
Lipophilicity Studies on Thiosemicarbazide Derivatives
by
 , ,
, ,
Agata Paneth
1,* ,
,
Anna Hawrył
2,
Tomasz Plech
1,
Mirosław Hawrył
2, 
Ryszard Świeboda
2,
Dominika Janowska
1,
Monika Wujec
1 and
Piotr Paneth
3 1
Department of Organic Chemistry, Medical University, Chodźki 4a, 20-093 Lublin, Poland
2
Department of Inorganic Chemistry, Medical University, Chodźki 4a, 20-093 Lublin, Poland
3
Institute of Applied Radiation Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(6), 952; https://doi.org/10.3390/molecules22060952
Submission received: 8 April 2017
/
Accepted: 1 June 2017
/
Published: 8 June 2017
(This article belongs to the Section Medicinal Chemistry)
Abstract
:The lipophilicity of two series of thiosemicarbazide derivatives was assessed by the RP-HPLC method with the RP-18 chromatographic column and the methanol–water mixture as the mobile phase. Distribution coefficients logPHPLC were compared to calculated values generated by commonly used AClogP software and quantum chemical calculations. The reliability of the predictions was evaluated using the correlation matrix and PCA. For 4-benzoylthiosemicarbazides, a high correlation between theoretical and experimental logP parameters was obtained using the XlogP3 algorithm, while for 4-aryl/(cyclohexyl)thiosemicarbazides, the XlogP2 parameter was strongly correlated with the experimentally obtained logP.
1. Introduction
In drug discovery projects, the ability to show a relationship between compounds’ molecular structures and their pharmacokinetic parameters, in vivo efficacy, or toxicity is paramount for the design of better analogues. To aid this understanding, taking measurements of lipophilicity expressed by the logarithm of the octanol–water partition coefficient logP or the distribution coefficient logD (if ionized molecular species are present) is common practice, as these parameters are considered the most important for rational drug design and deriving a quantitative structure activity relationship (QSAR), quantitative structure retention relationship (QSRR) or a quantitative structure property relationship (QSPR) [1,2,3,4,5]. Numerous literature reports relate lipophilicity to undesirable ADMET (absorption, disposition, metabolism, excretion, and toxicity) properties, including poor solubility, poor bioavailability, high protein binding, high affinity to microsomes and hepatocytes, and in vivo toxicological effects [6,7,8,9,10,11,12]. It is therefore recommended that the lipophilicity of drug candidates be determined to eliminate molecules with unfavorable pharmacokinetic and pharmacodynamic parameters before bioassay is undertaken [13].
In this respect, there is a strong interest in developing computational methods for rapid lipophilicity screening of potential drug candidates. In recent years, numerous theoretical methods have been developed for the prediction of lipophilicity. The nature of lipophilicity is complex, however, as the outcome of inter- and intra-molecular interactions is far from being precisely encoded in the various algorithms, and the reliability of the available software depends on the chemical structure and the inherent conception of the method [14]. Consequently, for new chemotypes synthesized, computed logP values may be misleading for modeling their biological activity or for estimating their permeability potential [15]. Therefore, the routine application of a theoretical approach requires a comparison of the results with the data obtained using experimental methods, with particular regard to cases where intramolecular H-bonding, conformational flexibility, and/or tautomerization is possible [16].
Recently, as part of our efforts to discover new molecules that might be used to combat clinically significant infections, for the first time we have documented inhibitory properties of 1,4-disubstituted thiosemicarbazides towards bacterial DNA gyrase and topoisomerase IV [17,18,19]. Unfortunately, results of the bacterial type IIA topoisomerases inhibition study did not parallel the antibacterial activities. A possible explanation is that tested thiosemicarbazides may have varied in cell membrane permeability. We have, therefore, focused our attention on finding a computational tool that would be applicable to rapid and accurate prediction of lipophilicity for thiosemicarbazide-based compounds. In this contribution, we present results of these investigations, which allowed us to conclude that XlogP3, XlogP2, and SMD/B3LYP/def2-TZVP calculations are promising for theoretical prediction of lipophilicity for this class of compounds.
2. Results and Discussion
2.1. The Relationship between the Retention Parameter logk and the Concentration of Organic Modifier φ
The chromatographic lipophilicity parameters logkw for thiosemicarbazide derivatives 1–17 were obtained by the extrapolation of the retention parameter logk to pure water, according to Equation (1):
where logkw is the value of the retention factor of a substance in pure water, S is the slope of the regression curve, and φ is the concentration of the organic modifier.
logk = logkw − S × φ
Raw data which were used for the determination of the dependence of logk for different concentrations of the organic modifier are included as supplementary material (Table S1). Seven standards with known logP were chromatographed under the same conditions and respective data are given in Table S2.
