In silico Study of the Pharmacologic Properties and Cytotoxicity Pathways in Cancer Cells of Various Indolylquinone Analogues of Perezone

 ,
,  ,
,
Abstract
:
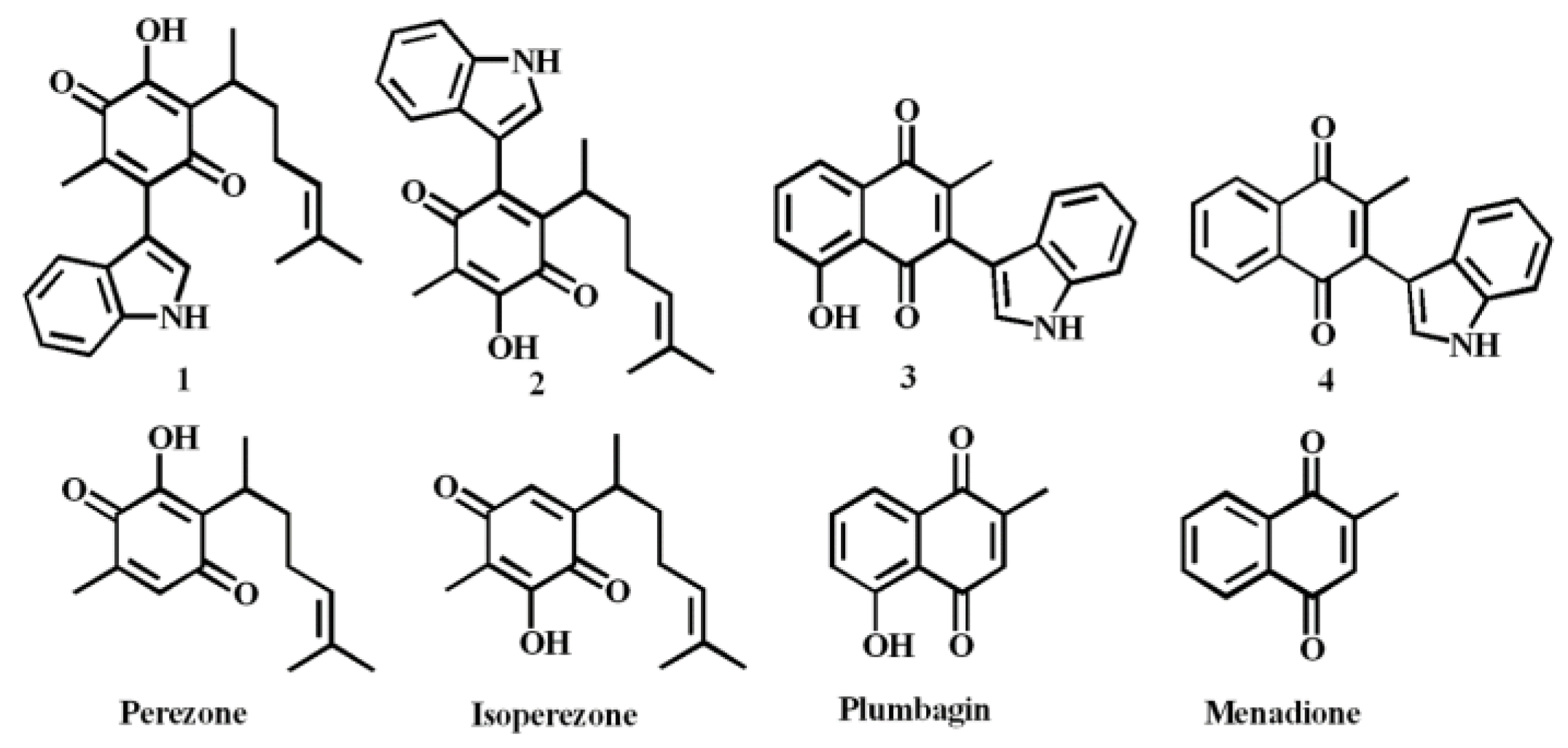
1. Introduction
