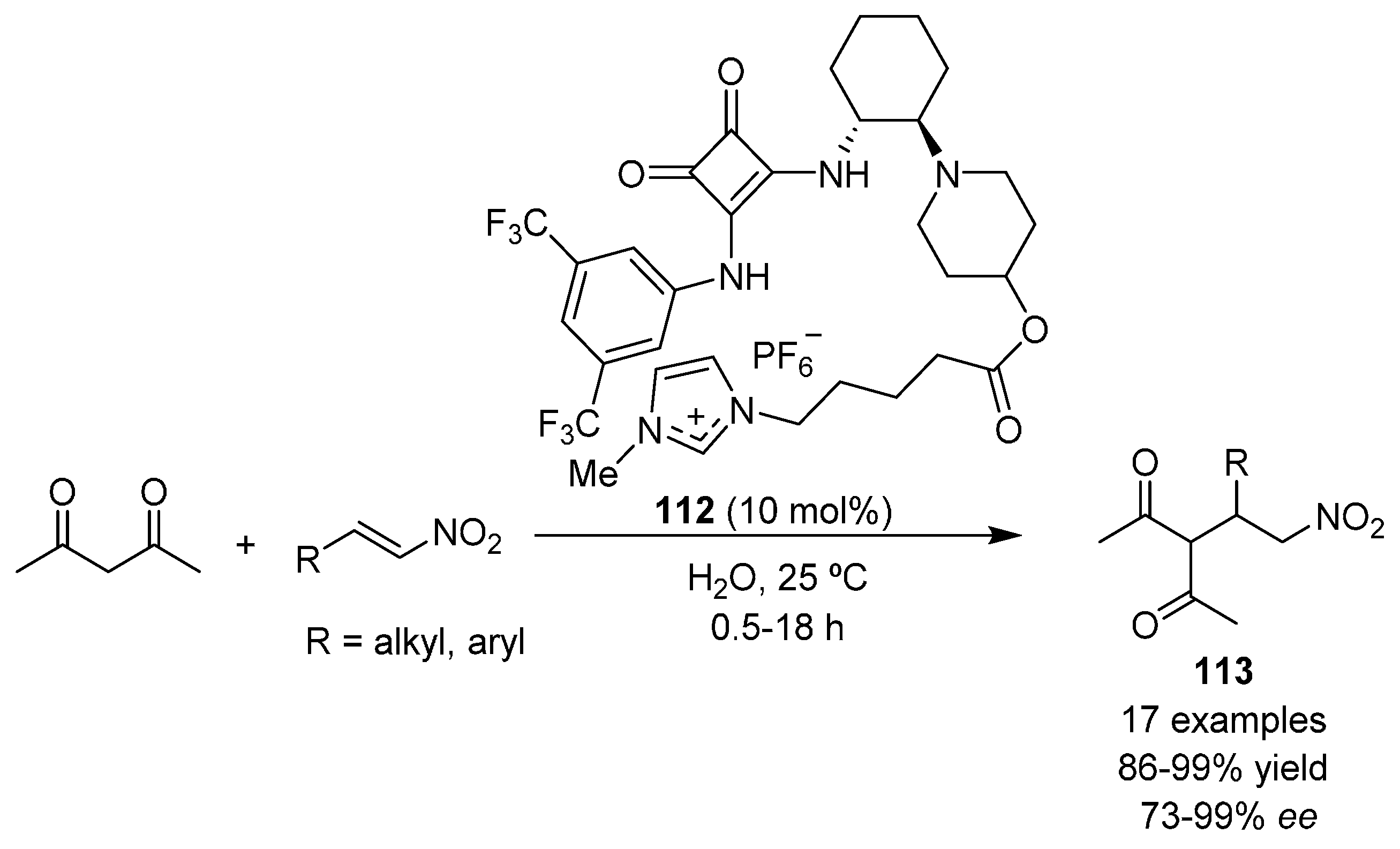
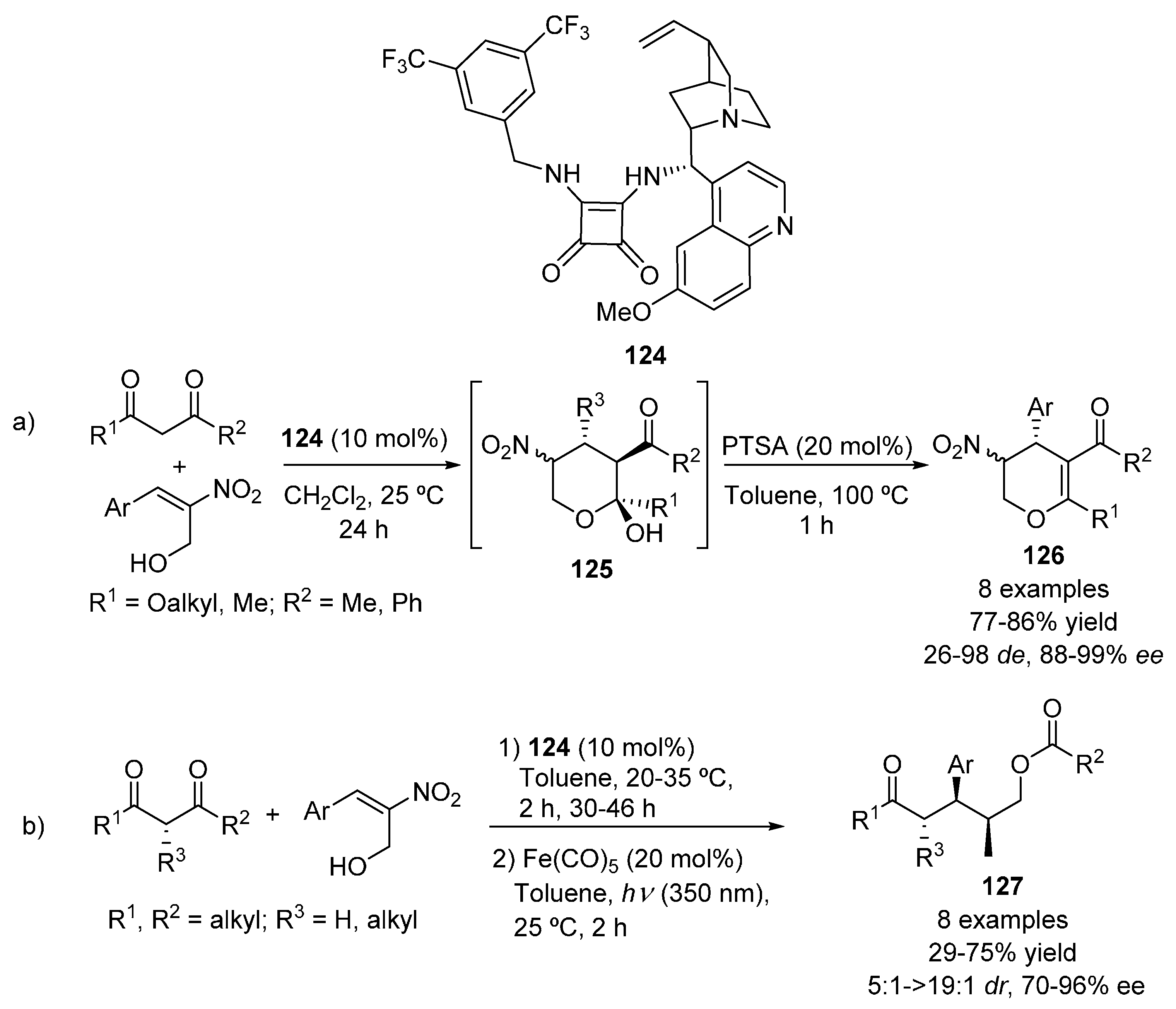
2.3. 1,3-Dicarbonyl Compounds
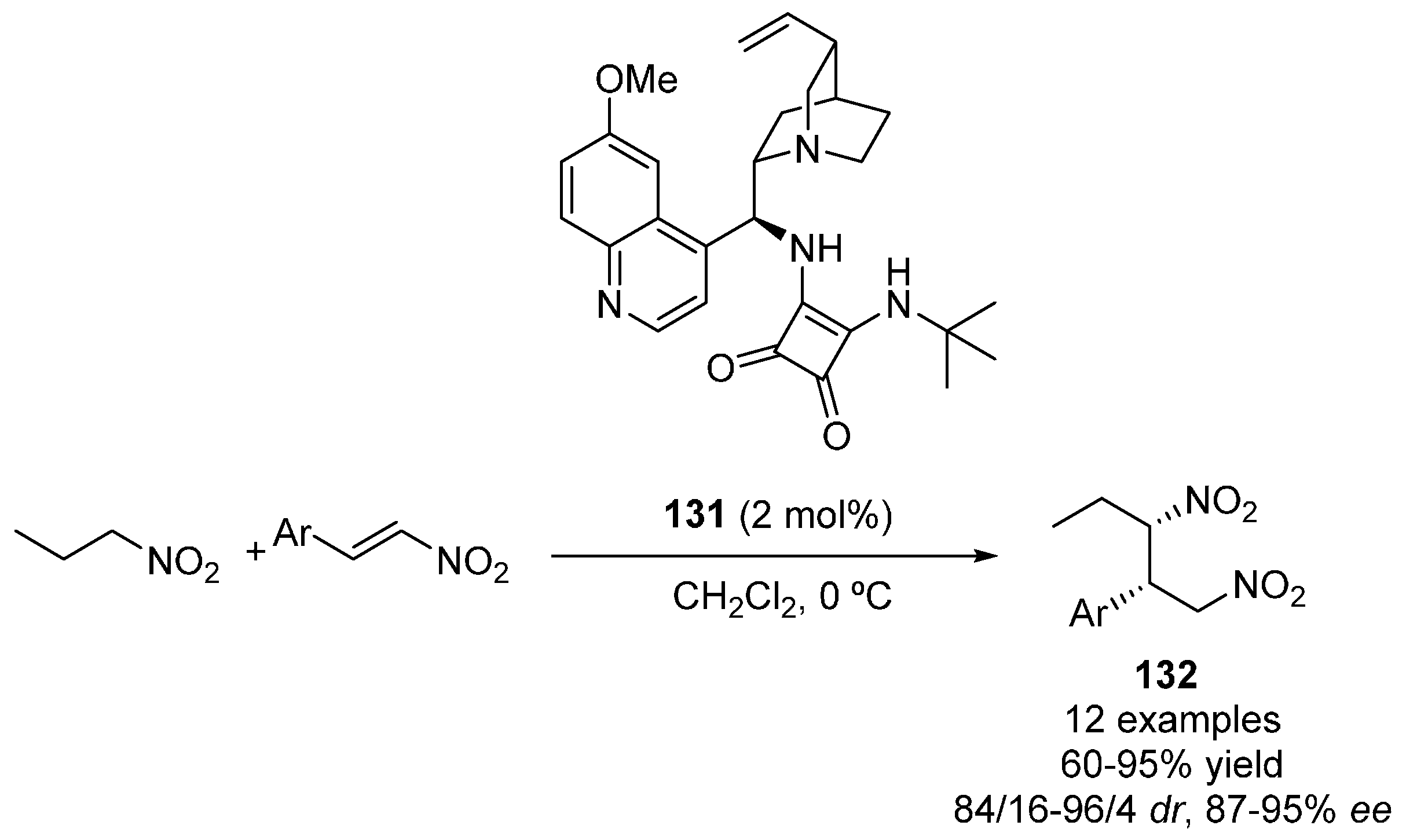
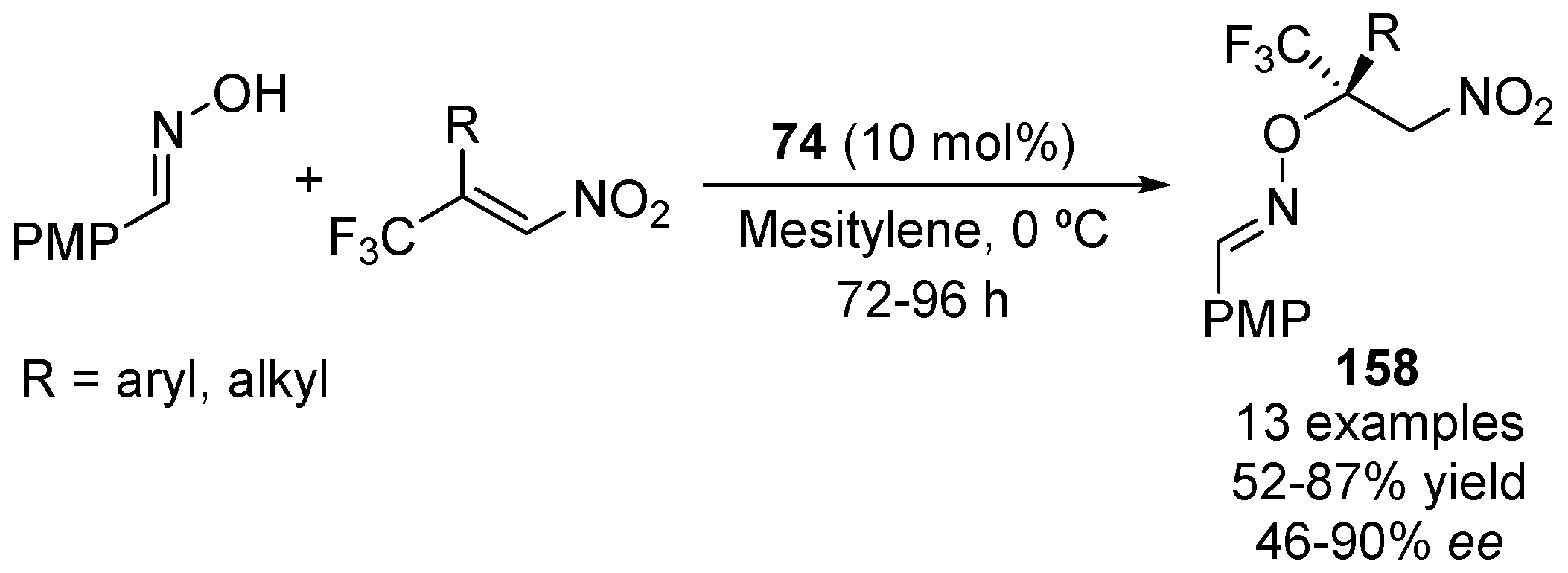
2.4. Nitroalkanes
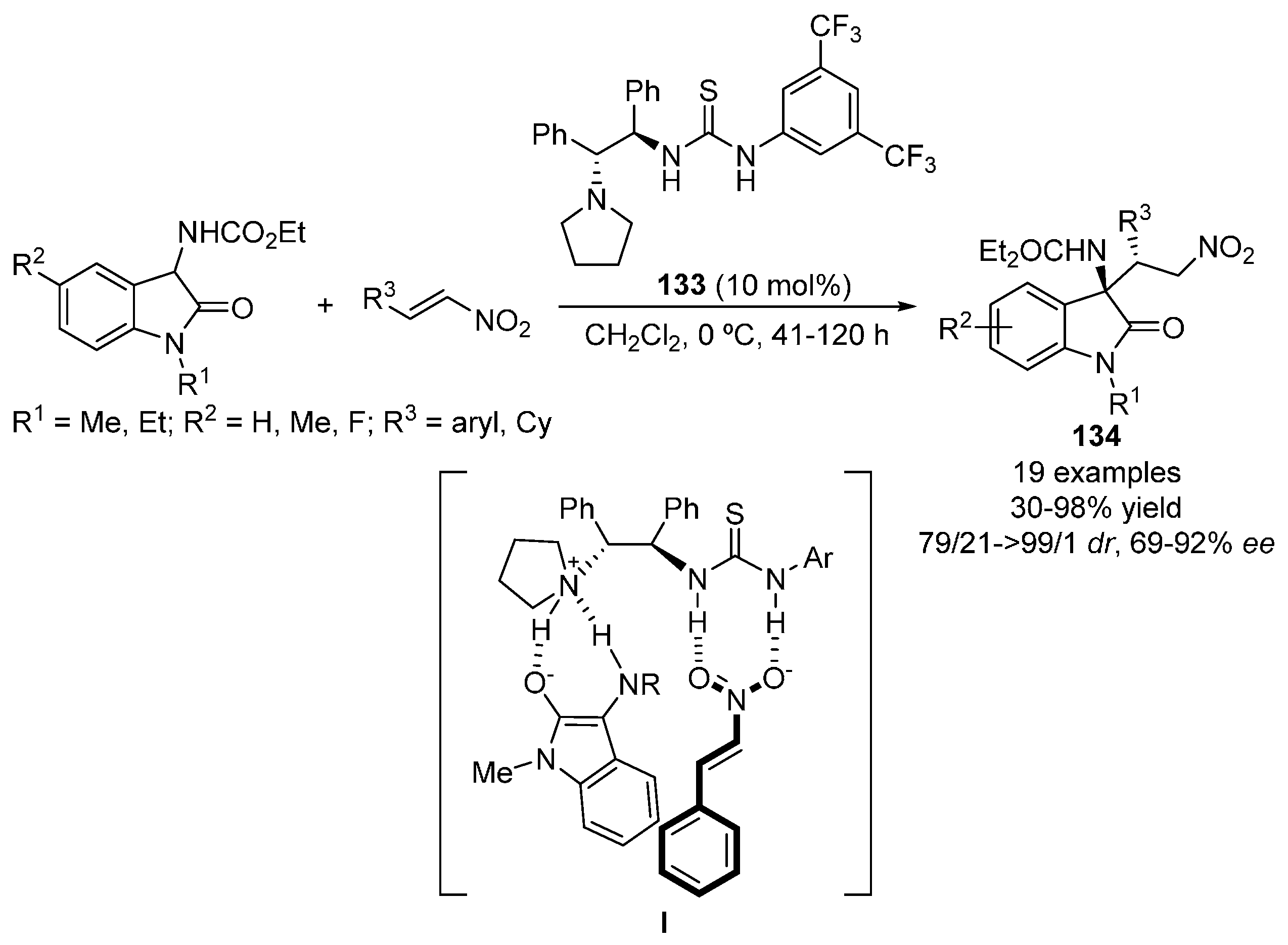
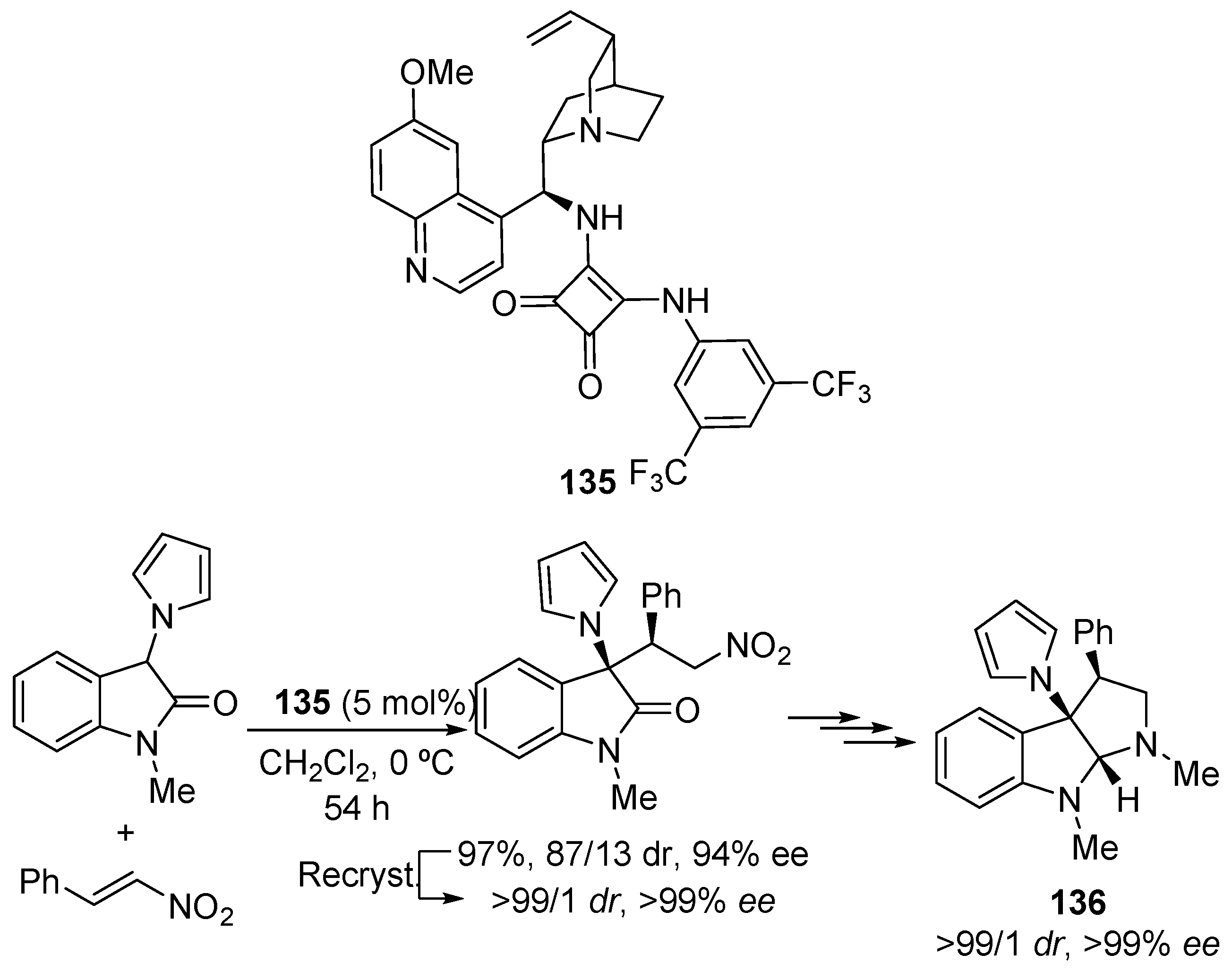
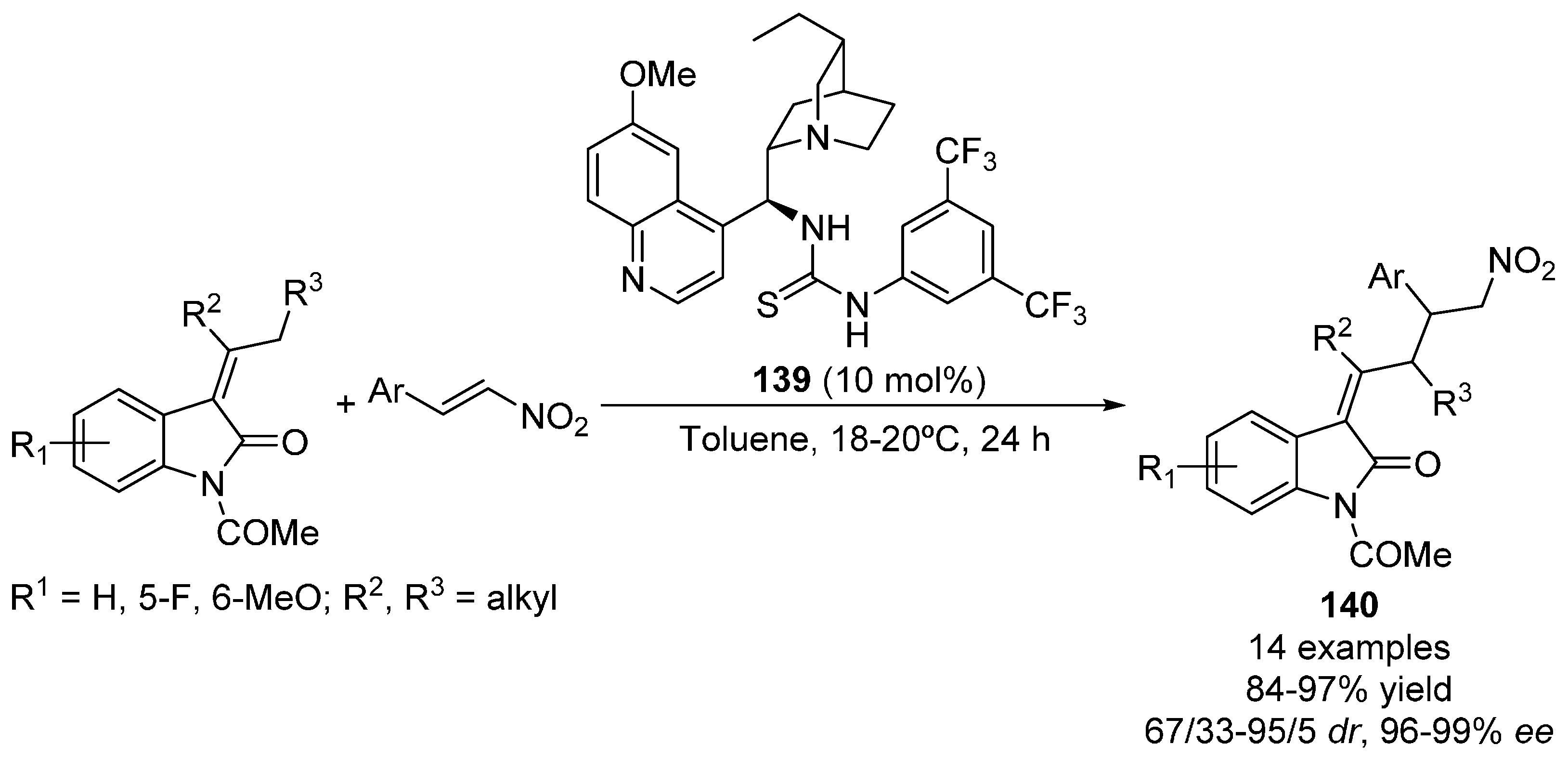
2.5. Heterocyclic Compounds
3. Nitrogen Nucleophiles
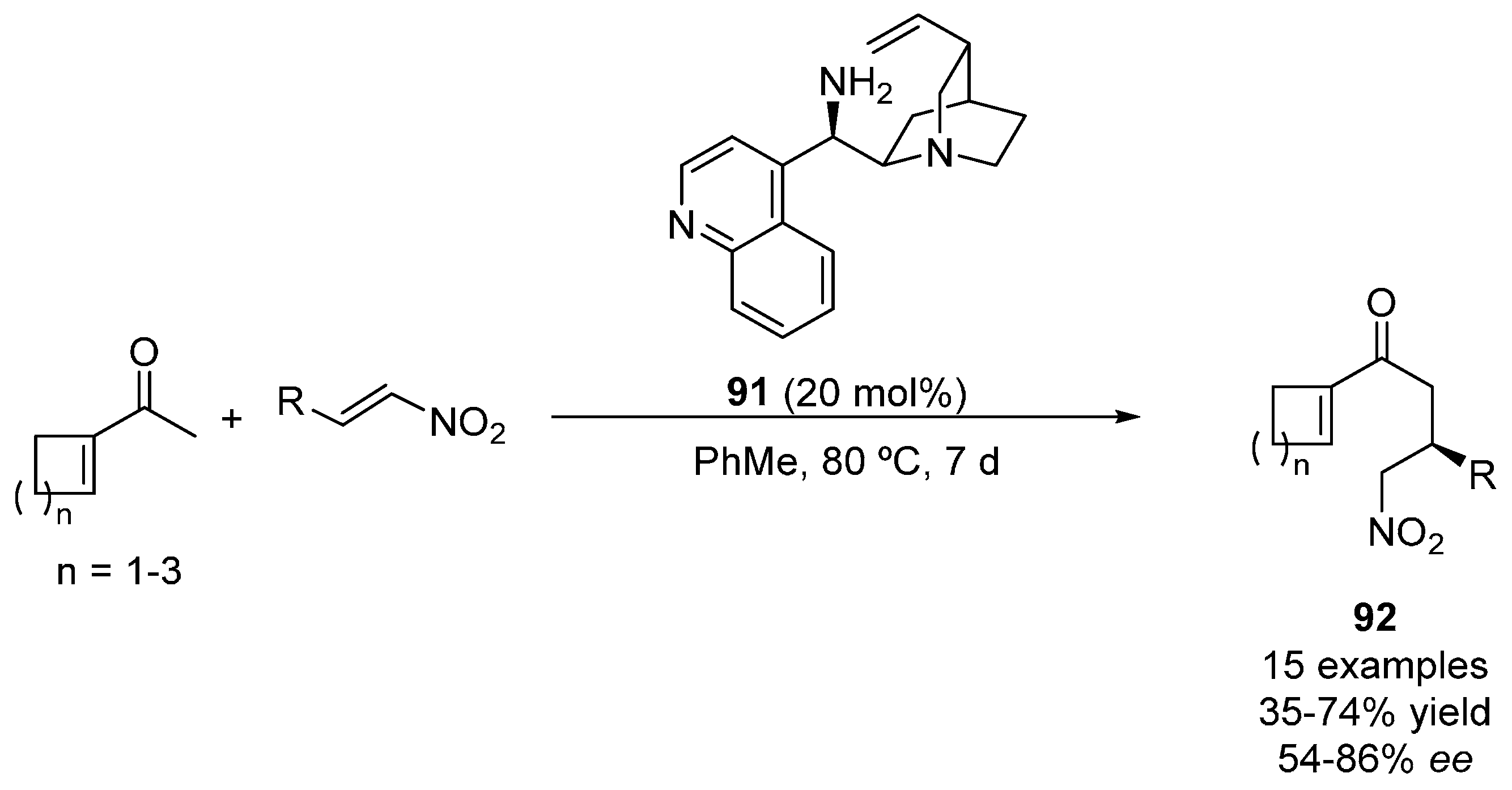
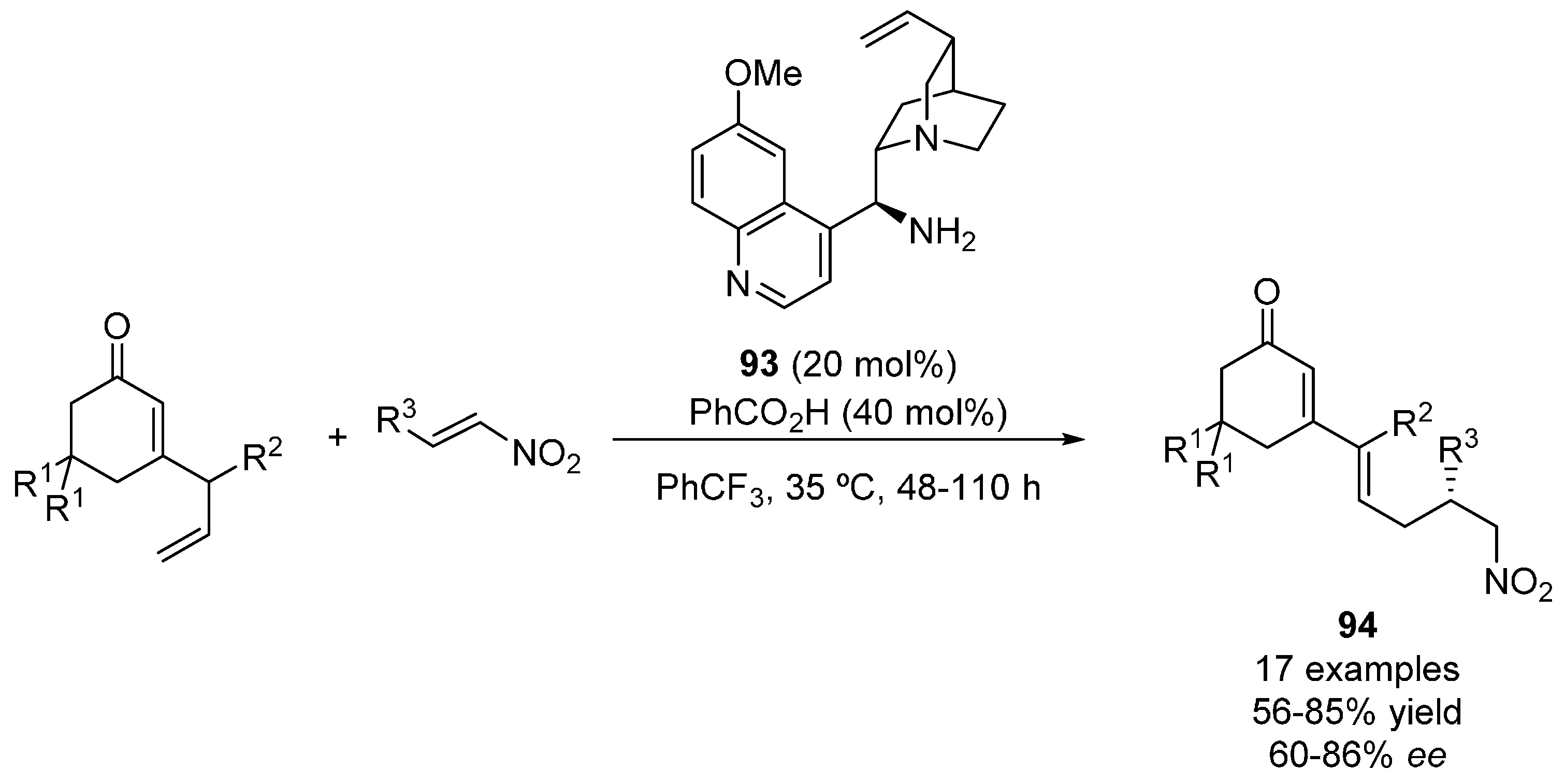
4. Oxygen Nucleophiles
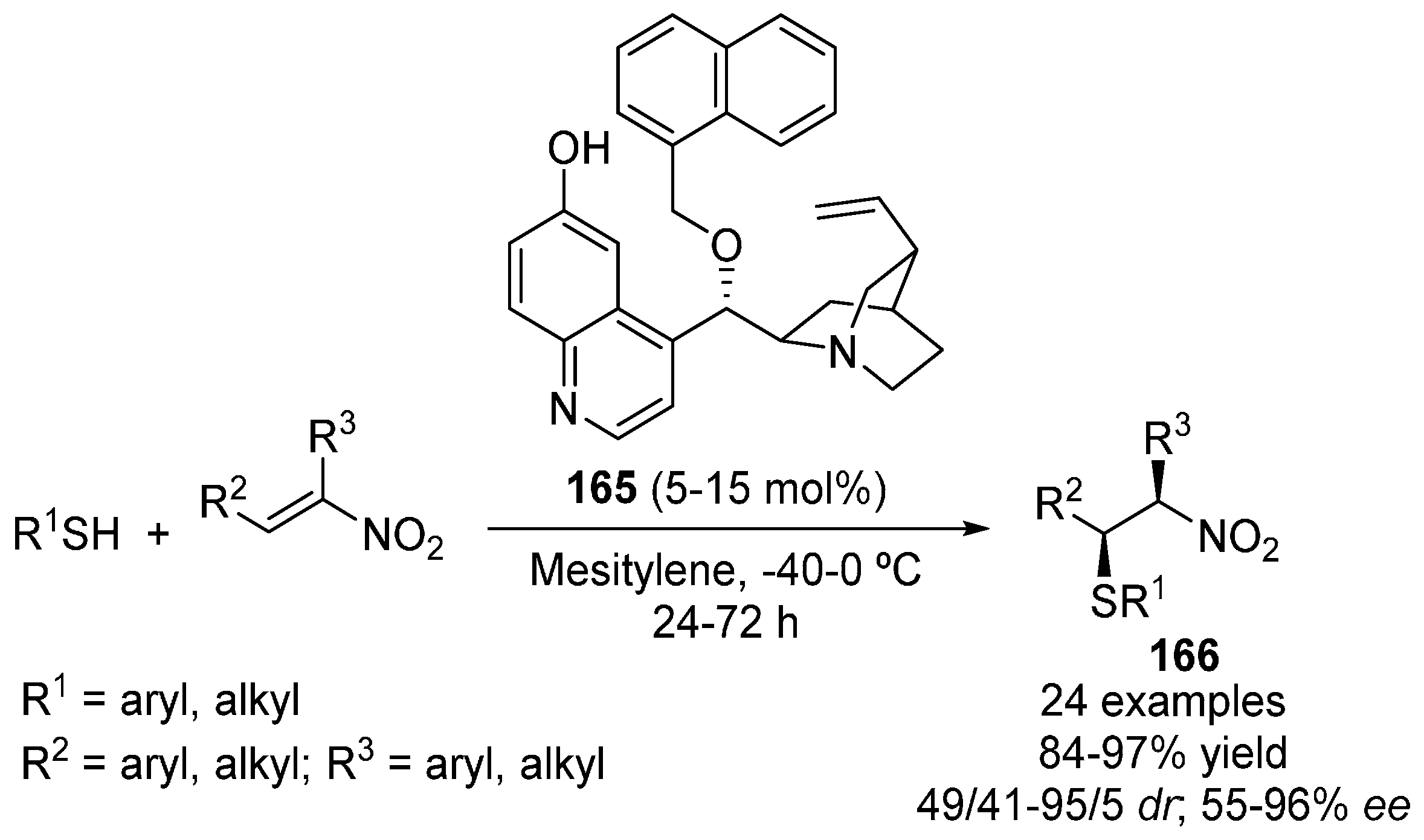
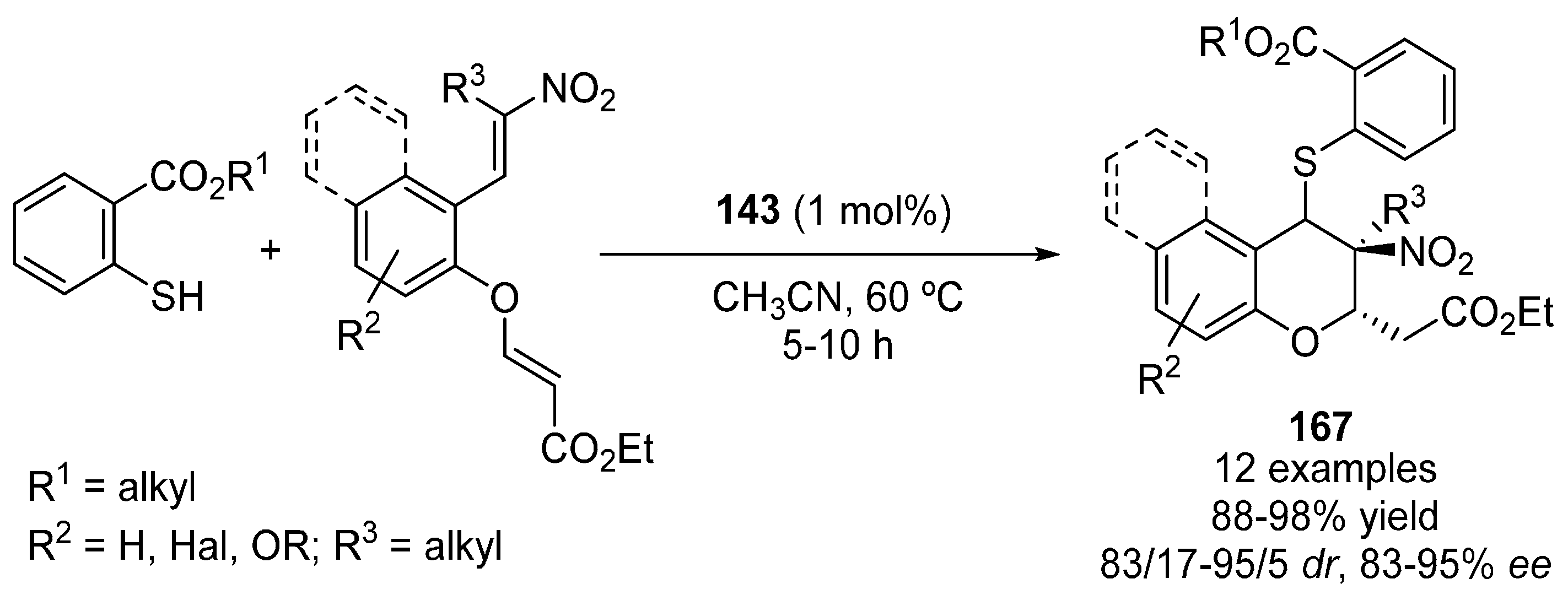
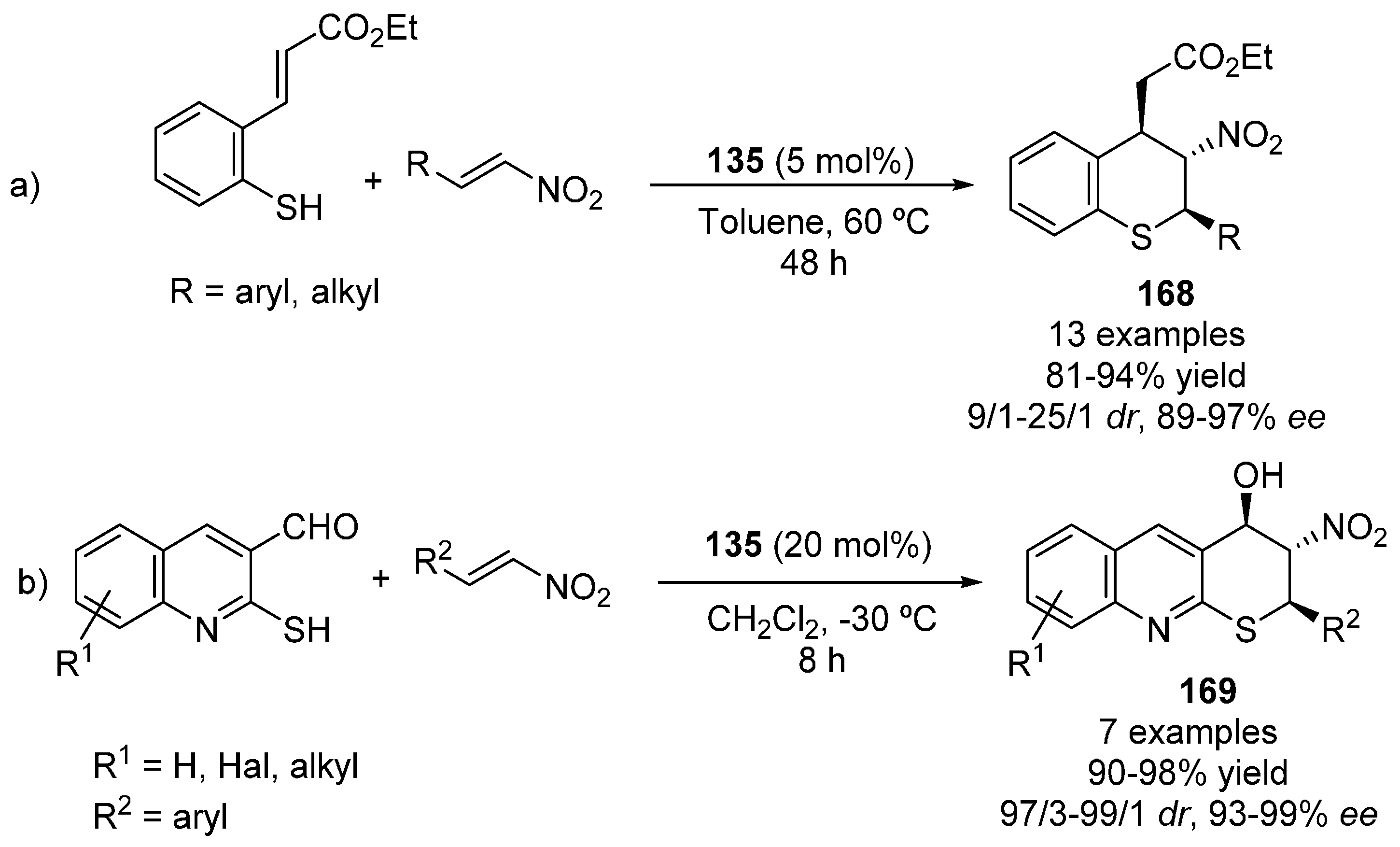
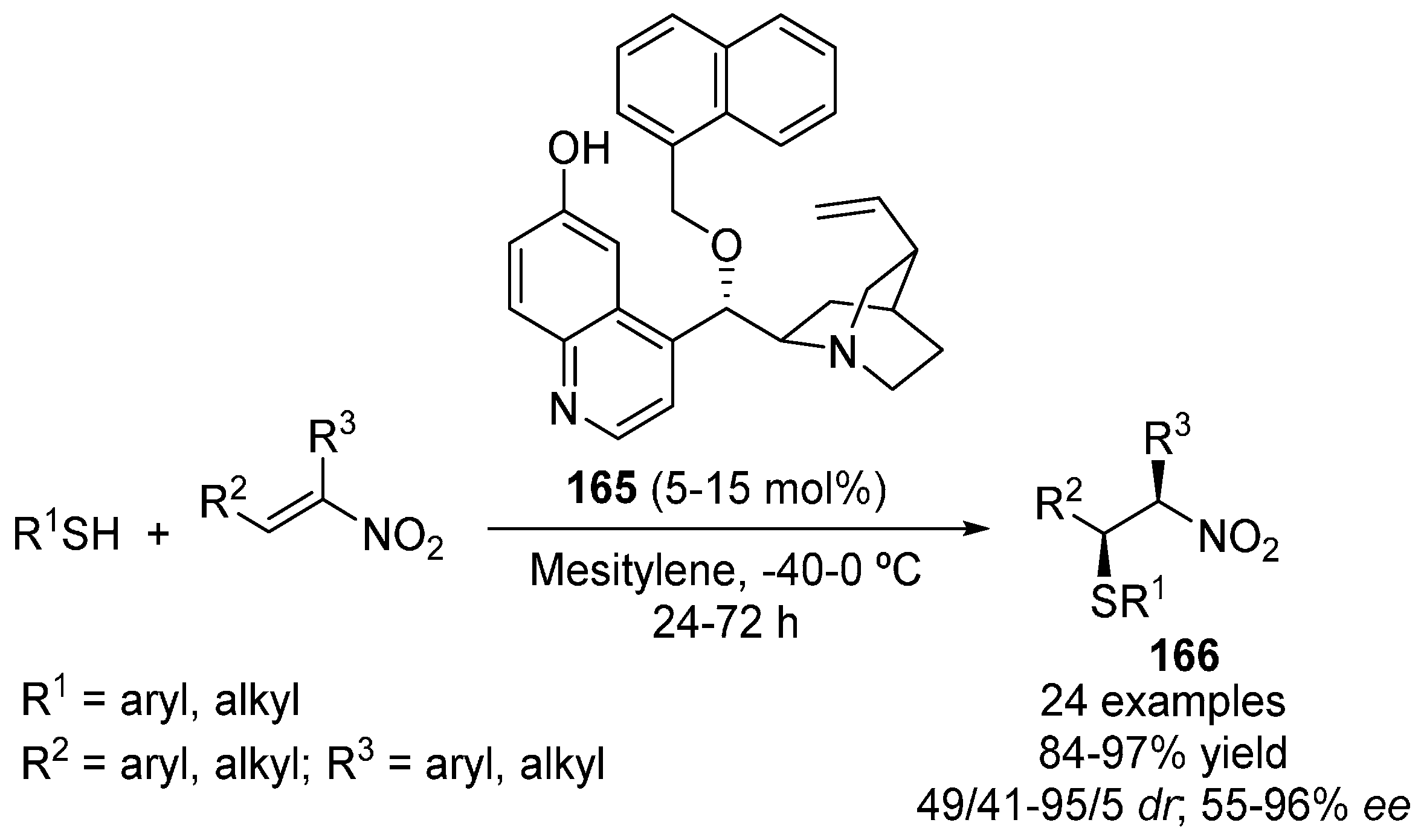
5. Sulfur Nucleophiles
6. Phosphorous Nucleophiles
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Atodiresei, I.; Vila, C.; Rueping, M. Asymmetric organocatalysis in continuous flow: Opportunities for impacting industrial catalysis. ACS Catal. 2015, 5, 1972–1985. [Google Scholar] [CrossRef]
- Krause, N.; Hoffmann-Roder, A. Recent advances in catalytic enantioselective Michael additions. Synthesis 2001, 2001, 0171–0196. [Google Scholar] [CrossRef]
- Jha, S.C.; Joshi, N.N. Catalytic, enantioselective Michael addition reactions. ARKIVOC 2002, viii, 167–196. [Google Scholar] [CrossRef]
- Christoffers, J.; Baro, A. Construction of quaternary stereocenters: New perspectives through enantioselective Michael reactions. Angew. Chem. Int. Ed. 2003, 42, 1688–1690. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Conjugate additions of nitroalkanes to electron-poor alkenes: Recent results. Chem. Rev. 2005, 105, 933–971. [Google Scholar] [CrossRef] [PubMed]
- Almasi, D.; Alonso, D.A.; Najera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar]
- Tsogoeva, S.B. Recent advances in asymmetric organocatalytic 1,4-conjugate additions. Eur. J. Org. Chem. 2007, 1701–1716. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hajiabbasi, P.; Hamidi, H. Recent developments in the asymmetric Michael addition for carbon-carbon bond formation. Curr. Org. Chem. 2014, 18, 489–511. [Google Scholar] [CrossRef]
- Nayak, S.; Chakroborty, S.; Bhakta, S.; Panda, P.; Mohapatra, S. Recent advances of organocatalytic enantioselective Michael-addition to chalcone. Res. Chem. Intermed. 2016, 42, 2731–2747. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael additions to nitroalkenes. Eur. J. Org. Chem. 2002, 1877–1894. [Google Scholar] [CrossRef]
- Sulzer-Mosse, S.; Alexakis, A. Chiral amines as organocatalysts for asymmetric conjugate addition to nitroolefins and vinyl sulfones via enamine activation. Chem. Commun. 2007, 43, 3123–3135. [Google Scholar] [CrossRef] [PubMed]
- Roca-López, D.; Sadaba, D.; Delso, I.; Herrera, R.P.; Tejero, T.; Merino, P. Asymmetric organocatalytic synthesis of γ-nitrocarbonyl compounds through Michael and Domino reaction. Tetrahedron Asymmetry 2010, 21, 2561–2601. [Google Scholar]
- Somanathan, R.; Chavez, D.; Servin, F.A.; Romero, J.A.; Navarrete, A.; Parra-Hake, M.; Aguirre, G.; Anaya, D.P.C.; González, J. Bifunctional organocatalysts in the asymmetric Michael additions of carbonylic compounds to nitroalkenes. Curr. Org. Chem. 2012, 16, 2440–2461. [Google Scholar] [CrossRef]
- Aitken, L.S.; Arezki, N.R.; Dell'Isola, A.; Cobb, A.J.A. Asymmetric organocatalysis and the nitro group functionality. Synthesis 2013, 45, 2627–2648. [Google Scholar]
- Bächle, F.; Duschmale, J.; Ebner, C.; Pfaltz, A.; Wennemers, H. Organocatalytic asymmetric conjugate addition of aldehydes to nitroolefins: Identification of catalytic intermediates and the stereoselectivity-determining step by ESI-MS. Angew. Chem. Int. Ed. 2013, 52, 12619–12623. [Google Scholar] [CrossRef] [PubMed]
- Isenegger, P.G.; Pfaltz, A. Mass spectrometric back reaction screening of quasi-enantiomeric products as a mechanistic tool. Chem. Rec. 2016, 16, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Patora-Komisarska, K.; Benohoud, M.; Ishikawa, H.; Seebach, D.; Hayashi, Y. Organocatalyzed Michael addition of aldehydes to nitro alkenes - Generally accepted mechanism revisited and revised. Helv. Chim. Acta 2011, 94, 719–745. [Google Scholar] [CrossRef]
- Seebach, D.; Sun, X.; Sparr, C.; Ebert, M.-O.; Schweizer, W.B.; Beck, A.K. 1,2-Oxazine N-oxides as catalyst resting states in Michael additions of aldehydes to nitro olefins organocatalyzed by α,α-diphenylprolinol trimethylsilyl ether. Helv. Chim. Acta 2012, 95, 1064–1078. [Google Scholar] [CrossRef]
- Bures, J.; Armstrong, A.; Blackmond, D.G. Mechanistic rationalization of organocatalyzed conjugate addition of linear aldehydes to nitro-olefins. J. Am. Chem. Soc. 2011, 133, 8822–8825. [Google Scholar] [CrossRef] [PubMed]
- Bures, J.; Armstrong, A.; Blackmond, D.G. Curtin-Hammett paradigm for stereocontrol in organocatalysis by diarylprolinol ether catalysts. J. Am. Chem. Soc. 2012, 134, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, G.; Rahaman, H.; Madarasz, Á.; Pápai, I.; Melarto, M.; Valkonen, A.; Pihko, P.M. Dihydrooxazine oxides as key intermediates in organocatalytic Michael additions of aldehydes to nitroalkenes. Angew. Chem. Int. Ed. 2012, 51, 13144–13148. [Google Scholar] [CrossRef] [PubMed]
- Duschmalé, J.; Wiest, J.; Wiesner, M.; Wennemers, H. Effects of internal and external carboxylic acids on the reaction pathway of organocatalytic 1,4-addition reactions between aldehydes and nitroolefins. Chem. Sci. 2013, 4, 1312–1318. [Google Scholar] [CrossRef]