2.2. The Calibration Equation logP vs. logkw
Subsequently, logPHPLC parameters for thiosemicarbazide derivatives 1–17 were determined, based on the correlation between the chromatographic lipophilicity index logkw and the octanol–water partition coefficient logPo–w obtained by the shake flask method for selected standards [21,22]:
logPo/w = a × logkw + b
Seven standards were analyzed in the same chromatographic conditions as for compounds 1–17. The logkw values of standards aniline (1.0186), benzene (2.2084), bromobenzene (3.0546), arametere (3.5502), toluene (2.8467), ethylbenzene (3.3754) and 2-hydroxyquinoline (1.3682) were obtained, based on the relationships between the retention parameters logk.
In order to calculate the lipophiliciy parameter logPo/w of thiosemicarbazide derivatives 1–17, the linear calibration curve was prepared:
logPo/w = 1.0239 (±0.03) × logkw − 0.1541 (0.08);
n = 7; r = 0.9976; se = 0.08; F = 1047.8
n = 7; r = 0.9976; se = 0.08; F = 1047.8
It should be noted that this calibration curve is based on compounds selected from the OECD approved standards, all of which are structurally different from the compounds studied herein. This approach has been used earlier [23,24].
The logk values of 1–17 were substituted into Equation (3) to obtain logPHPLC parameters. The values of calculated and experimental lipophilicity parameters are presented in Table 2.
2.3. Theoretical Calculation clogP
In the next step, the comparison between the experimental logPHPLC and the calculated values logP was carried out. For analysis, thiosemicarbazide derivatives 1–17 were divided into two groups; the first one (group A) included 4-benzoylothiosemicarbazides 1–10, and the second one (group B) included 4-aryl and 4-cyclohexylthiosemicarbazides 11–17. Based on the correlation matrices (Table 3 and Table 4) and PCA (Figure 1), it was found that for group A, the highest correlation was obtained using the XlogP3 program, while for group B the highest correlation was obtained using the XlogP2 program.
2.4. Correlation of Lipophilicity with Inhibitory Potency towards Bacterial Type IIA Topoisomerases
Lipophilicity is usually related to biological activity; the evidence for this is clearly explained in the frame of the lipid theory of Meyer and Overton [25], according to which logP is not only a function of the penetration and distribution of the drug, but also a function of its interaction with the molecular target. Therefore, the second part of our studies was dedicated to determining the role of lipophilicity on the inhibitory action of 1-hetaroyl-4-substituted-thiosemicarbazides against bacterial type IIA topoisomerases. Lead compounds from this series were reported to be potent and non-toxic inhibitors [17,18,19] (Table 5) and can be considered as a starting point for the development of improved antibacterial agents. Although a correlation of their inhibitory potency with hydrophobic/hydrophilic balance was suggested to exist, no such trend could be deduced from the comparison of their calculated logP values. A possible explanation for the lack of such correlation could be the use of unreliable research tools used for lipophilicity prediction. We therefore repeated logP calculations using the XlogP3 and XlogP2 programs, as these were recognized to be reliable tools for the prediction of lipophilicity for this class of compounds.
A set of seventeen 1-hetaroyl-4-substituted-thiosemicarbazides 1, 2, 18–32, previously tested as S. aureus DNA gyrase and topoisomerase IV (topo IV) inhibitors, was used for model generation. The analysis of calculated logP values for the compounds, however, leads to ambiguous conclusions. According to results collected in Table 5, within series of 4-benzoylthiosemicarbazides 1, 2, 18–20, the best inhibitory potency against DNA gyrase was found for 1 (IC50 14.59 μM), with XlogP3 = 2.59. The replacement of furan moiety in 1 with indole 18 or imidazole 20 leads to inactive compounds with a lipophilicity respectively higher (XlogP3 of 3.59) or lower (XlogP3 of 2.00) than that of 1, whereas, for instance, XlogP3 for inhibitors with pyrrole 2 and thiophene 19 substitution lie at 2.25 and 3.14, respectively. Again, 18 (XlogP3 of 3.59), 2 (XlogP3 of 2.25) and 20 (XlogP3 of 2.00) were found to have anti-topo IV activity, while other compounds of similar lipophilicity, 1 (XlogP3 of 2.59) and 19 (XlogP3 of 3.14), were inactive. No valid correlation between inhibitory activity of 4-arylthiosemicarbazides 21–32 against topo IV and their logP values expressed as XlogP2 was observed. For instance, within series 21–32, the best anti-topo IV activity was found for 21 (IC50 14 μM, XlogP2 of 2.82). The replacement of the 4-nitrophenyl group in 21 by o-fluoro-, p-fluorophenyl or 2,4-difluorophenyl groups, as in 22 (XlogP2 of 3.09), 23 (XlogP2 of 3.09) and 24 (XlogP2 of 3.25), leads to compounds with lipophilicity similar to 21 that, however, show only weak inhibitory potency. To conclude, although the most lipophilic of 4-benzoylthiosemicarbazides (18) inhibited topo IV most effectively, no linear relationship between the inhibitory potency of series 4-benzoylthiosemicarbazides against DNA gyrase and their lipophilicity was observed. No relationship between the inhibitory potency of series 4-arylthiosemicarbazides against bacterial topoisomerases and their lipophilicity was found. Virtually the same conclusion was reached by Tanitame et al. [26] in studies on the inhibitory activity of 5-vinylpyrazoles against bacterial DNA gyrase. The idea behind their concept was to design more potent 5-vinylpyrazoles by decreasing the lipophilicity of the parent compounds, while keeping the van der Waals interaction with the lipophilic area around Ile94 of DNA gyrase. Among the six analogs obtained, two compounds showed less potent inhibitory activity, while the bioactivity of others was similar or only modestly better. Finally, no linear relationship between the inhibitory potency of 5-vinylpyrazoles against DNA gyrase and their lipophilicity could be deduced.