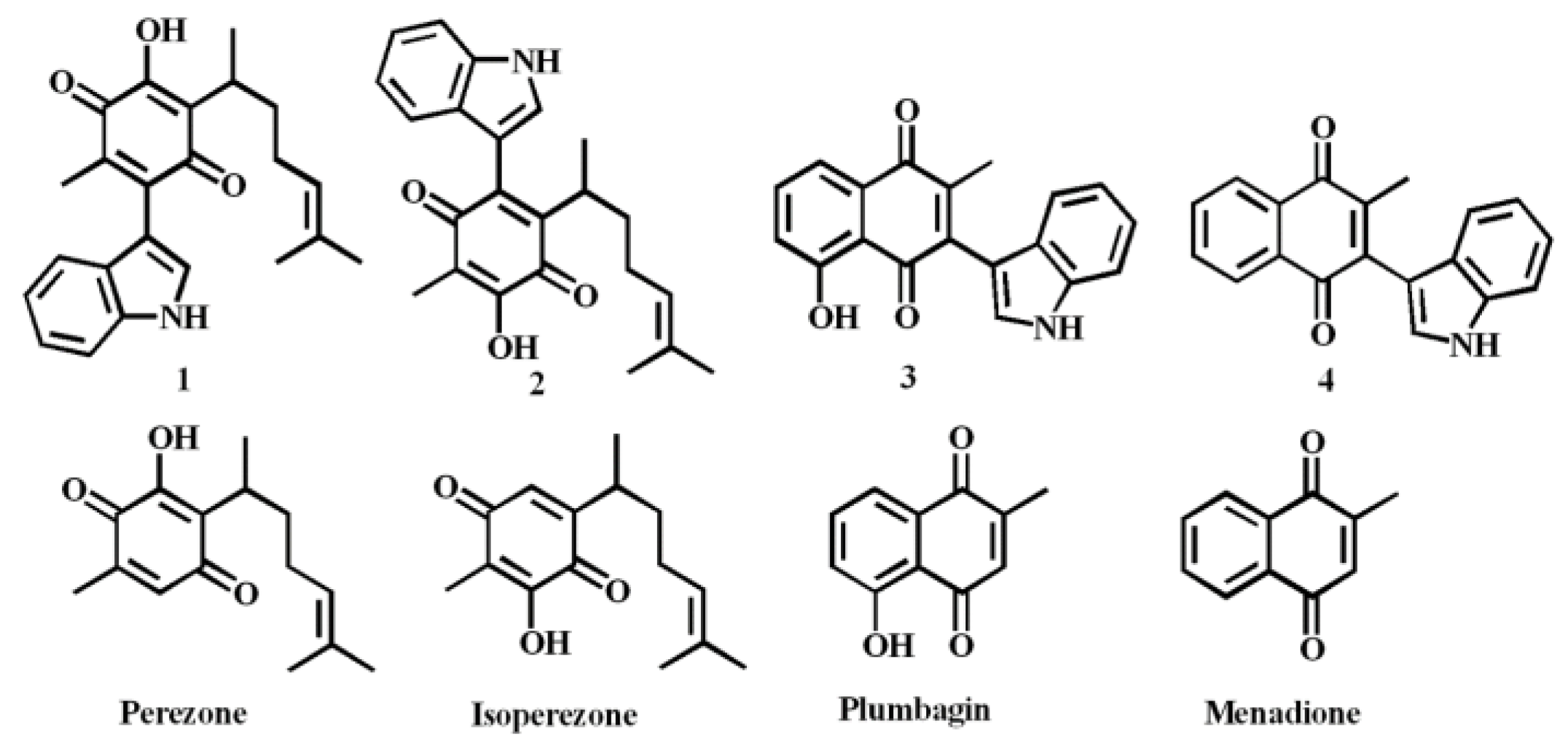
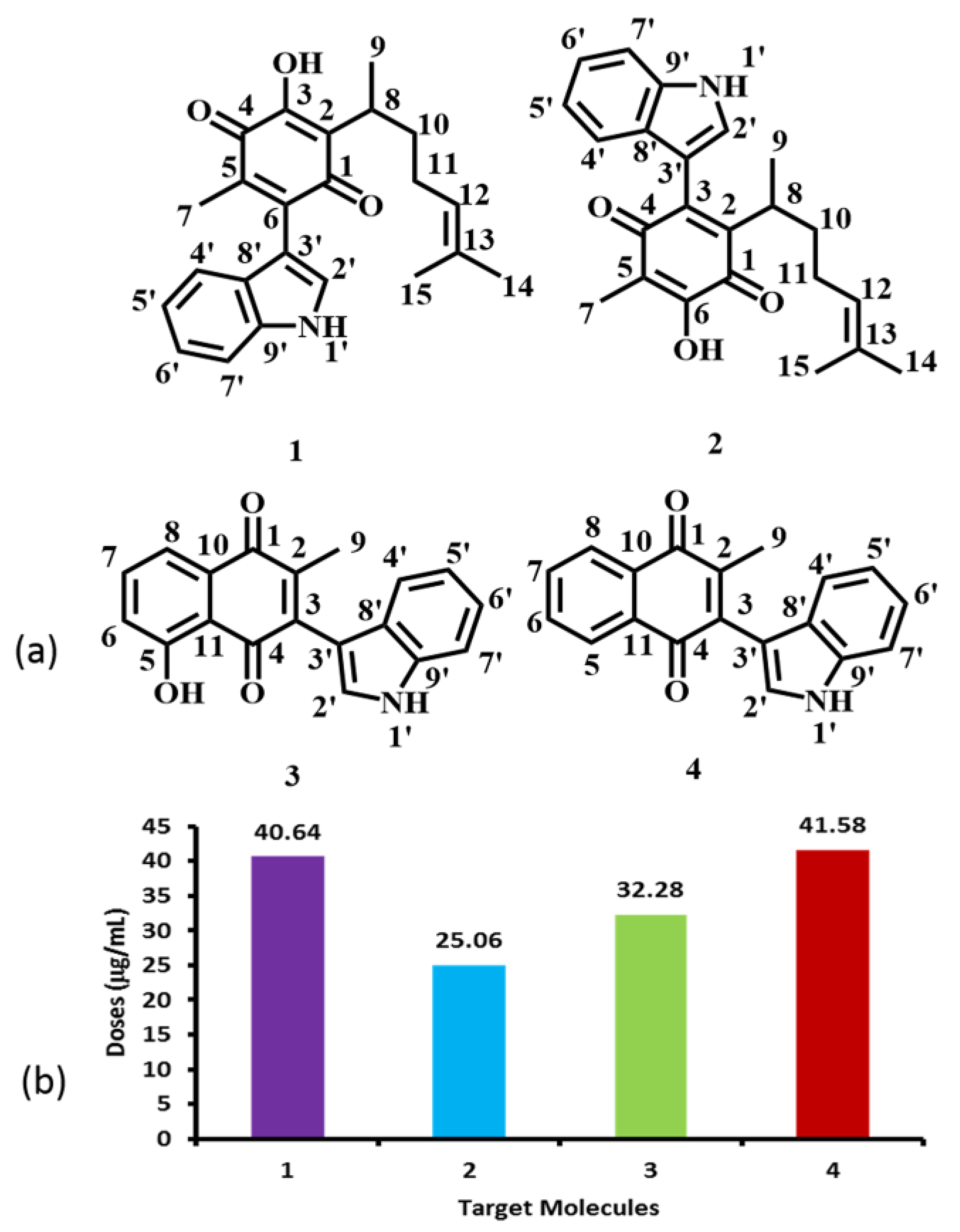
2. Results and Discussion
