Calculation of Five Thermodynamic Molecular Descriptors by Means of a General Computer Algorithm Based on the Group-Additivity Method: Standard Enthalpies of Vaporization, Sublimation and Solvation, and Entropy of Fusion of Ordinary Organic Molecules and Total Phase-Change Entropy of Liquid Crystals



Abstract
:1. Introduction
2. General Procedure
3. Results
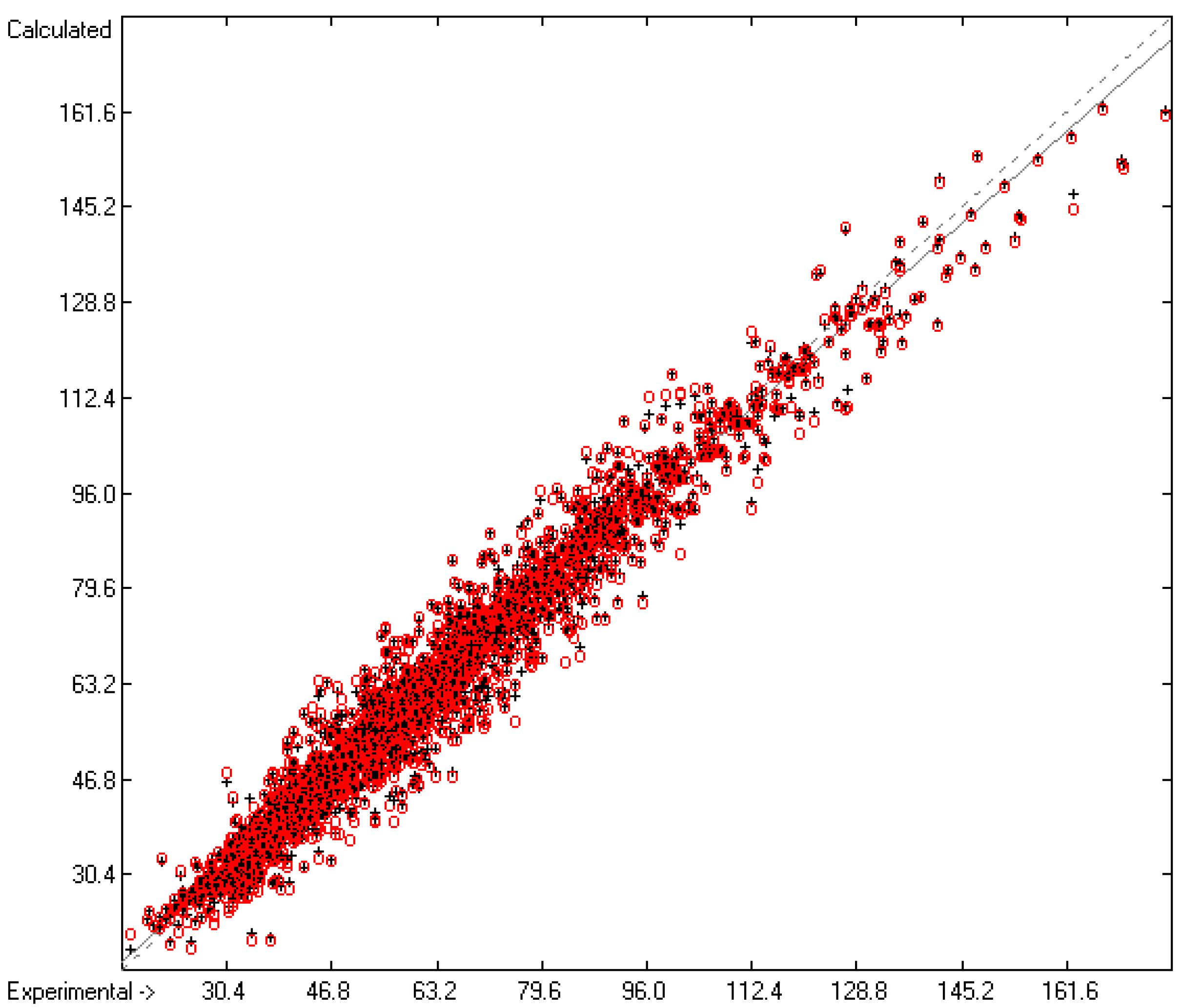
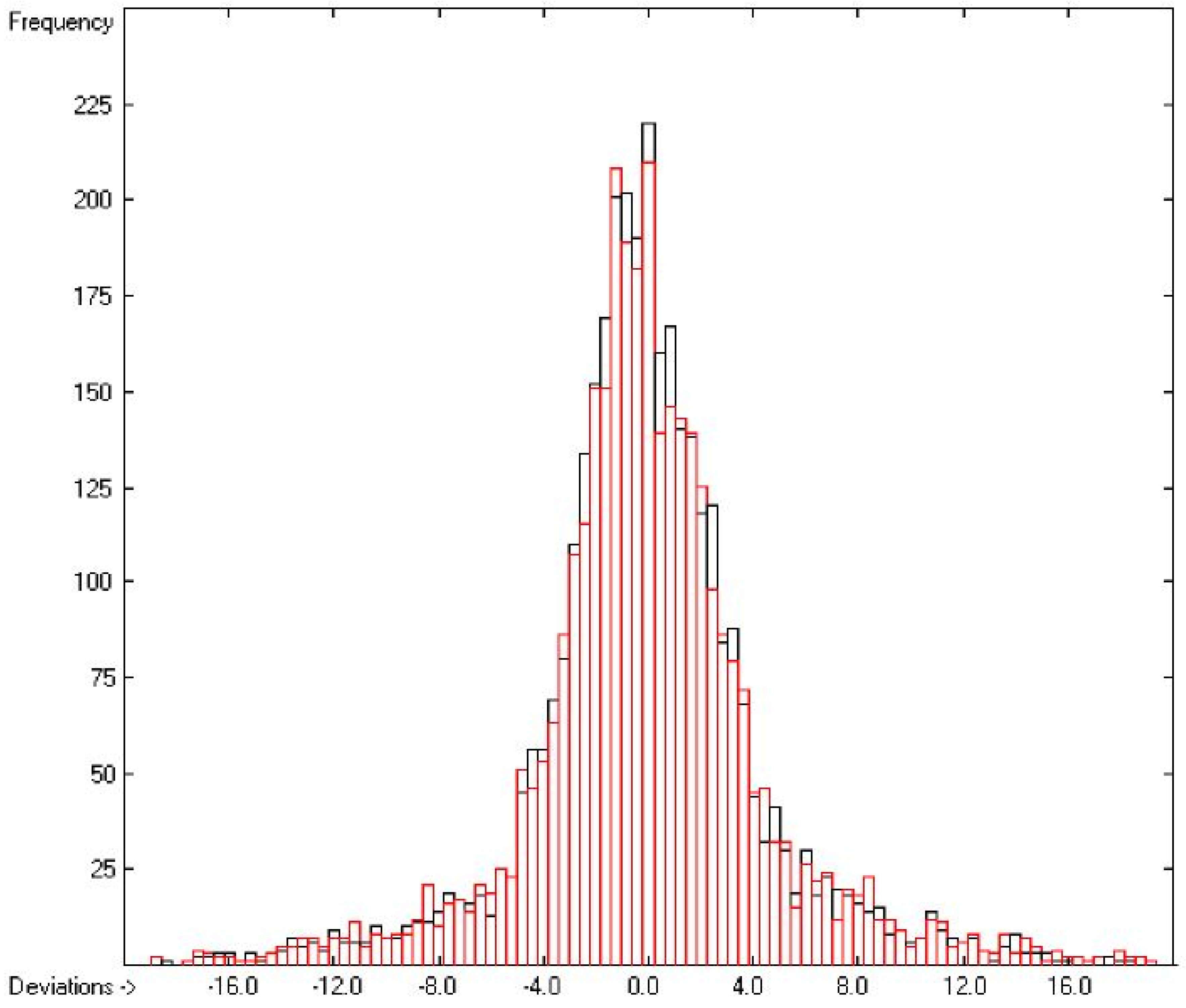
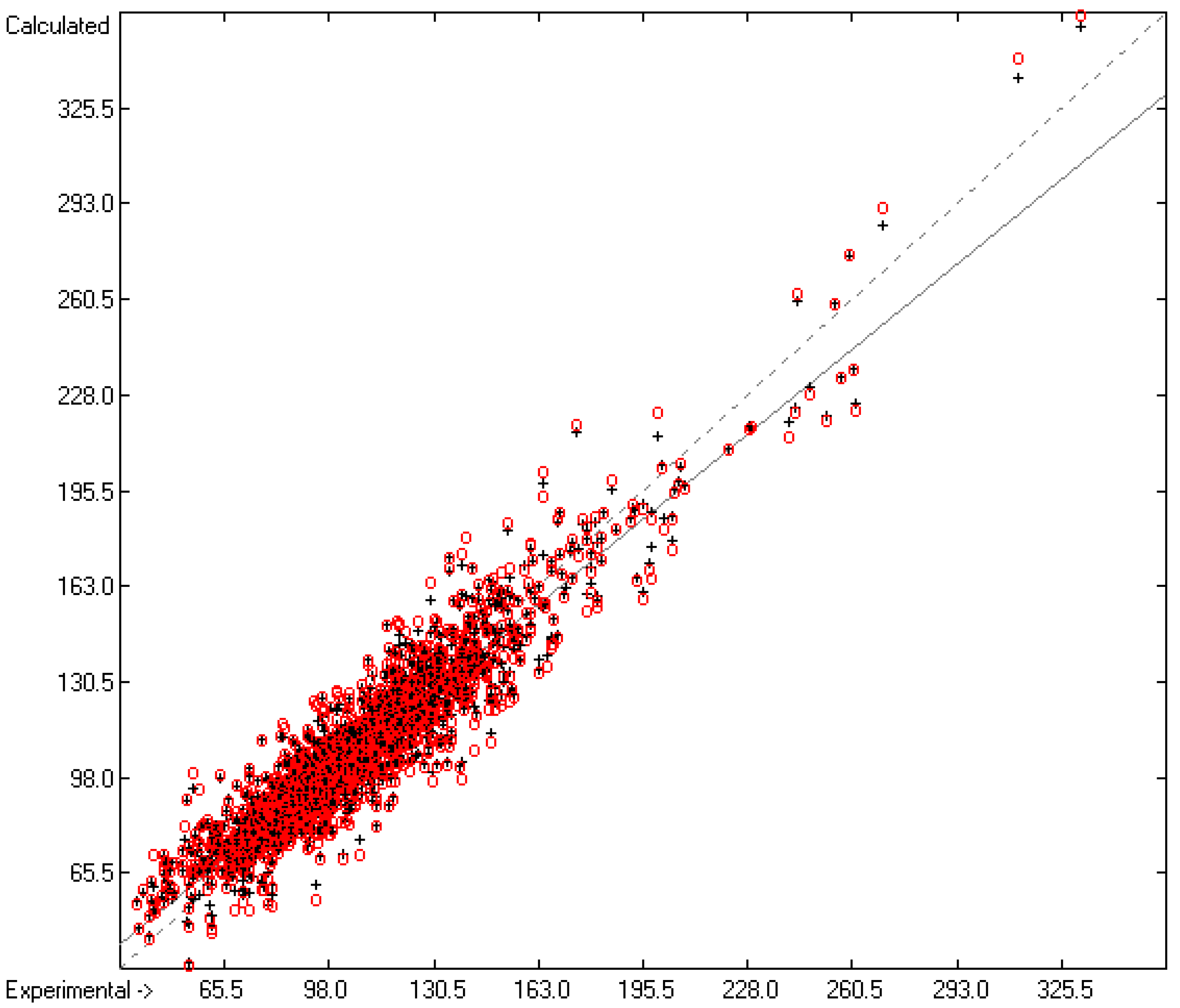
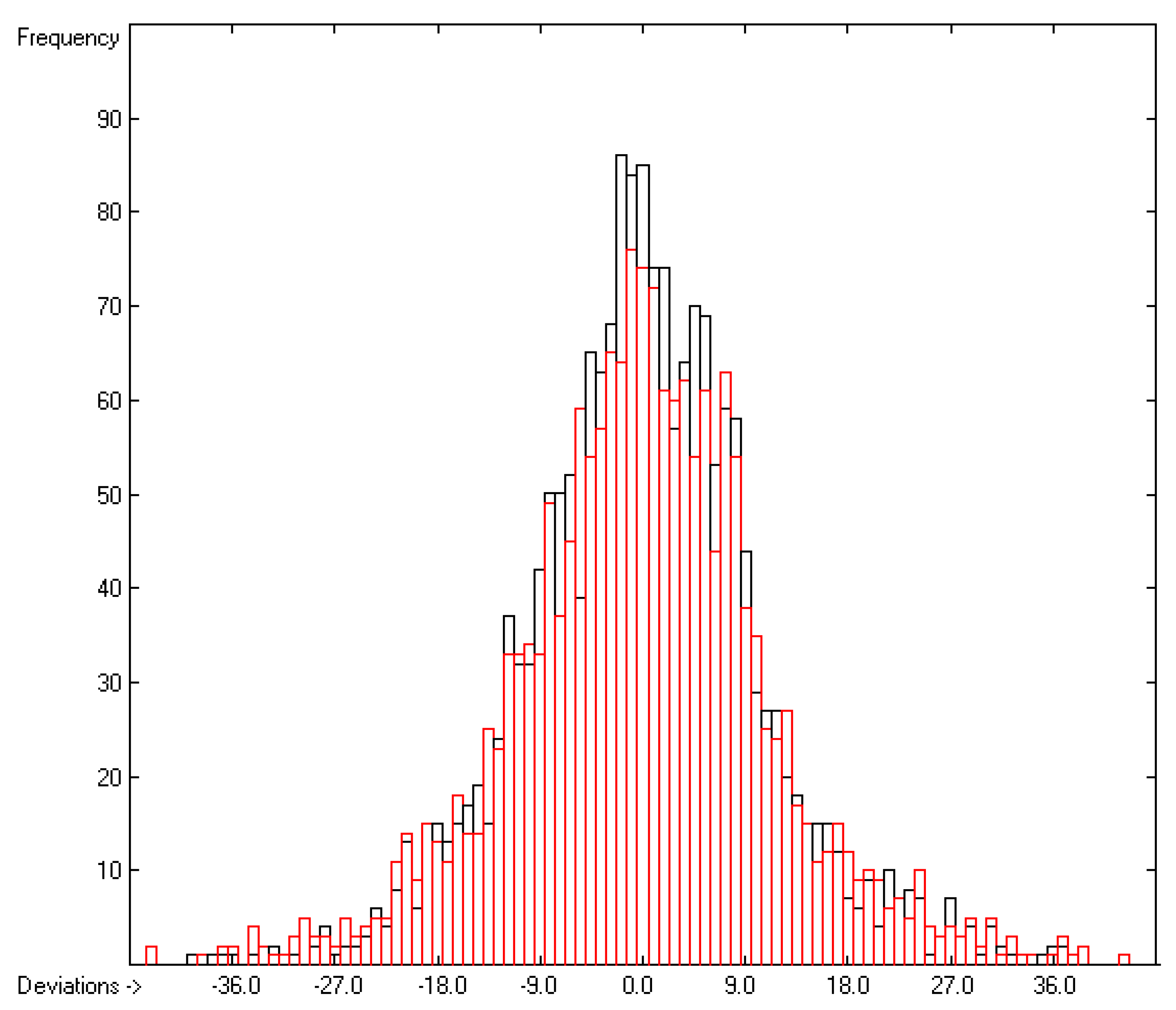
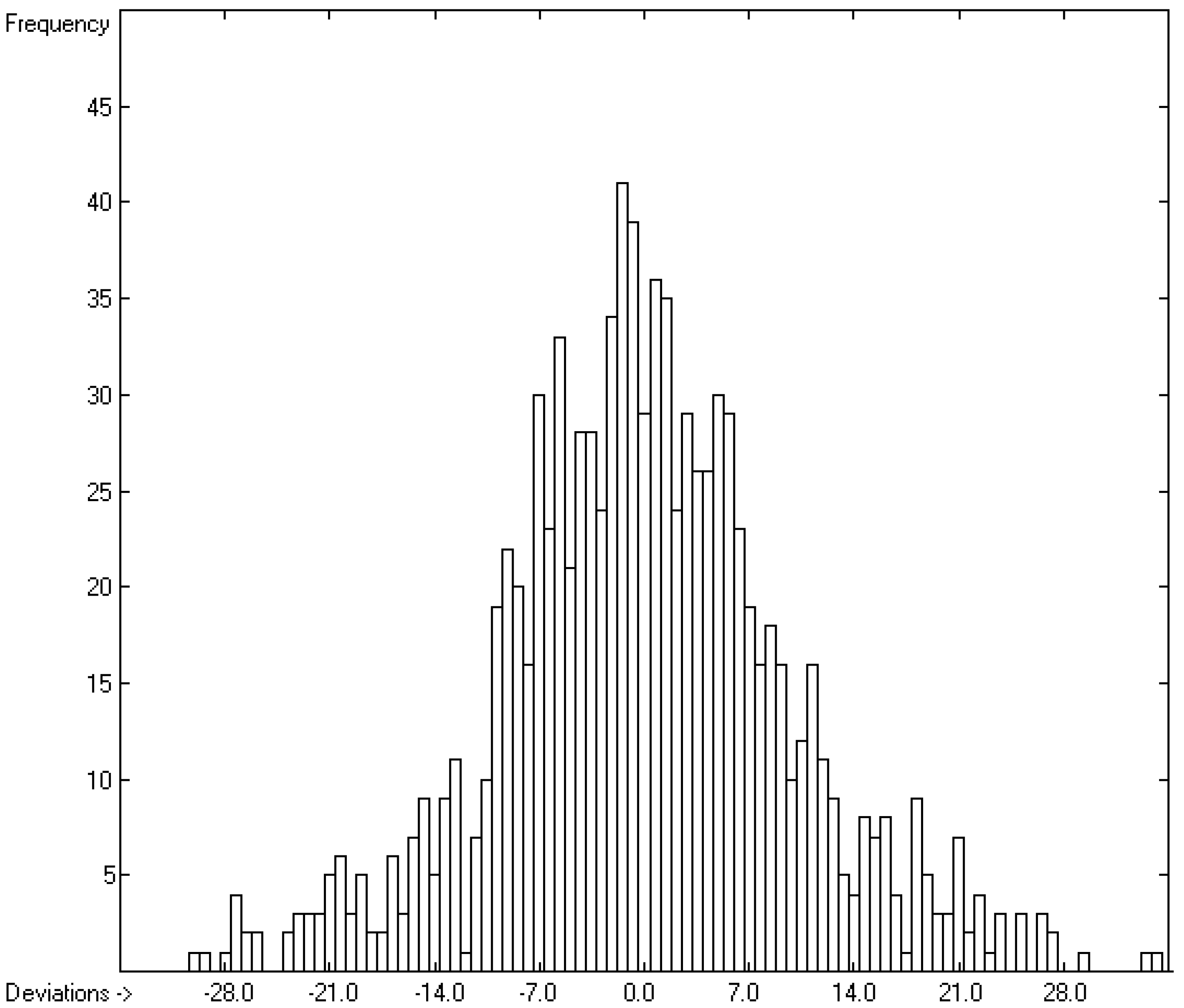
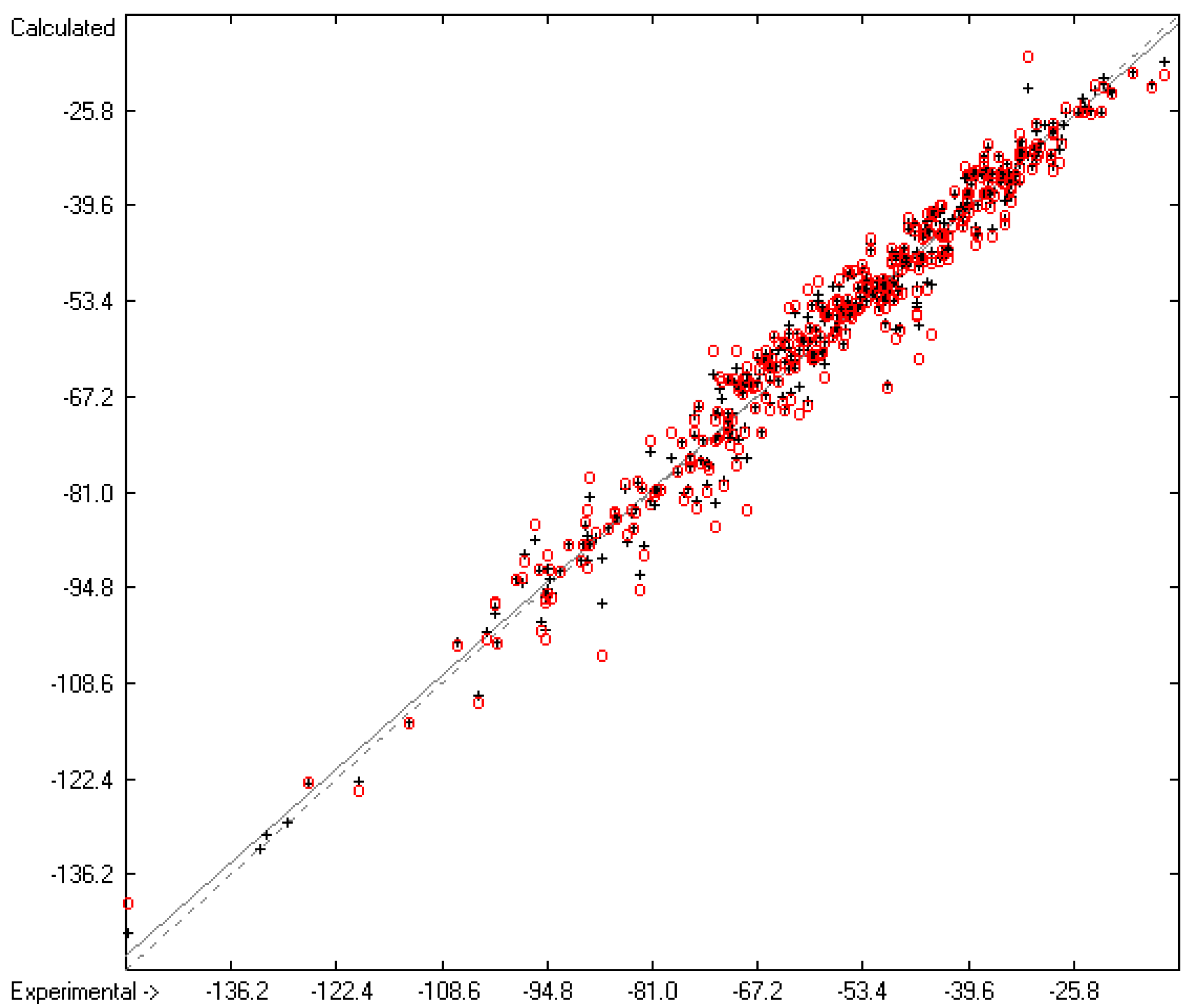
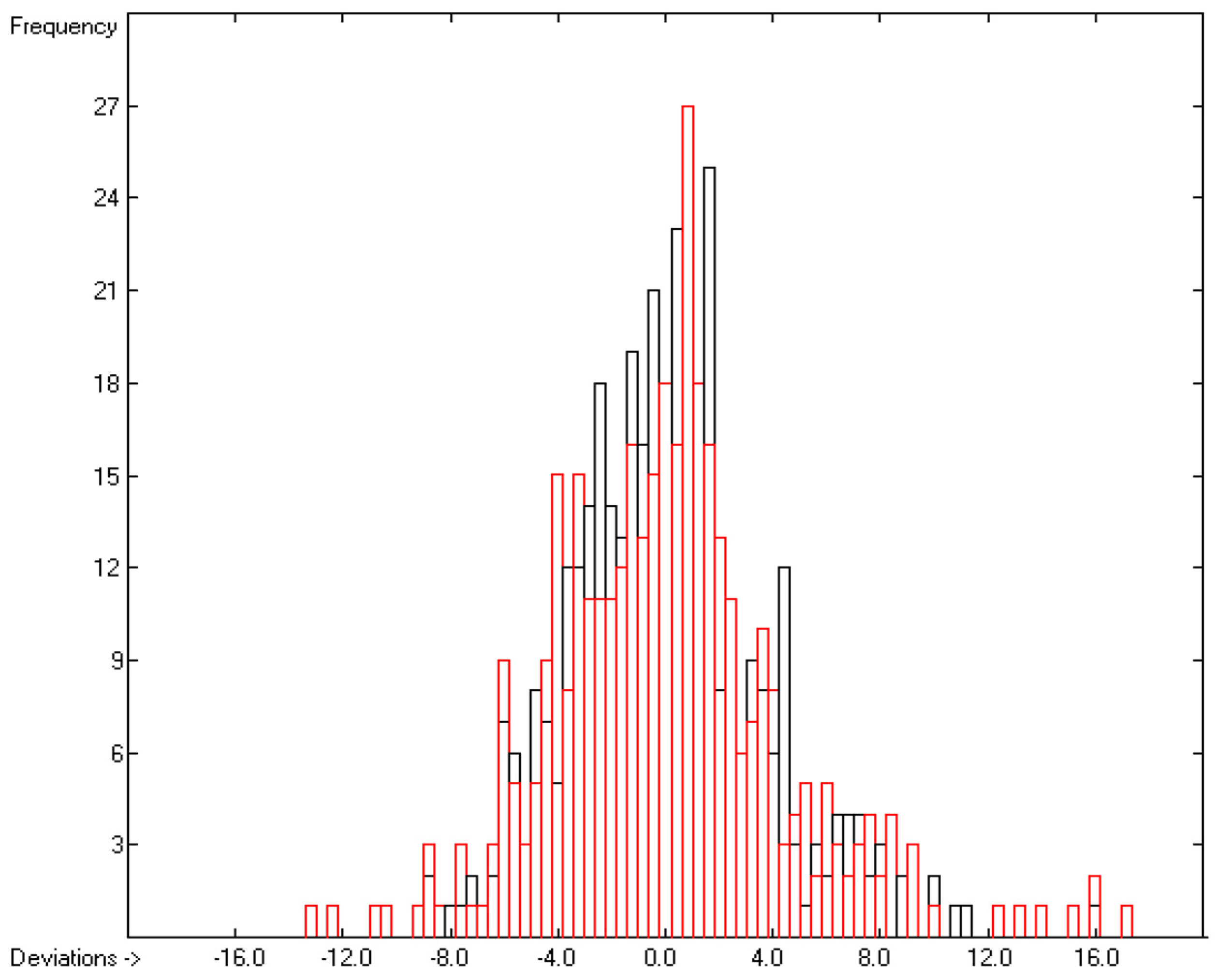
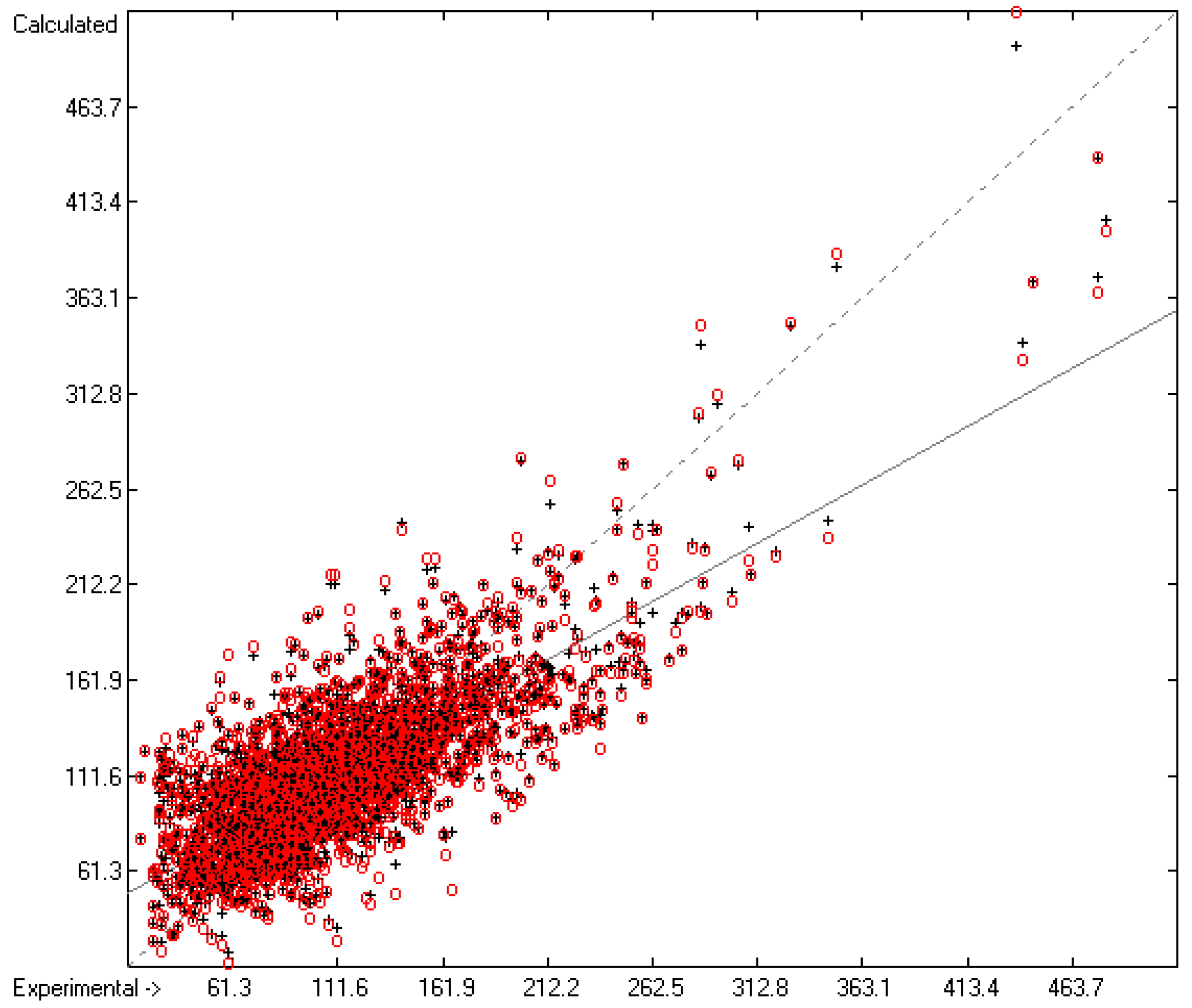