For comparison, we have tested quantum-chemical calculations of logP. From a thermodynamic point of view, the equilibrium constant K (at a given temperature and standard concentrations) is given by Equation:
where G is Gibbs free energy, R is the universal gas constant and T is the absolute temperature.
∆G = −RTlnK
It thus should be possible to evaluate logKo–w values by computing Gibbs free energies of a given molecule in aqueous and 1-octanol solutions. Expanding recent reports [27], we have tested three continuum solvent models (IEFPCM, CPCM, SMD) with the DFT B3LYP functional expressed in several basis sets (6-31+G(p,d), 6-311++G(d,p), aug-cc-pVDZ, def2-DZVP, def2-TZVP). Initially, RM1 optimized structures were reoptimized in the gas phase at the given theory level, and three types of calculations were then performed. Firstly, energies for the gas phase structures with inclusion of the solvent model were calculated. Secondly, reoptimization with the solvent model included was performed for both liquid phases. Finally, frequency calculations for the structures optimized with solvent models were carried out in order to calculate zero point energies (ZPEs) and thermal corrections to Gibbs free energies. We have found double-zeta basis sets to be inadequate, as they produced inverse solution stabilities compared with the experimental results. Among tested methods, only SMD/B3LYP/def-TZVP turned out to be promising in terms of speed of calculations and agreement of the results with the experimental data, with equation logP = 1.46·logKo–w + 1.21 (Figure 2) describing the correlation between the theoretical (Ko–w is the calculated equilibrium constant) and experimental results. However, slight deviation from linearity, the slope differing from unity, and the intercept differing from zero indicate that calibration is necessary for each class of studied compounds.
3. Materials and Methods
3.1. Chromatographic Analysis
The chromatographic analysis was performed using a liquid chromatograph equipped with an Elite LaChrom L-2130 gradient pump (Hitachi-Merck, Darmstadt, Germany), SPD-10AVP UVVIS detector (Shimadzu, Kyoto, Japan), and Rheodyne 7725i valve with a 20 μL loop. Methanolic solutions (5 μL, 0.1%) of selected samples were applied to the chromatographic column (RP-18 Waters Symmetry, 15 cm length, 4.6 mm i.d., 5 μm particle size) by use of an autosampler Hitachi L-2200 (LaChrom Elite, Hitachi-Merck, Darmstadt, Germany). The mobile phase, consisting of a methanol and water mixture, was degassed by use of the built-in membrane degasser. The analysis was performed with a flow rate of 1.0 mL/min in isocratic mode using various concentrations of organic modifier in binary polar mobile phases; percentages of methanol in water were 45–75% (v/v %), and changed by 5% per step. Chromatograms were detected at 254 nm and the temperature of the column was 25 °C. All experiments were repeated in triplicate and the final results were taken to be the arithmetic means. Dead time was measured by use of uracil (Calbiochem. Merck, Darmstadt, Germany). All experiments were performed at ambient temperature.
3.2. Standard Solutes
According to OECD guidelines [28], seven standards with known logPo/w values—aniline (0.9), benzene (2.1), bromobenzene (3.0), naphtalene (3.6), toluene (2.7), ethylbenzene (3.2) and 2-hydroxyquinoline (1.26)—were selected to create the correlation between the known logPo/w and the experimental chromatographic lipophilicity parameter logkW. The obtained calibration curve was used to calculate the logPo/w of compounds 1–17 under the same chromatographic conditions.
3.3. Statistical Analysis
All regression analyses were performed using statistical software (Statistica version 9.0 for Windows).