- Bures, J.; Armstrong, A.; Blackmond, D.G. Explaining anomalies in enamine catalysis: “Downstream species” as a new paradigm for stereocontrol. Acc. Chem. Res. 2016, 49, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Liu, S.-P.; Li, X.-M.; Yan, M.; Chan, A.S.C. Highly enantioselective conjugate addition of aldehydes to nitroolefins catalyzed by chiral bifunctional sulfamides. Chem. Commun. 2009, 45, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C.F., III. Amino acid catalyzed direct asymmetric aldol reactions: A bioorganic approach to catalytic asymmetric carbon-carbon bond-forming reactions. J. Am. Chem. Soc. 2001, 123, 5260–5267. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Pojarliev, P.; Martin, H.J. Efficient proline-catalyzed Michael additions of unmodified ketones to nitro olefins. Org. Lett. 2001, 3, 2423–2425. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, M.; Han, S. Highly efficient asymmetric Michael addition of aldehydes to nitroalkenes with 4,5-methano-l-proline as organocatalysts. Tetrahedron 2014, 70, 8380–8384. [Google Scholar] [CrossRef]
- An, Q.; Shen, J.; Butt, N.; Liu, D.; Liu, Y.; Zhang, W. Asymmetric domino double Michael addition of nitroolefins and aldehyde esters with trans-perhydroindolic acid as an organocatalyst. Synthesis 2013, 45, 1612–1623. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, J.; Wei, K. Aromatic l-prolinamide-catalyzed asymmetric Michael addition of aldehydes to nitroalkenes. Tetrahedron Asymmetry 2014, 25, 1599–1604. [Google Scholar] [CrossRef]
- Wang, Y.; Li, D.; Lin, J.; Wei, K. Organocatalytic asymmetric Michael addition of aldehydes and ketones to nitroalkenes catalyzed by adamantoyl l-prolinamide. RSC Adv. 2015, 5, 5863–5874. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, S.; Wei, K.; Lin, J. Epiandrosterone-derived prolinamide as an efficient asymmetric catalyst for Michael addition reactions of aldehydes to nitroalkenes. RSC Adv. 2014, 4, 30850–30856. [Google Scholar] [CrossRef]
- De la Torre, A.F.; Rivera, D.G.; Ferreira, M.A.B.; Correa, A.G.; Paixao, M.W. Multicomponent combinatorial development and conformational analysis of prolyl peptide-peptoid hybrid catalysts: Application in the direct asymmetric Michael addition. J. Org. Chem. 2013, 78, 10221–10232. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Clerget, M.; Gager, O.; Monteil, M.; Pirat, J.-L.; Migianu-Griffoni, E.; Deschamp, J.; Lecouvey, M. Novel easily recyclable bifunctional phosphonic acid carrying tripeptides for the stereoselective michael addition of aldehydes with nitroalkenes. Adv. Synth. Catal. 2016, 358, 34–40. [Google Scholar] [CrossRef]
- Kastl, R.; Arakawa, Y.; Duschmale, J.; Wiesner, M.; Wennemers, H. Peptide-catalyzed 1,4-addition reactions of aldehydes to nitroolefins. Chimia 2013, 67, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.L.; Dickmeiss, G.; Jiang, H.; Albrecht, L.; Jørgensen, K.A. The Diarylprolinol Silyl Ether System: A General Organocatalyst. Acc. Chem. Res. 2012, 45, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Meninno, S.; Lattanzi, A. Asymmetric organocatalysis mediated by α,α-l-diaryl prolinols: recent advances. Chem. Commun. 2013, 49, 3821–3832. [Google Scholar] [CrossRef] [PubMed]
- Candy, M.; Durand, T.; Galano, J.-M.; Oger, C. Total synthesis of the isoketal 5-D2-IsoK natural product based on organocatalysis. Eur. J. Org. Chem. 2016, 5813–5816. [Google Scholar] [CrossRef]
- Zheng, Y.; Ghazvini Zadeh, E.H.; Yuan, Y. One-pot, enantioselective synthesis of 2,3-dihydroazulen-6(1H)-one: A concise access to the core structure of cephalotaxus norditerpenes. Eur. J. Org. Chem. 2016, 2115–2119. [Google Scholar] [CrossRef]
- Zeng, X.; Ni, Q.; Raabe, G.; Enders, D. A branched domino reaction: Asymmetric organocatalytic two-component four-step synthesis of polyfunctionalized cyclohexene derivatives. Angew. Chem. Int. Ed. 2013, 52, 2977–2980. [Google Scholar] [CrossRef] [PubMed]
- Umemiya, S.; Sakamoto, D.; Kawauchi, G.; Hayashi, Y. Enantioselective total synthesis of beraprost using organocatalyst. Org. Lett. 2017, 19, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Szcześniak, P.; Staszewska-Krajewska, O.; Furman, B.; Młynarski, J. Asymmetric synthesis of cyclic nitrones via organocatalytic Michael addition of aldehydes to nitroolefins and subsequent reductive cyclization. Chem. Sel. 2017, 2, 2672–2678. [Google Scholar] [CrossRef]
- Keller, M.; Perrier, A.; Linhardt, R.; Travers, L.; Wittmann, S.; Caminade, A.-M.; Majoral, J.-P.; Reiser, O.; Ouali, A. Dendrimers or nanoparticles as supports for the design of efficient and recoverable organocatalysts? Adv. Synth. Catal. 2013, 355, 1748–1754. [Google Scholar] [CrossRef]
- Sagamanova, I.; Rodríguez-Escrich, C.; Molnár, I.G.; Sayalero, S.; Gilmour, R.; Pericàs, M.A. Translating the enantioselective Michael reaction to a continuous flow paradigm with an immobilized, fluorinated organocatalyst. ACS Catal. 2015, 5, 6241–6248. [Google Scholar] [CrossRef]
- Sebesta, R.; Latika, A. Enantioselective Michael additions of aldehydes to nitroalkenes catalyzed with ionically tagged organocatalyst. Cent. Eur. J. Chem. 2014, 12, 416–425. [Google Scholar] [CrossRef]
- Funabiki, K.; Ohta, M.; Sakaida, Y.; Oida, K.; Kubota, Y.; Matsui, M. High diastereoselectivity induced by a fluorous alkyl group in the asymmetric Michael reaction of nitroalkenes catalyzed by a prolinol methyl ether. Asian J. Org. Chem. 2013, 2, 1048–1054. [Google Scholar] [CrossRef]
- Kumar, T.P. Asymmetric Michael addition of aldehydes to nitroolefins catalyzed by a pyrrolidine-pyrazole. Tetrahedron Asymmetry 2014, 25, 1286–1291. [Google Scholar] [CrossRef]
- Kumar, T.P.; Abdul Sattar, M.; Prasad, S.S.; Haribabu, K.; Reddy, C.S. Enantioselective Michael addition of aldehydes to nitroolefins catalyzed by pyrrolidine-HOBt. Tetrahedron Asymmetry 2017, 28, 401–409. [Google Scholar] [CrossRef]