2.1. Molecular Reactivity
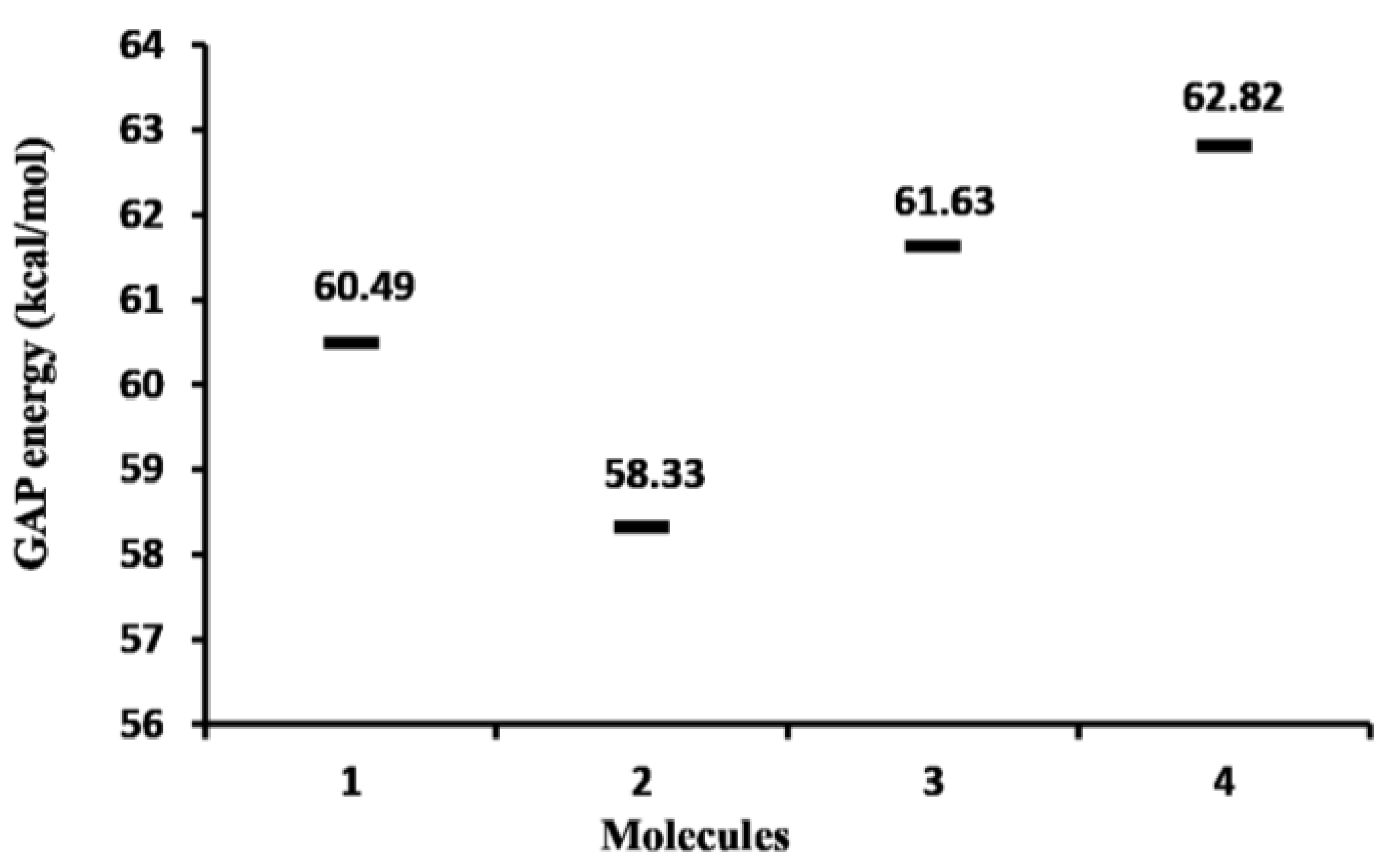
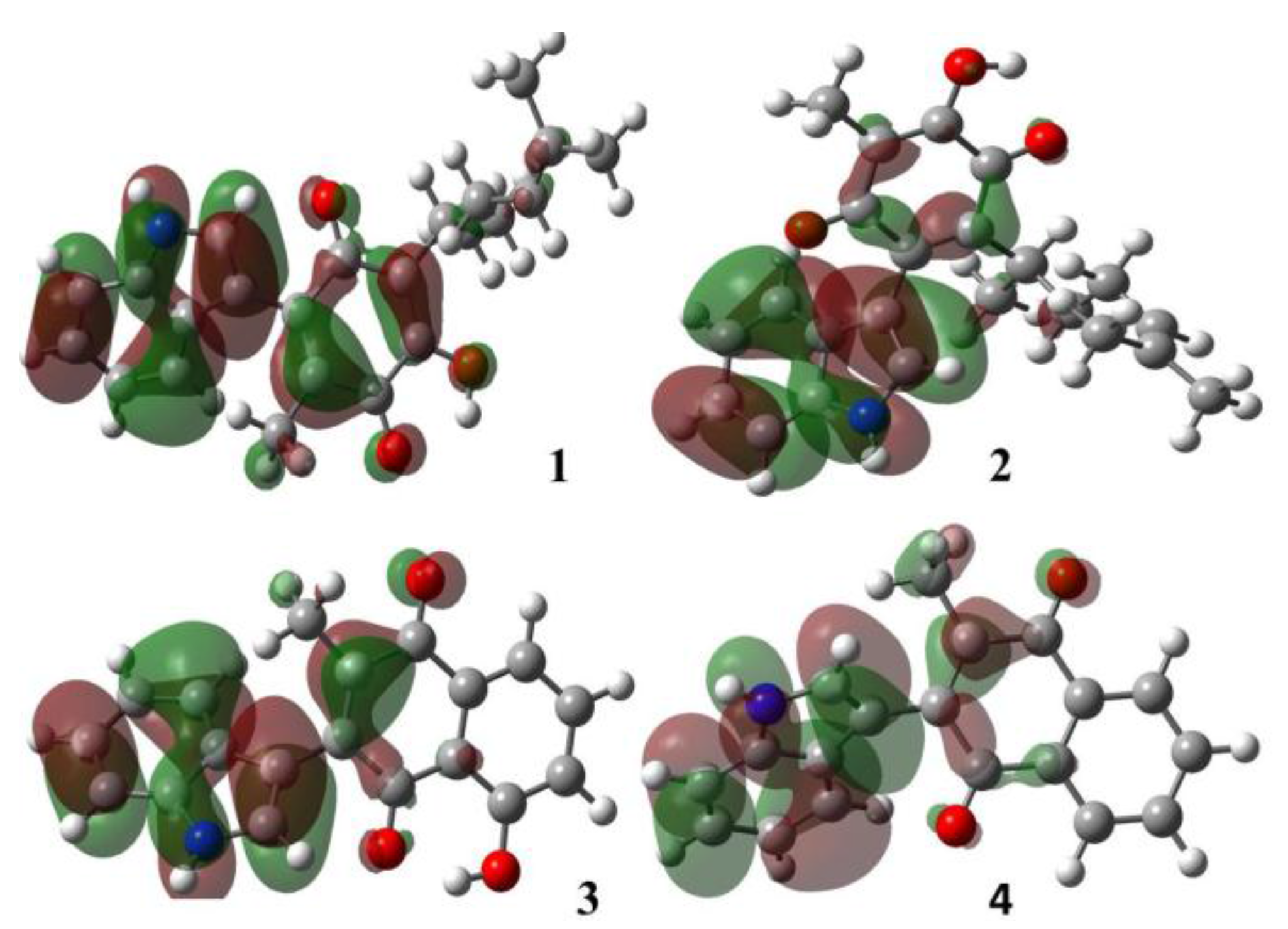