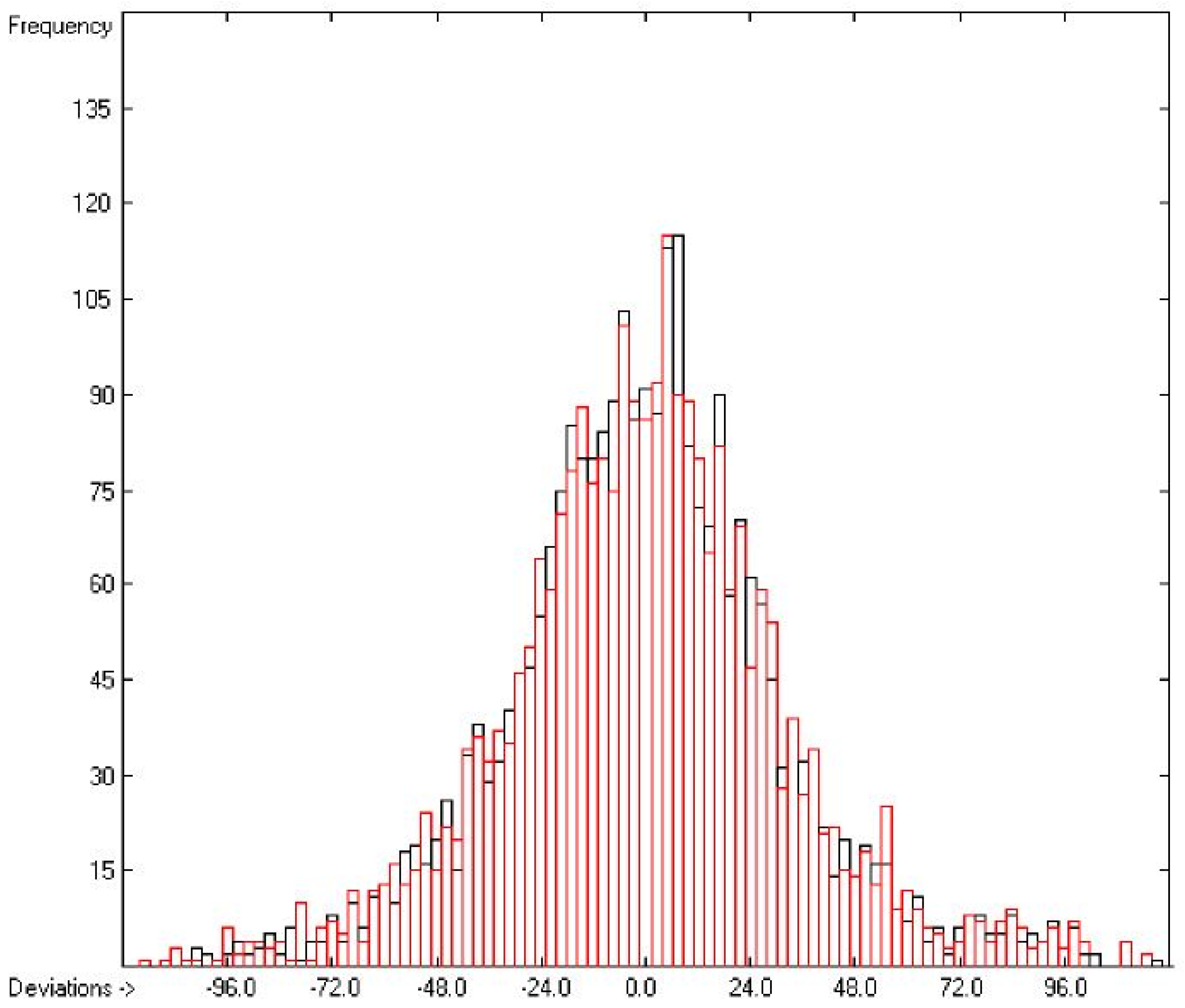
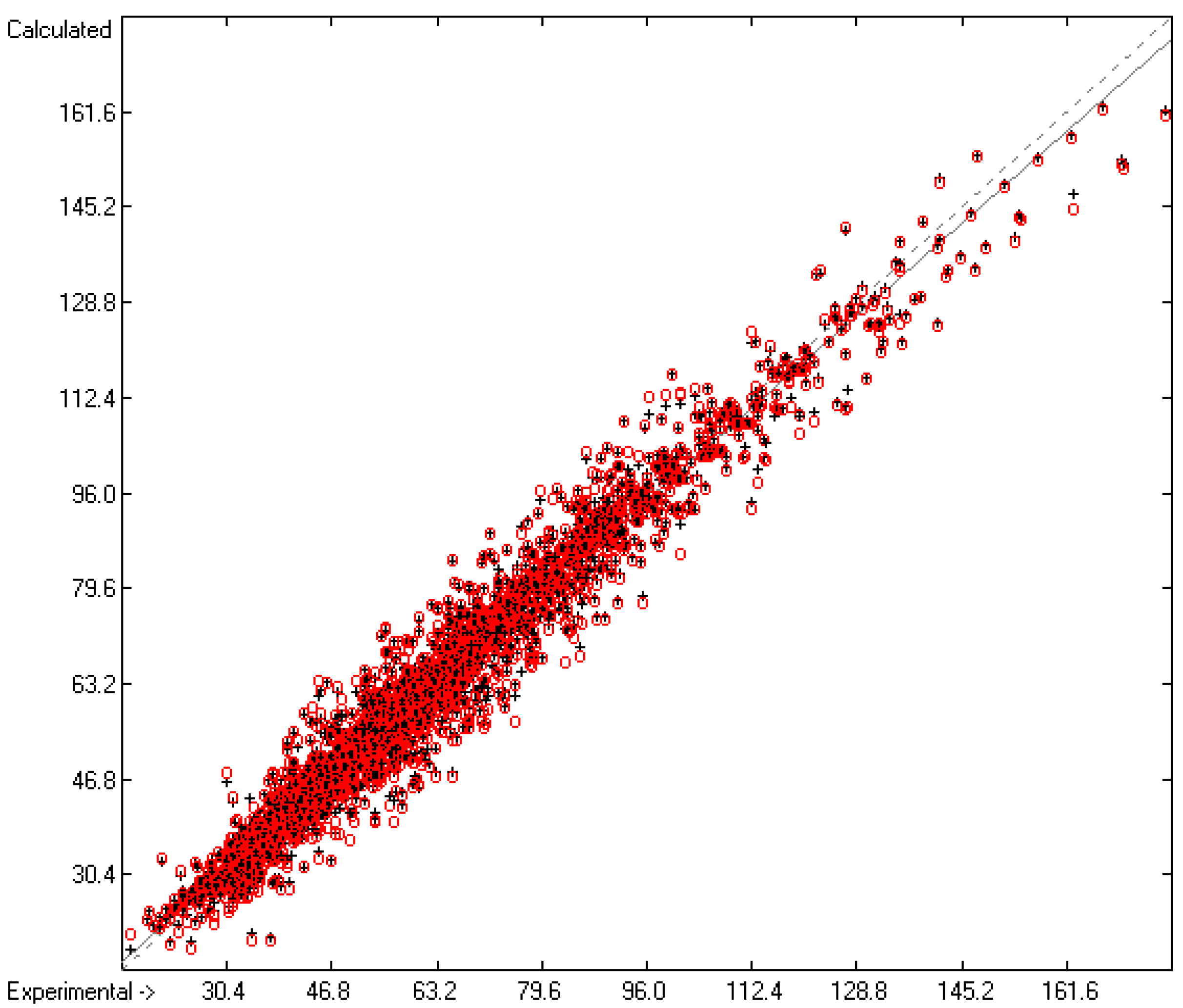
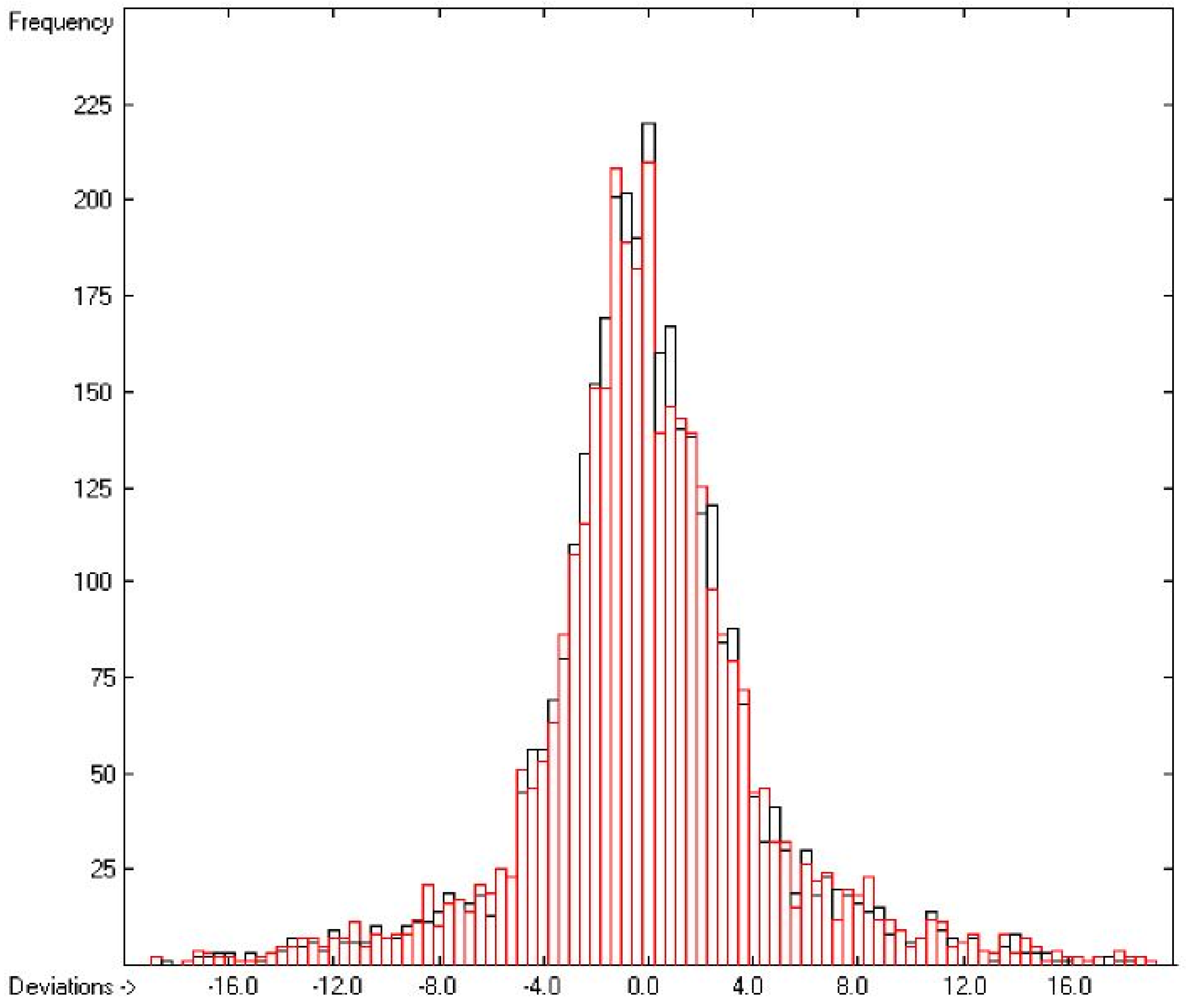
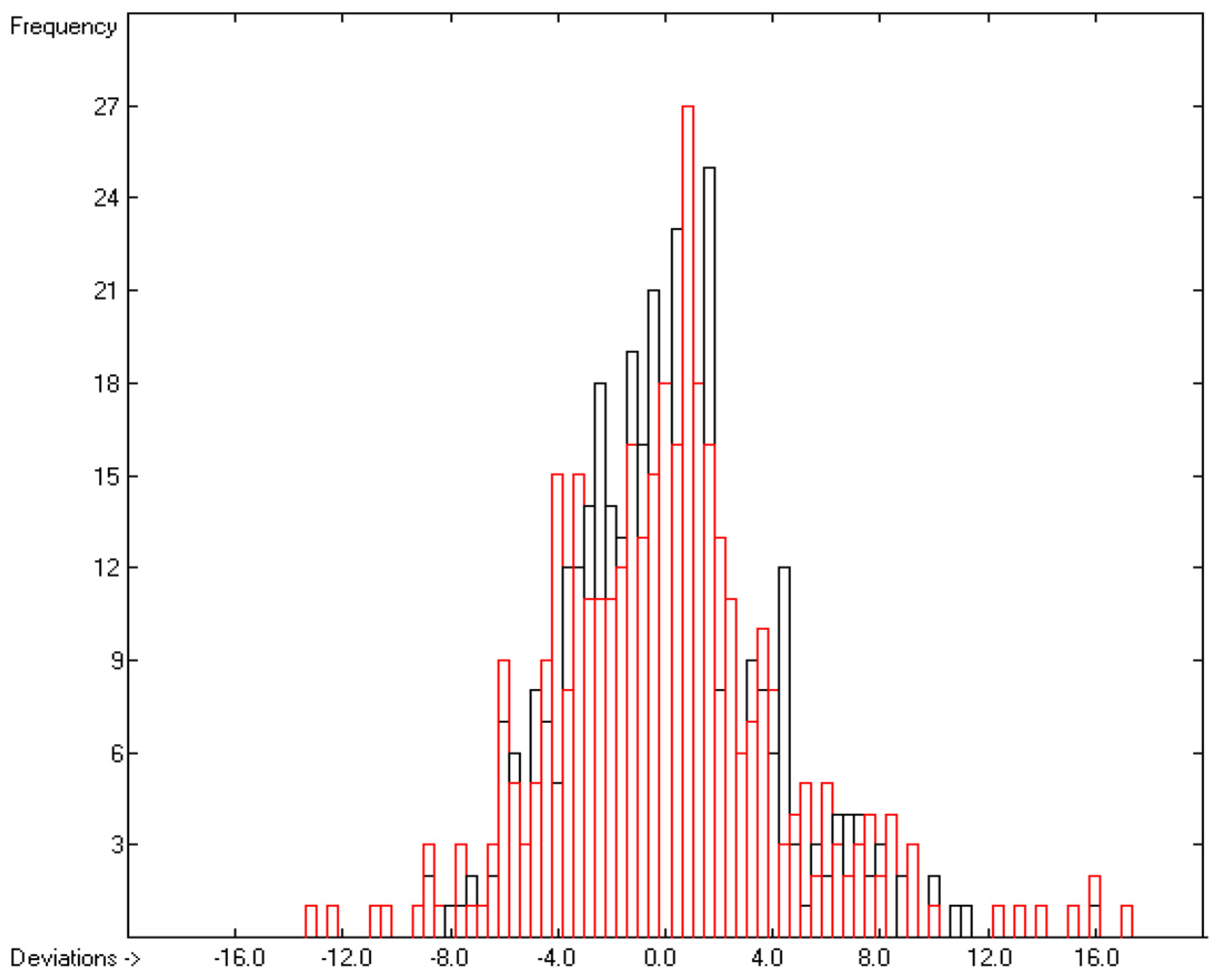
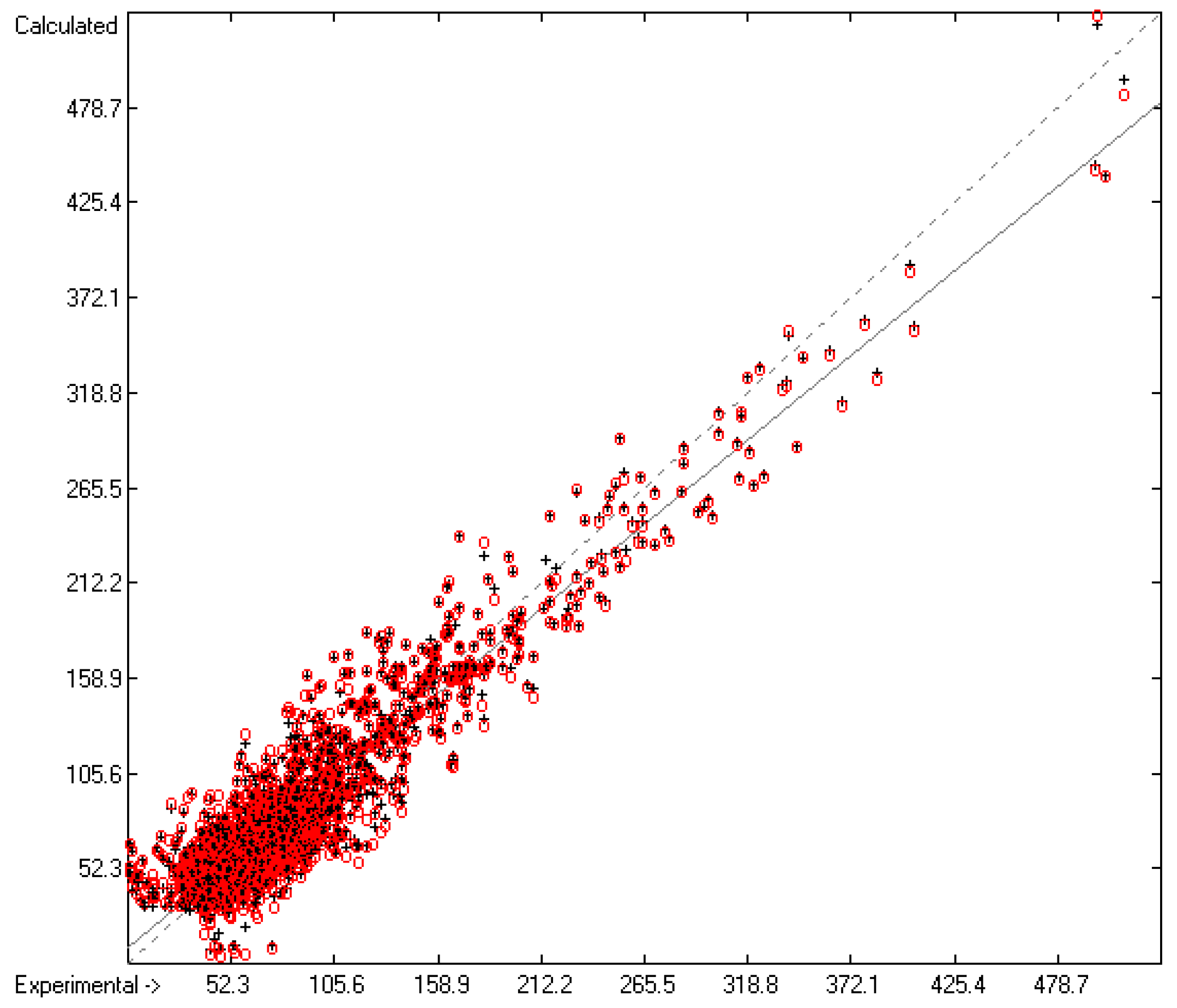
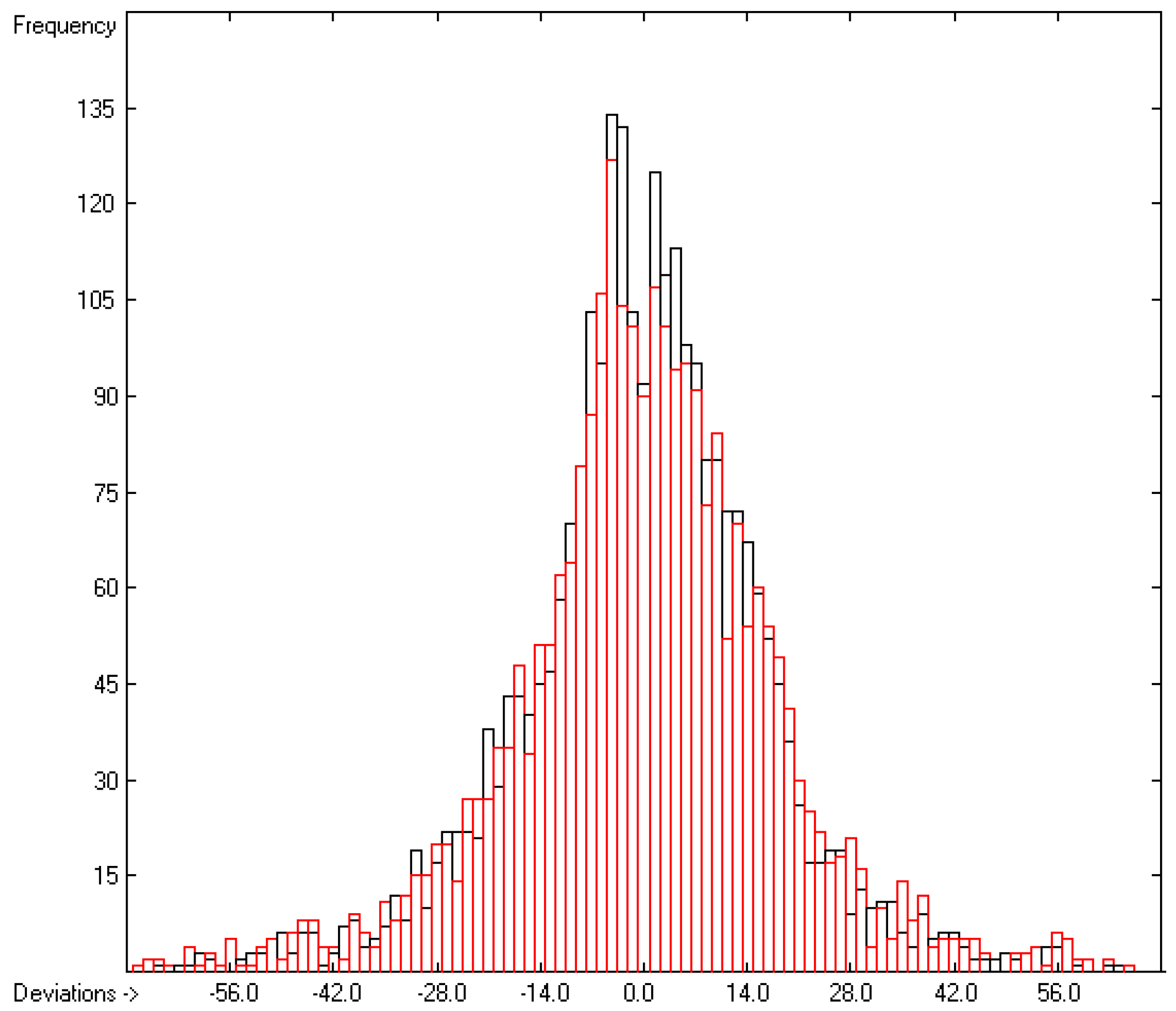
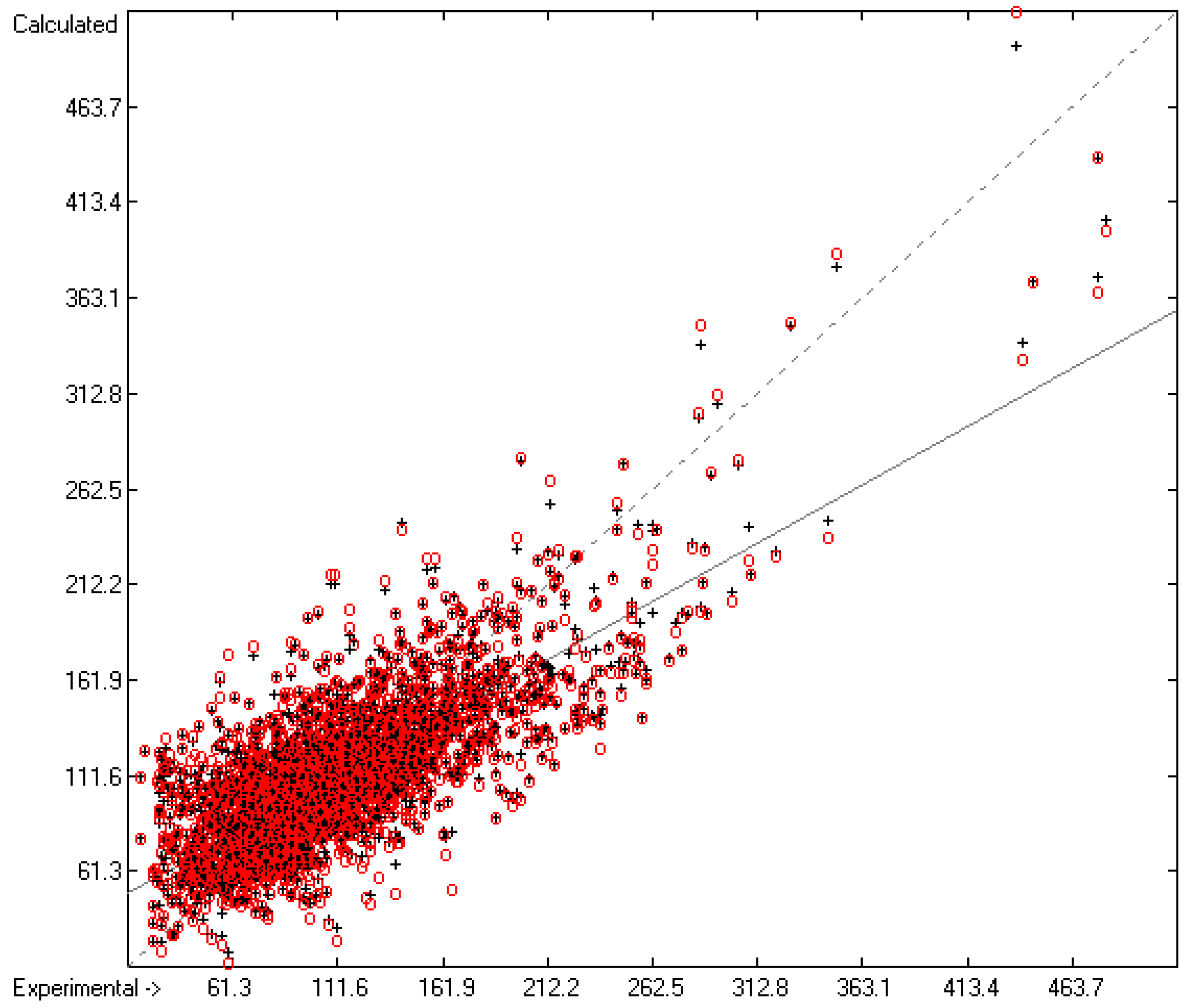
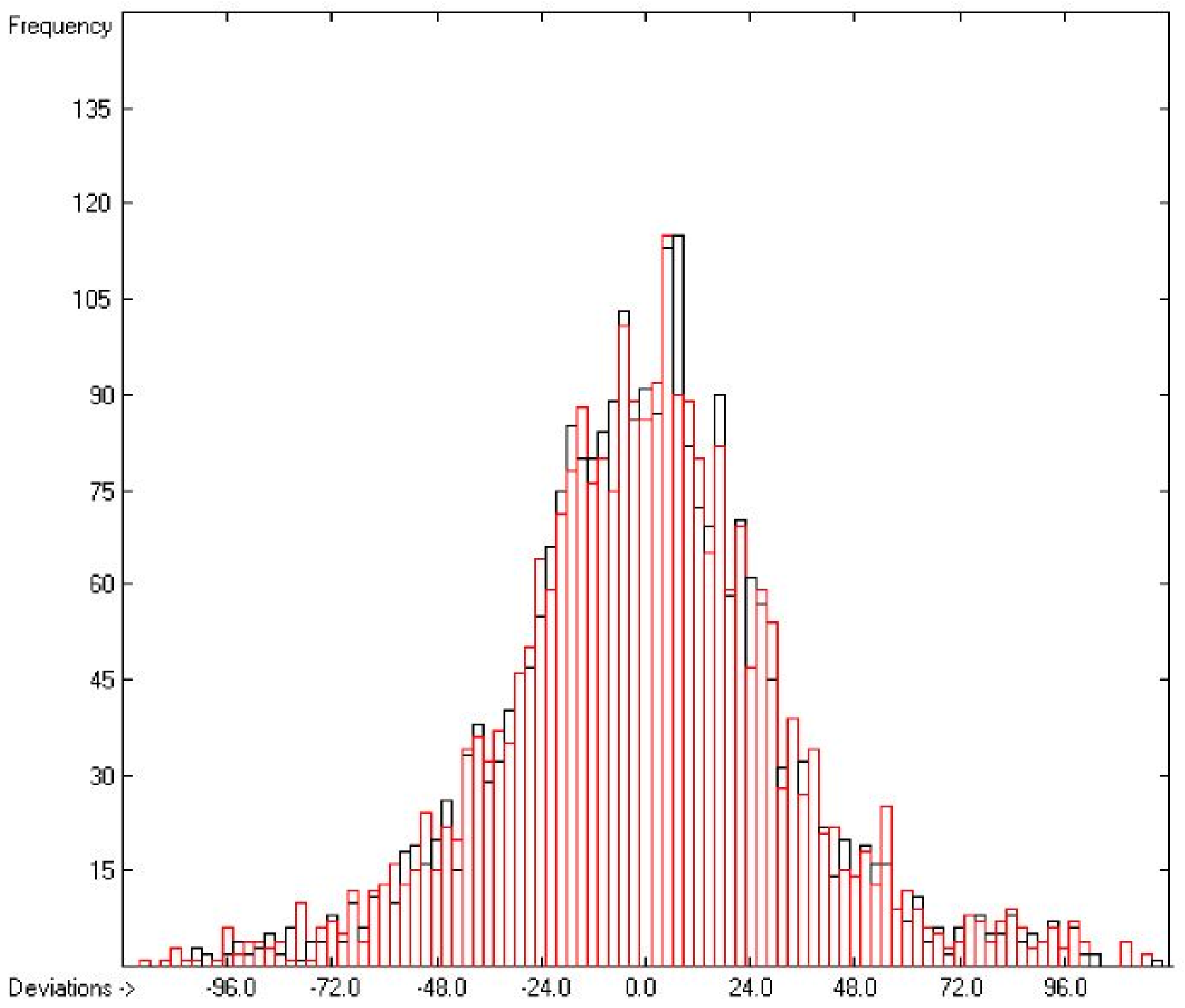
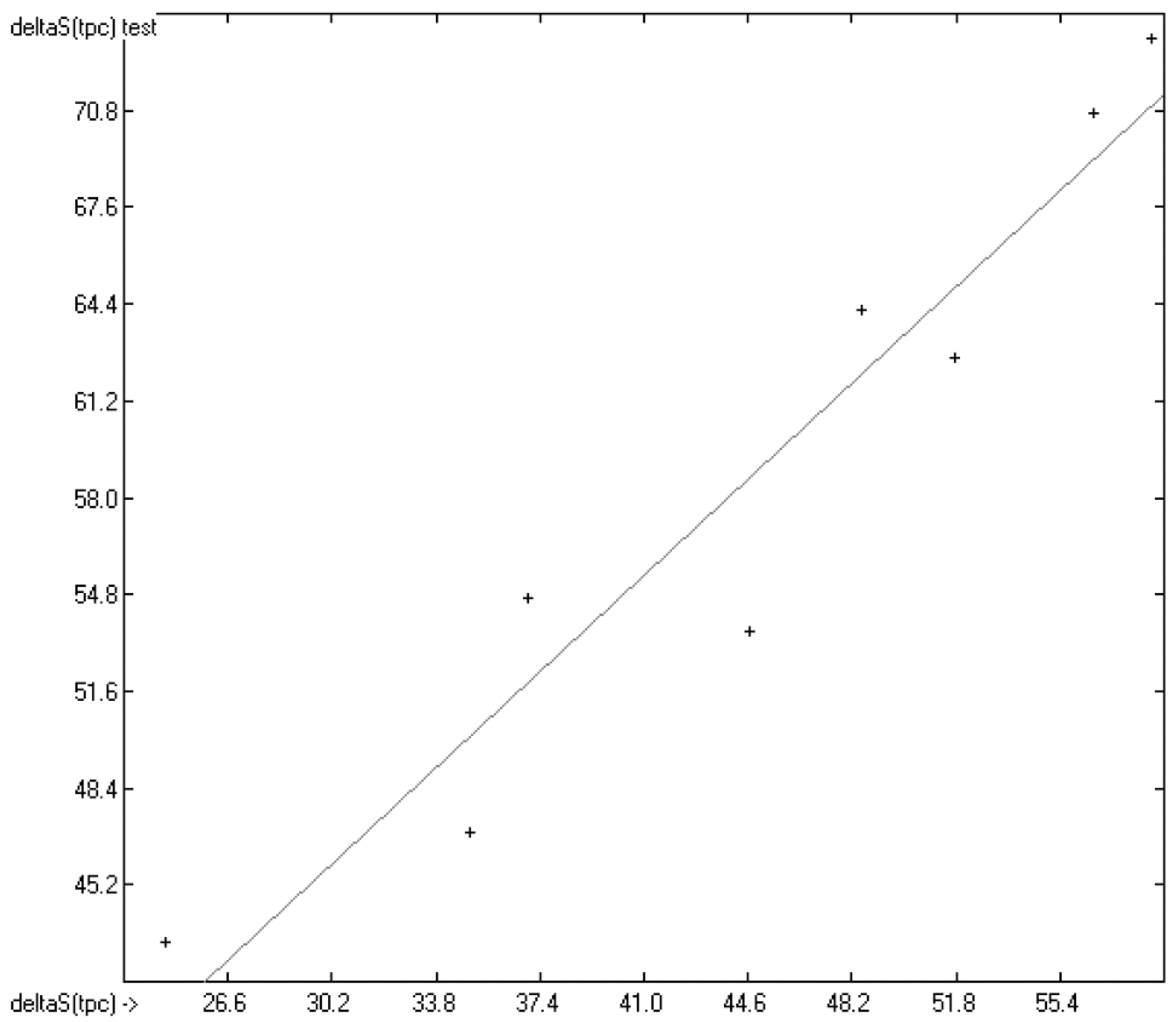
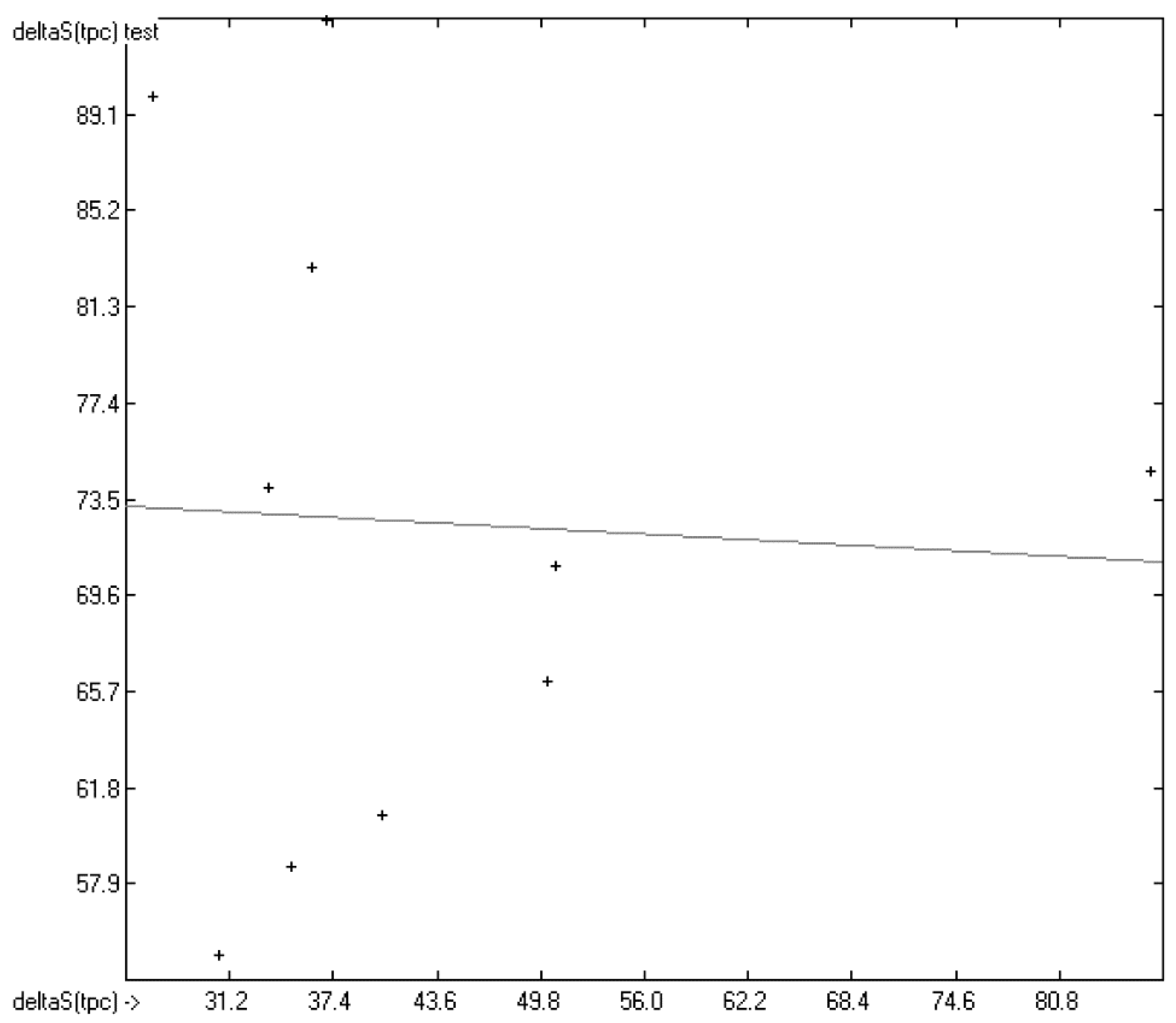
3.1. General Remarks
- (1)
- The experimental values of enthalpies and entropies are temperature-dependent. Any relationship within these properties or with other ones only make sense if they are referenced to the same temperature. The usual temperature of reference is 298.15 K, and thus it was ensured in this work that experimental data from literature were only accepted if they had been either measured at or adjusted to the standard temperatur of 298.15 K and standard pressure of 100 kPa.
- (2)
- All lists of molecules used in the atom-group parameters evaluations have been collected in standard SDF files, stored in the supplementary material, ready to be imported by external chemistry software. The supplementary material also provides the lists of results containing molecule names, experimental, training and cross-validation values. Beyond this, it also contains lists of experimental outliers.