- Hu, X.; Wei, Y.-F.; Wu, N.; Jiang, Z.; Liu, C.; Luo, R.-S. Indolinol-catalyzed asymmetric Michael reaction of aldehydes to nitroalkenes in brine. Tetrahedron Asymmetry 2016, 27, 420–427. [Google Scholar] [CrossRef]
- Kano, T.; Sugimoto, H.; Tokuda, O.; Maruoka, K. Unusual anti-selective asymmetric conjugate addition of aldehydes to nitroalkenes catalyzed by a biphenyl-based chiral secondary amine. Chem. Commun. 2013, 49, 7028–7030. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, D.; Xue, F.; Qin, Y. Synthesis of the atropurpuran A-ring via an organocatalytic asymmetric intramolecular Michael addition. Tetrahedron 2013, 69, 3141–3148. [Google Scholar] [CrossRef]
- He, J.; Chen, Q.; Ni, B. Highly efficient asymmetric organocatalytic Michael addition of α,α-disubstituted aldehydes to nitroolefins under solvent-free conditions. Tetrahedron Lett. 2014, 55, 3030–3032. [Google Scholar] [CrossRef]
- Kumar, T.P. Pyrrolidine-oxyimide catalyzed asymmetric Michael addition of α,α-disubstituted aldehydes to nitroolefins. Tetrahedron Asymmetry 2015, 26, 907–911. [Google Scholar] [CrossRef]
- Zlotin, S.G.; Kochetkov, S.V. C2-symmetric diamines and their derivatives as promising organocatalysts for asymmetric synthesis. Russ. Chem. Rev. 2015, 84, 1077–1099. [Google Scholar] [CrossRef]
- Servin, F.A.; Madrigal, D.; Romero, J.A.; Chávez, D.; Aguirre, G.; Anaya de Parrodi, C.; Somanathan, R. Synthesis of C2-symmetric 1,2-diamine-functionalized organocatalysts: Mimicking enzymes in enantioselective Michael addition reactions. Tetrahedron Lett. 2015, 56, 2355–2358. [Google Scholar] [CrossRef]
- Fernandes, T.A.; Vizcaíno-Milla, P.; Ravasco, J.M.J.M.; Ortega-Martinez, A.; Sansano, J.M.; Nájera, C.; Costa, P.R.R.; Fiser, B.; Gómez-Bengoa, E. Bifunctional primary amine 2-aminobenzimidazole organocatalyst anchored to trans-cyclohexane-1,2-diamine in enantioselective conjugate additions of aldehydes. Tetrahedron Asymmetry 2016, 27, 118–122. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Fiser, B.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Michael addition of isobutyraldehyde to nitroalkenes organocatalyzed by chiral primary amine-guanidines. Tetrahedron Asymmetry 2014, 25, 462–467. [Google Scholar] [CrossRef]
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071. [Google Scholar] [CrossRef] [PubMed]
- Tsakos, M.; Kokotos, C.G. Primary and secondary amine-(thio)ureas and squaramides and their applications in asymmetric organocatalysis. Tetrahedron 2013, 69, 10199–10222. [Google Scholar] [CrossRef]
- Fang, X.; Wang, C.-J. Recent advances in asymmetric organocatalysis mediated by bifunctional amine-thioureas bearing multiple hydrogen-bonding donors. Chem. Commun. 2015, 51, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-L.; Wei, Y.; Shi, M. Applications of chiral thiourea-amine/phosphine organocatalysts in catalytic asymmetric reactions. Chem. Cat. Chem. 2017, 9, 718–727. [Google Scholar] [CrossRef]
- Durmaz, M.; Sirit, A. Calixarene-based highly efficient primary amine-thiourea organocatalysts for asymmetric Michael addition of aldehydes to nitrostyrenes. Supramol. Chem. 2013, 25, 292–301. [Google Scholar] [CrossRef]
- Guo, X.-T.; Sha, F.; Wu, X.-Y. Highly enantioselective Michael addition of α,α-disubstituted aldehydes to nitroolefins. Res. Chem. Intermed. 2016, 42, 6373–6380. [Google Scholar] [CrossRef]
- Ji, Y.; Blackmond, D.G. The role of reversibility in the enantioselective conjugate addition of α,α-disubstituted aldehydes to nitro-olefins catalyzed by primary amine thioureas. Catal. Sci. Technol. 2014, 4, 3505–3509. [Google Scholar] [CrossRef]
- Rouf, A.; Tanyeli, C. Squaramide based organocatalysts in organic transformations. Curr. Org. Chem. 2016, 20, 2996–3013. [Google Scholar] [CrossRef]
- Han, X.; Zhou, H.-B.; Dong, C. Applications of chiral squaramides: From asymmetric organocatalysis to biologically active compounds. Chem. Rec. 2016, 16, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-W.; Liu, X.-F.; Sun, B.; Huang, X.-H.; Tao, J.-C. Chiral primary amine-squaramide catalyzed highly enantioselective Michael addition of isobutyraldehyde to nitroolefins. Synthesis 2017, 49, 1307–1314. [Google Scholar] [CrossRef]
- Frias, M.; Mas-Ballesté, R.; Arias, S.; Alvarado, C.; Alemán, J. Asymmetric synthesis of Rauhut-Currier type products by a regioselective Mukaiyama reaction under bifunctional catalysis. J. Am. Chem. Soc. 2017, 139, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Atsuki, S. Proline-catalyzed enantioselective Michael additions of ketones to nitrostyrene. Synlett 2002, 26–28. [Google Scholar] [CrossRef]
- Li, J.; Li, X.B.; Ma, S.S.; Liu, J.; Li, B.H.; Li, B.L. An ionic liquid containing l-proline moiety as highly efficient and recyclable chiral organocatalyst for Michael addition. Bull. Korean Chem. Soc. 2016, 37, 1259–1264. [Google Scholar] [CrossRef]
- Kumar, T.P.; Prasad, S.S.; Haribabu, K.; Kumar, V.N.; Reddy, C.S. Pyrrolidine-HOBt: An oxytriazole catalyst for the enantioselective Michael addition of cyclohexanone to nitroolefins in water. Tetrahedron Asymmetry 2016, 27, 1133–1138. [Google Scholar] [CrossRef]
- Kumar, T.P.; Radhika, L.; Haribabu, K.; Kumar, V.N. Pyrrolidine-oxyimides: new chiral catalysts for enantioselective Michael addition of ketones to nitroolefins in water. Tetrahedron Asymmetry 2014, 25, 1555–1560. [Google Scholar] [CrossRef]
- Singh, K.N.; Singh, P.; Kaur, A.; Singh, P.; Sharma, S.K.; Khullar, S.; Mandal, S.K. (2S)-2-[(Phenyl- sulfinyl)methyl]pyrrolidine-catalyzed efficient stereoselective Michael addition of cyclohexanone and cyclopentanone to nitroolefins. Synthesis 2013, 45, 1406–1413. [Google Scholar] [CrossRef]