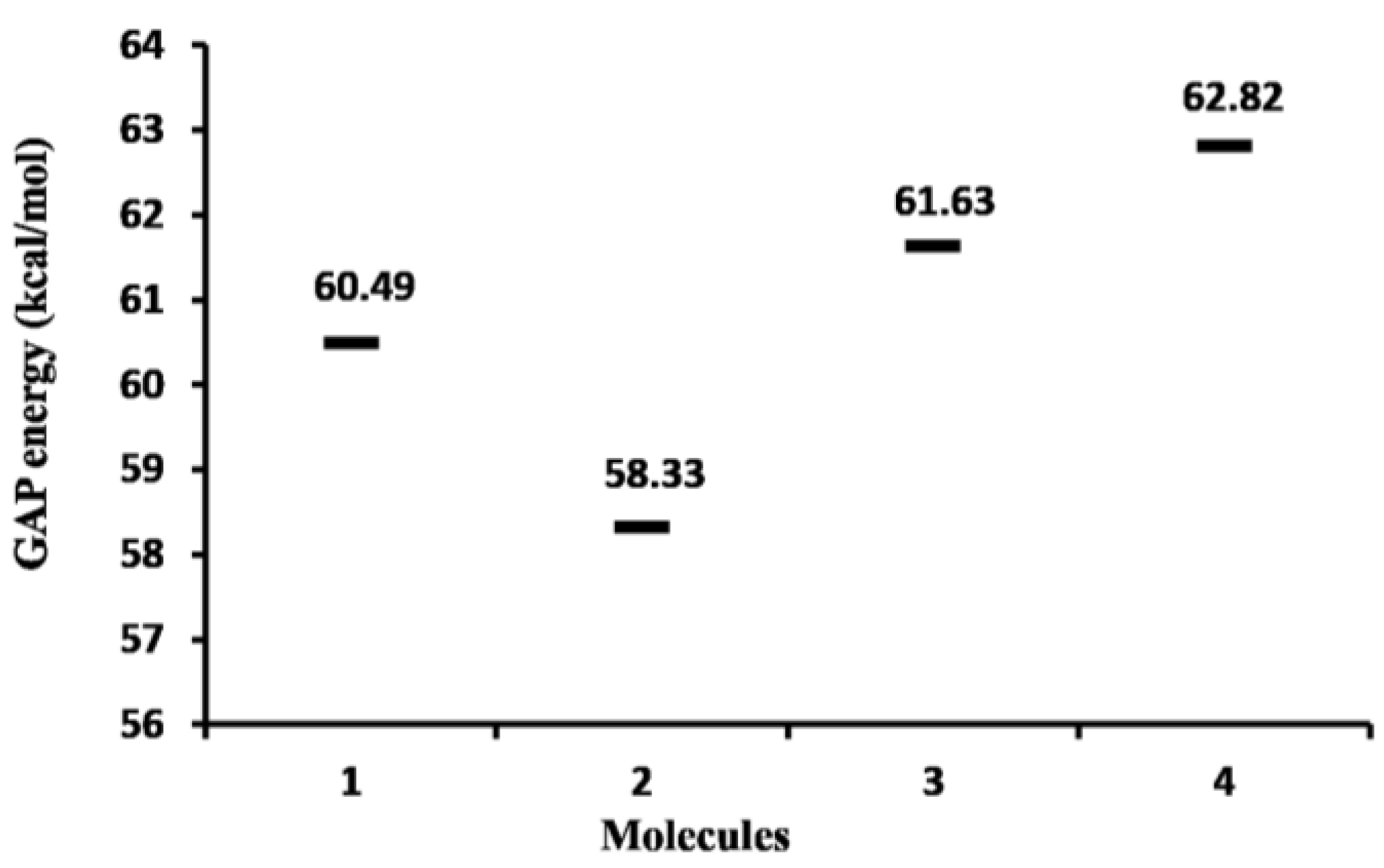
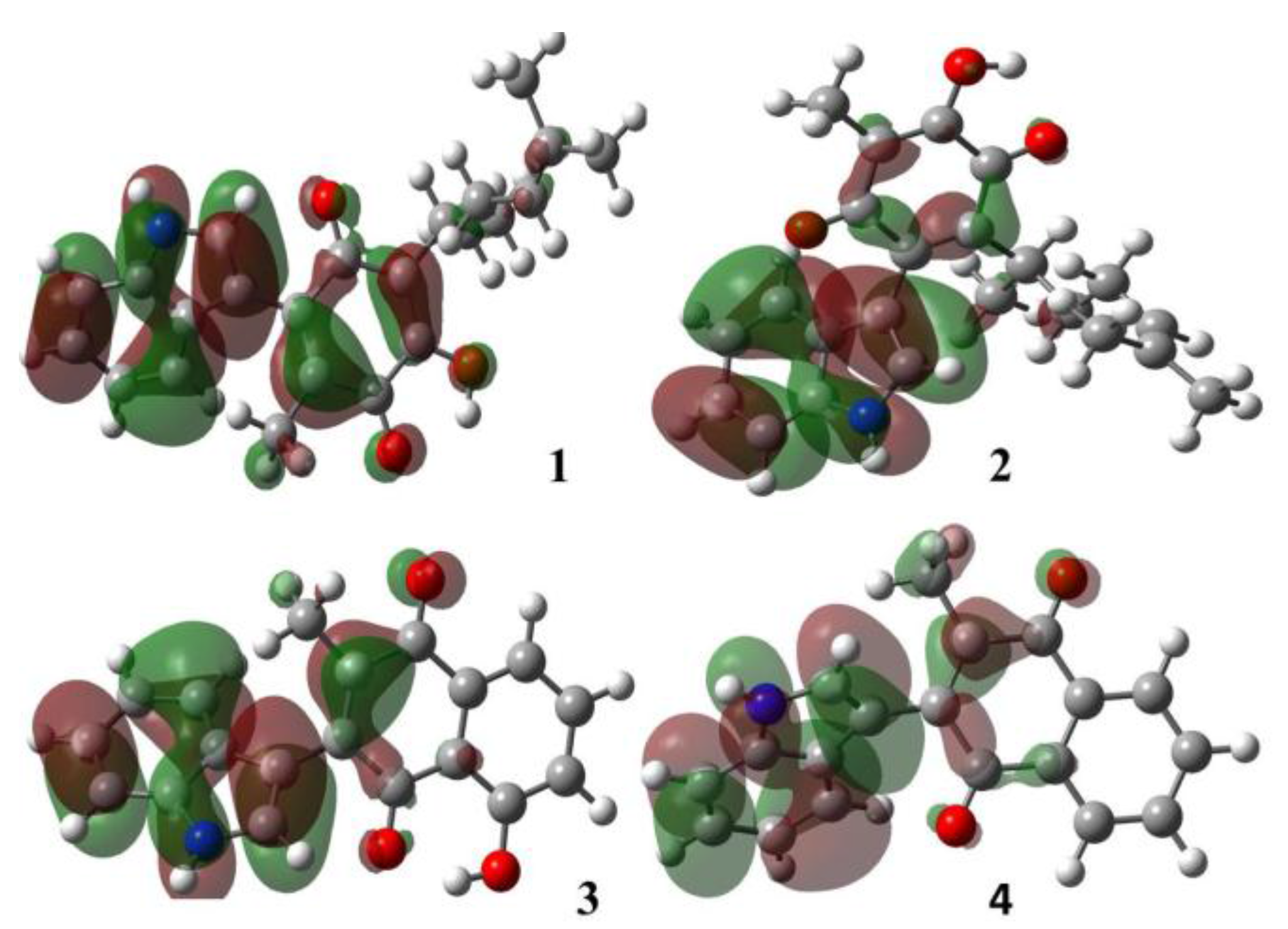
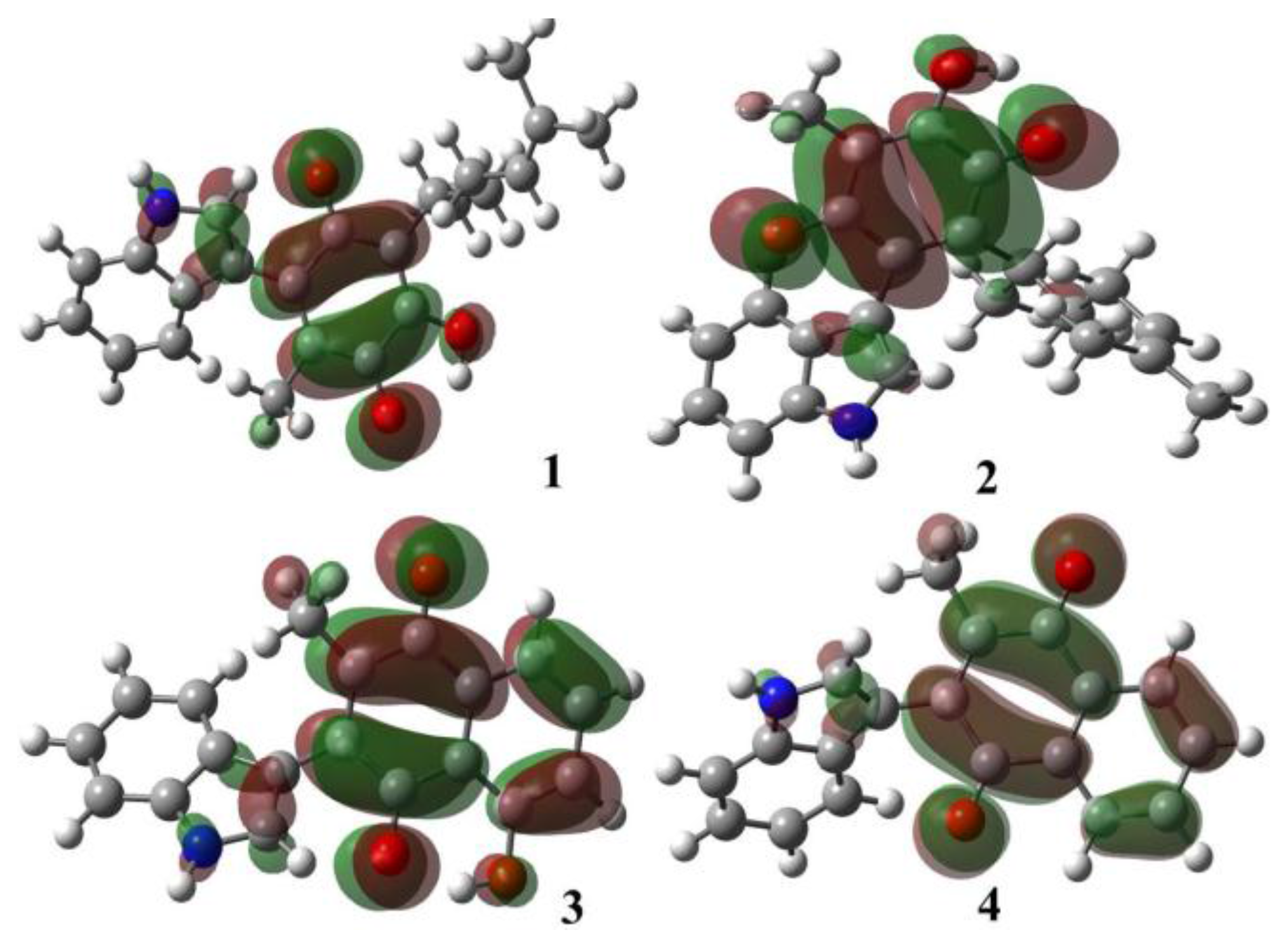