3.4. LogP Calculations
The theoretical partition coefficient values were calculated using the available ALOGPS 2.1 software program [29,30]. The calculations were based on the analysis of algorithm topology of the whole molecules of studied compounds (AClogP, AlogPs and MLOGP) and the analysis of individual atoms (ALOGP, XlogP2 and XlogP3).
3.5. Quantum-Chemical Calculations
Initial models were prepared and optimized by RM1 semiempirical parametrization [31] using Hyperchem (version 8.0.3, HyperCube Inc., Gainsville, FL, USA). They were subsequently reoptimized in the gas phase, IEFPCM [32], CPCM [33], and SMD [34] continuum models of the aqueous solution and 1-octanol at the DFT level using the B3LYP functional [35,36]. The performance of four basis sets (6-31+G(p,d), 6-311++G(d,p) [37,38,39], aug-cc-pVDZ [40,41,42,43], def2-DZVP [44]) have been tested. Finally, Gibbs free energies of the studied compounds were obtained from vibrational analysis. All these calculations were performed using a Gaussian computational package [45].
4. Conclusions
Distribution coefficients logP for two series of thiosemicarbazide derivatives were experimentally determined by the RP-HPLC method and correlated with those obtained using AClogP software and quantum-chemical calculations. For 4-benzoylthiosemicarbazides, the best results were achieved using the XlogP3 algorithm, while for derivatives of compounds 4-aryl/(cyclohexyl)thiosemicarbazides, XlogP2 parameters were strongly correlated with experimentally obtained logP. Among tested theory levels, only SMD/B3LYP/def2-TZVP results seem to be useful in theoretical prediction of lipophilicity for this class of compounds, although calibration is necessary.
Supplementary Materials
Supplementary materials are available online. Table S1: The values of the retention time (tr) and logk for different concentrations of methanol in water (ranging from 45% to 75%) obtained on RP-18 column; Table S2: Statistics and parameters of Equation (1) obtained for standards with known logP.
Acknowledgments
The project was supported by the grant number 2012/05/D/NZ7/02278 from the National Science Centre.
Author Contributions
A.P. and P.P. conceived and designed the experiments and wrote the paper; A.H., M.H. and R.Ś. performed the experiments and wrote the paper; T.P., M.W. and D.J. analyzed the data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hansch, C.; Leo, A. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Testa, B.; Crivori, P.; Reist, M.; Carrupt, P.-A. The influence of lipophilicity on the pharmacokinetic behavior of drugs: Concepts and examples. Perspect. Drug Discov. Des. 2000, 17, 179–211. [Google Scholar] [CrossRef]
- Lewis, M.L.; Cucurull-Sanchez, L. Structural pairwise comparisons of HLM stability of phenyl derivatives: Introduction of the Pfizer metabolism index (PMI) and metabolism-lipophilicity efficiency (MLE). J. Comput. Aided Mol. Des. 2009, 23, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Shamovsky, I.; Connolly, S.; David, L.; Ivanova, S.; Norden, B.; Springthorpe, B.; Urbahns, K. Overcoming undesirable hERG potency of chemokine receptor antagonists using baseline lipophilicity relationships. J. Med. Chem. 2008, 51, 1162–1178. [Google Scholar] [CrossRef] [PubMed]
- Briciu, R.D.; Kot-Wasik, A.; Wasik, A.; Namieśnik, J.; Sârbu, C. The lipophilicity of artificial and natural sweeteners estimated by reversed-phase thin-layer chromatography and computed by various methods. J. Chromatogr. A 2010, 1217, 3702–3706. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Yalkowsky, S.H. Estimation of the aqueous solubility I: Application to organic nonelectrolytes. J. Pharm. Sci. 2001, 90, 234–252. [Google Scholar] [CrossRef]
- Yoshida, F.; Topliss, J.G. QSAR model for drug human oral bioavailability. J. Med. Chem. 2000, 43, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Kanaoka, M. Computational prediction of the plasma protein-binding percent of diverse pharmaceutical compounds. J. Pharm. Sci. 2004, 93, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.P.; Barton, P.; Cockroft, S.L.; Wenlock, M.C.; Riley, R.J. The influence of nonspecific microsomal binding on apparent intrinsic clearance and its prediction from physicochemical properties. Drug Metab. Dispos. 2002, 30, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.P.; Barton, P.; Mohmed, S.; Riley, R.J. The binding of drugs to hepatocytes and its relationship to physicochemical properties. Drug Metab. Dispos. 2005, 33, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Alelyunasa, Y.W.; Pelosi-Kilbya, L.; Turcotteb, P.; Karyb, M.-B.; Spreena, R.C. A high throughput dried DMSO LogD lipophilicity measurement based on 96-well shake-flask and atmospheric pressure photoionization mass spectrometry detection. J. Chromatogr. A 2010, 1217, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Hollósy, F.; Seprödi, J.; Örfi, L.; Erös, D.; Kéri, G.; Idei, M. Evaluation of lipophilicity and antitumour activity of parallel carboxamide libraries. J. Chromatogr. B 2002, 780, 355–363. [Google Scholar] [CrossRef]