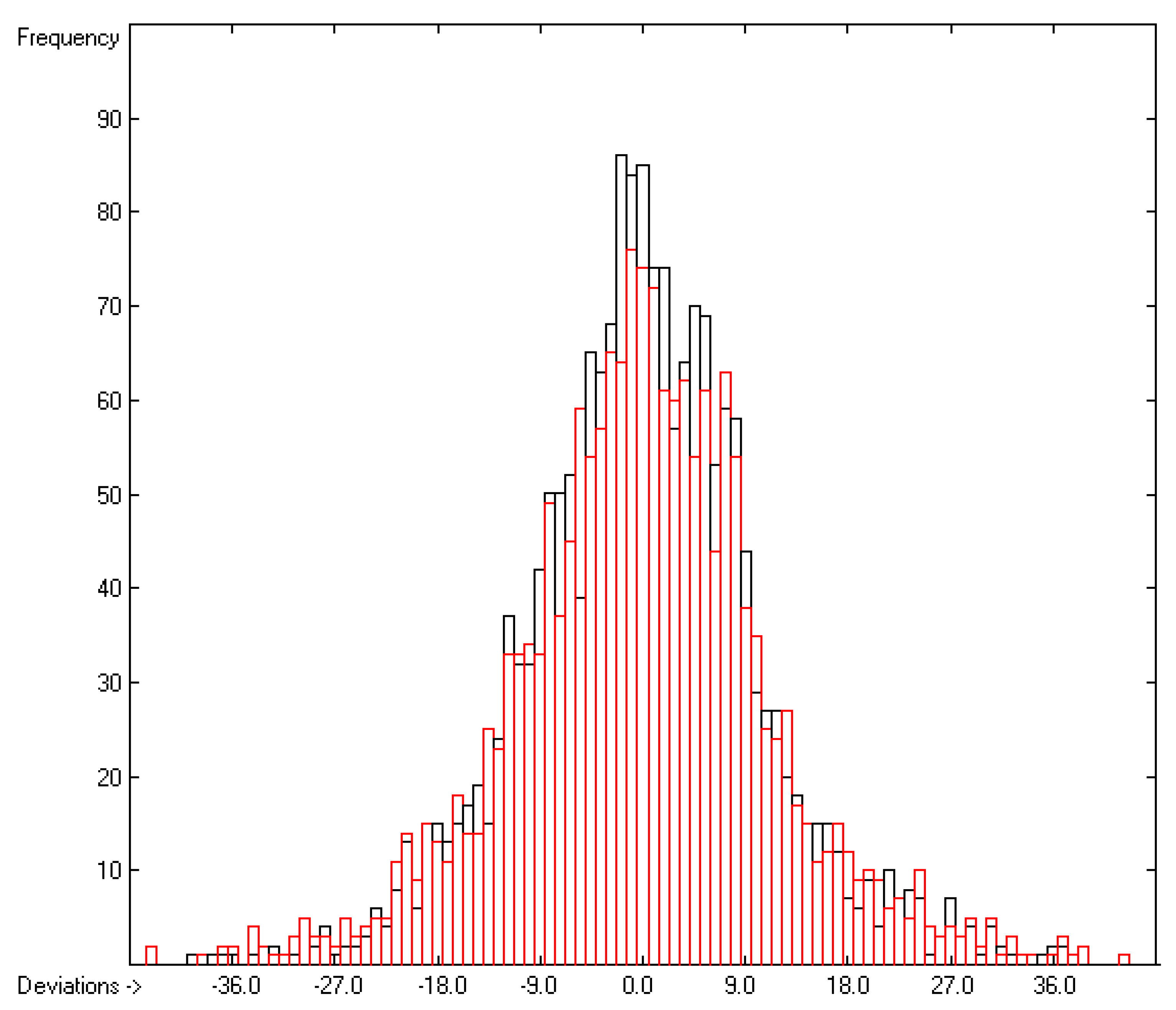
3.2. Enthalpy of Vaporization
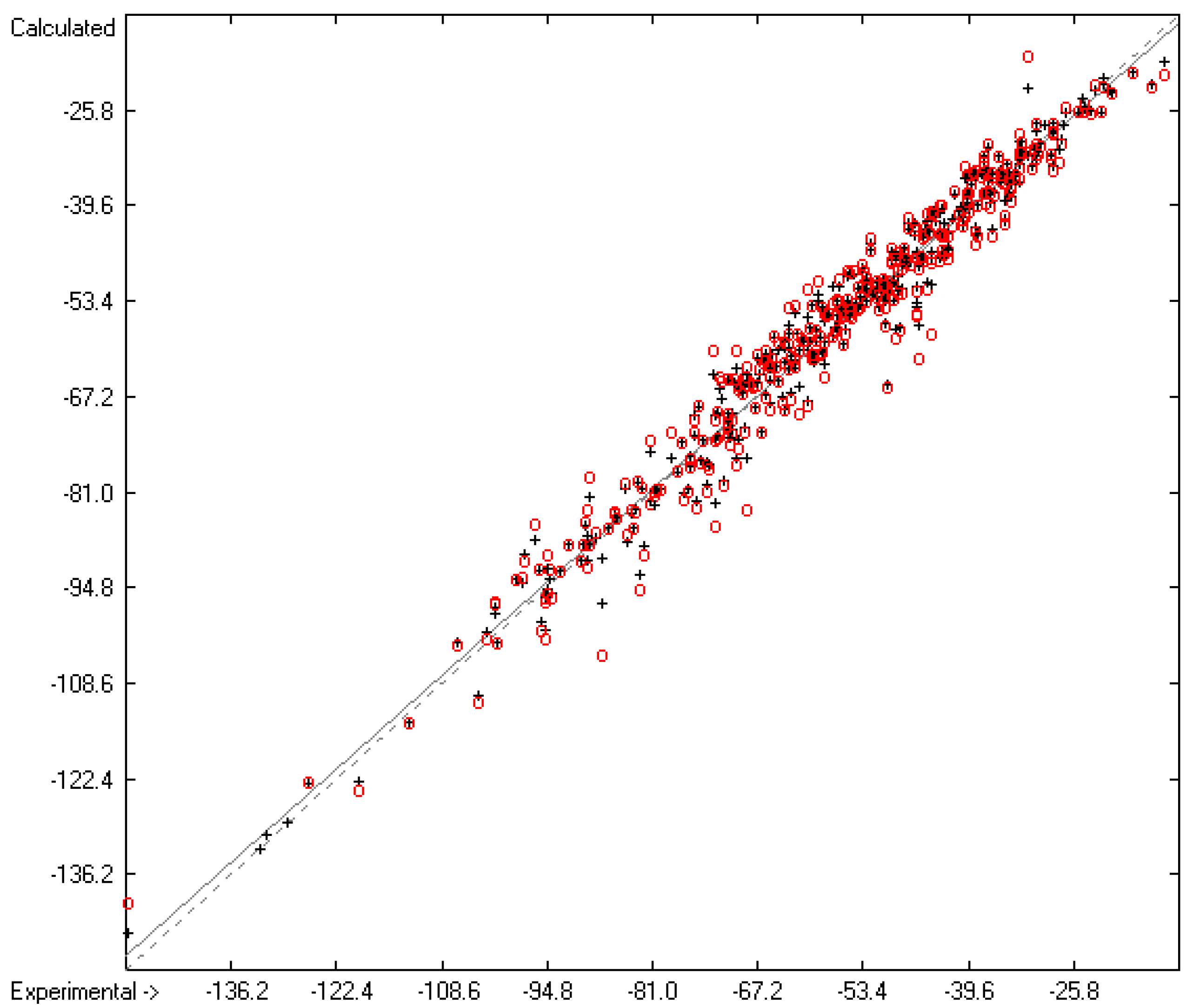
3.3. Enthalpy of Sublimation
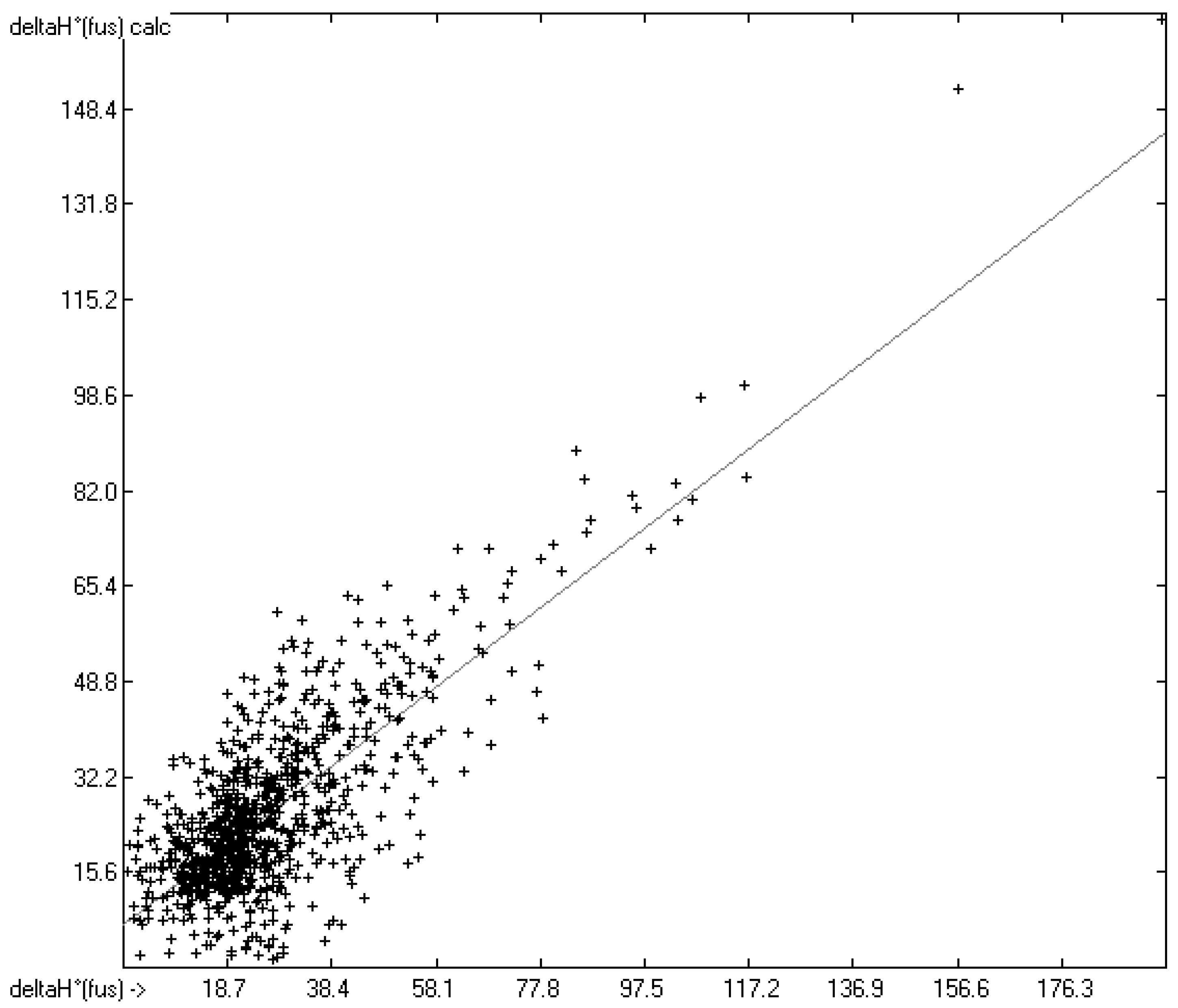
3.4. Enthalpy of Fusion
3.5. Enthalpy of Solvation
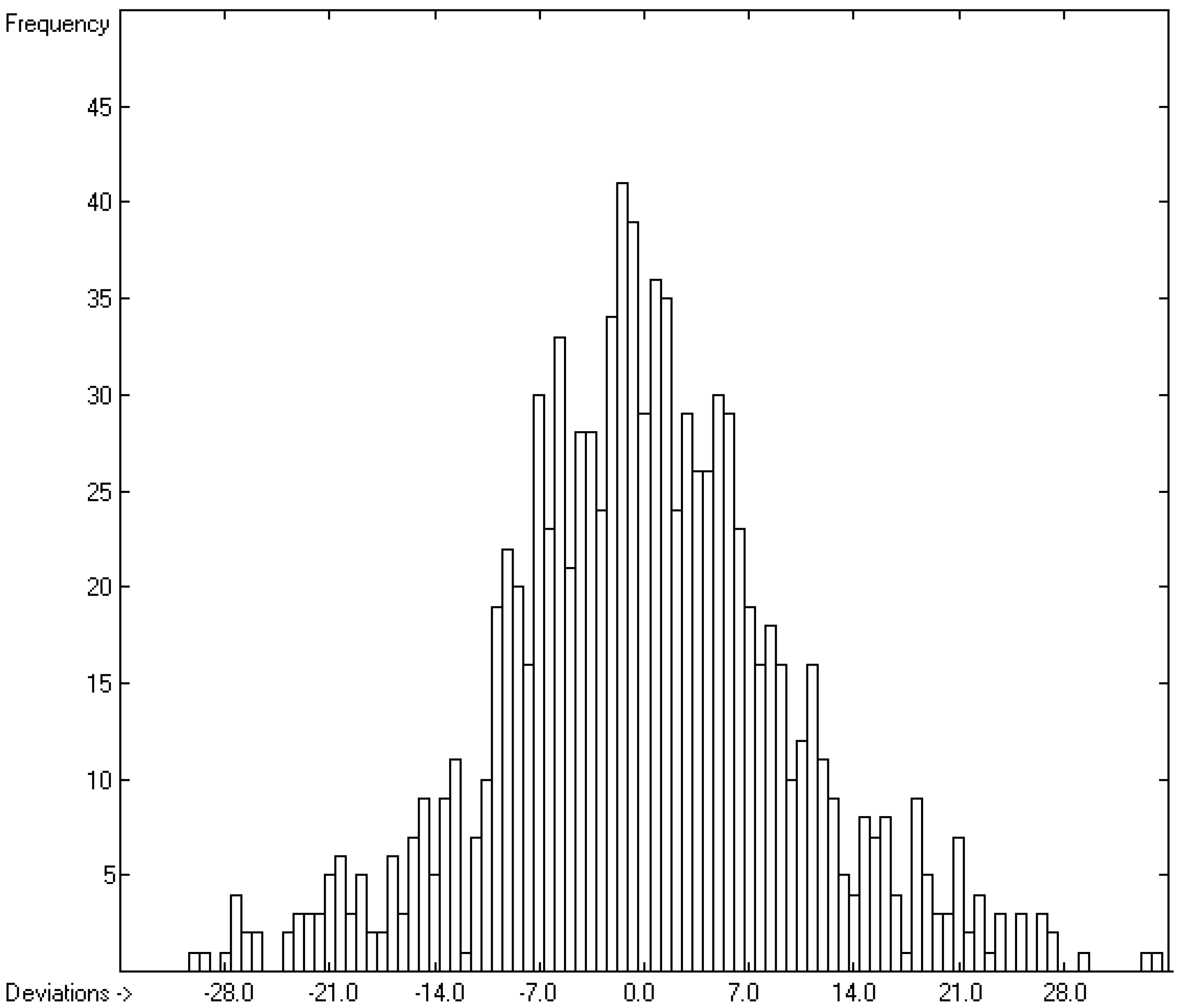
3.6. Entropy of Fusion
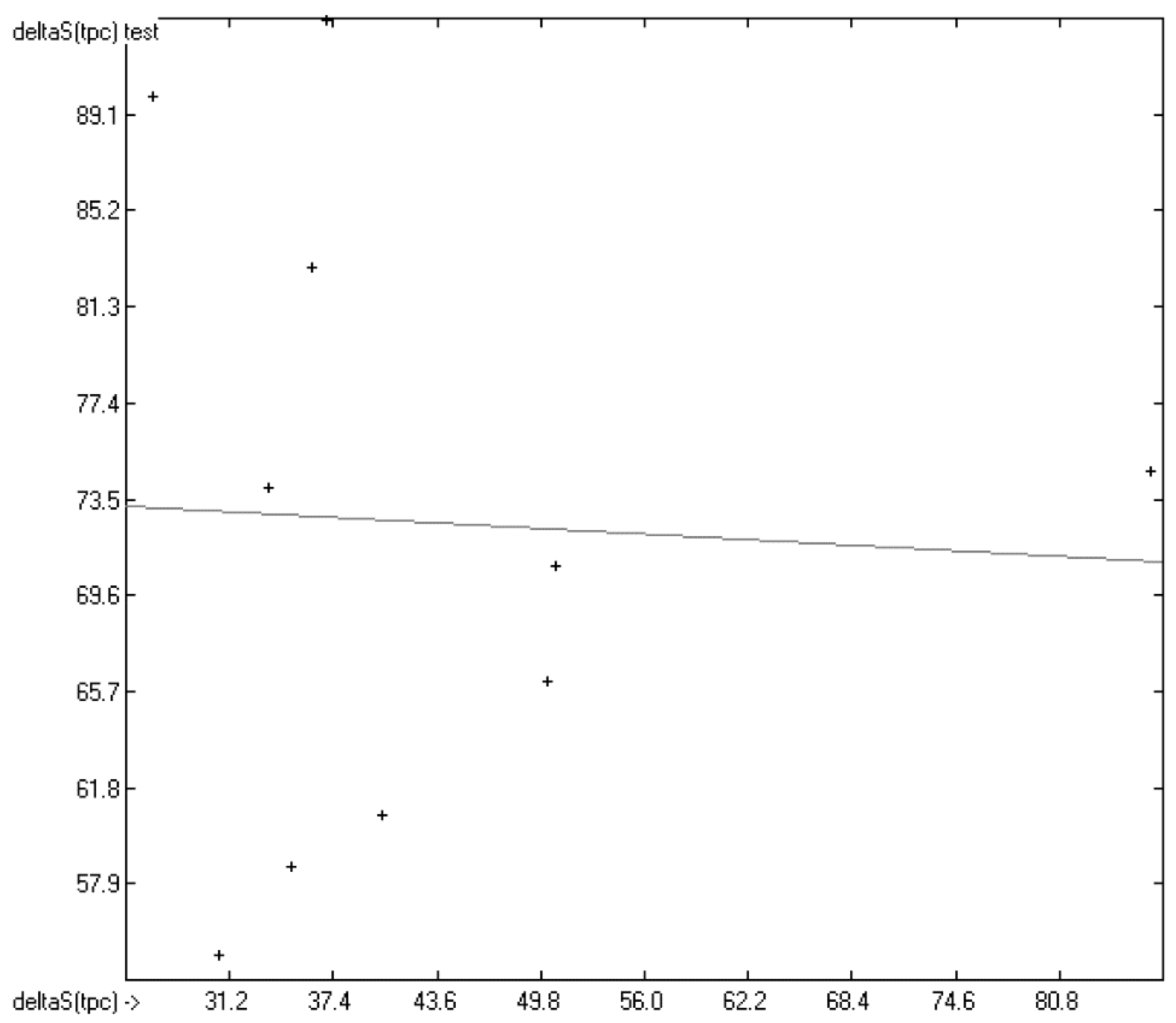
3.7. Total Phase-Change Entropy of Liquid Crystals
4. Conclusions
Supplementary Materials
Acknowledgments
Author contributions
Conflicts of Interest
References
- Naef, R. A Generally Applicable Computer Algorithm Based on the Group Additivity Method for the Calculation of Seven Molecular Descriptors: Heat of Combustion, LogPO/W, LogS, Refractivity, Polarizability, Toxicity and LogBB of Organic Compounds; Scope and Limits of Applicability. Molecules 2015, 20, 18279–18351. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships I. Partition coefficients as a measure of hydrophobicity. J. Comput. Chem. 1986, 7, 565–577. [Google Scholar] [CrossRef]
- Ghose, A.K.; Pritchett, A.; Crippen, G.M. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships III: Modeling hydrophobic interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.J.; Savchik, J.A. A new empirical Method to calculate Average Molecular Polarizabilities. J. Am. Chem. Soc. 1979, 101, 7206–7213. [Google Scholar] [CrossRef]
- Miller, K.J. Additivity methods in molecular polarizability. J. Am. Chem. Soc. 1990, 112, 8533–8542. [Google Scholar] [CrossRef]
- Sun, H. A universal molecular descriptor system for prediction of LogP, LogS, LogBB, and absorption. J. Chem. Inf. Comput. Sci. 2004, 44, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Acree, W.E., Jr.; Chickos, J.S. Phase Transition Enthalpy Measurements of Organic and Organometallic Compounds. Sublimation, Vaporization and Fusion Enthalpies from 1880 to 2010. J. Phys. Chem. Ref. Data 2010, 39, 043101. [Google Scholar] [CrossRef]
- Roux, M.V.; Temprado, M.; Chickos, J.; Nagano, Y. Critically Evaluated Thermo-chemical Properties of Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. Ref. Data 2008, 37, 1855. [Google Scholar] [CrossRef]
- Chickos, J.; Wang, T.; Sharma, E. Hypothetical Thermodynamic Properties: Vapor Pressures and Vaporization Enthalpies of the Even n-Alkanes from C40 to C76 at T = 298.15 K by Correlation-Gas Chromatography. Are the Vaporization Enthalpies a Linear Function of Carbon Number? J. Chem. Eng. Data 2008, 53, 481–491. [Google Scholar] [CrossRef]
- Chickos, J.; Lipkind, D. Hypothetical Thermodynamic Properties: Vapor Pressures and Vaporization Enthalpies of the Even n-Alkanes from C78 to C92 at T = 298.15 K by Correlation-Gas Chromatography. J. Chem. Eng. Data 2008, 53, 2432–2440. [Google Scholar] [CrossRef]
- Chickos, J.; Hanshaw, W. Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from C21 to C30 at T = 298.15 K by Correlation Gas Chromatography. J. Chem. Eng. Data 2004, 49, 77–85. [Google Scholar] [CrossRef]
- Chickos, J.; Hanshaw, W. Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from C31 to C38 at T = 298.15 K by Correlation Gas Chromatography. J. Chem. Eng. Data 2004, 49, 620–630. [Google Scholar] [CrossRef]
- Wilson, J.; Gobble, C.; Chickos, J. Vaporization, Sublimation, and Fusion Enthalpies of Some Saturated and Unsaturated Long Chain Fatty Acids by Correlation Gas Chromatography. J. Chem. Eng. Data 2015, 60, 202–212. [Google Scholar] [CrossRef]
- Abraham, M.H. Scales of Hydrogen-bonding: Their Construction and Application to Physicochemical and Biochemical Processes. Chem. Rev. 1993, 22, 73–83. [Google Scholar] [CrossRef]
- Abraham, M.H.; Chadha, H.S.; Whinting, G.S.; Mitchell, R.C. Hydrogen-bonding. 32. An Analysis of Water-Octanol and Water-Alkane Partitioning and the ΔlogP Parameter of Seiler. J. Pharm. Sci. 1994, 83, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.H.; Zissimos, A.M.; Acree, W.E. Partition of solutes from the gas phase and from water to wet and dry di-n-butyl Ether: A linear free energy relationship analysis. Phys. Chem. Chem. Phys. 2001, 3, 3732–3736. [Google Scholar] [CrossRef]
- Abraham, M.H.; Le, J. The Correlation and Prediction of the Solubility of Compounds in Water using an amended Solvation Energy Relationship. J. Pharm. Sci. 1999, 88, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Jover, J.; Bosque, R.; Sales, J. Determination of Abraham Solute Parameters from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Cabani, S.; Gianni, P.; Mollica, V.; Lepori, L. Group contributions to the thermodynamic properties of non-ionic organic solutes in dilute aqueous solution. J. Sol. Chem. 1981, 10, 563–595. [Google Scholar] [CrossRef]
- Chickos, J.S.; Acree, W.E. Jr.; Liebman, J.F. Estimating Solid-Liquid Phase Change Enthalpies and Entropies. J. Phys. Chem. Ref. Data 1999, 28, 1535–1673. [Google Scholar] [CrossRef]
- Acree, W.E., Jr.; Chickos, J.S. Phase Change Enthalpies and Entropies of Liquid Crystals. J. Phys. Chem. Ref. Data 2006, 35, 1051–1330. [Google Scholar] [CrossRef]
- Almeida, A.R.R.; Monte, M.J.S. Vapour pressures and phase transition properties of four substituted acetophenones. J. Chem. Thermodyn. 2016, 107, 42–50. [Google Scholar] [CrossRef]
- Gobble, C.; Vikman, J.; Chickos, J.S. Evaluation of the Vaporization Enthalpies and Liquid Vapor Pressures of (R)-Deprenyl, (S)-Benzphetamine, Alverine, and a Series of Aliphatic Tertiary Amines by Correlation Gas Chromatography at T/K = 298.15. J. Chem. Eng. Data 2014, 59, 2551–2562. [Google Scholar] [CrossRef]
- Miroshnichenko, E.A.; Kon’kova, T.S.; Pashchenko, L.L.; Matyushin, Y.N.; Inozemtsev, Y.O.; Tartakovskii, V.A. Energy characteristics of nitrooxazolidines and their radicals. Russ. Chem. Bull. Int. Ed. 2016, 65, 1876–1878. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Zaitseva, K.V.; Nagrimanov, R.N.; Solomonov, B.N.; Verevkin, S.P. Benchmark Thermondynamic Properties of Methyl- and Methoxy-Benzamides: Comprehensive Experimental and Theoretical Study. J. Phys. Chem. A 2016, 120, 8419–8429. [Google Scholar] [CrossRef] [PubMed]
- Gobble, C.; Gutterman, A.; Chickos, J.S. Some thermodynamic properties of benzocaine. Struct. Chem. 2013, 24, 1903–1907. [Google Scholar] [CrossRef]
- Keating, L.; Harris, H.H.; Chickos, J.S. Vapor pressures and vaporization enthalpy of (−) α-bisabolol and (dl) menthol by correlation gas chromatography. J. Chem. Thermodyn. 2017, 107, 18–25. [Google Scholar] [CrossRef]
- Sanchez-Buläs, T.; Cruz-Väsquez, O.; Hernändez-Obregon, J.; Rojas, A. Enthalpies of fusion, vaporisation and sublimation of crown ethers determined by thermogravimetry and differential scanning calorimetry. Thermochim. Acta 2017, 650, 123–133. [Google Scholar] [CrossRef]
- Panneerselvam, K.; Anthony, M.P.; Srinivasan, T.G.; Rao, P.R.V. Enthalpies of vaporization of N,N-dialkyl monamides at 298.15K. Thermochim. Acta 2009, 495, 1–4. [Google Scholar] [CrossRef]
- Gobble, C.; Walker, B.; Chickos, J.S. The Vaporization Enthalpy and Vapor Pressure of Fenpropidin and Phencyclidine (PCP) at T/K = 298.15 by Correlation Gas Chromatography. J. Chem. Eng. Data 2016, 61, 896–902. [Google Scholar] [CrossRef]
- Kozlovskiy, M.; Gobble, C.; Chickos, J.S. Vapor pressures and vaporization enthalpies of a series of esters used in flavors by correlation gas chromatography. J. Chem. Thermodyn. 2015, 86, 65–74. [Google Scholar] [CrossRef]
- Costa, J.C.S.; Lima, C.F.R.A.C.; Mendes, A.; Santos, L.M.N.B.F. Fluorination effect on the thermodynamic properties of long-chain hydrocarbons and alcohols. J. Chem. Thermodyn. 2016, 102, 378–385. [Google Scholar] [CrossRef]
- Simmons, D.; Chickos, J. Enthalpy of vaporization and vapor pressure of whiskey lactone and menthalactone by correlation gas chromatography. J. Chem. Thermodyn. 2017, 110, 65–70. [Google Scholar] [CrossRef]
- Oliveira, J.A.S.A.; Oliveira, T.S.M.; Gaspar, A.; Borges, F.; Ribeiro da Silva, M.D.M.C.; Monte, M.J.S. Study on the volatility of halogenated fluorenes. Chemosphere 2016, 157, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, R.; Chickos, J. An Examination of the Thermodynamics of Fusion, Vaporization, and Sublimation of Ibuprofen and Naproxen by Correlation Gas Chromatography. J. Pharm. Sci. 2012, 101, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Rath, N.; Gobble, C.; Chickos, J. Vaporization, Sublimation Enthalpy, and Crystal Structures of Imidazo[1,2-a]pyrazine and Phthalazine. J. Chem. Eng. Data 2016, 61, 370–379. [Google Scholar] [CrossRef]
- Goodrich, S.; Hasanovic, J.; Gobble, C.; Chickos, J.S. Vaporization Enthalpies and Vapor Pressures of Some Insect Pheromones by Correlation Gas Chromatography. J. Chem. Eng. Data 2016, 61, 1524–1530. [Google Scholar] [CrossRef]
- Freitas, V.L.S.; Silva, C.A.O.; Paiva, M.A.T.; Ribeiro da Silva, M.D.M.C. Energetic effects of alkyl groups (methyl and ethyl) on the nitrogen of the morpholine structure. J. Therm. Anal. Calorim. 2017, 121, 1059–1071. [Google Scholar] [CrossRef]
- Althoff, M.A.; Grieger, K.; Härtel, M.A.C.; Karaghiosoff, K.L.; Klapötke, T.M.; Metzulat, M. Application of the Transpiration Method to Determine the Vapor Pressure and Related Physico-Chemical Data of Low Volatile, Thermolabile, and Toxic Organo(thio)phosphates. J. Phys. Chem. A 2017, 121, 2603–2609. [Google Scholar] [CrossRef] [PubMed]
- Gobble, C.; Chickos, J.; Verevkin, S.P. Vapor Pressures and Vaporization Enthalpies of a Series of Dialkyl Phthalates by Correlation Gas Chromatography. J. Chem. Eng. Data 2014, 59, 1353–1365. [Google Scholar] [CrossRef]
- Brunetti, B.; Lapi, A.; Ciprioti, S.V. Thermodynamic study on six tricyclic nitrogen heterocyclic compounds by thermal analysis and effusion techniques. Thermochim. Acta 2016, 636, 71–84. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Kaliner, M.; Strassner, T.; Verevkin, S.P. Thermochemical properties of different 1-(R-phenyl)-1H-imidazoles. Fluid Phase Equilib. 2017, 433, 40–49. [Google Scholar] [CrossRef]
- Antón, V.; Artigas, H.; Muñoz-Embid, J.; Artal, M.; Lafuente, C. Thermophysical study of 2-acetylthiophene: Experimental and modelled results. Fluid Phase Equilib. 2017, 433, 126–134. [Google Scholar] [CrossRef]
- Portnova, S.V.; Krasnykh, E.L.; Levanova, S.V. Temperature Dependences of Saturated Vapor Pressure and the Enthalpy of Vaporization of n-Pentyl Esters of Dicarboxylic Acids. Russ. J. Phys. Chem. A 2016, 90, 990–993. [Google Scholar] [CrossRef]
- Lepori, L.; Matteoli, E.; Gianni, P. Vapor Pressure and Its Temperature Dependence of 28 Organic Compounds: Cyclic Amines, Cyclic Ethers, and Cyclic and Open Chain Secondary Alcohols. J. Chem. Eng. Data 2017, 62, 194–203. [Google Scholar] [CrossRef]
- Lima, C.F.R.A.C.; Rodrigues, A.S.M.C.; Santos, L.M.N.B.F. Effect of Confined Hindrance in Polyphenylbenzenes. J. Phys. Chem. A 2017, 121, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Abboud, J.-L.M.; Alkorta, I.; Davalos, J.Z.; Koppel, I.A.; Koppel, I.; Lenoir, D.; Martínez, S.; Mishima, M. The Thermodynamic Stability of Adamantylideneadamantane and Its Proton- and Electron-Exchanges. Comparison with Simple Alkenes. Bull. Chem. Soc. Jpn. 2016, 89, 762–769. [Google Scholar] [CrossRef]
- Silva, A.L.R.; Ribeira da Silva, M.D.M.C. Comprehensive Thermochemical Study of Cyclic Five- and Six-Membered N,N′-Thioureas. J. Chem. Eng. Data 2017. [Google Scholar] [CrossRef]
- Carvalho, T.M.T.; Amaral, L.M.P.F.; Morais, V.M.F.; Ribeiro da Silva, M.D.M.C. Energetic Effect of the Carboxylic Acid Functional Group in Indole Derivatives. J. Phys. Chem. A 2017, 121, 2980–2989. [Google Scholar] [CrossRef] [PubMed]
- Freitas, V.L.S.; Lima, A.C.M.O.; Sapei, E.; Ribeiro da Silva, M.D.M.C. Comprehensive thermophysical and thermochemical studies of vanillyl alcohol. J. Chem. Thermodyn. 2016, 102, 287–292. [Google Scholar] [CrossRef]
- Lopes, C.S.D.; Agapito, F.; Bernardes, C.E.S.; Minas da Piedade, M.E. Thermochemistry of 4-HOC6H4COR (R = H, CH3, C2H5, n-C3H7, n-C4H9, n-C5H11, and n-C6H13) compounds. J. Chem. Thermodyn. 2017, 104, 281–287. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Nagrimanov, R.N.; Solomonov, B.N.; Verevkin, S.P. Adamantanes: Benchmarking of thermochemical properties. J. Chem. Thermodyn. 2016, 101, 130–138. [Google Scholar] [CrossRef]
- Nagrimanov, R.N.; Solomonov, B.N.; Emel’yanenko, V.N.; Verevkin, S.P. Six-membered ring aliphatic compounds: A search for regularities in phase transitions. Thermochim. Acta 2016, 638, 80–88. [Google Scholar] [CrossRef]
- Blokhina, S.; Sharapova, A.; Ol’khovich, M.; Perlovich, G. Sublimation thermodynamics of four fluoroquinolone antimicrobial compounds. J. Chem. Thermodyn. 2017, 105, 37–43. [Google Scholar] [CrossRef]
- Flores, H.; Ledo, J.M.; Hernandez-Pérez, J.M.; Camarillo, E.A.; Sandoval-Lira, J.; Amador, M.P. Thermochemical and theoretical study of 2-oxazolidinone and 3-acetyl-2-oxazolidinone. J. Chem. Thermodyn. 2016, 102, 386–391. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Nagrimanov, R.N.; Verevkin, S.P. Benchmarking thermochemical experiments and calculations of nitrogen-containing substituted adamantanes. J. Thermal. Anal. Calorim. 2017, 128, 1535. [Google Scholar] [CrossRef]
- Oliveira, J.A.S.A.; Freitas, V.L.S.; Notario, R.; da Silva, M.D.M.C.R.; Monte, M.J.S. Thermodynamic properties of 2,7-di-tert-butylfluorene—An experimental and computational study. J. Chem. Thermodyn. 2016, 101, 115–122. [Google Scholar] [CrossRef]