- Wagner, M.; Contie, Y.; Ferroud, C.; Revial, G. Enantioselective aldol reactions and Michael additions using proline derivatives as organocatalysts. Int. J. Org. Chem. 2014, 4, 55–67. [Google Scholar] [CrossRef]
- Mahato, C.K.; Kundu, M.; Pramanik, A. Solvent free, fast and asymmetric Michael additions of ketones to nitroolefins using chiral pyrrolidine–pyridone conjugate bases as organocatalysts. Tetrahedron Asymmetry 2017, 28, 511–515. [Google Scholar] [CrossRef]
- Nakashima, K.; Hirashima, S.-I.; Kawada, M.; Koseki, Y.; Tada, N.; Itoh, A.; Miura, T. Pyrrolidine-diaminomethylenemalononitrile organocatalyst for Michael additions of carbonyl compounds to nitro alkenes under solvent-free conditions. Tetrahedron Lett. 2014, 55, 2703–2706. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, M.; Liu, J.-T. Enantioselective Michael addition of cyclic ketones to nitroolefins catalyzed by a novel fluorine-insertion organic catalyst. Tetrahedron Asymmetry 2014, 25, 212–218. [Google Scholar] [CrossRef]
- Kaplanaris, N.; Koutoulogenis, G.; Raftopoulou, M.; Kokotos, C.G. 4-Fluoro and 4-hydroxy pyrrolidine-thioxotetrahydropyrimidinones: Organocatalysts for green asymmetric transformations in brine. J. Org. Chem. 2015, 80, 5464–5473. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, Y.C.; Zhao, J.Q.; Yang, Q.S.; Ma, Y.; Cao, X.H. The highly enantioselective bifunctional organocatalysts for the Michael addition of cyclohexanone to nitro olefins. Russ. J. Gen. Chem. 2016, 86, 1381–1388. [Google Scholar] [CrossRef]
- Siyutkin, D.E.; Kucherenko, A.S.; Frolova, L.L.; Kuchin, A.V.; Zlotin, S.G. N-Pyrrolidine-2-ylmethyl)-2-hydroxy-3-aminopinanes as novel organocatalysts for asymmetric conjugate additions of ketones to α-nitroalkenes. Tetrahedron Asymmetry 2013, 24, 776–779. [Google Scholar] [CrossRef]
- Kumar, T.P.; Balaji, S.V. Sugar amide-pyrrolidine catalyst for the asymmetric Michael addition of ketones to nitroolefins. Tetrahedron Asymmetry 2014, 25, 473–477. [Google Scholar] [CrossRef]
- Kamal, A.; Sathish, M.; Srinivasulu, V.; Chetna, J.; Chandra Shekar, K.; Nekkanti, S.; Tangella, Y.; Shankaraiah, N. Asymmetric Michael addition of ketones to nitroolefins: Pyrrolidinyl- oxazole-carboxamides as new efficient organocatalysts. Org. Biomol. Chem. 2014, 12, 8008–8018. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, S.; Kumar, C.P.; Kumar, T.P.; Haribabu, K.; Jagadeesh, B.; Lakshmi, J.K.; Mainkar, P.S. Peptidomimetic organocatalysts: efficient Michael addition of ketones onto nitroolefins with very low catalyst loading. RSC Adv. 2014, 4, 30325–30331. [Google Scholar] [CrossRef]
- Zhao, H.-W.; Li, H.-L.; Yue, Y.-Y.; Sheng, Z.-H. Diastereoselective and enantioselective Michael addition reactions of ketones and aldehydes to nitro olefins catalyzed by C2-symmetric axially-unfixed biaryl-based organocatalysts derived from enantiopure α-proline. Eur. J. Org. Chem. 2013, 2013, 1740–1748. [Google Scholar] [CrossRef]
- Zhong, J.; Guan, Z.; He, Y.-H. A novel organocatalyst for direct asymmetric Michael additions of cyclohexanone to nitroolefins. Catal. Commun. 2013, 32, 18–22. [Google Scholar] [CrossRef]
- Zhang, R.; Yin, G.; Li, Y.; Yan, X.; Chen, L. Resin-immobilized pyrrolidine-based chiral organocatalysts for asymmetric Michael additions of ketones and aldehydes to nitroolefins. RSC Adv. 2015, 5, 3461–3464. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Liu, L. Efficient enantioselective Michael addition of nitroalkenes catalyzed by a surfactant-type bifunctional thiourea organocatalyst in the presence of water. Lett. Org. Chem. 2012, 9, 51–55. [Google Scholar] [CrossRef]
- Ban, S.-R.; Zhu, X.-X.; Zhang, Z.-P.; Xie, H.-Y.; Li, Q.-S. Benzoylthiourea-pyrrolidine as another bifunctional organocatalyst: Highly enantioselective Michael addition of cyclohexanone to nitroolefins. Eur. J. Org. Chem. 2013, 2013, 2977–2980. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Ban, S.-R.; Yang, M.-C.; Li, Q.-S. Highly enantioselective Michael addition of cyclohexanone to nitroolefins catalyzed by pyrrolidine-based bifunctional benzoylthiourea in water. Chirality 2016, 28, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Z.; Li, S.; Qiu, Y.; Liu, B.; Song, Z.; Liu, Z. Synthesis of novel thiazoline catalysts and their application in Michael addition reaction. Chem. Res. Chin. Univ. 2016, 32, 373–379. [Google Scholar] [CrossRef]
- Lu, J.; Chen, W.-Y. Asymmetric Michael addition of ketones to nitroolefins catalyzed by a novel chiral pyrrolidine-thiourea in the ionic liquid. Bull. Korean Chem. Soc. 2013, 34, 3179–3180. [Google Scholar] [CrossRef]
- Yu, L.; Li, P. New simple primary amine-thiourea organocatalysts and their application in asymmetric conjugate addition. Tetrahedron Lett. 2014, 55, 3697–3700. [Google Scholar] [CrossRef]
- Orlandi, S.; Pozzi, G.; Ghisetti, M.; Benaglia, M. Synthesis and catalytic activity of fluorous chiral primary amine-thioureas. New J. Chem. 2013, 37, 4140–4147. [Google Scholar] [CrossRef]
- Capitta, F.; Frongia, A.; Ollivier, J.; Aitken, D.J.; Secci, F.; Piras, P.P.; Guillot, R. Enantioselective organocatalyzed desymmetrization of 3-substituted cyclobutanones through Michael addition to nitroalkenes. Synlett 2015, 26, 123–126. [Google Scholar]
- Guang, J.; Zhao, J.C.-G. Organocatalyzed asymmetric Michael reaction of β-aryl-α-ketophosphonates and nitroalkenes. Tetrahedron Lett. 2013, 54, 5703–5706. [Google Scholar] [CrossRef] [PubMed]