2.1.1. Molecular Orbital Analysis
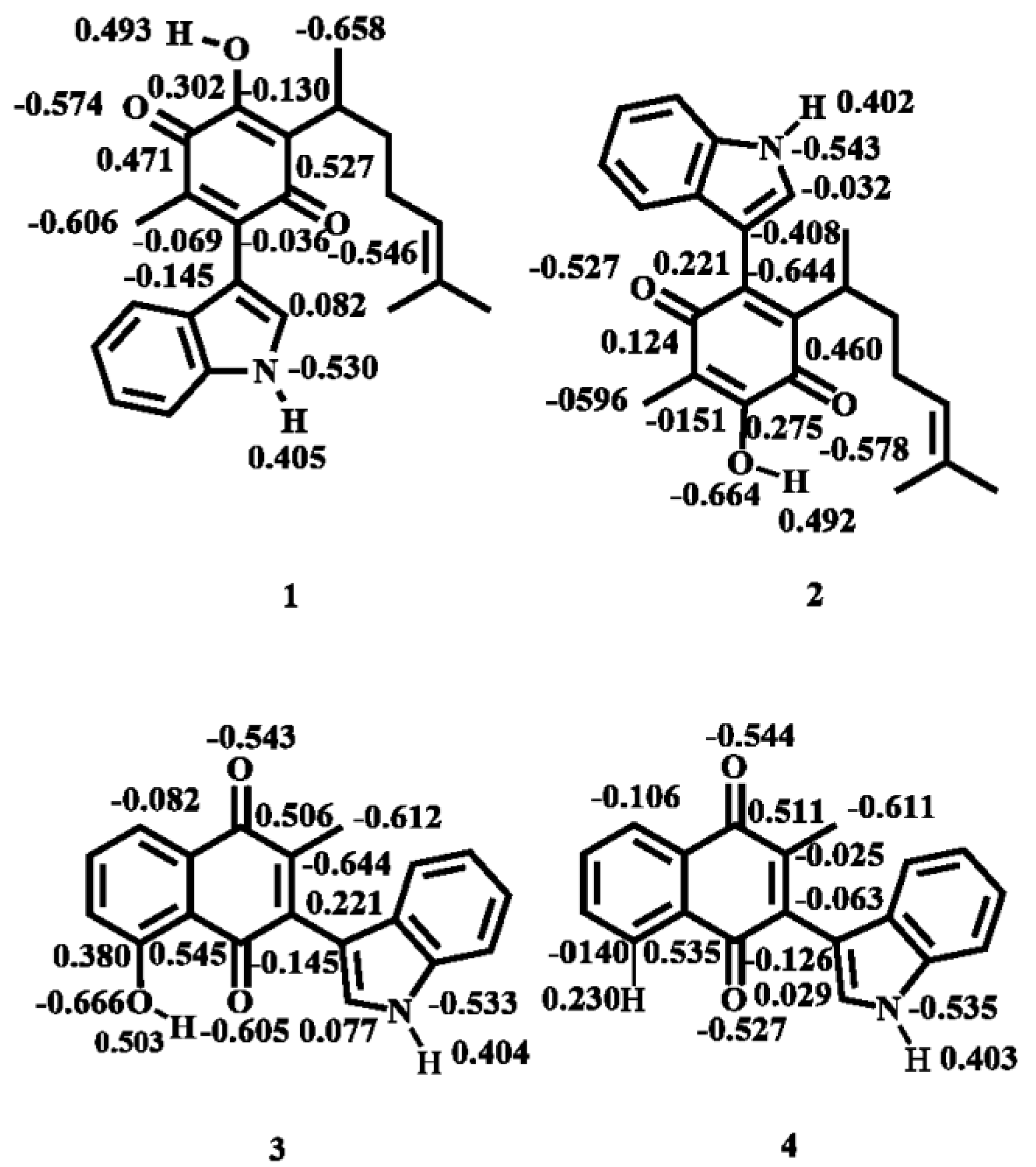
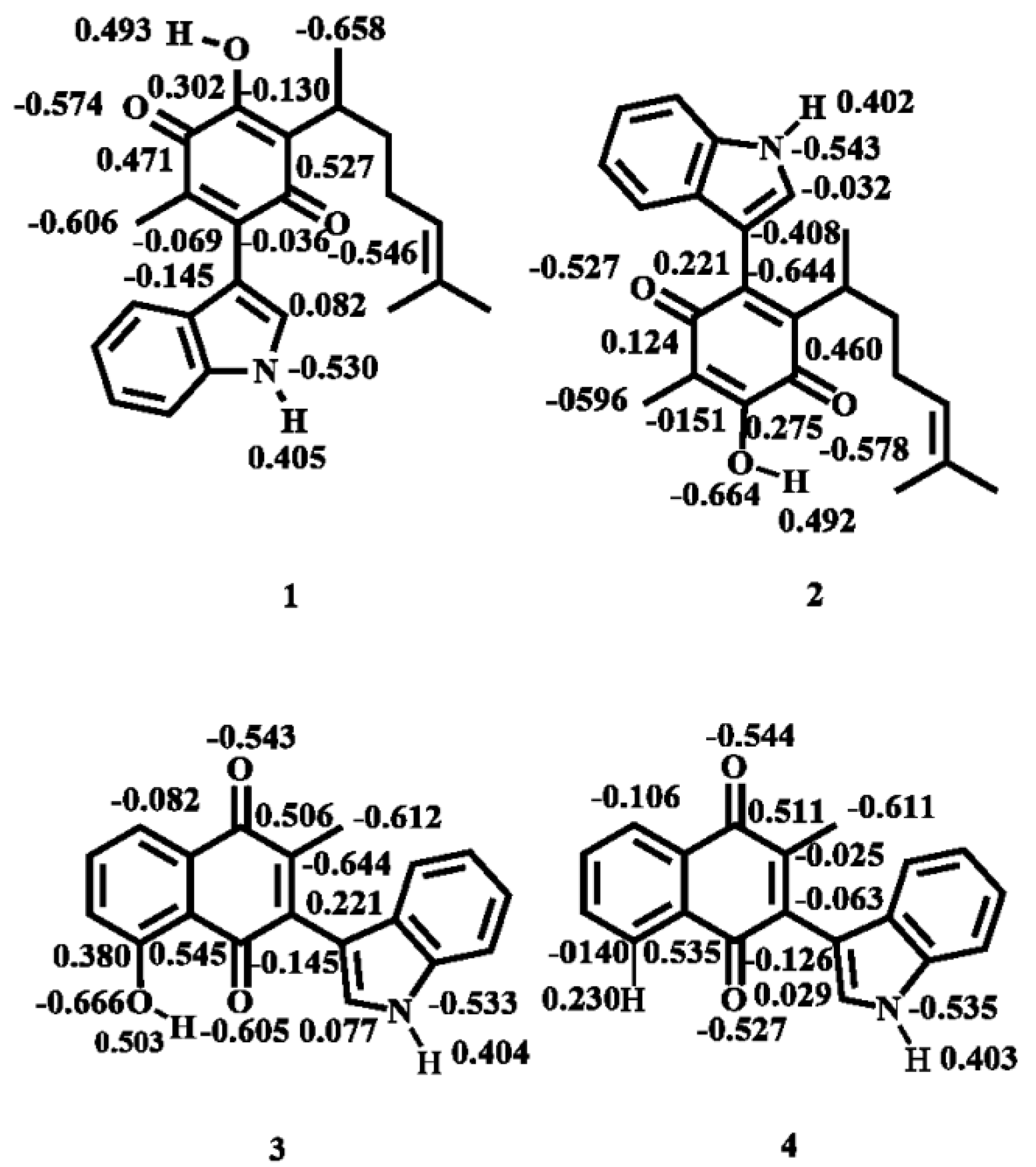