- Carvalho, T.M.T.; Amaral, L.M.P.F.; Morais, V.M.F.; Ribeiro da Silva, M.D.M.C. Calorimetric and computational studies for three nitroimidazole isomers. J. Chem. Thermodyn. 2016, 105, 267–275. [Google Scholar] [CrossRef]
- Chickos, J.S.; Acree, W.E., Jr. Total phase change entropies and enthalpies. An update on fusion enthalpies and their estimation. Thermochim. Acta 2009, 495, 5–13. [Google Scholar] [CrossRef]
- Wang, L.; Xing, C.; Zhao, L.; Xu, L.; Liu, G. Measurement and correlation of solubility of 2-chloro-3-(trifluoromethyl)pyridine in pure solvents and ethanol + n-propanol mixtures. J. Mol. Liq. 2017, 238, 470–477. [Google Scholar] [CrossRef]
- Guenthner, A.J.; Ramirez, S.M.; Ford, M.D.; Soto, D.; Boatz, J.A.; Ghiassi, K.B.; Mabry, J.M. Organic Crystal Engineering of Thermosetting Cyanate Ester Monomers: Influence of Structure on Melting Point. Cryst. Growth Des. 2016, 16, 4082–4093. [Google Scholar] [CrossRef]
- Trache, D.; Khimeche, K.; Dahmani, A. Study of (Solid–Liquid) Phase Equilibria for Mixtures of Energetic Material Stabilizers and Prediction for Their Subsequent Performance. Int. J. Thermophys. 2013, 34, 226–239. [Google Scholar] [CrossRef]
- Eckert, K.-A.; Dasgupta, S.; Selge, B.; Ay, P. Solid liquid phase diagrams of binary fatty acid mixtures—Palmitic/stearic with oleic/linoleic/linolenic acid mixture. Thermochim. Acta 2016, 630, 50–63. [Google Scholar] [CrossRef]
- Blokhina, S.; Sharapova, A.; Ol’khovich, M.; Volkova, T.; Perlovich, G. Studying the sublimation thermodynamics of ethionamide and pyridinecarbothioamide isomers by transpiration method. Thermochim. Acta 2015, 622, 97–102. [Google Scholar] [CrossRef]
- Forte, A.; Melo, C.I.; Bogel-Lukasik, R.; Bogel-Lukasik, E. A favourable solubility of isoniazid, an antitubercular antibiotic drug, in alternative solvents. Fluid Phase Equil. 2012, 318, 89–95. [Google Scholar] [CrossRef]
- Carletta, A.; Meinguet, C.; Wouters, J.; Tilborg, A. Solid-State Investigation of Polymorphism and Tautomerism of Phenylthiazole-thione: A Combined Crystallographic, Calorimetric, and Theoretical Survey. Cryst. Growth Des. 2015, 15, 2461–2473. [Google Scholar] [CrossRef]
- Leitner, J.; Jurik, S. DSC study and thermodynamic modelling of the system paracetamol–o-acetylsalicylic acid. J. Therm. Anal. Calorim. 2017. [Google Scholar] [CrossRef]
- Mintz, C.; Clark, M.; Acree, W.E., Jr.; Abraham, M.H. Enthalpy of Solvation Correlations for Gaseous Solutes Dissolved in Water and in 1-Octanol Based on the Abraham Model. J. Chem. Inf. Model. 2007, 47, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Catalan, J.; Couto, A.; Gomez, J.; Saiz, J.L.; Laynez, J. Towards a solvent acidity scale: The calorimetry of the N-methyl imidazole probe. J. Chem. Soc. Perkin Trans. 2 1992, 7, 1181–1185. [Google Scholar] [CrossRef]
- Spencer, J.N.; Hovick, J.W. Solvation of urea and methyl-substituted ureas by water and DMF. Can. J. Chem. 1988, 66, 562–565. [Google Scholar] [CrossRef]
- Gatta, G.D.; Badea, E. Thermodynamics of Solvation of Urea and Some Monosubstituted N-Alkylureas in Water at 298.15 K. J. Chem. Eng. Data 2007, 52, 419–425. [Google Scholar] [CrossRef]
- Rouw, A.; Somsen, G. Solvation and Hydrophobic Hydration of Alkyl-substituted Ureas and Amides in N,N-Dimethylformamide + Water Mixtures. J. Chem. Soc. Faraday Trans. 1 1982, 78, 3397–3408. [Google Scholar] [CrossRef]
- Badea, E.; della Gatta, G.; Jozwiak, M.; Giancola, C. Hydration of Thiourea and Mono-, Di-, and Tetra-N-Alkylthioureas at Infinite Dilution: A Thermodynamic Study at a Temperature of 298.15 K. J. Chem. Eng. Data 2011, 56, 4778–4785. [Google Scholar] [CrossRef]
- Stimson, E.R.; Schrier, E.E. Calorimetric Investigation of Salt-Amide Interactions in Aqueous Solution. J. Chem. Eng. Data 1974, 19, 354–358. [Google Scholar] [CrossRef]
- Batov, D.V.; Zaichikov, A.M. Group Contributions to the Enthalpy Characteristics of Solutions of Formic and Acetic Acid Amides in Water-1,2-Propanediol Mixtures. Russ. J. Gen. Chem. 2003, 73, 511–518. [Google Scholar] [CrossRef]
- Starzewski, P.; Wadsö, I.; Zielenkiewicz, W. Enthalpies of vaporization of some N-alkylamides at 298.15 K. J. Chem. Thermodyn. 1984, 16, 331–334. [Google Scholar] [CrossRef]
- Morgan, K.M.; Kopp, D.A. Solvent effects on the stability of simple secondary amides. J. Chem. Soc. Perkin Trans. 2 1998, 2759–2763. [Google Scholar] [CrossRef]
- Teplitsky, A.B.; Glukhova, O.T.; Sukhodub, L.F.; Yanson, I.K.; Zielenkiewicz, A.; Zielenkiewicz, W.; Kosinski, J.; Wierzchowski, K.L. Thermochemistry of aqueous Solutions of alkylated Nucleic Acid Bases. IV. Enthalpies of 5-Alkyluracils. Biophys. Chem. 1982, 15, 139–147. [Google Scholar] [CrossRef]
- Zielenkiewicz, W.; Szterner, P.; Kaminski, M. Vapor Pressures, Molar Enthalpies of Sublimation, and Molar Enthalpies of Solution in Water of Selected Amino Derivatives of Uracil and 5-Nitrouracil. J. Chem. Eng. Data 2003, 48, 1132–1136. [Google Scholar] [CrossRef]
- Zielenkiewicz, W.; Szterner, P. Vapor Pressures, Molar Enthalpies of Sublimation, and Molar Enthalpies of Solution in Water of 5-(Trifluoromethyl)uracil. J. Chem. Eng. Data 2004, 49, 1197–1200. [Google Scholar] [CrossRef]
- Zielenkiewicz, W.; Szterner, P. Thermodynamic Investigation of Uracil and Its Halo Derivatives. Enthalpies of Solution and Solvation in Methanol. J. Chem. Eng. Data 2005, 50, 1139–1143. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Fang, B.; Guo, N.; Xiao, Y.; Hao, H.; Bao, Y.; Huang, X. Solubility and dissolution thermodynamic properties of 2-Cyano-4′-methylbiphenyl in binary solvent mixtures. J. Mol. Liq. 2017, 236, 298–307. [Google Scholar] [CrossRef]
- Zhang, Q.-A.; Du, C.-J. Solubility of cyclohexyl-phosphoramidic acid diphenyl ester in selected solvents. J. Mol. Liq. 2015, 211, 527–533. [Google Scholar] [CrossRef]
- Zhao, F.-Q.; Pei, C.; Hu, R.-Z.; Yang, L.; Zhang, Z.-Z.; Zhou, Y.-S.; Yang, X.-W.; Yin, G.; Gao, S.L.; Shi, Q.-Z. Thermochemical properties and non-isothermal decomposition reaction kinetics of 3,4-dinitrofurazanfuroxan (DNTF). J. Hazard. Mat. 2004, A113, 67–71. [Google Scholar]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | 8.61 | 3581 | 3581 | |
| 2 | B | C3 | 21.55 | 2 | 2 |
| 3 | B | N2Cl | 33.19 | 1 | 1 |
| 4 | B | NCl2 | 28.59 | 1 | 1 |
| 5 | B | O2Cl | 28.23 | 2 | 2 |
| 6 | B | OCl2 | 25.53 | 1 | 1 |
| 7 | B | S3 | 76.74 | 4 | 4 |
| 8 | C sp3 | H3C | 3.07 | 5380 | 2388 |
| 9 | C sp3 | H3N | 15.65 | 242 | 133 |
| 10 | C sp3 | H3N(+) | 31.33 | 2 | 2 |
| 11 | C sp3 | H3O | 16.71 | 372 | 263 |
| 12 | C sp3 | H3S | 14.44 | 31 | 25 |
| 13 | C sp3 | H3P | 9.04 | 6 | 4 |
| 14 | C sp3 | H3Si | 5.87 | 136 | 53 |
| 15 | C sp3 | H2BC | −3.07 | 6 | 2 |
| 16 | C sp3 | H2C2 | 4.67 | 10,588 | 2030 |
| 17 | C sp3 | H2CN | 15.00 | 430 | 243 |
| 18 | C sp3 | H2CN(+) | 29.15 | 10 | 9 |
| 19 | C sp3 | H2CO | 15.79 | 1147 | 779 |
| 20 | C sp3 | H2CS | 15.50 | 159 | 101 |
| 21 | C sp3 | H2CP | 6.67 | 6 | 2 |
| 22 | C sp3 | H2CF | 6.20 | 11 | 11 |
| 23 | C sp3 | H2CCl | 14.13 | 76 | 65 |
| 24 | C sp3 | H2CBr | 16.69 | 24 | 21 |
| 25 | C sp3 | H2CJ | 20.90 | 29 | 26 |
| 26 | C sp3 | H2CSi | 2.01 | 134 | 54 |
| 27 | C sp3 | H2N2 | 28.27 | 5 | 3 |
| 28 | C sp3 | H2NO | 20.46 | 4 | 4 |
| 29 | C sp3 | H2O2 | 27.43 | 19 | 16 |
| 30 | C sp3 | H2OS | 22.40 | 1 | 1 |
| 31 | C sp3 | H2OF | 18.90 | 1 | 1 |
| 32 | C sp3 | H2OCl | 23.06 | 2 | 2 |
| 33 | C sp3 | H2OSi | 10.30 | 1 | 1 |
| 34 | C sp3 | H2S2 | 24.08 | 2 | 2 |
| 35 | C sp3 | H2SSi | 6.66 | 9 | 9 |
| 36 | C sp3 | H2Si2 | 2.87 | 2 | 1 |
| 37 | C sp3 | HC3 | 3.54 | 939 | 615 |
| 38 | C sp3 | HC2N | 12.69 | 75 | 64 |
| 39 | C sp3 | HC2N(+) | 28.39 | 3 | 3 |
| 40 | C sp3 | HC2O | 14.99 | 243 | 203 |
| 41 | C sp3 | HC2S | 13.61 | 26 | 22 |
| 42 | C sp3 | HC2Si | 7.20 | 6 | 4 |
| 43 | C sp3 | HC2F | 5.96 | 7 | 6 |
| 44 | C sp3 | HC2Cl | 9.66 | 40 | 38 |
| 45 | C sp3 | HC2Br | 12.12 | 21 | 16 |
| 46 | C sp3 | HC2J | 18.79 | 4 | 4 |
| 47 | C sp3 | HCN2(+) | 47.10 | 3 | 3 |
| 48 | C sp3 | HCO2 | 25.39 | 25 | 22 |
| 49 | C sp3 | HCOCl | 20.93 | 1 | 1 |
| 50 | C sp3 | HCF2 | 7.10 | 15 | 14 |
| 51 | C sp3 | HCFCl | 12.61 | 15 | 15 |
| 52 | C sp3 | HCCl2 | 16.96 | 23 | 22 |
| 53 | C sp3 | HCClBr | 18.23 | 1 | 1 |
| 54 | C sp3 | HNO2 | 32.31 | 1 | 1 |
| 55 | C sp3 | HO3 | 37.33 | 4 | 4 |
| 56 | C sp3 | HOF2 | 17.06 | 7 | 7 |
| 57 | C sp3 | HOFCl | 20.49 | 1 | 1 |
| 58 | C sp3 | HSiCl2 | 23.89 | 1 | 1 |
| 59 | C sp3 | C4 | 1.92 | 335 | 274 |
| 60 | C sp3 | C3N | 12.60 | 28 | 23 |
| 61 | C sp3 | C3N(+) | 26.15 | 4 | 4 |
| 62 | C sp3 | C3O | 12.21 | 135 | 116 |
| 63 | C sp3 | C3S | 13.69 | 18 | 16 |
| 64 | C sp3 | C3F | 2.94 | 31 | 19 |
| 65 | C sp3 | C3Cl | 7.77 | 8 | 6 |
| 66 | C sp3 | C3Br | 11.95 | 3 | 3 |
| 67 | C sp3 | C3J | 19.63 | 2 | 2 |
| 68 | C sp3 | C2NO | 20.34 | 1 | 1 |
| 69 | C sp3 | C2NF | 8.88 | 1 | 1 |
| 70 | C sp3 | C2O2 | 23.16 | 35 | 27 |
| 71 | C sp3 | C2OF | 18.38 | 3 | 3 |
| 72 | C sp3 | C2F2 | 4.75 | 328 | 70 |
| 73 | C sp3 | C2FCl | 8.73 | 5 | 5 |
| 74 | C sp3 | C2Cl2 | 13.35 | 5 | 5 |
| 75 | C sp3 | CN3(+) | 46.89 | 3 | 3 |
| 76 | C sp3 | CNF2 | 15.25 | 15 | 6 |
| 77 | C sp3 | CNF2(+) | 30.77 | 3 | 2 |
| 78 | C sp3 | CN2F(+) | 28.25 | 4 | 3 |
| 79 | C sp3 | CO3 | 28.48 | 6 | 6 |
| 80 | C sp3 | COF2 | 13.65 | 36 | 30 |
| 81 | C sp3 | COCl2 | 20.61 | 4 | 4 |
| 82 | C sp3 | CSF2 | 12.70 | 2 | 1 |
| 83 | C sp3 | CF3 | 2.96 | 147 | 90 |
| 84 | C sp3 | CF2Cl | 6.64 | 10 | 9 |
| 85 | C sp3 | CF2Br | 9.02 | 5 | 4 |
| 86 | C sp3 | CFCl2 | 13.41 | 7 | 7 |
| 87 | C sp3 | CFClBr | 17.37 | 1 | 1 |
| 88 | C sp3 | CCl3 | 17.43 | 22 | 21 |
| 89 | C sp3 | NF3 | 14.48 | 5 | 4 |
| 90 | C sp3 | NF3(+) | −1.76 | 2 | 1 |
| 91 | C sp3 | N3F(+) | 32.36 | 1 | 1 |
| 92 | C sp3 | O4 | 38.15 | 2 | 2 |
| 93 | C sp3 | O2F2 | 24.80 | 14 | 2 |
| 94 | C sp3 | OF3 | 9.71 | 9 | 7 |
| 95 | C sp3 | OF2Cl | 17.84 | 2 | 2 |
| 96 | C sp3 | OCl3 | 27.40 | 2 | 2 |
| 97 | C sp3 | PF3 | 2.73 | 2 | 1 |
| 98 | C sp2 | H2=C | 2.17 | 182 | 170 |
| 99 | C sp2 | HC=C | 5.03 | 1314 | 694 |
| 100 | C sp2 | HC=N | 8.81 | 15 | 15 |
| 101 | C sp2 | HC=O | 11.44 | 122 | 122 |
| 102 | C sp2 | H=CN | 17.18 | 103 | 57 |
| 103 | C sp2 | H=CO | 10.25 | 35 | 32 |
| 104 | C sp2 | H=CS | 8.20 | 49 | 35 |
| 105 | C sp2 | H=CSi | 10.77 | 4 | 4 |
| 106 | C sp2 | H=CF | −0.09 | 1 | 1 |
| 107 | C sp2 | H=CCl | 10.38 | 8 | 6 |
| 108 | C sp2 | H=CBr | 13.73 | 1 | 1 |
| 109 | C sp2 | HN=N | 30.13 | 39 | 39 |
| 110 | C sp2 | HN=O | 34.46 | 6 | 6 |
| 111 | C sp2 | H=NO | 14.07 | 1 | 1 |
| 112 | C sp2 | H=NS | 18.07 | 2 | 2 |
| 113 | C sp2 | HO=O | 18.86 | 14 | 12 |
| 114 | C sp2 | C2=C | 5.27 | 220 | 190 |
| 115 | C sp2 | C2=N | 8.22 | 15 | 14 |
| 116 | C sp2 | C2=O | 13.59 | 149 | 140 |
| 117 | C sp2 | C=CN | 15.36 | 14 | 10 |
| 118 | C sp2 | C=CO | 12.54 | 39 | 31 |
| 119 | C sp2 | C2=S | 71.29 | 2 | 2 |
| 120 | C sp2 | C=CS | 9.45 | 29 | 24 |
| 121 | C sp2 | C=CF | 2.72 | 11 | 5 |
| 122 | C sp2 | C=CCl | 5.83 | 8 | 5 |
| 123 | C sp2 | C=CBr | 15.79 | 1 | 1 |
| 124 | C sp2 | =CN2 | 9.12 | 3 | 2 |
| 125 | C sp2 | CN=N | 28.80 | 16 | 16 |
| 126 | C sp2 | CN=N(+) | 11.32 | 2 | 2 |
| 127 | C sp2 | CN=O | 35.35 | 47 | 47 |
| 128 | C sp2 | C=NO | 22.79 | 5 | 5 |
| 129 | C sp2 | CN=S | 18.27 | 3 | 2 |
| 130 | C sp2 | C=NS | 17.49 | 1 | 1 |
| 131 | C sp2 | C=NCl | 11.93 | 1 | 1 |
| 132 | C sp2 | =CNCl | 22.67 | 2 | 1 |
| 133 | C sp2 | CO=O | 17.20 | 684 | 594 |
| 134 | C sp2 | =COS | 17.48 | 1 | 1 |
| 135 | C sp2 | C=OS | 12.33 | 9 | 9 |
| 136 | C sp2 | =COF | 15.53 | 1 | 1 |
| 137 | C sp2 | C=OCl | 15.41 | 11 | 9 |
| 138 | C sp2 | C=OBr | 22.28 | 3 | 3 |
| 139 | C sp2 | C=OJ | 25.82 | 2 | 2 |
| 140 | C sp2 | =CF2 | −0.26 | 3 | 3 |
| 141 | C sp2 | =CFCl | 9.81 | 3 | 2 |
| 142 | C sp2 | =CCl2 | 17.52 | 6 | 5 |
| 143 | C sp2 | N2=N | 29.25 | 2 | 2 |
| 144 | C sp2 | N2=O | 35.05 | 3 | 3 |
| 145 | C sp2 | N=NS | 13.50 | 5 | 5 |
| 146 | C sp2 | NO=O | 33.48 | 3 | 3 |
| 147 | C sp2 | =NOCl | 24.27 | 1 | 1 |
| 148 | C sp2 | NS=S | 44.39 | 2 | 2 |
| 149 | C sp2 | O2=O | 31.57 | 13 | 13 |
| 150 | C sp2 | O=OCl | 22.73 | 2 | 2 |
| 151 | C sp2 | S2=S | 34.03 | 1 | 1 |
| 152 | C aromatic | H:C2 | 4.64 | 4749 | 928 |
| 153 | C aromatic | H:C:N | 11.74 | 118 | 70 |
| 154 | C aromatic | H:C:N(+) | 22.04 | 2 | 1 |
| 155 | C aromatic | H:N2 | 15.36 | 7 | 5 |
| 156 | C aromatic | :C3 | 6.67 | 233 | 69 |
| 157 | C aromatic | C:C2 | 5.29 | 1053 | 618 |
| 158 | C aromatic | C:C:N | 9.94 | 38 | 30 |
| 159 | C aromatic | :C2N | 14.44 | 140 | 115 |
| 160 | C aromatic | :C2N(+) | 24.38 | 33 | 31 |
| 161 | C aromatic | :C2:N | 10.60 | 21 | 14 |
| 162 | C aromatic | :C2O | 8.04 | 443 | 253 |
| 163 | C aromatic | :C2S | 9.47 | 30 | 25 |
| 164 | C aromatic | :C2Si | 4.67 | 10 | 8 |
| 165 | C aromatic | :C2F | 4.45 | 143 | 72 |
| 166 | C aromatic | :C2Cl | 9.43 | 429 | 146 |
| 167 | C aromatic | :C2Br | 12.49 | 149 | 69 |
| 168 | C aromatic | :C2J | 19.48 | 29 | 26 |
| 169 | C aromatic | :CN:N | 16.72 | 2 | 2 |
| 170 | C aromatic | :C:NO | 13.67 | 4 | 3 |
| 171 | C aromatic | :C:NF | 14.34 | 1 | 1 |
| 172 | C aromatic | :C:NCl | 15.74 | 3 | 3 |
| 173 | C aromatic | :C:NBr | 25.24 | 1 | 1 |
| 174 | C aromatic | N:N2 | 20.19 | 5 | 2 |
| 175 | C aromatic | :N2O | 16.44 | 2 | 2 |
| 176 | C sp | H#C | 2.42 | 15 | 14 |
| 177 | C sp | C#C | 6.05 | 62 | 33 |
| 178 | C sp | =C2 | 5.50 | 4 | 4 |
| 179 | C sp | C#N | 17.38 | 72 | 70 |