- Saidalimu, I.; Fang, X.; Lv, W.; Yang, X.; He, X.; Zhang, J.; Wu, F. Organocatalytic asymmetric Michael addition/carbon-carbon bond cleavage of trifluoromethyl α-fluorinated gem-diols to nitroolefins. Adv. Synth. Catal. 2013, 355, 857–863. [Google Scholar] [CrossRef]
- Ansari, S.; Raabe, G.; Enders, D. Asymmetric Michael addition of 1,3-bis(phenylthio)propan-2-one to nitroalkenes employing Takemoto's thiourea catalyst. Monatsh. Chem. 2013, 144, 641–646. [Google Scholar] [CrossRef]
- Corbett, M.T.; Xu, Q.; Johnson, J.S. Trisubstituted 2-trifluoromethyl pyrrolidines via catalytic asymmetric Michael addition/reductive cyclization. Org. Lett. 2014, 16, 2362–2365. [Google Scholar] [CrossRef] [PubMed]
- Ramachary, D.B.; Shruthi, K.S. Asymmetric synthesis of tetrahydroquinolines through supra-molecular organocatalysis. Org. Biomol. Chem. 2014, 12, 4300–4304. [Google Scholar] [CrossRef] [PubMed]
- Preegel, G.; Noole, A.; Ilmarinen, K.; Jarving, I.; Kanger, T.; Pehk, T.; Lopp, M. Enantioselective organocatalytic Michael addition of cyclopentane-1,2-diones to nitroolefins. Synthesis 2014, 46, 2595–2600. [Google Scholar]
- Urruzuno, I.; Mugica, O.; Oiarbide, M.; Palomo, C. Bifunctional bronsted base catalyst enables regio-, diastereo-, and enantioselective Cα-alkylation of β-tetralones and related aromatic-ring-fused cycloalkanones. Angew. Chem. Int. Ed. 2017, 56, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Kasaplar, P.; Rodriguez-Escrich, C.; Pericàs, M.A. Continuous flow, highly enantioselective Michael additions catalyzed by a PS-supported squaramide. Org. Lett. 2013, 15, 3498–3501. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Planes, L.; Rodríguez-Escrich, C.; Pericàs, M.A. Removing the superfluous: A supported squaramide catalyst with a minimalistic linker applied to the enantioselective flow synthesis of pyranonaphthoquinones. Catal. Sci. Technol. 2016, 6, 4686–4689. [Google Scholar] [CrossRef]
- Klare, H.; Neudoerfl, J.M.; Goldfuss, B. New hydrogen-bonding organocatalysts: chiral cyclophosphazanes and phosphorus amides as catalysts for asymmetric Michael additions. Beilstein J. Org. Chem. 2014, 10, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.A.; Navarrate, A.; Servin, F.A.; Madrigal, D.; Cooksy, A.L.; Aguirre, G.; Chavez, D.; Somanathan, R. Oxygen-chlorine interactions in the transition state of asymmetric Michael additions of carbonyl compounds to β-nitrostyrene. Tetrahedron Asymmetry 2014, 25, 997–1001. [Google Scholar] [CrossRef]
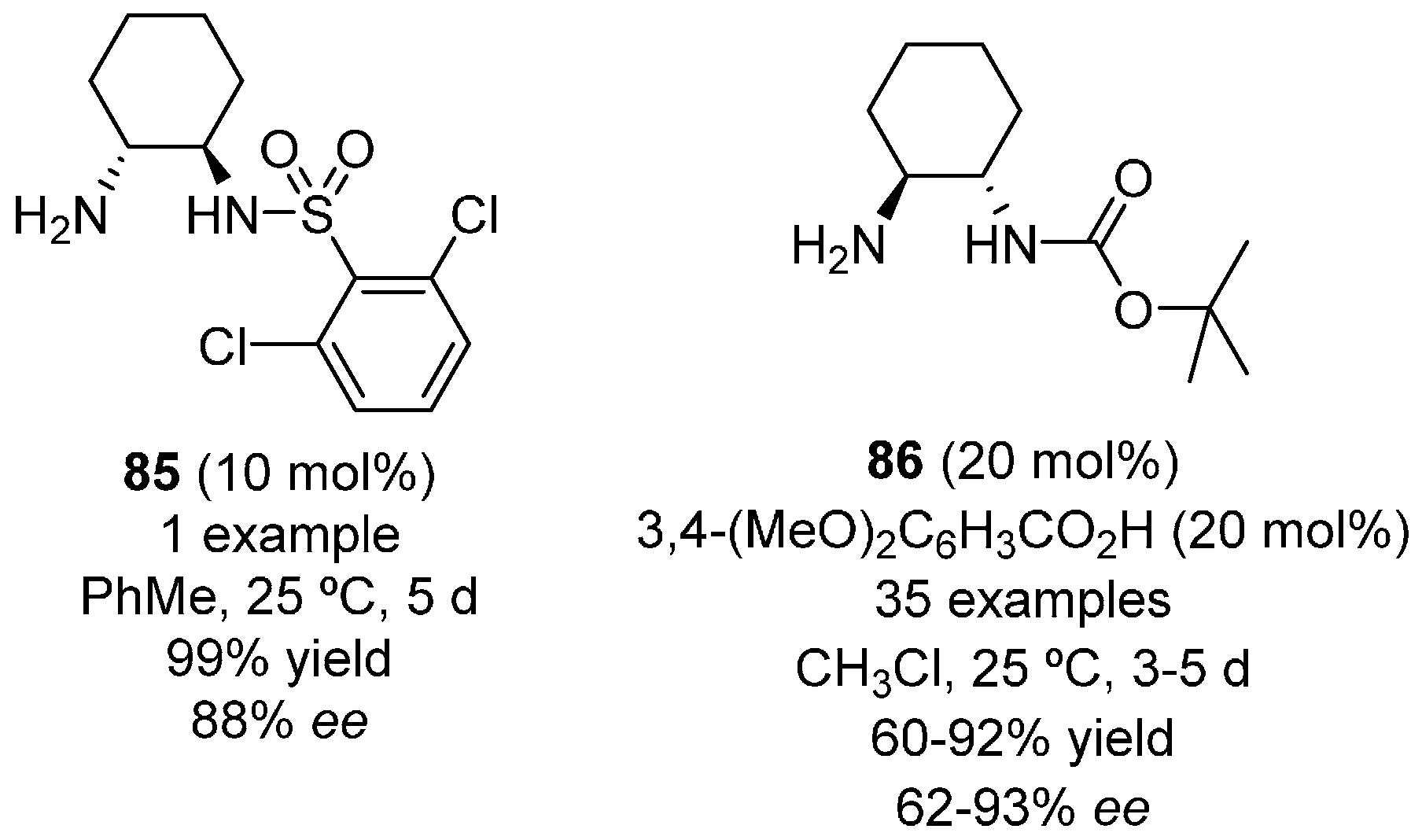
- Flores-Ferrandiz, J.; Stiven, A.; Sotorrios, L.; Gomez-Bengoa, E.; Chinchilla, R. Enantioselective addition of aryl ketones and acetone to nitroalkenes organocatalyzed by carbamate-monoprotected cyclohexa-1,2-diamines. Tetrahedron Asymmetry 2015, 26, 970–979. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Luo, W.; Lin, L.; Feng, X. Asymmetric Organocatalytic Michael/Michael/Henry Sequence to Construct Cyclohexanes with Six Vicinal Stereogenic Centers. Synlett 2017, 28, 966–969. [Google Scholar]
- Avila-Ortiz, C.G.; Lopez-Ortiz, M.; Vega-Penaloza, A.; Regla, I.; Juaristi, E. Use of (R)-mandelic acid as chiral co-catalyst in the Michael addition reaction organocatalyzed by (1S,4S)-2-Tosyl-2,5-diazabicyclo[2.2.1]heptane under solvent-free conditions. Asymmetric Catal. 2015, 2, 37–44. [Google Scholar] [CrossRef]
- Chen, L.-Y.; Guillarme, S.; Whiting, A.; Saluzzo, C. Asymmetric Michael addition of acetone to β-nitrostyrenes catalyzed by novel organocatalysts derived from d-isomannide or l-isoidide. ARKIVOC 2014, iv, 215–227. [Google Scholar]
- Nath, U.; Banerjee, A.; Ghosh, B.; Pan, S.C. Organocatalytic asymmetric Michael addition of 1-acetylcyclohexene and 1-acetylcyclopentene to nitroolefins. Org. Biomol. Chem. 2015, 13, 7076–7083. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Feng, X.; Yin, X.; Chen, Y.-C. Direct remote asymmetric bisvinylogous 1,4-additions of cyclic 2,5-dienones to nitroalkenes. Org. Lett. 2014, 16, 2370–2373. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-T.; Zhang, L.; You, S.-L. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev. 2016, 45, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-G.; Liu, X.-J.; Zhao, Q.-C.; Zheng, C.; Wang, S.-B.; You, S.-L. Asymmetric dearomatization of β-naphthols through a bifunctional-thiourea-catalyzed Michael reaction. Angew. Chem. Int. Ed. 2015, 54, 14929–14932. [Google Scholar] [CrossRef] [PubMed]
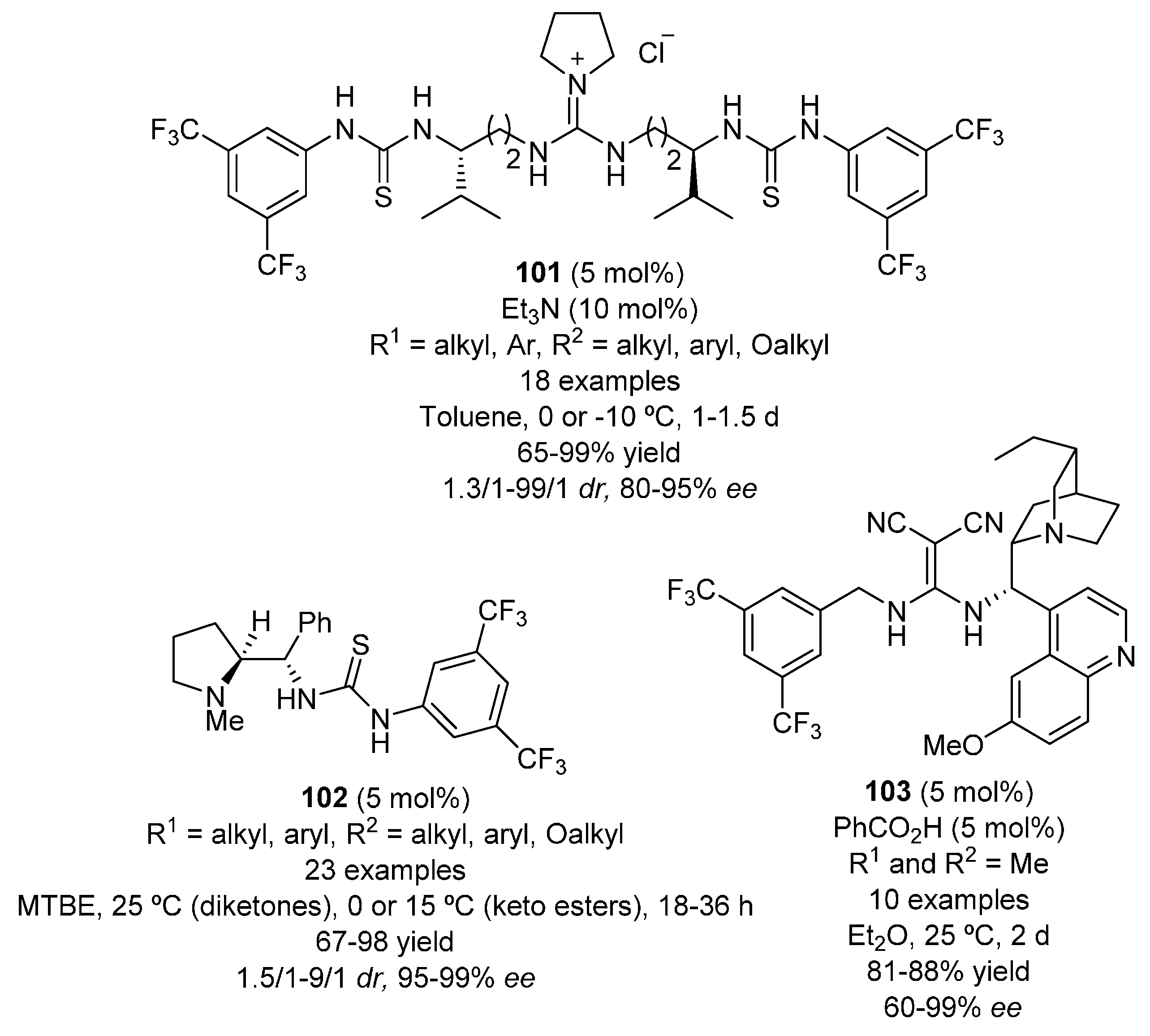
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Isik, M.; Tanyeli, C. Cu-catalyzed selective mono-N-pyridylation: Direct access to 2-aminoDMAP/sulfonamides as bifunctional organocatalysts. J. Org. Chem. 2013, 78, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Isik, M.; Unver, M.Y.; Tanyeli, C. Modularly evolved 2-aminoDMAP/squaramides as highly active bifunctional organocatalysts in Michael addition. J. Org. Chem. 2015, 80, 828–835. [Google Scholar] [CrossRef] [PubMed]
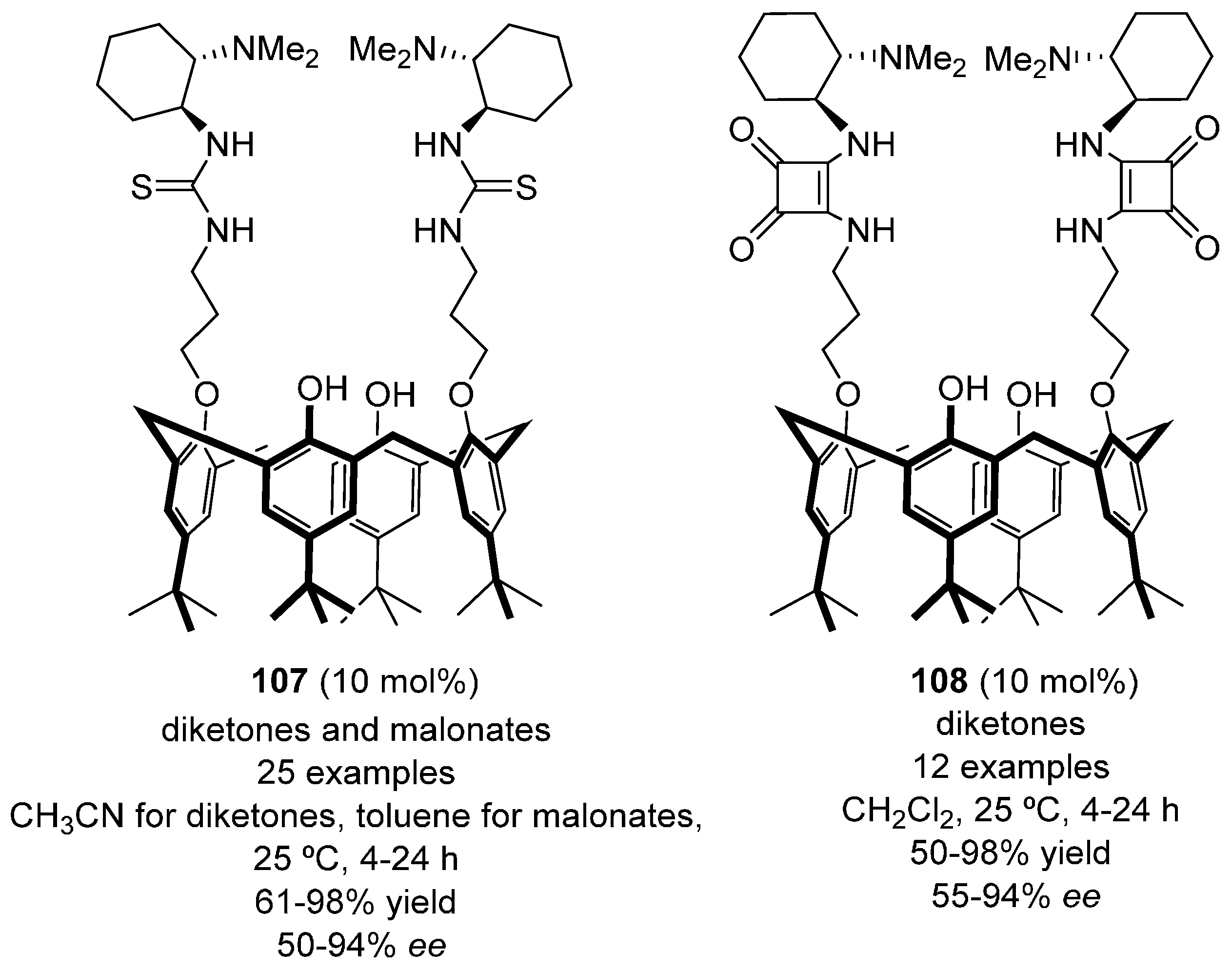
- Liu, B.; Han, X.; Dong, Z.; Lv, H.; Zhou, H.-B.; Dong, C. Highly enantioselective Michael addition of 1,3-dicarbonyl compounds to nitroalkenes catalyzed by designer chiral BINOl-quinine-squaramide: Efficient access to optically active nitro-alkanes and their isoxazole derivatives. Tetrahedron Asymmetry 2013, 24, 1276–1280. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Swain, M.; Reddy, S.M.; Yadav, J.S. Enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinone and 1,3-dicarbonyls to β-nitroalkenes catalyzed by a novel bifunctional rosin-indane amine thiourea catalyst. RSC Adv. 2013, 3, 8756–8765. [Google Scholar] [CrossRef]