- Testa, B.; Carrupt, P.-A.; Gaillard, P.; Tsai, R.-S. Lipophilicity in Drug Action and Toxicology; Pliska, V., Testa, B., van de Waterbeemd, H., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA (VCH): Weinheim, Germany, 1996; pp. 49–71. [Google Scholar]
- Tetko, I.V.; Poda, G.I.; Ostermann, C.; Mannhold, R. Accurate in silico logP predictions: One can’t embrace the unembraceable. QSAR Comb. Sci. 2009, 28, 845–849. [Google Scholar] [CrossRef]
- Lombardo, F.; Shalaeva, M.Y.; Tupper, K.A.; Gao, F.; Abraham, M.H. ElogPoct: A tool for lipophilicity determination in drug discovery. J. Med. Chem. 2000, 43, 2922–2928. [Google Scholar] [CrossRef] [PubMed]
- Paneth, A.; Stączek, P.; Plech, T.; Strzelczyk, A.; Dzitko, K.; Wujec, M.; Kuśmierz, E.; Kosikowska, U.; Grzegorczyk, A.; Paneth, P. Biological evaluation and molecular modelling study of thiosemicarbazide derivatives as bacterial type IIA topoisomerases inhibitors. J. Enzyme Inhib. Med. Chem. 2016, 31, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Siwek, A.; Stą̨czek, P.; Wujec, M.; Stefańska, J.; Kosikowska, U.; Malm, A.; Jankowski, S.; Paneth, P. Biological and docking studies of topoisomerase IV inhibition by thiosemicarbazides. J. Mol. Model. 2011, 17, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Siwek, A.; Stą̨czek, P.; Stefańska, J. Synthesis and structure-activity relationship studies of 4-arylthiosemicarbazides as topoisomerase IV inhibitors with Gram-positive antibacterial activity. Search for molecular basis of antibacterial activity of thiosemicarbazides. Eur. J. Med. Chem. 2011, 46, 5717–5726. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, H.H. A reexamination of the Hammet equation. Chem. Rev. 1953, 53, 191–261. [Google Scholar] [CrossRef]
- Leśniewska, M.A.; Gdaniec, Z.; Musialska, I. Calculation procedures and HPLC method for analysis of the lipophilicity of acyclovir esters. Drug Dev. Ind. Pharm. 2014, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rabtti, E.H.M.A.; Natic, M.M.; Milojkovic-Opsenica, D.M.; Trifkovic, J.D.; Vuckovic, I.M.; Vajs, V.E.; Tešic, Ž.L. RP TLC based lipophilicity assessment of some natural and synthetic coumarins. J. Braz. Chem. Soc. 2012, 23, 522–530. [Google Scholar] [CrossRef]
- Hawrył, A.M.; Popiołek, Ł.P.; Hawrył, M.A.; Świeboda, R.S.; Niejedli, M.A. Chromatographic and calculation methods for analysis of the lipophilicity of newly synthesized thiosemicarbazides and their cyclic analogues 1,2,4-triazol-3-thiones. J. Braz. Chem. Soc. 2015, 26, 1617–1624. [Google Scholar] [CrossRef]
- Pachuta-Stec, A.; Hawrył, A.M.; Wróbel, A.; Hawrył, M.A.; Pitucha, M. Chromatographic evaluation of the lipophilic properties of some 1,2,4-triazole with potential antitumour activity. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 1199–1206. [Google Scholar] [CrossRef]
- Meyer, H. Zur theorie der alkoholnarkose, I. Mit welch eigenshaft der anasthetika bedingt ihre narkotische Wirkung? Arch. Exp. Path. Pharmakol. (Naunyn Schmiedebergs) 1899, 42, 109–137. [Google Scholar] [CrossRef]
- Tanitame, A.; Oyamada, Y.; Ofuji, K.; Suzuki, K.; Ito, H.; Kawasaki, M.; Wachi, M.; Yamagishi, J.-I. Potent DNA gyrase inhibitors; novel 5-vinylpyrazole analogues with Gram-positive antibacterial activity. Bioorg. Med. Chem. Lett. 2004, 14, 2863–2866. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-S.; Lin, S.-T. Prediction of pH Effect on the octanol−water partition coefficient of ionizable pharmaceuticals. Ind. Eng. Chem. Res. 2016, 55, 9284–9294. [Google Scholar] [CrossRef]
- Organisation for Economic Co-operation and Development (OECD). Test No. 117: Partition Coefficient (n-Octanol/Water), HPLC method. In OECD Guidelines for the Testing of Chemicals; Section 1; OECD Publishing: Paris, French, 2004. [Google Scholar]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aid. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Virtual Computational Chemistry Labolatory. Available online: http://www.vcclab.org (accessed on 27 May 2017).