| 180 | C sp | #CCl | 9.31 | 3 | 2 |
| 181 | C sp | =N=O | 10.44 | 6 | 5 |
| 182 | C sp | =N=S | 23.08 | 3 | 3 |
| 183 | N sp3 | H2C | 2.30 | 78 | 58 |
| 184 | N sp3 | H2C(pi) | 8.05 | 61 | 59 |
| 185 | N sp3 | H2N | 19.23 | 8 | 7 |
| 186 | N sp3 | H2S | 28.18 | 2 | 2 |
| 187 | N sp3 | HC2 | −11.34 | 59 | 56 |
| 188 | N sp3 | HC2(pi) | −1.94 | 27 | 26 |
| 189 | N sp3 | HC2(2pi) | −2.43 | 21 | 21 |
| 190 | N sp3 | HCN | −0.76 | 3 | 2 |
| 191 | N sp3 | HCN(pi) | −13.33 | 3 | 3 |
| 192 | N sp3 | HCN(2pi) | 4.97 | 1 | 1 |
| 193 | N sp3 | HCS(pi) | 5.34 | 7 | 7 |
| 194 | N sp3 | HCSi | −4.02 | 6 | 6 |
| 195 | N sp3 | HSi2 | 1.94 | 1 | 1 |
| 196 | N sp3 | BC2 | −31.30 | 3 | 2 |
| 197 | N sp3 | C3 | −30.50 | 111 | 101 |
| 198 | N sp3 | C3(pi) | −25.56 | 37 | 31 |
| 199 | N sp3 | C3(2pi) | −22.95 | 52 | 50 |
| 200 | N sp3 | C3(3pi) | −27.03 | 13 | 13 |
| 201 | N sp3 | C2N | −19.64 | 4 | 3 |
| 202 | N sp3 | C2N(+) | 0.00 | 1 | 1 |
| 203 | N sp3 | C2N(pi) | −27.16 | 3 | 2 |
| 204 | N sp3 | C2N(+)(pi) | 3.24 | 4 | 4 |
| 205 | N sp3 | C2N(2pi) | −24.28 | 4 | 4 |
| 206 | N sp3 | C2N(3pi) | −26.84 | 2 | 2 |
| 207 | N sp3 | C2O | 8.24 | 1 | 1 |
| 208 | N sp3 | C2P | −17.98 | 5 | 2 |
| 209 | N sp3 | C2Si | −19.79 | 12 | 8 |
| 210 | N sp3 | CN2(2pi) | −36.43 | 1 | 1 |
| 211 | N sp3 | CN2(+)(2pi) | 16.44 | 1 | 1 |
| 212 | N sp3 | CF2 | −4.56 | 2 | 2 |
| 213 | N sp3 | CF2(pi) | −12.61 | 1 | 1 |
| 214 | N sp3 | CSi2 | −17.81 | 1 | 1 |
| 215 | N sp3 | Si3 | −1.79 | 1 | 1 |
| 216 | N sp2 | H=C | 1.29 | 2 | 2 |
| 217 | N sp2 | C=C | −10.46 | 85 | 82 |
| 218 | N sp2 | C=N | −5.89 | 19 | 10 |
| 219 | N sp2 | C=N(+) | −2.79 | 15 | 13 |
| 220 | N sp2 | =CN | 18.81 | 9 | 9 |
| 221 | N sp2 | =CO | 10.27 | 17 | 14 |
| 222 | N sp2 | =CF | 0.00 | 1 | 1 |
| 223 | N sp2 | N=N | 15.91 | 5 | 3 |
| 224 | N sp2 | O=O | 0.59 | 7 | 7 |
| 225 | N aromatic | :C2 | −5.10 | 104 | 78 |
| 226 | N aromatic | :C:N | 5.35 | 8 | 4 |
| 227 | N(+) sp3 | C2NO(-) | 0.00 | 1 | 1 |
| 228 | N(+) sp2 | CO=O(-) | −2.09 | 78 | 56 |
| 229 | N(+) sp2 | C=NO(-) | −19.89 | 3 | 3 |
| 230 | N(+) sp2 | NO=O(-) | 0.35 | 6 | 5 |
| 231 | N(+) sp2 | O2=O(-) | 9.02 | 17 | 11 |
| 232 | N(+) aromatic | :C2O(-) | 0.00 | 1 | 1 |
| 233 | N(+) sp | C#C(-) | −8.48 | 2 | 2 |
| 234 | N(+) sp | =N2(-) | 5.96 | 12 | 10 |
| 235 | O | HC | 14.55 | 322 | 288 |
| 236 | O | HC(pi) | 20.98 | 174 | 157 |
| 237 | O | HN | 0.00 | 1 | 1 |
| 238 | O | HN(pi) | 19.03 | 2 | 2 |
| 239 | O | HO | 23.75 | 5 | 5 |
| 240 | O | HSi | 26.41 | 1 | 1 |
| 241 | O | BC | −17.91 | 5 | 3 |
| 242 | O | C2 | −17.86 | 424 | 270 |
| 243 | O | C2(pi) | −13.29 | 744 | 629 |
| 244 | O | C2(2pi) | −7.15 | 145 | 120 |
| 245 | O | CN(pi) | 0.00 | 7 | 7 |
| 246 | O | CN(+)(pi) | 2.17 | 17 | 11 |
| 247 | O | CN(2pi) | −2.82 | 9 | 9 |
| 248 | O | CO | −8.76 | 54 | 20 |
| 249 | O | CS | 2.45 | 18 | 9 |
| 250 | O | CP | −2.71 | 104 | 42 |
| 251 | O | CP(pi) | 1.25 | 7 | 5 |
| 252 | O | CSi | −11.39 | 79 | 29 |
| 253 | O | CSi(pi) | −14.85 | 37 | 13 |
| 254 | O | N2(2pi) | −0.72 | 3 | 3 |
| 255 | O | OSi | 4.23 | 9 | 4 |
| 256 | O | P2 | 16.68 | 1 | 1 |
| 257 | O | Si2 | −6.52 | 15 | 4 |
| 258 | P3 | C3 | −6.83 | 3 | 3 |
| 259 | P3 | C2O | 2.71 | 1 | 1 |
| 260 | P3 | N3 | −7.09 | 1 | 1 |
| 261 | P3 | N2Cl | 10.64 | 1 | 1 |
| 262 | P3 | O3 | −4.07 | 1 | 1 |
| 263 | P4 | HO2=O | 9.23 | 2 | 2 |
| 264 | P4 | CO2=O | 5.40 | 3 | 3 |
| 265 | P4 | O3=O | −3.86 | 16 | 15 |
| 266 | P4 | O3=S | 1.10 | 9 | 9 |
| 267 | P4 | O2=OS | 1.77 | 4 | 4 |
| 268 | P4 | O2S=S | 1.73 | 8 | 8 |
| 269 | S2 | HC | 1.49 | 33 | 29 |
| 270 | S2 | HC(pi) | 6.23 | 1 | 1 |
| 271 | S2 | HP | 23.50 | 3 | 3 |
| 272 | S2 | BC | −24.53 | 12 | 4 |
| 273 | S2 | C2 | −10.51 | 67 | 65 |
| 274 | S2 | C2(pi) | −2.71 | 23 | 22 |
| 275 | S2 | C2(2pi) | 0.53 | 44 | 44 |
| 276 | S2 | CS | −0.35 | 16 | 8 |
| 277 | S2 | CS(pi) | 2.39 | 2 | 1 |
| 278 | S2 | CP | −1.99 | 9 | 9 |
| 279 | S2 | Si2 | −3.40 | 1 | 1 |
| 280 | S4 | C2=O | 22.60 | 4 | 4 |
| 281 | S4 | C2=O2 | 27.80 | 9 | 9 |
| 282 | S4 | C2F2 | −5.92 | 1 | 1 |
| 283 | S4 | CN=O2 | 1.94 | 9 | 9 |
| 284 | S4 | C=O2S | 37.54 | 2 | 1 |
| 285 | S4 | O2=O | −3.83 | 5 | 5 |
| 286 | S4 | O2=O2 | 4.79 | 4 | 4 |
| 287 | Si | H3C | 0.00 | 1 | 1 |
| 288 | Si | H2CN | 2.20 | 1 | 1 |
| 289 | Si | HC3 | −4.21 | 24 | 24 |
| 290 | Si | HC2O | 2.36 | 2 | 1 |
| 291 | Si | HC2S | 0.00 | 2 | 1 |
| 292 | Si | HCO2 | 8.33 | 5 | 1 |
| 293 | Si | HN3 | 8.01 | 2 | 2 |
| 294 | Si | C4 | −0.57 | 21 | 20 |
| 295 | Si | C3N | −1.80 | 18 | 14 |
| 296 | Si | C3O | 0.35 | 6 | 6 |
| 297 | Si | C2O2 | 5.64 | 18 | 11 |
| 298 | Si | CO3 | −2.40 | 26 | 26 |
| 299 | Si | O4 | −16.14 | 6 | 6 |
| 300 | H | H Acceptor | −12.45 | 16 | 16 |
| 301 | Alkane | No. of C atoms | 0.09 | 3072 | 286 |
| 302 | Unsaturated HC | No. of C atoms | −0.07 | 4100 | 413 |
| A | Based on | Valid groups | 185 | 3581 | |
| B | Goodness of fit | R2 | 0.9678 | 3460 | |
| C | Deviation | Average | 2.99 | 3460 | |
| D | Deviation | Standard | 4.30 | 3460 | |
| E | K-fold cv | K | 10 | 3386 | |
| F | Goodness of fit | Q2 | 0.9641 | 3386 | |
| G | Deviation | Average (cv) | 3.14 | 3386 | |
| H | Deviation | Standard (cv) | 4.56 | 3386 |
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | 21.03 | 1960 | 1960 | |
| 2 | B | C3 | 65.82 | 2 | 2 |
| 3 | C sp3 | H3C | 5.99 | 1322 | 623 |
| 4 | C sp3 | H3N | 26.96 | 143 | 87 |
| 5 | C sp3 | H3N(+) | 98.98 | 1 | 1 |
| 6 | C sp3 | H3O | 28.51 | 181 | 122 |
| 7 | C sp3 | H3S | 30.06 | 7 | 6 |
| 8 | C sp3 | H2C2 | 6.88 | 2602 | 508 |
| 9 | C sp3 | H2CN | 21.98 | 224 | 116 |
| 10 | C sp3 | H2CN(+) | 27.46 | 13 | 11 |
| 11 | C sp3 | H2CO | 29.62 | 242 | 134 |
| 12 | C sp3 | H2CS | 23.29 | 50 | 31 |
| 13 | C sp3 | H2CF | 15.91 | 1 | 1 |
| 14 | C sp3 | H2CCl | 17.59 | 3 | 3 |
| 15 | C sp3 | H2CBr | 22.76 | 5 | 4 |
| 16 | C sp3 | H2CJ | 21.83 | 3 | 2 |
| 17 | C sp3 | H2N2 | 43.95 | 18 | 6 |
| 18 | C sp3 | H2NCl | 36.29 | 1 | 1 |
| 19 | C sp3 | H2O2 | 53.35 | 25 | 13 |
| 20 | C sp3 | H2OS | 54.78 | 1 | 1 |
| 21 | C sp3 | H2S2 | 47.45 | 6 | 4 |
| 22 | C sp3 | HBC2 | −36.17 | 3 | 1 |
| 23 | C sp3 | HC3 | 2.28 | 509 | 190 |
| 24 | C sp3 | HC2N | 14.28 | 34 | 30 |
| 25 | C sp3 | HC2N(+) | 21.01 | 9 | 9 |
| 26 | C sp3 | HC2O | 24.27 | 82 | 47 |
| 27 | C sp3 | HC2S | 17.59 | 14 | 11 |
| 28 | C sp3 | HC2F | 5.18 | 1 | 1 |
| 29 | C sp3 | HC2Cl | 11.49 | 7 | 2 |
| 30 | C sp3 | HC2Br | −0.95 | 1 | 1 |
| 31 | C sp3 | HCN2 | 39.48 | 8 | 2 |
| 32 | C sp3 | HCN2(+) | 39.93 | 2 | 2 |
| 33 | C sp3 | HCNO | 34.73 | 2 | 1 |
| 34 | C sp3 | HCNS | 20.56 | 2 | 1 |
| 35 | C sp3 | HCO2 | 39.96 | 3 | 3 |
| 36 | C sp3 | HCF2 | −0.19 | 1 | 1 |
| 37 | C sp3 | HCCl2 | 15.78 | 1 | 1 |
| 38 | C sp3 | HN3(+) | 37.31 | 1 | 1 |
| 39 | C sp3 | HO3 | 72.23 | 3 | 3 |
| 40 | C sp3 | C4 | −4.25 | 209 | 137 |
| 41 | C sp3 | C3N | 5.87 | 18 | 13 |
| 42 | C sp3 | C3N(+) | 18.44 | 14 | 11 |
| 43 | C sp3 | C3O | 15.18 | 40 | 31 |
| 44 | C sp3 | C3S | 6.40 | 5 | 5 |
| 45 | C sp3 | C3F | 1.89 | 3 | 3 |
| 46 | C sp3 | C3Cl | −8.06 | 1 | 1 |
| 47 | C sp3 | C3Br | 2.34 | 1 | 1 |
| 48 | C sp3 | C2N2(+) | 34.78 | 7 | 6 |
| 49 | C sp3 | C2O2 | 39.73 | 8 | 8 |
| 50 | C sp3 | C2S2 | 37.28 | 4 | 1 |
| 51 | C sp3 | C2F2 | 7.07 | 62 | 8 |
| 52 | C sp3 | CN3(+) | 43.89 | 19 | 12 |
| 53 | C sp3 | CN2F(+) | 25.98 | 1 | 1 |
| 54 | C sp3 | CO3 | 57.42 | 2 | 2 |
| 55 | C sp3 | CF3 | −4.71 | 27 | 23 |
| 56 | C sp3 | CCl3 | 16.10 | 3 | 2 |
| 57 | C sp3 | N3F(+) | 44.00 | 1 | 1 |
| 58 | C sp3 | O4 | 73.43 | 1 | 1 |
| 59 | C sp2 | H2=C | 7.97 | 12 | 12 |
| 60 | C sp2 | HC=C | 5.10 | 452 | 213 |
| 61 | C sp2 | HC=N | 35.49 | 21 | 19 |
| 62 | C sp2 | HC=N(+) | 72.64 | 7 | 7 |
| 63 | C sp2 | H=CN | 32.79 | 83 | 69 |
| 64 | C sp2 | HC=O | 20.74 | 15 | 15 |
| 65 | C sp2 | H=CO | 16.89 | 16 | 14 |
| 66 | C sp2 | H=CS | 15.22 | 49 | 36 |
| 67 | C sp2 | HN=N | 55.52 | 19 | 18 |
| 68 | C sp2 | HN=O | 35.41 | 4 | 3 |
| 69 | C sp2 | H=NO | 40.91 | 1 | 1 |
| 70 | C sp2 | H=NS | 33.85 | 2 | 2 |
| 71 | C sp2 | C2=C | 3.91 | 78 | 61 |
| 72 | C sp2 | C2=N | 30.47 | 35 | 26 |
| 73 | C sp2 | C2=N(+) | 13.76 | 5 | 5 |
| 74 | C sp2 | C=CN | 26.81 | 57 | 48 |
| 75 | C sp2 | C=CN(+) | 41.65 | 7 | 7 |
| 76 | C sp2 | C2=O | 15.10 | 200 | 161 |
| 77 | C sp2 | C=CO | 22.08 | 40 | 31 |
| 78 | C sp2 | C2=S | 18.21 | 3 | 3 |
| 79 | C sp2 | C=CS | 15.64 | 36 | 27 |
| 80 | C sp2 | C=CF | 16.81 | 2 | 2 |
| 81 | C sp2 | C=CCl | 11.02 | 9 | 5 |
| 82 | C sp2 | C=CBr | 34.06 | 2 | 2 |
| 83 | C sp2 | C=CJ | 32.46 | 1 | 1 |
| 84 | C sp2 | =CN2 | 64.94 | 6 | 6 |
| 85 | C sp2 | =CN2(+) | 60.65 | 4 | 4 |
| 86 | C sp2 | CN=N | 54.51 | 27 | 25 |
| 87 | C sp2 | CN=N(+) | 44.16 | 3 | 3 |
| 88 | C sp2 | CN=O | 39.66 | 234 | 194 |
| 89 | C sp2 | C=NO | 42.74 | 2 | 2 |
| 90 | C sp2 | CN=S | 39.85 | 8 | 7 |
| 91 | C sp2 | C=NS | 34.89 | 1 | 1 |
| 92 | C sp2 | =CNS(+) | 41.29 | 2 | 2 |
| 93 | C sp2 | =CNCl | 38.14 | 4 | 3 |
| 94 | C sp2 | CO=O | 34.06 | 424 | 345 |
| 95 | C sp2 | CO=O(-) | 80.89 | 22 | 22 |
| 96 | C sp2 | C=OCl | 29.03 | 1 | 1 |
| 97 | C sp2 | CS=S | 56.97 | 3 | 3 |
| 98 | C sp2 | N2=N | 80.72 | 4 | 4 |
| 99 | C sp2 | N2=N(+) | 65.95 | 6 | 5 |
| 100 | C sp2 | N2=O | 59.57 | 76 | 70 |
| 101 | C sp2 | N2=S | 66.62 | 29 | 29 |
| 102 | C sp2 | N=NS | 51.62 | 22 | 22 |
| 103 | C sp2 | NO=O | 52.79 | 8 | 8 |
| 104 | C sp2 | =NO2 | 61.12 | 1 | 1 |
| 105 | C sp2 | N=OS | 48.27 | 1 | 1 |
| 106 | C sp2 | NO=S | 58.04 | 11 | 11 |
| 107 | C sp2 | =NOS | 52.75 | 1 | 1 |
| 108 | C sp2 | NS=S | 60.83 | 5 | 3 |
| 109 | C sp2 | =NS2 | 64.37 | 1 | 1 |
| 110 | C sp2 | O2=O | 41.40 | 7 | 7 |
| 111 | C sp2 | =OS2 | 41.22 | 2 | 2 |
| 112 | C sp2 | OS=S | 73.06 | 1 | 1 |
| 113 | C sp2 | S2=S | 49.39 | 5 | 5 |
| 114 | C aromatic | H:C2 | 5.36 | 7115 | 1269 |
| 115 | C aromatic | H:C:N | 18.20 | 150 | 96 |
| 116 | C aromatic | H:C:N(+) | 28.26 | 48 | 28 |
| 117 | C aromatic | H:N2 | 23.27 | 7 | 5 |
| 118 | C aromatic | B:C2 | −25.04 | 3 | 1 |
| 119 | C aromatic | :C3 | 5.51 | 454 | 155 |
| 120 | C aromatic | C:C2 | 3.12 | 1684 | 835 |
| 121 | C aromatic | C:C:N | 11.10 | 80 | 48 |
| 122 | C aromatic | C:C:N(+) | 16.04 | 33 | 21 |
| 123 | C aromatic | :C2N | 22.21 | 354 | 258 |
| 124 | C aromatic | :C2N(+) | 28.67 | 169 | 134 |
| 125 | C aromatic | :C2:N | 17.03 | 79 | 61 |
| 126 | C aromatic | :C2:N(+) | 18.05 | 35 | 20 |
| 127 | C aromatic | :C2O | 20.46 | 617 | 387 |
| 128 | C aromatic | :C2P | −1.63 | 12 | 4 |
| 129 | C aromatic | :C2S | 16.31 | 80 | 64 |
| 130 | C aromatic | :C2F | 4.45 | 77 | 42 |
| 131 | C aromatic | :C2Cl | 12.48 | 424 | 166 |
| 132 | C aromatic | :C2Br | 14.66 | 63 | 43 |
| 133 | C aromatic | :C2J | 20.68 | 31 | 27 |
| 134 | C aromatic | :C2Si | 4.80 | 10 | 2 |
| 135 | C aromatic | C:N2 | 28.80 | 4 | 2 |
| 136 | C aromatic | :CN:N | 29.72 | 11 | 9 |
| 137 | C aromatic | :CN:N(+) | 33.74 | 3 | 2 |
| 138 | C aromatic | :C:NO | 41.44 | 13 | 12 |
| 139 | C aromatic | :C:NO(+) | 33.50 | 5 | 5 |
| 140 | C aromatic | :C:NCl | 21.70 | 18 | 13 |
| 141 | C aromatic | :C:NBr | 31.31 | 3 | 2 |
| 142 | C aromatic | N:N2 | 43.11 | 13 | 8 |
| 143 | C aromatic | :N2O | 39.92 | 3 | 1 |
| 144 | C aromatic | :N2S | 36.08 | 3 | 3 |
| 145 | C aromatic | :N2Cl | 35.90 | 3 | 3 |
| 146 | C sp | =C2 | 6.39 | 3 | 2 |
| 147 | C sp | C#C | 3.24 | 14 | 7 |
| 148 | C sp | C#N | 16.49 | 96 | 67 |
| 149 | C sp | C#N(+) | 11.33 | 4 | 3 |
| 150 | C sp | #CS | 28.03 | 2 | 2 |
| 151 | C sp | N#N | 47.80 | 1 | 1 |
| 152 | C sp | #NP | 12.53 | 3 | 1 |
| 153 | N sp3 | H2C | 5.03 | 23 | 12 |
| 154 | N sp3 | H2C(pi) | 6.38 | 223 | 199 |
| 155 | N sp3 | H2N | 17.97 | 10 | 8 |
| 156 | N sp3 | H2S | 41.98 | 1 | 1 |
| 157 | N sp3 | HC2 | −23.83 | 14 | 13 |
| 158 | N sp3 | HC2(pi) | −13.51 | 72 | 55 |
| 159 | N sp3 | HC2(2pi) | −20.10 | 200 | 165 |
| 160 | N sp3 | HCN | −0.15 | 2 | 1 |
| 161 | N sp3 | HCN(pi) | 6.71 | 14 | 9 |
| 162 | N sp3 | HCN(2pi) | −6.84 | 25 | 25 |
| 163 | N sp3 | HCS(pi) | −15.10 | 20 | 20 |
| 164 | N sp3 | C3 | −51.07 | 16 | 11 |
| 165 | N sp3 | C3(pi) | −53.90 | 59 | 49 |
| 166 | N sp3 | C3(2pi) | −60.80 | 72 | 54 |
| 167 | N sp3 | C3(3pi) | −61.26 | 18 | 14 |
| 168 | N sp3 | C2N(pi) | −7.05 | 6 | 3 |
| 169 | N sp3 | C2N(+)(pi) | −5.52 | 24 | 9 |
| 170 | N sp3 | C2N(2pi) | −36.36 | 4 | 4 |
| 171 | N sp3 | C2N(+)(2pi) | −20.13 | 1 | 1 |
| 172 | N sp3 | C2N(3pi) | −54.74 | 3 | 3 |
| 173 | N sp3 | C2S | −49.13 | 4 | 2 |
| 174 | N sp3 | C2F(2pi) | −64.78 | 1 | 1 |
| 175 | N sp3 | CN2(pi) | 30.74 | 4 | 3 |
| 176 | N sp3 | CN2(2pi) | −49.40 | 3 | 3 |
| 177 | N sp3 | CN2(+)(2pi) | 3.72 | 1 | 1 |
| 178 | N sp3 | CNF(2pi) | −34.74 | 5 | 4 |
| 179 | N sp2 | C=C | −32.77 | 79 | 74 |
| 180 | N sp2 | C=N | −4.54 | 13 | 9 |
| 181 | N sp2 | C=N(+) | −15.43 | 5 | 5 |
| 182 | N sp2 | =CN | −4.63 | 38 | 36 |
| 183 | N sp2 | =CN(+) | 36.68 | 1 | 1 |
| 184 | N sp2 | C=O | −12.04 | 9 | 9 |
| 185 | N sp2 | C=P | −49.18 | 1 | 1 |
| 186 | N sp2 | =CO | −16.24 | 18 | 13 |
| 187 | N sp2 | =CS | −26.78 | 10 | 8 |
| 188 | N sp2 | N=N | 12.19 | 21 | 13 |
| 189 | N sp2 | N=O | 0.00 | 10 | 6 |
| 190 | N sp2 | =NO | −6.67 | 2 | 1 |
| 191 | N aromatic | :C2 | −14.01 | 208 | 145 |
| 192 | N aromatic | :C:N | −4.98 | 4 | 2 |
| 193 | N(+) sp3 | H3C | 2.77 | 13 | 13 |
| 194 | N(+) sp3 | H2C2 | −82.36 | 3 | 3 |
| 195 | N(+) sp2 | C=CO(-) | −68.61 | 7 | 7 |
| 196 | N(+) sp2 | C=NO | −26.37 | 10 | 5 |
| 197 | N(+) sp2 | C=NO(-) | −11.30 | 3 | 3 |
| 198 | N(+) sp2 | CO=O(-) | −4.38 | 270 | 163 |