- Horitsugi, N.; Kojima, K.; Yasui, K.; Sohtome, Y.; Nagasawa, K. Asymmetric Michael reaction of nitroolefins with β-dicarbonyl compounds catalysed by 1,3-diamine-tethered guanidine-thiourea bifunctional organocatalysts. Asian J. Org. Chem. 2014, 3, 445–448. [Google Scholar] [CrossRef]
- Vinayagam, P.; Vishwanath, M.; Kesavan, V. New class of bifunctional thioureas from l-proline: Highly enantioselective Michael addition of 1,3-dicarbonyls to nitroolefins. Tetrahedron Asymmetry 2014, 25, 568–577. [Google Scholar] [CrossRef]
- Hirashima, S.-i.; Nakashima, K.; Fujino, Y.; Arai, R.; Sakai, T.; Kawada, M.; Koseki, Y.; Murahashi, M.; Tada, N.; Itoh, A.; et al. Cinchona-diaminomethylenemalononitrile organocatalyst for asymmetric conjugate addition of 1,3-diketone to nitroalkene. Tetrahedron Lett. 2014, 55, 4619–4622. [Google Scholar] [CrossRef]
- Ashokkumar, V.; Siva, A. Cinchona alkaloid-based chiral catalysts act as highly efficient multifunctional organocatalysts for the asymmetric conjugate addition of malonates to nitroolefins. Org. Biomol. Chem. 2015, 13, 10216–10225. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, M.; Tataroglu, A.; Yilmaz, H.; Sirit, A. Calixarene-derived chiral tertiary amine-thiourea organocatalyzed asymmetric Michael additions of acetyl acetone and dimethyl malonate to nitroolefins. Tetrahedron Asymmetry 2016, 27, 148–156. [Google Scholar] [CrossRef]
- Vural, U.; Durmaz, M.; Sirit, A. A novel calix[4]arene-based bifunctional squaramide organocatalyst for enantioselective Michael addition of acetylacetone to nitroolefins. Org. Chem. Front. 2016, 3, 730–736. [Google Scholar] [CrossRef]
- Bae, H.Y.; Song, C.E. Unprecedented hydrophobic amplification in noncovalent organocatalysis "on water": Hydrophobic chiral squaramide catalyzed Michael addition of malonates to nitroalkenes. ACS Catal. 2015, 5, 3613–3619. [Google Scholar] [CrossRef]
- Sim, J.H.; Song, C.E. Water-enabled catalytic asymmetric Michael reactions of unreactive nitroalkenes: One-pot synthesis of chiral GABA-analogs with all-carbon quaternary stereogenic centers. Angew. Chem. Int. Ed. 2017, 56, 1835–1839. [Google Scholar] [CrossRef] [PubMed]
- Tukhvatshin, R.S.; Kucherenko, A.S.; Nelyubina, Y.V.; Zlotin, S.G. Tertiary amine-derived ionic liquid-supported squaramide as a recyclable organocatalyst for noncovalent "on water" catalysis. ACS Catal. 2017, 7, 2981–2989. [Google Scholar] [CrossRef]
- Du, H.; Rodriguez, J.; Bugaut, X.; Constantieux, T. Organocatalytic enantio- and diastereoselective conjugate addition to nitroolefins: When β-ketoamides surpass β-ketoesters. Chem. Eur. J. 2014, 20, 8458–8466. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, Y.; Fritz, S.P.; Wennemers, H. Organocatalytic stereoselective synthesis of acyclic γ-nitrothioesters with all-carbon quaternary stereogenic centers. J. Org. Chem. 2014, 79, 3937–3945. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Kim, S.T.; Hwang, G.-S.; Ryu, D.H. l-Proline derived bifunctional organocatalysts: Enantioselective Michael addition of dithiomalonates to trans-β-nitroolefins. J. Org. Chem. 2016, 81, 3263–3274. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Cho, S.M.; Hwang, G.-S.; Ryu, D.H. Construction of 3,4-dihydrocoumarin derivatives with adjacent quaternary and tertiary stereocenters: Organocatalytic asymmetric Michael addition of 2-oxochroman-3-carboxylate esters to trans-β-nitroolefins. Adv. Synth. Catal. 2017, 359, 163–167. [Google Scholar] [CrossRef]
- Kwiatkowski, J.; Lu, Y. Highly enantioselective Michael addition of 2-fluoro-1,3-diketones to nitroalkenes. Eur. J. Org. Chem. 2015, 320–324. [Google Scholar] [CrossRef]
- Cosimi, E.; Saadi, J.; Wennemers, H. Stereoselective synthesis of α-fluoro-γ-nitro thioesters under organocatalytic conditions. Org. Lett. 2016, 18, 6014–6017. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, J.; Lu, Y. Highly enantioselective preparation of fluorinated phosphonates by Michael addition of α-fluoro-β-ketophosphonates to nitroalkenes. Asian J. Org. Chem. 2014, 3, 458–461. [Google Scholar] [CrossRef]
- Jeong, H.J.; Kim, D.Y. Organocatalytic asymmetric Michael addition of α-fluoro β-ketophosphonate to nitroalkenes. Bull. Korean Chem. Soc. 2015, 36, 2936–2939. [Google Scholar] [CrossRef]
- Urbanietz, G.; Atodiresei, I.; Enders, D. Asymmetric synthesis of functionalized dihydro- and tetrahydropyrans via an organocatalytic domino Michael-hemiacetalization reaction. Synthesis 2014, 46, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Quintard, A.; Rodriguez, J. Organo- and iron(0) catalysis for an enantioselective Michael addition-hemiketalization-fragmentation sequence to protected ω-hydroxy-nitroketones. Adv. Synth. Catal. 2016, 358, 3362–3367. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Raabe, G.; Enders, D. Organocatalytic one-pot 1,4-/1,6-/1,2-addition sequence for the stereocontrolled formation of six consecutive stereocenters. Chem. Commun. 2015, 51, 2270–2272. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, J.; Lu, Y. Asymmetric Michael addition of α-fluoro-α-nitro esters to nitroolefins: Towards synthesis of α-fluoro-α-substituted amino acids. Org. Biomol. Chem. 2015, 13, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Kanberoglu, E.; Tanyeli, C. Enantioselective Michael addition of nitroalkanes to nitroalkenes catalyzed by chiral bifunctional quinine-based squaramides. Asian J. Org. Chem. 2016, 5, 114–119. [Google Scholar] [CrossRef]
- Trost, B.M.; Brennan, M.K. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis 2009, 3003–3025. [Google Scholar] [CrossRef]