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401–146404. [Google Scholar] [CrossRef] [PubMed]
- Baronel, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti conelation energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistentmolecular orbital methods: 9. Extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods. 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; Defrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. 23. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Lynch, B.J.; Fast, P.L.; Harris, M.; Truhlar, D.G. Adiabatic connection for kinetics. J. Phys. Chem. A 2000, 104, 4811–4815. [Google Scholar] [CrossRef]
- Fast, P.L.; Sanchez, M.L.; Truhlar, D.G. Multi-coefficient Gaussian-3 method for calculating potential energy surfaces. Chem. Phys. Lett. 1999, 306, 407–410. [Google Scholar] [CrossRef]
- Môller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Gonzalez-Lafont, A.; Truong, T.N.; Truhlar, D.G. Direct dynamics calculations with neglect of diatomic differential overlap molecular orbital theory with specific reaction parameters. J. Phys. Chem. 1991, 95, 4618–4627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence,triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Not Available. |
Figure 1.
The PCA graphs for calculated and experimental lipophilicity parameters of 4-benzoylthiosemicarbazides (group A) and 4-aryl/(cyclohexyl)thiosemicarbazides (group B).
Figure 1.
The PCA graphs for calculated and experimental lipophilicity parameters of 4-benzoylthiosemicarbazides (group A) and 4-aryl/(cyclohexyl)thiosemicarbazides (group B).

Figure 2.
Correlation of experimental logP with theoretical results obtained at the SMD/B3LYP/def2-TZVP level.
Figure 2.
Correlation of experimental logP with theoretical results obtained at the SMD/B3LYP/def2-TZVP level.


{kind=link}
{kind=link}
Table 1.
Parameters of Equation (1). Obtained F values were higher than F-critical in each case.
| Compound | logkw | −S | r | n | F | SD of Estimation | |
|---|---|---|---|---|---|---|---|
| 1 | 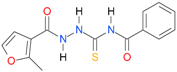 | 2.9992 | 4.3720 | 0.9986 | 7 | 1796.1 | 0.027 |
| 2 | 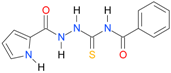 | 2.5105 | 4.1416 | 0.9983 | 7 | 1487.0 | 0.028 |
| 3 |  | 4.2230 | 6.1166 | 0.9983 | 7 | 1440.4 | 0.043 |
| 4 | 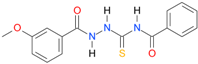 | 3.3130 | 4.8205 | 0.9984 | 7 | 1598.1 | 0.032 |
| 5 | 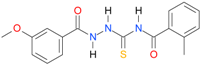 | 3.4487 | 4.8562 | 0.9987 | 7 | 1864.2 | 0.030 |
| 6 |  | 3.7864 | 5.1137 | 0.9976 | 7 | 1049.6 | 0.042 |
| 7 | 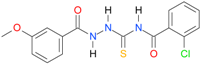 | 3.8283 | 5.6579 | 0.9934 | 7 | 372.8 | 0.078 |
| 8 | 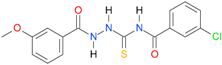 | 3.7422 | 5.0742 | 0.9972 | 7 | 901.4 | 0.045 |
| 9 | 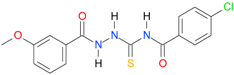 | 3.9422 | 5.3417 | 0.9974 | 7 | 975.2 | 0.045 |
| 10 |  | 3.2498 | 5.7291 | 0.9838 | 7 | 150.3 | 0.124 |
| 11 | 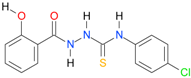 | 3.3324 | 4.8778 | 0.9983 | 7 | 2138.6 | 0.028 |
| 12 | 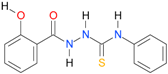 | 2.4813 | 4.1962 | 0.9992 | 7 | 3260.5 | 0.019 |
| 13 | 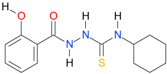 | 3.2386 | 4.7076 | 0.9987 | 7 | 1871.6 | 0.029 |
| 14 | 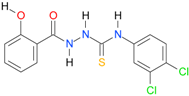 | 4.1453 | 5.6538 | 0.9980 | 7 | 1251.7 | 0.042 |
| 15 |  | 3.9451 | 5.5432 | 0.9964 | 7 | 690.2 | 0.056 |
| 16 | 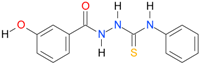 | 2.0687 | 4.1968 | 0.9993 | 7 | 3508.6 | 0.019 |
| 17 |  | 2.7583 | 4.3297 | 0.9993 | 7 | 3507.0 | 0.019 |
Table 2.