| 199 | N(+) sp2 | =CO2(-) | 2.17 | 5 | 5 |
| 200 | N(+) sp2 | NO=O(-) | 0.15 | 28 | 12 |
| 201 | N(+) sp2 | O2=O(-) | 6.00 | 14 | 6 |
| 202 | N(+) aromatic | H:C2 | −46.79 | 6 | 6 |
| 203 | N(+) aromatic | :C2O(-) | −7.10 | 56 | 40 |
| 204 | N(+) sp | C#C(-) | −14.36 | 3 | 3 |
| 205 | N(+) sp | #CO(-) | 0.00 | 4 | 3 |
| 206 | N(+) sp | =N2(-) | 19.14 | 2 | 2 |
| 207 | O | HC | 4.49 | 143 | 92 |
| 208 | O | HC(pi) | 8.19 | 560 | 470 |
| 209 | O | HN(pi) | 2.28 | 4 | 3 |
| 210 | O | HO | 29.95 | 4 | 4 |
| 211 | O | C2 | −39.23 | 94 | 37 |
| 212 | O | C2(pi) | −31.33 | 292 | 201 |
| 213 | O | C2(2pi) | −24.06 | 147 | 121 |
| 214 | O | CN(pi) | 0.00 | 2 | 1 |
| 215 | O | CN(+)(pi) | 0.00 | 14 | 6 |
| 216 | O | CN(2pi) | 4.91 | 1 | 1 |
| 217 | O | CO(pi) | −27.16 | 8 | 6 |
| 218 | O | CP(pi) | −16.12 | 3 | 1 |
| 219 | O | N2(2pi) | 5.87 | 4 | 4 |
| 220 | O | N2(+)(2pi) | 6.27 | 5 | 5 |
| 221 | P3 | C3 | 16.70 | 2 | 2 |
| 222 | P3 | S3 | −66.68 | 1 | 1 |
| 223 | P4 | C3=N | 0.00 | 1 | 1 |
| 224 | P4 | C3=O | −30.50 | 1 | 1 |
| 225 | P4 | C3=S | 46.30 | 1 | 1 |
| 226 | P4 | O3=O | 0.00 | 1 | 1 |
| 227 | S2 | HC | −2.58 | 1 | 1 |
| 228 | S2 | HC(pi) | 18.47 | 2 | 2 |
| 229 | S2 | C2 | −22.69 | 19 | 12 |
| 230 | S2 | C2(pi) | −15.86 | 34 | 29 |
| 231 | S2 | C2(2pi) | −7.94 | 59 | 49 |
| 232 | S2 | CN(pi) | 25.96 | 1 | 1 |
| 233 | S2 | CN(2pi) | −6.82 | 6 | 6 |
| 234 | S2 | CS(pi) | −6.16 | 8 | 4 |
| 235 | S2 | CP(pi) | 0.00 | 3 | 1 |
| 236 | S2 | N2 | −2.00 | 1 | 1 |
| 237 | S2 | N2(2pi) | 21.36 | 2 | 2 |
| 238 | S2 | NS | 1.00 | 2 | 1 |
| 239 | S4 | C2=O | −5.89 | 2 | 2 |
| 240 | S4 | C2=O2 | −4.26 | 27 | 27 |
| 241 | S4 | CN=O2 | 9.20 | 20 | 20 |
| 242 | Si | C4 | 2.02 | 1 | 1 |
| 243 | Si | C3Si | −0.67 | 2 | 1 |
| 244 | H | H Acceptor | −8.63 | 107 | 89 |
| 245 | Alkane | No. of C atoms | −0.53 | 849 | 59 |
| 246 | Unsaturated HC | No. of C atoms | −0.10 | 2679 | 148 |
| A | Based on | Valid groups | 154 | 1960 | |
| B | Goodness of fit | R2 | 0.8887 | 1866 | |
| C | Deviation | Average | 7.81 | 1866 | |
| D | Deviation | Standard | 10.33 | 1866 | |
| E | K-fold cv | K | 10 | 1791 | |
| F | Goodness of fit | Q2 | 0.8657 | 1791 | |
| G | Deviation | Average (cv) | 8.56 | 1791 | |
| H | Deviation | Standard (cv) | 11.39 | 1791 |
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | −13.33 | 436 | 436 | |
| 2 | C sp3 | H3C | −4.44 | 483 | 265 |
| 3 | C sp3 | H3N | −31.51 | 47 | 28 |
| 4 | C sp3 | H3N(+) | −31.22 | 1 | 1 |
| 5 | C sp3 | H3O | −15.38 | 34 | 29 |
| 6 | C sp3 | H3S | −12.79 | 7 | 4 |
| 7 | C sp3 | H2C2 | −3.86 | 506 | 186 |
| 8 | C sp3 | H2CN | −31.29 | 55 | 37 |
| 9 | C sp3 | H2CN(+) | −22.60 | 2 | 2 |
| 10 | C sp3 | H2CO | −15.26 | 178 | 90 |
| 11 | C sp3 | H2CS | −12.03 | 9 | 6 |
| 12 | C sp3 | H2CF | −6.02 | 1 | 1 |
| 13 | C sp3 | H2CCl | −8.52 | 15 | 11 |
| 14 | C sp3 | H2CBr | −11.73 | 1 | 1 |
| 15 | C sp3 | H2CJ | −13.80 | 2 | 2 |
| 16 | C sp3 | H2O2 | −14.86 | 1 | 1 |
| 17 | C sp3 | HC3 | −2.51 | 45 | 35 |
| 18 | C sp3 | HC2N | −29.99 | 6 | 5 |
| 19 | C sp3 | HC2N(+) | −20.74 | 1 | 1 |
| 20 | C sp3 | HC2O | −14.95 | 32 | 29 |
| 21 | C sp3 | HC2F | −5.77 | 1 | 1 |
| 22 | C sp3 | HC2Cl | −8.53 | 1 | 1 |
| 23 | C sp3 | HC2J | −14.39 | 1 | 1 |
| 24 | C sp3 | HCF2 | −5.07 | 3 | 3 |
| 25 | C sp3 | HCCl2 | −11.02 | 5 | 4 |
| 26 | C sp3 | C4 | 0.43 | 10 | 10 |
| 27 | C sp3 | C3N | −24.37 | 3 | 3 |
| 28 | C sp3 | C3O | −16.23 | 6 | 6 |
| 29 | C sp3 | C3Cl | −1.29 | 1 | 1 |
| 30 | C sp3 | C3Br | 1.24 | 1 | 1 |
| 31 | C sp3 | C3J | −7.51 | 1 | 1 |
| 32 | C sp3 | C2F2 | −5.12 | 2 | 2 |
| 33 | C sp3 | COF2 | 0.74 | 1 | 1 |
| 34 | C sp3 | CF3 | −2.85 | 11 | 9 |
| 35 | C sp3 | CF2Cl | −3.44 | 3 | 2 |
| 36 | C sp3 | CFCl2 | −12.04 | 1 | 1 |
| 37 | C sp3 | CCl3 | −12.64 | 2 | 2 |
| 38 | C sp2 | H2=C | −2.93 | 15 | 13 |
| 39 | C sp2 | HC=C | −2.16 | 26 | 20 |
| 40 | C sp2 | HC=O | −16.45 | 9 | 9 |
| 41 | C sp2 | H=CN | −13.78 | 17 | 13 |
| 42 | C sp2 | H=CO | −10.21 | 1 | 1 |
| 43 | C sp2 | H=CS | −6.13 | 2 | 1 |
| 44 | C sp2 | H=CCl | −7.34 | 5 | 3 |
| 45 | C sp2 | HN=N | −10.70 | 2 | 2 |
| 46 | C sp2 | HN=O | −33.05 | 4 | 4 |
| 47 | C sp2 | HO=O | −14.45 | 7 | 7 |
| 48 | C sp2 | C2=C | 1.28 | 11 | 11 |
| 49 | C sp2 | C=CN | −15.51 | 2 | 2 |
| 50 | C sp2 | C=CN(+) | −39.48 | 1 | 1 |
| 51 | C sp2 | C2=O | −17.65 | 20 | 20 |
| 52 | C sp2 | C=CF | −6.97 | 2 | 2 |
| 53 | C sp2 | C=CCl | −31.39 | 1 | 1 |
| 54 | C sp2 | C=CBr | −28.79 | 1 | 1 |
| 55 | C sp2 | C=CJ | −31.42 | 1 | 1 |
| 56 | C sp2 | =CN2 | −32.45 | 3 | 3 |
| 57 | C sp2 | CN=O | −39.35 | 30 | 30 |
| 58 | C sp2 | =CNCl | −30.33 | 1 | 1 |
| 59 | C sp2 | CO=O | −17.24 | 63 | 52 |
| 60 | C sp2 | =CF2 | 0.44 | 3 | 2 |
| 61 | C sp2 | =CCl2 | −11.89 | 2 | 2 |
| 62 | C sp2 | N2=O | −35.29 | 25 | 25 |
| 63 | C sp2 | N2=S | −41.79 | 6 | 6 |
| 64 | C aromatic | H:C2 | −2.84 | 437 | 100 |
| 65 | C aromatic | H:C:N | −14.82 | 29 | 18 |
| 66 | C aromatic | :C3 | −3.23 | 13 | 6 |
| 67 | C aromatic | C:C2 | −1.72 | 90 | 63 |
| 68 | C aromatic | C:C:N | −15.13 | 7 | 6 |
| 69 | C aromatic | :C2N | −10.35 | 13 | 13 |
| 70 | C aromatic | :C2N(+) | −21.83 | 6 | 6 |
| 71 | C aromatic | :C2:N | −15.19 | 1 | 1 |
| 72 | C aromatic | :C2O | −9.63 | 21 | 17 |
| 73 | C aromatic | :C2F | −1.79 | 1 | 1 |
| 74 | C aromatic | :C2Cl | −3.91 | 37 | 19 |
| 75 | C aromatic | :C2Br | −5.99 | 1 | 1 |
| 76 | C aromatic | :CN:N | −16.20 | 1 | 1 |
| 77 | C sp | H#C | −1.37 | 1 | 1 |
| 78 | C sp | C#C | 0.00 | 1 | 1 |
| 79 | C sp | C#N | −17.66 | 15 | 12 |
| 80 | N sp3 | H2C | −2.40 | 25 | 20 |
| 81 | N sp3 | H2C(pi) | −16.13 | 32 | 30 |
| 82 | N sp3 | HC2 | 24.30 | 6 | 6 |
| 83 | N sp3 | HC2(pi) | 11.97 | 26 | 22 |
| 84 | N sp3 | HC2(2pi) | 3.09 | 21 | 12 |
| 85 | N sp3 | C3 | 57.51 | 5 | 5 |
| 86 | N sp3 | C3(pi) | 52.51 | 10 | 9 |
| 87 | N sp3 | C3(2pi) | 36.53 | 13 | 8 |
| 88 | N sp2 | C=C | −19.81 | 2 | 2 |
| 89 | N aromatic | :C2 | 5.38 | 19 | 19 |
| 90 | N(+) sp2 | CO=O(-) | 8.85 | 11 | 11 |
| 91 | O | HC | −17.23 | 61 | 50 |
| 92 | O | HC(pi) | −18.29 | 32 | 26 |
| 93 | O | HO | −22.54 | 2 | 1 |
| 94 | O | C2 | 8.60 | 68 | 39 |
| 95 | O | C2(pi) | 10.97 | 56 | 49 |
| 96 | O | C2(2pi) | 9.97 | 2 | 2 |
| 97 | S2 | HC | 1.98 | 4 | 4 |
| 98 | S2 | C2 | 6.62 | 3 | 3 |
| 99 | S2 | C2(2pi) | 0.00 | 1 | 1 |
| 100 | S2 | CS | 2.30 | 4 | 2 |
| 101 | S4 | C2=O | −33.00 | 1 | 1 |
| 102 | H | H Acceptor | 10.02 | 2 | 2 |
| 103 | Alkane | No. of C atoms | 0.96 | 142 | 23 |
| 104 | Unsaturated HC | No. of C atoms | 0.25 | 307 | 37 |
| A | Based on | Valid groups | 61 | 436 | |
| B | Goodness of fit | R2 | 0.9731 | 388 | |
| C | Deviation | Average | 2.68 | 388 | |
| D | Deviation | Standard | 3.53 | 388 | |
| E | K-fold cv | K | 10 | 373 | |
| F | Goodness of fit | Q2 | 0.9546 | 373 | |
| G | Deviation | Average (cv) | 3.22 | 373 | |
| H | Deviation | Standard (cv) | 4.34 | 373 |
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | 31.12 | 2809 | 2809 | |
| 2 | B | C3 | 12.34 | 2 | 2 |
| 3 | B | CO2 | 51.11 | 5 | 5 |
| 4 | C sp3 | H3B | −4.93 | 3 | 1 |
| 5 | C sp3 | H2BC | 4.93 | 3 | 1 |
| 6 | C sp3 | H3C | 1.90 | 2944 | 1402 |
| 7 | C sp3 | H3N | 15.63 | 279 | 149 |
| 8 | C sp3 | H3N(+) | 7.07 | 2 | 2 |
| 9 | C sp3 | H3O | 14.42 | 366 | 232 |
| 10 | C sp3 | H3P | 21.07 | 3 | 3 |
| 11 | C sp3 | H3S | 12.93 | 35 | 31 |
| 12 | C sp3 | H3Si | 8.19 | 283 | 46 |
| 13 | C sp3 | H2C2 | 8.46 | 8600 | 1239 |
| 14 | C sp3 | H2CN | 14.85 | 505 | 257 |
| 15 | C sp3 | H2CN(+) | 19.09 | 29 | 21 |
| 16 | C sp3 | H2CO | 14.52 | 952 | 473 |
| 17 | C sp3 | H2CP | 17.50 | 3 | 2 |
| 18 | C sp3 | H2CS | 16.77 | 166 | 83 |
| 19 | C sp3 | H2CF | 12.36 | 1 | 1 |
| 20 | C sp3 | H2CCl | 10.67 | 30 | 24 |
| 21 | C sp3 | H2CBr | 11.79 | 24 | 17 |
| 22 | C sp3 | H2CJ | 3.10 | 2 | 2 |
| 23 | C sp3 | H2CSi | 8.50 | 62 | 20 |
| 24 | C sp3 | H2N2 | 5.03 | 20 | 11 |
| 25 | C sp3 | H2NO | 8.98 | 8 | 7 |
| 26 | C sp3 | H2NS | 43.70 | 4 | 4 |
| 27 | C sp3 | H2O2 | 22.34 | 23 | 14 |
| 28 | C sp3 | H2S2 | 29.21 | 7 | 5 |
| 29 | C sp3 | H2SCl | 22.89 | 1 | 1 |
| 30 | C sp3 | H2Si2 | 12.02 | 6 | 3 |
| 31 | C sp3 | HC3 | 0.64 | 817 | 388 |
| 32 | C sp3 | HC2N | 18.09 | 117 | 103 |
| 33 | C sp3 | HC2N(+) | −9.91 | 16 | 16 |
| 34 | C sp3 | HC2O | 10.63 | 357 | 226 |
| 35 | C sp3 | HC2S | 9.80 | 18 | 13 |
| 36 | C sp3 | HC2F | 8.23 | 2 | 2 |
| 37 | C sp3 | HC2Cl | 10.38 | 22 | 10 |
| 38 | C sp3 | HC2Br | 8.94 | 5 | 4 |
| 39 | C sp3 | HC2Si | −14.02 | 1 | 1 |
| 40 | C sp3 | HCN2 | 1.21 | 2 | 1 |
| 41 | C sp3 | HCNO | 23.14 | 7 | 6 |
| 42 | C sp3 | HCNS | 23.70 | 1 | 1 |
| 43 | C sp3 | HCO2 | 19.18 | 30 | 26 |
| 44 | C sp3 | HCOCl | 19.13 | 2 | 1 |
| 45 | C sp3 | HCF2 | 4.20 | 4 | 4 |
| 46 | C sp3 | HCFCl | −10.16 | 1 | 1 |
| 47 | C sp3 | HCCl2 | 9.01 | 10 | 9 |
| 48 | C sp3 | HCClBr | −3.80 | 1 | 1 |
| 49 | C sp3 | C4 | −0.23 | 435 | 256 |
| 50 | C sp3 | C3N | 14.87 | 22 | 20 |
| 51 | C sp3 | C3N(+) | 12.86 | 6 | 5 |
| 52 | C sp3 | C3O | 4.63 | 81 | 74 |
| 53 | C sp3 | C3S | 16.54 | 6 | 6 |
| 54 | C sp3 | C3F | 18.64 | 14 | 12 |
| 55 | C sp3 | C3Cl | 9.23 | 14 | 9 |
| 56 | C sp3 | C3Br | 3.44 | 2 | 2 |
| 57 | C sp3 | C3J | 31.10 | 1 | 1 |
| 58 | C sp3 | C2N2 | 52.69 | 3 | 2 |
| 59 | C sp3 | C2N2(+) | 4.24 | 7 | 6 |
| 60 | C sp3 | C2NO | 34.66 | 1 | 1 |
| 61 | C sp3 | C2NF | 47.27 | 1 | 1 |
| 62 | C sp3 | C2NCl(+) | 13.35 | 1 | 1 |
| 63 | C sp3 | C2O2 | 13.44 | 47 | 29 |
| 64 | C sp3 | C2S2 | 10.13 | 1 | 1 |
| 65 | C sp3 | C2F2 | −0.09 | 262 | 37 |
| 66 | C sp3 | C2Cl2 | 10.32 | 9 | 7 |
| 67 | C sp3 | CN3(+) | 7.29 | 6 | 5 |
| 68 | C sp3 | CNF2 | 6.86 | 7 | 3 |
| 69 | C sp3 | COF2 | −3.57 | 4 | 3 |
| 70 | C sp3 | CS3 | 30.56 | 4 | 1 |
| 71 | C sp3 | CSF2 | 41.61 | 2 | 1 |
| 72 | C sp3 | CSCl2 | 46.90 | 2 | 2 |
| 73 | C sp3 | CF3 | 3.38 | 91 | 76 |
| 74 | C sp3 | CF2Cl | −1.55 | 6 | 5 |
| 75 | C sp3 | CF2Br | 8.94 | 4 | 3 |
| 76 | C sp3 | CFCl2 | −6.89 | 3 | 2 |
| 77 | C sp3 | CCl3 | 0.92 | 17 | 16 |
| 78 | C sp3 | NF3 | 11.04 | 1 | 1 |
| 79 | C sp3 | O2F2 | 20.23 | 1 | 1 |
| 80 | C sp3 | OF3 | 2.25 | 2 | 2 |
| 81 | C sp3 | SF3 | 24.96 | 4 | 4 |
| 82 | C sp3 | SCl3 | 46.90 | 1 | 1 |
| 83 | C sp3 | SiCl3 | 14.20 | 1 | 1 |
| 84 | C sp2 | H2=C | 5.49 | 84 | 76 |
| 85 | C sp2 | HC=C | 2.46 | 607 | 323 |
| 86 | C sp2 | HC=N | −0.81 | 48 | 40 |
| 87 | C sp2 | H=CN | 3.18 | 44 | 37 |
| 88 | C sp2 | HC=O | 8.29 | 18 | 18 |
| 89 | C sp2 | H=CO | 5.29 | 19 | 17 |
| 90 | C sp2 | H=CS | −1.85 | 43 | 33 |
| 91 | C sp2 | H=CCl | 10.11 | 3 | 3 |
| 92 | C sp2 | H=CSi | 2.92 | 3 | 3 |
| 93 | C sp2 | HN=N | 9.78 | 30 | 22 |
| 94 | C sp2 | HN=O | −10.25 | 3 | 3 |
| 95 | C sp2 | H=NO | 21.94 | 1 | 1 |
| 96 | C sp2 | H=NS | 1.04 | 4 | 4 |
| 97 | C sp2 | HO=O | 14.63 | 2 | 2 |
| 98 | C sp2 | C2=C | 0.30 | 212 | 166 |
| 99 | C sp2 | C2=N | 7.33 | 35 | 33 |
| 100 | C sp2 | C2=N(+) | 2.31 | 1 | 1 |
| 101 | C sp2 | C=CN | −2.70 | 51 | 45 |
| 102 | C sp2 | C=CN(+) | 0.00 | 2 | 1 |
| 103 | C sp2 | C2=O | 1.57 | 386 | 298 |
| 104 | C sp2 | C=CO | 5.58 | 70 | 52 |
| 105 | C sp2 | C=CS | 0.18 | 38 | 25 |
| 106 | C sp2 | C=CCl | 3.68 | 20 | 13 |
| 107 | C sp2 | C=CBr | 45.90 | 1 | 1 |
| 108 | C sp2 | =CN2 | 12.85 | 17 | 17 |
| 109 | C sp2 | =CN2(+) | 6.14 | 1 | 1 |
| 110 | C sp2 | CN=N | 1.47 | 25 | 19 |
| 111 | C sp2 | =CNO | −1.47 | 6 | 4 |
| 112 | C sp2 | CN=O | 0.63 | 366 | 234 |
| 113 | C sp2 | C=NO | 9.33 | 5 | 5 |
| 114 | C sp2 | C=NS | 7.20 | 7 | 7 |
| 115 | C sp2 | CN=S | −2.87 | 10 | 8 |
| 116 | C sp2 | =CNCl | 11.25 | 1 | 1 |
| 117 | C sp2 | CO=O | 5.68 | 718 | 546 |
| 118 | C sp2 | CO=O(-) | −16.84 | 19 | 19 |
| 119 | C sp2 | C=OF | 9.78 | 3 | 2 |
| 120 | C sp2 | C=OCl | 14.97 | 2 | 1 |
| 121 | C sp2 | C=OS | 16.72 | 1 | 1 |
| 122 | C sp2 | =CS2 | −7.29 | 12 | 2 |
| 123 | C sp2 | =CSCl | 2.93 | 3 | 2 |
| 124 | C sp2 | =CSBr | −4.03 | 1 | 1 |
| 125 | C sp2 | =CF2 | 11.60 | 3 | 2 |
| 126 | C sp2 | =CFCl | 1.87 | 1 | 1 |
| 127 | C sp2 | =CCl2 | 5.32 | 9 | 8 |
| 128 | C sp2 | =CBr2 | 46.05 | 1 | 1 |
| 129 | C sp2 | N2=N | 11.87 | 9 | 9 |
| 130 | C sp2 | N2=O | −3.48 | 90 | 84 |
| 131 | C sp2 | N=NO | 3.41 | 1 | 1 |
| 132 | C sp2 | N2=S | 0.55 | 32 | 31 |
| 133 | C sp2 | N=NS | −3.08 | 23 | 23 |
| 134 | C sp2 | NO=O | 0.38 | 62 | 60 |
| 135 | C sp2 | N=OS | 20.86 | 2 | 2 |
| 136 | C sp2 | NO=S | −2.08 | 8 | 8 |
| 137 | C sp2 | NS=S | 25.24 | 3 | 3 |
| 138 | C sp2 | =NS2 | −12.86 | 2 | 2 |
| 139 | C sp2 | O2=O | −9.60 | 10 | 10 |
| 140 | C sp2 | =OS2 | 6.53 | 1 | 1 |
| 141 | C aromatic | B:C2 | −47.51 | 5 | 5 |
| 142 | C aromatic | H:C2 | 2.57 | 8600 | 1498 |
| 143 | C aromatic | H:C:N | 1.17 | 108 | 68 |
| 144 | C aromatic | H:N2 | −1.12 | 5 | 3 |
| 145 | C aromatic | :C3 | −1.60 | 481 | 153 |
| 146 | C aromatic | C:C2 | −2.58 | 2198 | 1062 |
| 147 | C aromatic | C:C:N | 5.44 | 46 | 38 |
| 148 | C aromatic | :C2N | −0.38 | 524 | 389 |
| 149 | C aromatic | :C2:N | −5.26 | 33 | 20 |
| 150 | C aromatic | :C2N(+) | 4.26 | 203 | 144 |
| 151 | C aromatic | :C2O | 2.82 | 853 | 532 |
| 152 | C aromatic | :C2P | −2.68 | 12 | 5 |
| 153 | C aromatic | :C2S | 0.30 | 98 | 73 |
| 154 | C aromatic | :C2Si | 3.80 | 45 | 21 |
| 155 | C aromatic | :C2F | 4.24 | 150 | 69 |
| 156 | C aromatic | :C2Cl | 5.68 | 860 | 318 |
| 157 | C aromatic | :C2Br | 4.73 | 92 | 57 |
| 158 | C aromatic | :C2J | 6.30 | 26 | 19 |
| 159 | C aromatic | :CN:N | 5.87 | 28 | 27 |
| 160 | C aromatic | :CN:N(+) | 0.05 | 2 | 1 |
| 161 | C aromatic | :C:NO | 3.76 | 9 | 7 |