- Saraswat, P.; Jeyabalan, G.; Hassan, M.Z.; Rahman, M.U.; Nyola, N.K. Review of synthesis and various biological activities of spiro heterocyclic compounds comprising oxindole and pyrrolidine moieties. Synth. Commun. 2016, 46, 1643–1664. [Google Scholar] [CrossRef]
- Kaur, M.; Singh, M.; Chadha, N.; Silakari, O. Oxindole: A chemical prism carrying plethora of therapeutic benefits. Eur. J. Med. Chem. 2016, 123, 858–894. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Chen, H.; Wold, E.A.; Shi, P.-Y.; Zhou, J. Therapeutic potential of spirooxindoles as antiviral agents. ACS Infect. Dis. 2016, 2, 382–392. [Google Scholar] [CrossRef] [PubMed]
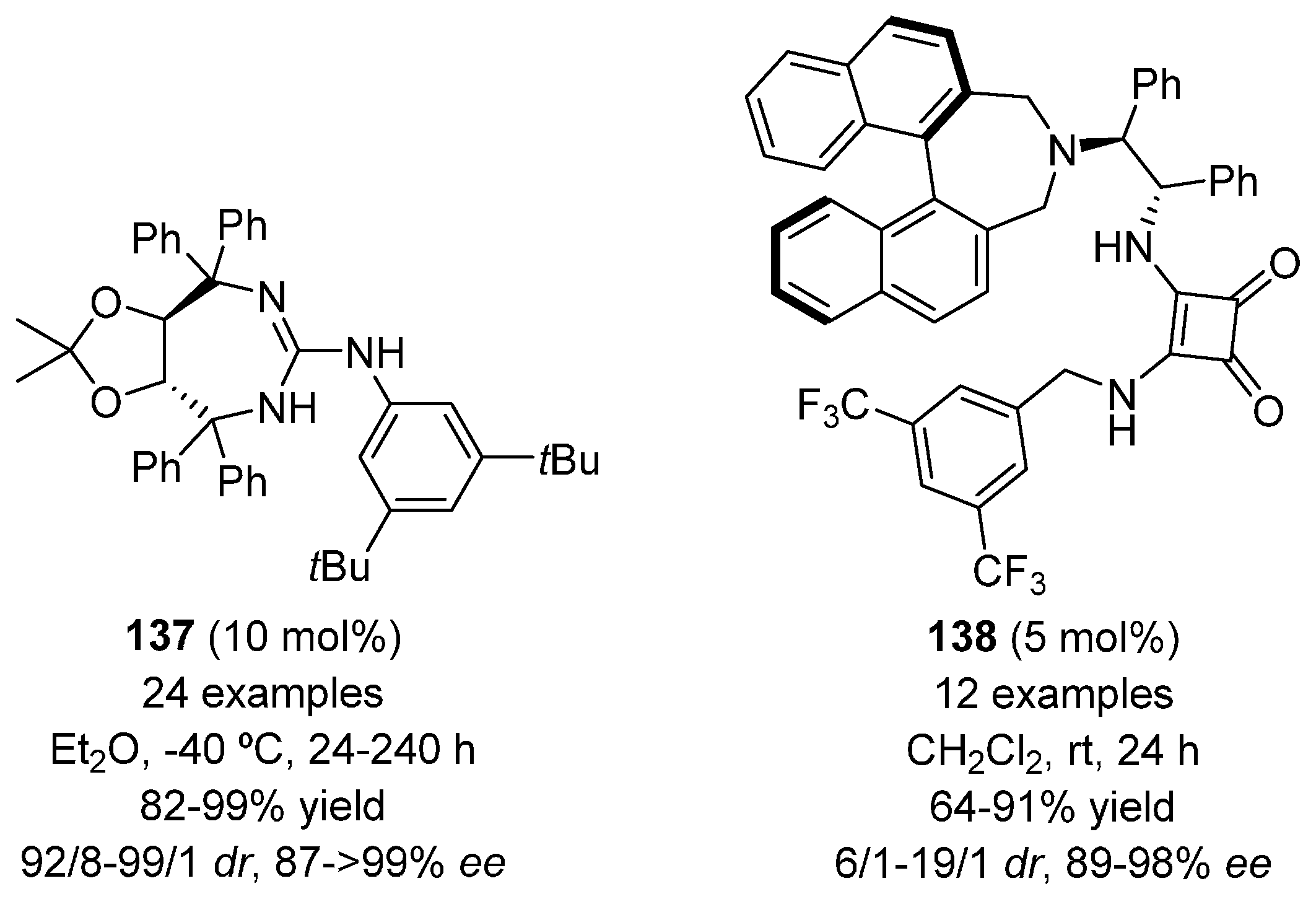
- Cui, B.-D.; Han, W.-Y.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Enantioselective synthesis of quaternary 3-aminooxindoles via organocatalytic asymmetric Michael addition of 3-monosubstituted 3-aminooxindoles to nitroolefins. J. Org. Chem. 2013, 78, 8833–8839. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.-D.; You, Y.; Zhao, J.-Q.; Zuo, J.; Wu, Z.-J.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. 3-Pyrrolyl-oxindoles as efficient nucleophiles for organocatalytic asymmetric synthesis of structurally diverse 3,3'-disubstituted oxindole derivatives. Chem. Commun. 2015, 51, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Bao, X.; Ma, Y.; Song, Y.; Qu, J.; Wang, B. Novel tartrate-derived guanidine-catalyzed highly enantio- and diastereoselective Michael addition of 3-substituted oxindoles to nitroolefins. Chem. Commun. 2014, 50, 5760–5762. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Kwon, S.J.; Kim, D.Y. Diastereo- and enantioselective conjugate addition of 3-chlorooxindoles to nitroalkenes catalyzed by binaphthyl-modified organocatalyst. Bull. Korean Chem. Soc. 2015, 36, 1516–1519. [Google Scholar] [CrossRef]
- Rassu, G.; Zambrano, V.; Pinna, L.; Curti, C.; Battistini, L.; Sartori, A.; Pelosi, G.; Zanardi, F.; Casiraghi, G. Direct regio-, diastereo-, and enantioselective vinylogous Michael addition of prochiral 3-alkylideneoxindoles to nitroolefins. Adv. Synth. Catal. 2013, 355, 1881–1886. [Google Scholar] [CrossRef]
- Rassu, G.; Zambrano, V.; Pinna, L.; Curti, C.; Battistini, L.; Sartori, A.; Pelosi, G.; Casiraghi, G.; Zanardi, F. Direct and enantioselective vinylogous Michael addition of α-alkylidenepyrazolinones to nitroolefins catalyzed by dual Cinchona alkaloid thioureas. Adv. Synth. Catal. 2014, 356, 2330–2336. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Enders, D. Asymmetric synthesis of pyrazoles and pyrazolones employing the reactivity of pyrazolin-5-one derivatives. Chem. Commun. 2015, 51, 12890–12907. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.P.; Ellman, J.A. Catalytic enantioselective addition of pyrazol-5-ones to trisubstituted nitroalkenes with an N-sulfinylurea organocatalyst. Adv. Synth. Catal. 2016, 358, 1713–1718. [Google Scholar] [CrossRef]
- Lai, X.; Zha, G.; Liu, W.; Xu, Y.; Sun, P.; Xia, T.; Shen, Y. Enantioselective Michael addition of pyrazolin-5-ones to β-CF3-β-disubstituted nitroalkenes catalyzed by squaramide organocatalyst. Synlett 2016, 27, 1983–1988. [Google Scholar] [CrossRef]
- Qiao, B.; An, Y.; Liu, Q.; Yang, W.; Liu, H.; Shen, J.; Yan, L.; Jiang, Z. Organocatalytic asymmetric Michael addition of 5H-oxazol-4-ones to nitroolefins. Org. Lett. 2013, 15, 2358–2361. [Google Scholar] [CrossRef] [PubMed]
- Diosdado, S.; Etxabe, J.; Izquierdo, J.; Landa, A.; Mielgo, A.; Olaizola, I.; Lopez, R.; Palomo, C. Catalytic enantioselective synthesis of tertiary thiols from 5H-thiazol-4-ones and nitroolefins: Bifunctional ureidopeptide-based Brönsted base catalysis. Angew. Chem. Int. Ed. 2013, 52, 11846–11851. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Zhao, X.; Liu, H.; Ye, X.; Li, Y.; Jiang, Z. Organocatalytic asymmetric conjugate addition of diaryloxazolidin-2,4-diones to nitroolefins: an efficient approach to chiral α-aryl-α-hydroxy carboxylic acids. Org. Chem. Front. 2016, 3, 470–474. [Google Scholar] [CrossRef]
- Jiao, L.; Bu, L.; Ye, X.; Zhao, X.; Jiang, Z. Catalytic asymmetric conjugate addition and sulfenylation of diarylthiazolidin-2,4-diones. J. Org. Chem. 2016, 81, 9620–9629. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ray, B.; Rathi, P.; Mukherjee, S. Catalytic asymmetric direct vinylogous Michael addition of γ-aryl-substituted deconjugated butenolides to nitroolefins and N-phenylmaleimide. Synthesis 2013, 45, 1641–1646. [Google Scholar] [CrossRef]
- Fang, X.; Dong, X.-Q.; Wang, C.-J. Highly efficient organocatalytic asymmetric Michael addition of homoserine lactone derived cyclic imino esters to nitroolefins. Tetrahedron Lett. 2014, 55, 5660–5662. [Google Scholar] [CrossRef]
- Han, X.; Chen, F.; Ye, C.; Wang, Y. Enantioselective conjugate addition of azlactones to nitroolefins with a thiourea-based bifunctional organocatalyst. Synlett 2017, 28, 989–993. [Google Scholar] [CrossRef]
- Luo, J.; Wang, H.; Zhong, F.; Kwiatkowski, J.; Xu, L.-W.; Lu, Y. Highly diastereoselective and enantioselective direct Michael addition of phthalide derivatives to nitroolefins. Chem. Commun. 2013, 49, 5775–5777. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qian, P.; Yang, J.; Chen, S.; Hu, Y.; Wu, P.; Wang, W.; Zhang, W.; Zhang, S. Organocatalytic enantioselective Michael addition of cyclic hemiacetals to nitroolefins: A facile access to chiral substituted 5- and 6-membered cyclic ethers. Org. Biomol. Chem. 2015, 13, 4769–4775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-P.; Dong, N.; Li, X.; Cheng, J.-P. Highly enantioselective Michael addition reactions of 2-substituted benzofuran-3(2H)-ones to nitroolefins. Org. Biomol. Chem. 2015, 13, 9943–9947. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, R.; Pahari, P.; Mal, D. Phthalides and phthalans: Synthetic methodologies and their applications in the total synthesis. Chem. Rev. 2014, 114, 6213–6284. [Google Scholar] [CrossRef] [PubMed]