The values of calculated and experimental lipophilicity parameters of thiosemicarbazides 1–17.
Table 2.
The values of calculated and experimental lipophilicity parameters of thiosemicarbazides 1–17.
| Compound | AlogPs | AclogP | milogP | AlogP | MlogP | XlogP2 | XlogP3 | logkw | logPHPLC |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.90 | 1.70 | 1.28 | 1.80 | 1.33 | 1.62 | 2.59 | 2.9992 | 3.0469 |
| 2 | 1.66 | 1.06 | 1.27 | 1.94 | 1.06 | 1.59 | 2.25 | 2.5105 | 2.5254 |
| 3 | 2.86 | 2.59 | 2.23 | 2.95 | 3.20 | 3.43 | 3.95 | 4.2300 | 4.3606 |
| 4 | 2.36 | 2.17 | 2.15 | 2.53 | 2.42 | 2.70 | 3.10 | 3.3130 | 3.3819 |
| 5 | 2.41 | 2.49 | 2.55 | 3.02 | 2.66 | 3.14 | 3.46 | 3.4487 | 3.5267 |
| 6 | 2.41 | 2.49 | 2.57 | 3.02 | 2.66 | 3.14 | 3.46 | 3.7864 | 3.8871 |
| 7 | 3.00 | 2.79 | 2.78 | 3.20 | 2.93 | 3.32 | 3.73 | 3.8283 | 3.9318 |
| 8 | 2.93 | 2.79 | 2.80 | 3.20 | 2.93 | 3.32 | 3.73 | 3.7422 | 3.8399 |
| 9 | 2.88 | 2.79 | 2.83 | 3.20 | 2.93 | 3.32 | 3.73 | 3.9422 | 4.0534 |
| 10 | 2.62 | 1.59 | 1.53 | 2.81 | 2.57 | 2.43 | 3.06 | 3.2498 | 3.3144 |
| 11 | 2.82 | 2.66 | 3.31 | 3.08 | 3.39 | 3.41 | 3.52 | 3.3324 | 3.4026 |
| 12 | 1.95 | 2.05 | 2.63 | 2.41 | 2.86 | 2.78 | 3.21 | 2.4813 | 2.4942 |
| 13 | 1.72 | 2.05 | 3.48 | 2.69 | 2.44 | 3.05 | 3.14 | 3.2386 | 3.3025 |
| 14 | 3.32 | 3.27 | 3.91 | 3.74 | 3.64 | 4.03 | 4.15 | 4.1453 | 4.2702 |
| 15 | 3.02 | 2.97 | 3.69 | 3.56 | 3.64 | 3.63 | 3.88 | 3.9451 | 4.0565 |
| 16 | 1.99 | 2.05 | 1.64 | 2.41 | 2.35 | 2.36 | 2.34 | 2.0687 | 2.0538 |
| 17 | 1.74 | 2.05 | 2.48 | 2.69 | 1.93 | 2.62 | 2.59 | 2.7583 | 2.7898 |
Table 3.
The correlation matrix for logPcalc vs. logPHPLC (group A: 4-benzoylthiosemicarbazides).
| AlogPs | AclogP | milogP | AlogP | MlogP | XlogP2 | XlogP3 | logkw | logPHPLC | |
|---|---|---|---|---|---|---|---|---|---|
| AlogPs | 1.0000 | 0.7241 | 0.7833 | 0.9335 | 0.9132 | 0.8906 | 0.8518 | 0.7676 | 0.7676 |
| AclogP | 1.0000 | 0.9500 | 0.8541 | 0.8641 | 0.9348 | 0.9403 | 0.9052 | 0.9052 | |
| milogP | 1.0000 | 0.8993 | 0.8226 | 0.9282 | 0.8755 | 0.8013 | 0.8013 | ||
| AlogP | 1.0000 | 0.9425 | 0.9529 | 0.9177 | 0.8361 | 0.8361 | |||
| MlogP | 1.0000 | 0.9632 | 0.9667 | 0.9251 | 0.9251 | ||||
| XlogP2 | 1.0000 | 0.9776 | 0.9265 | 0.9265 | |||||
| XlogP3 | 1.0000 | 0.9716 | 0.9716 | ||||||
| logkw | 1.0000 | 1.0000 | |||||||
| logPHPLC | 1.0000 | 1.0000 |
Table 4.
The correlation matrix for logPcalc vs. logPHPLC (group B: 4-aryl/(cyclohexyl)thiosemicarbazides).
Table 4.