| 162 | C aromatic | :C:NS | 2.70 | 2 | 1 |
| 163 | C aromatic | :C:NCl | 9.38 | 8 | 8 |
| 164 | C aromatic | N:N2 | −9.59 | 85 | 40 |
| 165 | C aromatic | :N2O | −5.16 | 4 | 2 |
| 166 | C aromatic | :N2S | −2.43 | 5 | 5 |
| 167 | C aromatic | :N2Cl | 19.07 | 8 | 7 |
| 168 | C sp | H#C | 2.83 | 26 | 23 |
| 169 | C sp | C#C | −0.52 | 183 | 83 |
| 170 | C sp | =C2 | 7.54 | 4 | 4 |
| 171 | C sp | C#N | 2.66 | 120 | 94 |
| 172 | C sp | #CSi | 3.40 | 3 | 2 |
| 173 | C sp | N#N | −16.19 | 1 | 1 |
| 174 | C sp | =N2 | 23.07 | 1 | 1 |
| 175 | C sp | #NO | 6.78 | 10 | 4 |
| 176 | C sp | =N=O | 14.08 | 6 | 3 |
| 177 | N sp3 | H2C | 9.39 | 34 | 21 |
| 178 | N sp3 | H2C(pi) | 7.89 | 190 | 160 |
| 179 | N sp3 | H2N | 0.92 | 5 | 5 |
| 180 | N sp3 | H2P | −16.37 | 1 | 1 |
| 181 | N sp3 | H2S | 10.07 | 7 | 7 |
| 182 | N sp3 | HC2 | −1.65 | 20 | 20 |
| 183 | N sp3 | HC2(pi) | −9.81 | 190 | 133 |
| 184 | N sp3 | HC2(2pi) | 4.73 | 204 | 169 |
| 185 | N sp3 | HCN | −5.80 | 4 | 3 |
| 186 | N sp3 | HCN(pi) | −2.85 | 8 | 6 |
| 187 | N sp3 | HCN(+)(pi) | 16.06 | 4 | 2 |
| 188 | N sp3 | HCN(2pi) | 0.95 | 12 | 11 |
| 189 | N sp3 | HCO(pi) | 30.19 | 1 | 1 |
| 190 | N sp3 | HCP | −6.83 | 2 | 2 |
| 191 | N sp3 | HCS | 17.10 | 2 | 2 |
| 192 | N sp3 | HCS(pi) | 9.38 | 22 | 22 |
| 193 | N sp3 | HSi2 | 1.67 | 7 | 2 |
| 194 | N sp3 | C3 | −32.04 | 41 | 37 |
| 195 | N sp3 | C3(pi) | −17.08 | 137 | 97 |
| 196 | N sp3 | C3(2pi) | −12.64 | 136 | 108 |
| 197 | N sp3 | C3(3pi) | 4.26 | 22 | 20 |
| 198 | N sp3 | C2N | −18.10 | 3 | 3 |
| 199 | N sp3 | C2N(pi) | −6.67 | 7 | 5 |
| 200 | N sp3 | C2N(+)(pi) | 20.95 | 32 | 17 |
| 201 | N sp3 | C2N(2pi) | −3.87 | 15 | 14 |
| 202 | N sp3 | C2N(3pi) | 1.17 | 6 | 6 |
| 203 | N sp3 | C2N(+)(2pi) | −0.16 | 12 | 12 |
| 204 | N sp3 | C2O | −41.10 | 5 | 5 |
| 205 | N sp3 | C2O(pi) | 9.25 | 39 | 15 |
| 206 | N sp3 | C2O(2pi) | 29.03 | 1 | 1 |
| 207 | N sp3 | C2P | 7.24 | 1 | 1 |
| 208 | N sp3 | C2S | −25.22 | 3 | 3 |
| 209 | N sp3 | C2S(pi) | −22.07 | 1 | 1 |
| 210 | N sp3 | C2S(2pi) | −6.25 | 3 | 3 |
| 211 | N sp3 | CF2 | −2.10 | 6 | 2 |
| 212 | N(+) sp3 | H2C2 | 4.33 | 19 | 19 |
| 213 | N(+) sp3 | C3O(-) | −33.09 | 1 | 1 |
| 214 | N sp2 | H=C | 16.94 | 3 | 3 |
| 215 | N sp2 | C=C | −7.28 | 122 | 101 |
| 216 | N sp2 | C=N | −11.24 | 64 | 32 |
| 217 | N sp2 | C=N(+) | 10.95 | 10 | 7 |
| 218 | N sp2 | =CN | −0.51 | 38 | 31 |
| 219 | N sp2 | =CO | 0.98 | 32 | 31 |
| 220 | N sp2 | =CS | −4.17 | 3 | 2 |
| 221 | N sp2 | N=N | −0.32 | 10 | 6 |
| 222 | N sp2 | N=O | 18.24 | 4 | 2 |
| 223 | N aromatic | :C2 | 5.43 | 222 | 128 |
| 224 | N aromatic | :C:N | −4.60 | 6 | 3 |
| 225 | N(+) sp2 | C=NO(-) | −19.90 | 4 | 4 |
| 226 | N(+) sp2 | CO=O(-) | 1.45 | 248 | 163 |
| 227 | N(+) sp2 | =CO2(-) | −3.88 | 1 | 1 |
| 228 | N(+) sp2 | NO=O(-) | −1.33 | 48 | 31 |
| 229 | N(+) sp2 | O2=O(-) | 1.85 | 7 | 5 |
| 230 | N(+) sp | C#C(-) | 10.24 | 1 | 1 |
| 231 | N(+) sp | =N2(-) | 2.76 | 6 | 3 |
| 232 | O | HC | −2.00 | 452 | 254 |
| 233 | O | HC(pi) | 3.39 | 478 | 400 |
| 234 | O | HN | 0.63 | 36 | 12 |
| 235 | O | HN(pi) | −1.02 | 19 | 19 |
| 236 | O | HP | −8.39 | 2 | 1 |
| 237 | O | HS | 60.03 | 5 | 2 |
| 238 | O | BC | 0.00 | 5 | 5 |
| 239 | O | BN | 0.00 | 5 | 5 |
| 240 | O | C2 | −4.67 | 357 | 135 |
| 241 | O | C2(pi) | −5.72 | 740 | 513 |
| 242 | O | C2(2pi) | −3.04 | 267 | 217 |
| 243 | O | CN | −20.33 | 4 | 4 |
| 244 | O | CN(pi) | 0.00 | 1 | 1 |
| 245 | O | CN(2pi) | 1.82 | 12 | 11 |
| 246 | O | CN(+)(pi) | 0.47 | 7 | 5 |
| 247 | O | CO | 1.80 | 8 | 4 |
| 248 | O | CP | −6.11 | 47 | 25 |
| 249 | O | CP(pi) | 6.35 | 20 | 17 |
| 250 | O | CS(pi) | 1.11 | 3 | 3 |
| 251 | O | CSi | −12.94 | 5 | 2 |
| 252 | O | N2(2pi) | |||
| 253 | O | N2(+)(2pi) | 0.00 | 1 | 1 |
| 254 | O | Si2 | 2.53 | 84 | 24 |
| 255 | P3 | C3 | −6.01 | 3 | 2 |
| 256 | P4 | C3=O | −6.07 | 1 | 1 |
| 257 | P4 | C=OF2 | −1.93 | 1 | 1 |
| 258 | P4 | C=OFCl | −4.92 | 1 | 1 |
| 259 | P4 | C=OCl2 | 6.84 | 1 | 1 |
| 260 | P4 | N2O=O | 6.11 | 1 | 1 |
| 261 | P4 | NO2=O | −7.48 | 1 | 1 |
| 262 | P4 | NOS=S | 6.11 | 1 | 1 |
| 263 | P4 | O3=O | −5.29 | 2 | 2 |
| 264 | P4 | O3=S | −3.13 | 13 | 12 |
| 265 | P4 | CO2=O | 0.00 | 1 | 1 |
| 266 | P4 | CO2=S | 7.66 | 2 | 2 |
| 267 | P4 | O2S=S | −5.52 | 7 | 7 |
| 268 | S2 | HC | −0.29 | 19 | 19 |
| 269 | S2 | HC(pi) | −11.91 | 2 | 2 |
| 270 | S2 | C2 | −10.10 | 74 | 47 |
| 271 | S2 | C2(pi) | 1.44 | 44 | 37 |
| 272 | S2 | C2(2pi) | 8.54 | 74 | 60 |
| 273 | S2 | CN | 0.00 | 3 | 3 |
| 274 | S2 | CN(pi) | 5.57 | 1 | 1 |
| 275 | S2 | CS | 1.49 | 8 | 4 |
| 276 | S2 | CS(pi) | 0.18 | 6 | 4 |
| 277 | S2 | CP | 0.00 | 8 | 8 |
| 278 | S2 | N2(2pi) | −3.71 | 1 | 1 |
| 279 | S4 | C2=O | −10.46 | 6 | 4 |
| 280 | S4 | C2=O2 | −10.18 | 22 | 22 |
| 281 | S4 | CN=O2 | 1.23 | 31 | 31 |
| 282 | S4 | CO=O2 | 0.00 | 8 | 5 |
| 283 | S4 | C=OS | 4.07 | 2 | 2 |
| 284 | S4 | N2=O2 | 4.49 | 2 | 2 |
| 285 | Si | H3C | 0.00 | 1 | 1 |
| 286 | Si | HC2O | −77.65 | 3 | 3 |
| 287 | Si | HCO2 | 18.28 | 1 | 1 |
| 288 | Si | C4 | −12.05 | 23 | 18 |
| 289 | Si | C3O | −15.58 | 14 | 9 |
| 290 | Si | C3Cl | −8.02 | 2 | 2 |
| 291 | Si | C3Si | −6.42 | 6 | 3 |
| 292 | Si | C2N2 | 0.00 | 7 | 2 |
| 293 | Si | C2O2 | 1.03 | 75 | 18 |
| 294 | Si | C2Cl2 | −1.79 | 2 | 2 |
| 295 | Si | C2Si2 | −10.09 | 34 | 5 |
| 296 | Si | CCl3 | 4.64 | 8 | 7 |
| 297 | Si | O4 | 13.30 | 1 | 1 |
| 298 | H | H Acceptor | 6.31 | 153 | 128 |
| 299 | Angle60 | 0.54 | 120 | 33 | |
| 300 | Angle90 | 2.37 | 138 | 29 | |
| 301 | Angle102 | 0.12 | 1131 | 342 | |
| 302 | Endocyclic bonds | No. of single bonds | −4.42 | 5302 | 680 |
| A | Based on | Valid groups | 188 | 2809 | |
| B | Goodness of fit | R2 | 0.8875 | 2701 | |
| C | Deviation | Average | 12.33 | 2701 | |
| D | Deviation | Standard | 16.72 | 2701 | |
| E | K-fold cv | K | 10 | 2637 | |
| F | Goodness of fit | Q2 | 0.8727 | 2637 | |
| G | Deviation | Average (cv) | 13.23 | 2637 | |
| H | Deviation | Standard (cv) | 17.93 | 2637 |
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | 60.14 | 2686 | 2686 | |
| 2 | C sp3 | H3C | 5.33 | 5873 | 2490 |
| 3 | C sp3 | H3N | 16.05 | 12 | 6 |
| 4 | C sp3 | H3O | 2.66 | 195 | 172 |
| 5 | C sp3 | H3Si | 3.08 | 110 | 5 |
| 6 | C sp3 | H2C2 | 4.04 | 30,650 | 2478 |
| 7 | C sp3 | H2CN | −1.70 | 286 | 114 |
| 8 | C sp3 | H2CO | −0.01 | 3584 | 1901 |
| 9 | C sp3 | H2CS | −8.01 | 68 | 42 |
| 10 | C sp3 | H2CCl | −27.41 | 2 | 2 |
| 11 | C sp3 | H2CBr | −10.24 | 3 | 3 |
| 12 | C sp3 | H2CJ | 30.88 | 1 | 1 |
| 13 | C sp3 | H2CSi | −2.48 | 6 | 3 |
| 14 | C sp3 | HC3 | −9.84 | 1088 | 414 |
| 15 | C sp3 | HC2N | −17.47 | 4 | 4 |
| 16 | C sp3 | HC2O | −19.96 | 428 | 324 |
| 17 | C sp3 | HC2S | −42.59 | 18 | 18 |
| 18 | C sp3 | HC2Cl | −12.96 | 53 | 53 |
| 19 | C sp3 | HC2Br | 6.97 | 4 | 4 |
| 20 | C sp3 | HCO2 | 7.19 | 34 | 28 |
| 21 | C sp3 | HCF2 | −21.83 | 11 | 11 |
| 22 | C sp3 | C4 | −0.53 | 212 | 120 |
| 23 | C sp3 | C3O | 12.06 | 10 | 10 |
| 24 | C sp3 | C3F | −25.29 | 2 | 2 |
| 25 | C sp3 | C2F2 | 4.67 | 272 | 57 |
| 26 | C sp3 | CSF2 | −1.17 | 5 | 5 |
| 27 | C sp3 | CF3 | −8.30 | 67 | 54 |
| 28 | C sp3 | OF3 | 24.11 | 2 | 2 |
| 29 | C sp3 | SF3 | −196.06 | 1 | 1 |
| 30 | C sp2 | H2=C | 14.81 | 58 | 56 |
| 31 | C sp2 | HC=C | −2.97 | 946 | 440 |
| 32 | C sp2 | HC=N | −2.07 | 922 | 704 |
| 33 | C sp2 | HC=N(+) | 32.39 | 9 | 9 |
| 34 | C sp2 | HC=O | 15.32 | 6 | 6 |
| 35 | C sp2 | H=CN | −16.69 | 43 | 41 |
| 36 | C sp2 | H=CO | −2.30 | 28 | 28 |
| 37 | C sp2 | H=CS | −4.67 | 2 | 2 |
| 38 | C sp2 | H=NS | 74.91 | 1 | 1 |
| 39 | C sp2 | C2=C | −13.21 | 186 | 160 |
| 40 | C sp2 | C2=N | 9.17 | 17 | 17 |
| 41 | C sp2 | C2=O | 2.80 | 266 | 202 |
| 42 | C sp2 | C=CN | 2.69 | 28 | 21 |
| 43 | C sp2 | C=CO | −53.38 | 21 | 21 |
| 44 | C sp2 | C=CS | −5.66 | 340 | 150 |
| 45 | C sp2 | C=CF | 31.70 | 10 | 5 |
| 46 | C sp2 | CN=N | −13.68 | 15 | 15 |
| 47 | C sp2 | CN=O | −1.75 | 326 | 171 |
| 48 | C sp2 | C=NO | −39.68 | 45 | 30 |
| 49 | C sp2 | CN=S | −6.95 | 8 | 6 |
| 50 | C sp2 | C=NS | 38.49 | 105 | 77 |
| 51 | C sp2 | =CNS | −47.14 | 22 | 11 |
| 52 | C sp2 | CO=O | 8.07 | 3115 | 1580 |
| 53 | C sp2 | =COS | 128.10 | 5 | 5 |
| 54 | C sp2 | C=OS | 5.46 | 91 | 81 |
| 55 | C sp2 | =CSCl | 15.27 | 9 | 9 |
| 56 | C sp2 | =CSJ | 10.36 | 2 | 2 |
| 57 | C sp2 | N=NS | −11.16 | 72 | 72 |
| 58 | C sp2 | NO=O | 38.80 | 6 | 6 |
| 59 | C sp2 | =NOS | 96.96 | 24 | 12 |
| 60 | C sp2 | O2=O | 26.06 | 3 | 3 |
| 61 | C aromatic | H:C2 | 3.37 | 28,602 | 2538 |
| 62 | C aromatic | H:C:N | −0.02 | 151 | 82 |
| 63 | C aromatic | H:C:N(+) | −9.49 | 12 | 6 |
| 64 | C aromatic | :C3 | −8.40 | 322 | 107 |
| 65 | C aromatic | C:C2 | −9.58 | 7933 | 2410 |
| 66 | C aromatic | C:C:N | −38.40 | 89 | 61 |
| 67 | C aromatic | :C2N | −13.66 | 1866 | 1124 |
| 68 | C aromatic | :C2N(+) | −5.68 | 135 | 119 |
| 69 | C aromatic | :C2:N | 16.73 | 34 | 33 |
| 70 | C aromatic | :C2O | −4.24 | 5711 | 2230 |
| 71 | C aromatic | :C2S | −29.84 | 116 | 105 |
| 72 | C aromatic | :C2Si | 10.60 | 4 | 2 |
| 73 | C aromatic | :C2F | 4.38 | 525 | 266 |
| 74 | C aromatic | :C2Cl | −3.87 | 197 | 151 |
| 75 | C aromatic | :C2Br | 2.55 | 24 | 23 |
| 76 | C aromatic | :C2J | −35.42 | 9 | 9 |
| 77 | C aromatic | C:N2 | −43.07 | 27 | 21 |
| 78 | C aromatic | :C:NCl | −51.42 | 2 | 2 |
| 79 | C aromatic | N:N2 | −17.88 | 6 | 3 |
| 80 | C aromatic | :N2O | −31.16 | 4 | 4 |
| 81 | C sp | H#C | 15.40 | 1 | 1 |
| 82 | C sp | C#C | −1.90 | 929 | 304 |
| 83 | C sp | =C2 | −15.98 | 9 | 9 |
| 84 | C sp | C#N | 4.72 | 229 | 212 |
| 85 | C sp | #CO | 29.96 | 2 | 1 |
| 86 | C sp | =N=O | 0.85 | 3 | 2 |
| 87 | C sp | =N=S | 15.48 | 42 | 42 |
| 88 | C sp | #NS | 7.49 | 26 | 26 |
| 89 | N sp3 | H2C | −12.08 | 5 | 5 |
| 90 | N sp3 | H2C(pi) | −66.66 | 6 | 6 |
| 91 | N sp3 | HC2(pi) | 18.61 | 17 | 9 |
| 92 | N sp3 | HC2(2pi) | −4.58 | 233 | 143 |
| 93 | N sp3 | HCN(pi) | −6.87 | 6 | 3 |
| 94 | N sp3 | HCN(2pi) | 42.99 | 12 | 12 |
| 95 | N sp3 | HCS(pi) | 157.30 | 1 | 1 |
| 96 | N sp3 | C3 | −75.12 | 10 | 10 |
| 97 | N sp3 | C3(pi) | −20.84 | 64 | 33 |
| 98 | N sp3 | C3(2pi) | 8.12 | 34 | 25 |
| 99 | N sp3 | C3(3pi) | 29.75 | 24 | 14 |
| 100 | N sp2 | C=C | 14.07 | 1014 | 778 |
| 101 | N sp2 | C=N | 9.88 | 722 | 295 |
| 102 | N sp2 | C=N(+) | 8.87 | 32 | 32 |
| 103 | N sp2 | =CN | −40.91 | 206 | 94 |
| 104 | N sp2 | =CO | 33.53 | 26 | 26 |
| 105 | N aromatic | :C2 | 18.59 | 169 | 125 |
| 106 | N aromatic | :C:N | 17.07 | 12 | 3 |
| 107 | N(+) sp2 | CO=O(-) | 0.77 | 94 | 78 |
| 108 | N(+) sp2 | C=CO(-) | −3.27 | 9 | 9 |
| 109 | N(+) sp2 | C=NO(-) | 0.00 | 32 | 32 |
| 110 | N(+) aromatic | :C2O(-) | 23.39 | 6 | 6 |
| 111 | O | HC | 20.86 | 186 | 70 |
| 112 | O | HC(pi) | 16.46 | 202 | 156 |
| 113 | O | C2 | 1.72 | 100 | 57 |
| 114 | O | C2(pi) | −0.12 | 3901 | 2018 |
| 115 | O | C2(2pi) | −2.52 | 2419 | 1340 |
| 116 | O | CN(2pi) | −4.06 | 26 | 26 |
| 117 | S2 | HC(pi) | −10.11 | 2 | 2 |
| 118 | S2 | C2 | 12.90 | 18 | 18 |
| 119 | S2 | C2(pi) | 14.58 | 55 | 42 |
| 120 | S2 | C2(2pi) | 15.10 | 379 | 314 |
| 121 | S4 | CN=O2 | −36.49 | 1 | 1 |
| 122 | Si | C3Si | 0.00 | 10 | 5 |
| 123 | Si | C2Si2 | −3.55 | 45 | 5 |
| 124 | H | H Acceptor | −17.84 | 151 | 107 |
| 125 | Angle60 | 0.00 | 0 | 0 | |
| 126 | Angle90 | 0.00 | 0 | 0 | |
| 127 | Angle102 | 7.37 | 513 | 138 | |
| 128 | Endocyclic bonds | No of single bonds | −1.14 | 3024 | 309 |
| A | Based on | Valid groups | 108 | 2686 | |
| B | Goodness of fit | R2 | 0.6094 | 2663 | |
| C | Deviation | Average | 23.83 | 2663 | |
| D | Deviation | Standard | 31.62 | 2663 | |
| E | K-fold cv | K | 10 | 2643 | |
| F | Goodness of fit | Q2 | 0.5804 | 2643 | |
| G | Deviation | Average (cv) | 24.65 | 2643 | |
| H | Deviation | Standard (cv) | 32.79 | 2643 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naef, R.; Acree, W.E. Calculation of Five Thermodynamic Molecular Descriptors by Means of a General Computer Algorithm Based on the Group-Additivity Method: Standard Enthalpies of Vaporization, Sublimation and Solvation, and Entropy of Fusion of Ordinary Organic Molecules and Total Phase-Change Entropy of Liquid Crystals. Molecules 2017, 22, 1059. https://doi.org/10.3390/molecules22071059
Naef R, Acree WE. Calculation of Five Thermodynamic Molecular Descriptors by Means of a General Computer Algorithm Based on the Group-Additivity Method: Standard Enthalpies of Vaporization, Sublimation and Solvation, and Entropy of Fusion of Ordinary Organic Molecules and Total Phase-Change Entropy of Liquid Crystals. Molecules. 2017; 22(7):1059. https://doi.org/10.3390/molecules22071059
Chicago/Turabian StyleNaef, Rudolf, and William E. Acree. 2017. "Calculation of Five Thermodynamic Molecular Descriptors by Means of a General Computer Algorithm Based on the Group-Additivity Method: Standard Enthalpies of Vaporization, Sublimation and Solvation, and Entropy of Fusion of Ordinary Organic Molecules and Total Phase-Change Entropy of Liquid Crystals" Molecules 22, no. 7: 1059. https://doi.org/10.3390/molecules22071059
APA StyleNaef, R., & Acree, W. E. (2017). Calculation of Five Thermodynamic Molecular Descriptors by Means of a General Computer Algorithm Based on the Group-Additivity Method: Standard Enthalpies of Vaporization, Sublimation and Solvation, and Entropy of Fusion of Ordinary Organic Molecules and Total Phase-Change Entropy of Liquid Crystals. Molecules, 22(7), 1059. https://doi.org/10.3390/molecules22071059