- Vicario, J.L.; Badia, D.; Carrillo, L. Organocatalytic enantioselective Michael and hetero-Michael reactions. Synthesis 2007, 2065–2092. [Google Scholar] [CrossRef]
- Enders, D.; Wang, C.; Liebich, J.X. Organocatalytic Asymmetric Aza-Michael Additions. Chem. Eur. J. 2009, 15, 11058–11076. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, P.; Choy, P.Y.; Chan, A.S.C.; Kwong, F.Y. Advances and applications in organocatalytic asymmetric aza-michael addition. ChemCatChem 2012, 4, 917–925. [Google Scholar] [CrossRef]
- Sanchez-Rosello, M.; Acena, J.L.; Simon-Fuentes, A.; del Pozo, C. A general overview of the organocatalytic intramolecular aza-MICHAEL reaction. Chem. Soc. Rev. 2014, 43, 7430–7453. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Hajiabbasi, P. Recent advances in C-heteroatom bond forming by asymmetric Michael addition. Mol. Diversity 2014, 18, 411–439. [Google Scholar] [CrossRef] [PubMed]
- Alcaine, A.; Marques-Lopez, E.; Herrera, R.P. Synthesis of interesting β-nitrohydrazides through a thiourea organocatalyzed aza-Michael addition. RSC Adv. 2014, 4, 9856–9865. [Google Scholar] [CrossRef]
- Ma, S.; Wu, L.; Liu, M.; Huang, Y.; Wang, Y. Asymmetric aza-Michael additions of 4-nitrophthalimide to nitroalkenes and preliminary study of the products for herbicidal activities. Tetrahedron 2013, 69, 2613–2618. [Google Scholar] [CrossRef]
- Chen, S.-W.; Zhang, G.-C.; Lou, Q.-X.; Cui, W.; Zhang, S.-S.; Hu, W.-H.; Zhao, J.-L. Organocatalytic enantioselective aza-Michael reaction of benzotriazole to β,β-disubstituted nitroalkenes. ChemCatChem 2015, 7, 1935–1938. [Google Scholar] [CrossRef]
- Kriis, K.; Melnik, T.; Lips, K.; Juhanson, I.; Kaabel, S.; Jarving, I.; Kanger, T. Asymmetric synthesis of 2,3,4-trisubstituted piperidines. Synthesis 2017, 49, 604–614. [Google Scholar]
- Liu, F.-L.; Chen, J.-R.; Feng, B.; Hu, X.-Q.; Ye, L.-H.; Lu, L.-Q.; Xiao, W.-J. Enantioselective organocatalytic oxa-Michael addition of oximes to β-CF3-β-disubstituted nitroalkenes: Efficient synthesis of β-amino-α-trifluoromethyl alcohols. Org. Biomol. Chem. 2014, 12, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Biswas, A.; Molleti, N.; Singh, V.K. Enantioselective synthesis of highly substituted chromans via the oxa-Michael-Michael cascade reaction with a bifunctional organocatalyst. J. Org. Chem. 2015, 80, 11115–11122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Chen, Q.; Su, J.; Jia, F.; Qiu, S.; Ma, M.; Sun, Q.; Yan, W.; Wang, K.; Wang, R. Highly enantioselective cascade reaction catalyzed by squaramides: The synthesis of CF3-containing chromanes. Org. Lett. 2015, 17, 3826–3829. [Google Scholar] [CrossRef] [PubMed]
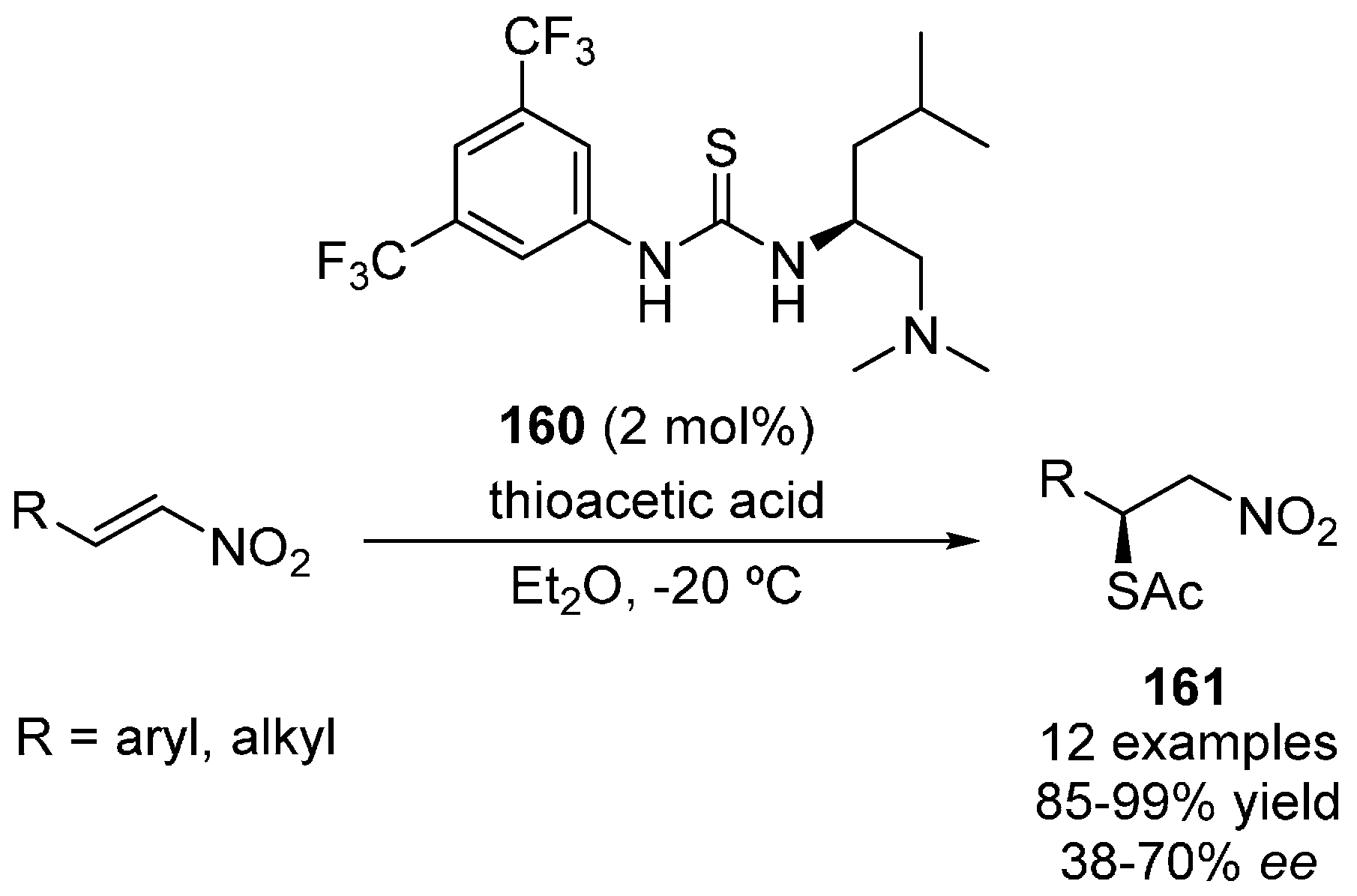
- Wang, R.; Xu, J. Enantioselective sulfur-Michael addition of thioacetic acid to nitroalkenes catalyzed by bifunctional amine-thiourea catalysts. Helv. Chim. Acta 2014, 97, 1700–1707. [Google Scholar] [CrossRef]
- Phelan, J.P.; Patel, E.J.; Ellman, J.A. Catalytic enantioselective addition of thioacids to trisubstituted nitroalkenes. Angew. Chem. Int. Ed. 2014, 53, 11329–11332. [Google Scholar] [CrossRef] [PubMed]
- Pei, Q.-L.; Han, W.-Y.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Organocatalytic diastereo- and enantioselective sulfa-Michael addition to α,β-disubstituted nitroalkenes. Tetrahedron 2013, 69, 5367–5373. [Google Scholar] [CrossRef]
- Yang, W.; Yang, Y.; Du, D.-M. Squaramide-tertiary amine catalyzed asymmetric cascade sulfa-Michael/Michael addition via dynamic kinetic resolution: Access to highly functionalized chromans with three contiguous stereocenters. Org. Lett. 2013, 15, 1190–1193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Du, D. Enantioselective construction of functionalized thiochromans via squaramide-catalyzed asymmetric cascade sulfa-Michael/Michael addition. Chin. J. Chem. 2014, 32, 853–858. [Google Scholar] [CrossRef]
- Ping, X.-N.; Wei, P.-S.; Zhu, X.-Q.; Xie, J.-W. Catalyst-controlled switch in diastereoselectivities: Enantioselective construction of functionalized 3,4-dihydro-2H-thiopyrano[2,3-b]quinolines with three contiguous stereocenters. J. Org. Chem. 2017, 82, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Kuchurov, I.V.; Nigmatov, A.G.; Kryuchkova, E.V.; Kostenko, A.A.; Kucherenko, A.S.; Zlotin, S.G. Stereodivergent Michael addition of diphenyl phosphite to α-nitroalkenes in the presence of squaramide-derived tertiary amines: An enantioselective organocatalytic reaction in supercritical carbon dioxide. Green Chem. 2014, 16, 1521–1526. [Google Scholar] [CrossRef]




































































© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso, D.A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Recent Advances in Asymmetric Organocatalyzed Conjugate Additions to Nitroalkenes. Molecules 2017, 22, 895. https://doi.org/10.3390/molecules22060895
Alonso DA, Baeza A, Chinchilla R, Gómez C, Guillena G, Pastor IM, Ramón DJ. Recent Advances in Asymmetric Organocatalyzed Conjugate Additions to Nitroalkenes. Molecules. 2017; 22(6):895. https://doi.org/10.3390/molecules22060895
Chicago/Turabian StyleAlonso, Diego A., Alejandro Baeza, Rafael Chinchilla, Cecilia Gómez, Gabriela Guillena, Isidro M. Pastor, and Diego J. Ramón. 2017. "Recent Advances in Asymmetric Organocatalyzed Conjugate Additions to Nitroalkenes" Molecules 22, no. 6: 895. https://doi.org/10.3390/molecules22060895