The correlation matrix for logPcalc vs. logPHPLC (group B: 4-aryl/(cyclohexyl)thiosemicarbazides).
| AlogPs | AclogP | milogP | AlogP | MlogP | XlogP2 | XlogP3 | logkw | logPHPLC | |
|---|---|---|---|---|---|---|---|---|---|
| AlogPs | 1.0000 | 0.9809 | 0.6650 | 0.9168 | 0.9309 | 0.8909 | 0.8564 | 0.8014 | 0.8014 |
| AclogP | 1.0000 | 0.7475 | 0.9725 | 0.8927 | 0.9373 | 0.8881 | 0.8839 | 0.8839 | |
| milogP | 1.0000 | 0.8199 | 0.7237 | 0.9251 | 0.9135 | 0.9520 | 0.9521 | ||
| AlogP | 1.0000 | 0.8142 | 0.9450 | 0.8769 | 0.9510 | 0.9510 | |||
| MlogP | 1.0000 | 0.8805 | 0.9201 | 0.7670 | 0.7670 | ||||
| XlogP2 | 1.0000 | 0.9712 | 0.9650 | 0.9650 | |||||
| XlogP3 | 1.0000 | 0.9136 | 0.9136 | ||||||
| logkw | 1.0000 | 1.0000 | |||||||
| logPHPLC | 1.0000 | 1.0000 |
Table 5.
Structures and inhibitory potency of thiosemicarbazides 18–34 towards bacterial topoisomerases.
Table 5.
Structures and inhibitory potency of thiosemicarbazides 18–34 towards bacterial topoisomerases.
| Compound | XlogP3 | XlogP2 | Inhibitory Potency IC50 [μM] | ||
|---|---|---|---|---|---|
| DNA Gyrase | Topo IV | ||||
| 1 [16] | 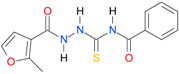 | 2.59 | n.d. * | 14.59 | n.a. ** |
| 2 [16] | 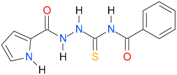 | 2.25 | n.d. | 93.30 | 41.04 |
| 18 [17] |  | 3.59 | n.d. | n.d. | 14 |
| 19 [16] | 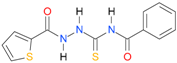 | 3.14 | n.d. | 83.63 | n.a. |
| 20 [17] | 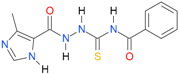 | 2.00 | n.d. | n.a. | 90.00 |
| 21 [18] | 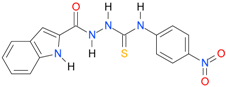 | n.d. | 2.82 | n.a. | 14.00 |
| 22 [16] | 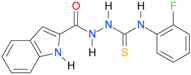 | n.d. | 3.09 | n.a. | 63.47 |
| 23 [16] | 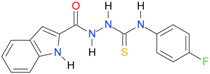 | n.d. | 3.09 | 127.68 | 267.04 |
| 24 [18] | 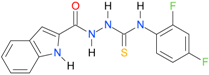 | n.d. | 3.25 | n.a. | 295.00 |
| 25 [16] | 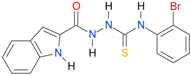 | n.d. | 3.73 | n.a. | n.a. |
| 26 [18] | 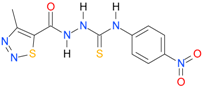 | n.d. | 2.25 | n.a. | n.a. |
| 27 [16] |  | n.d. | 1.46 | 64.21 | n.a. |
| 28 [18] |  | n.d. | 3.16 | n.a. | 403.00 |
| 29 [18] | 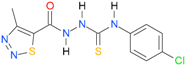 | n.d. | 2.98 | n.a. | n.a. |
| 30 [18] |  | n.d. | 2.52 | n.a. | n.a. |
| 31 [18] | 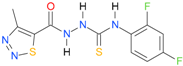 | n.d. | 2.68 | n.a. | n.a. |
| 32 [18] | 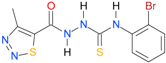 | n.d. | 3.16 | n.a. | n.a. |
* n.d.—not determined, ** n.a.—no activity.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Paneth, A.; Hawrył, A.; Plech, T.; Hawrył, M.; Świeboda, R.; Janowska, D.; Wujec, M.; Paneth, P. Lipophilicity Studies on Thiosemicarbazide Derivatives. Molecules 2017, 22, 952. https://doi.org/10.3390/molecules22060952
AMA Style
Paneth A, Hawrył A, Plech T, Hawrył M, Świeboda R, Janowska D, Wujec M, Paneth P. Lipophilicity Studies on Thiosemicarbazide Derivatives. Molecules. 2017; 22(6):952. https://doi.org/10.3390/molecules22060952
Chicago/Turabian StylePaneth, Agata, Anna Hawrył, Tomasz Plech, Mirosław Hawrył, Ryszard Świeboda, Dominika Janowska, Monika Wujec, and Piotr Paneth. 2017. "Lipophilicity Studies on Thiosemicarbazide Derivatives" Molecules 22, no. 6: 952. https://doi.org/10.3390/molecules22060952