2.1.2. Atomic Charges
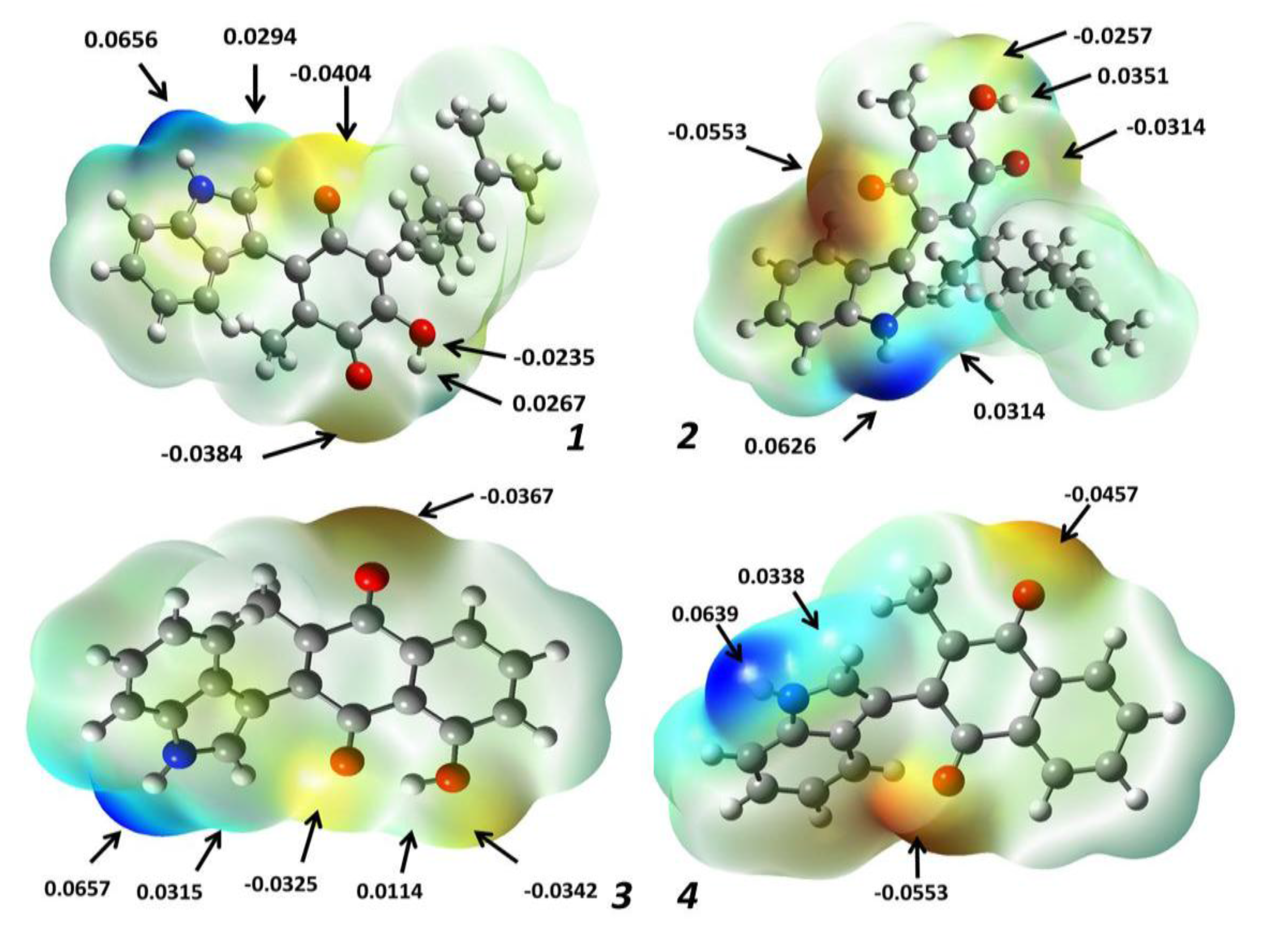
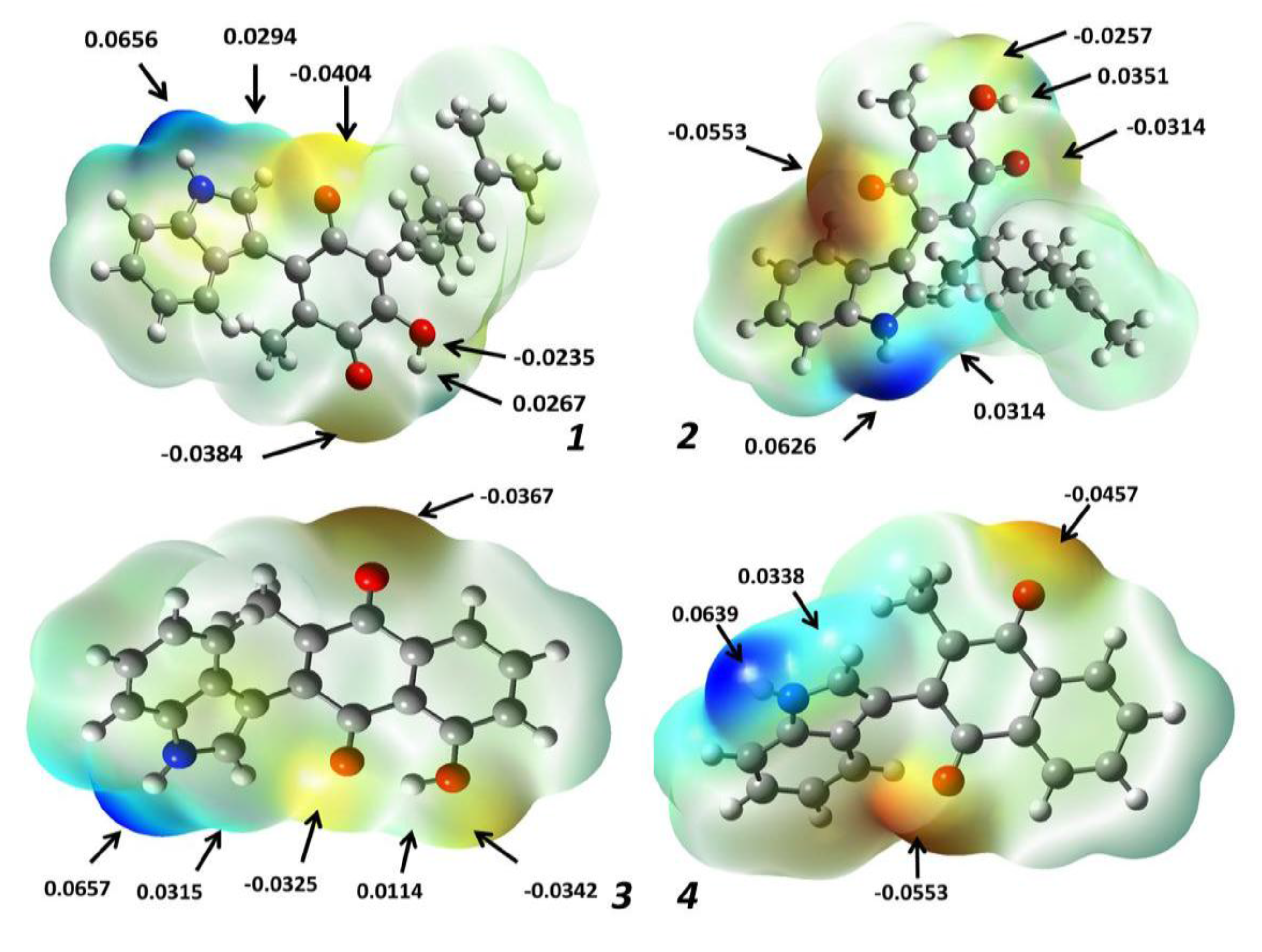
2.1.3. Molecular Electrostatic Potential Maps
2.2. Toxicological and Physicochemical Properties Prediction
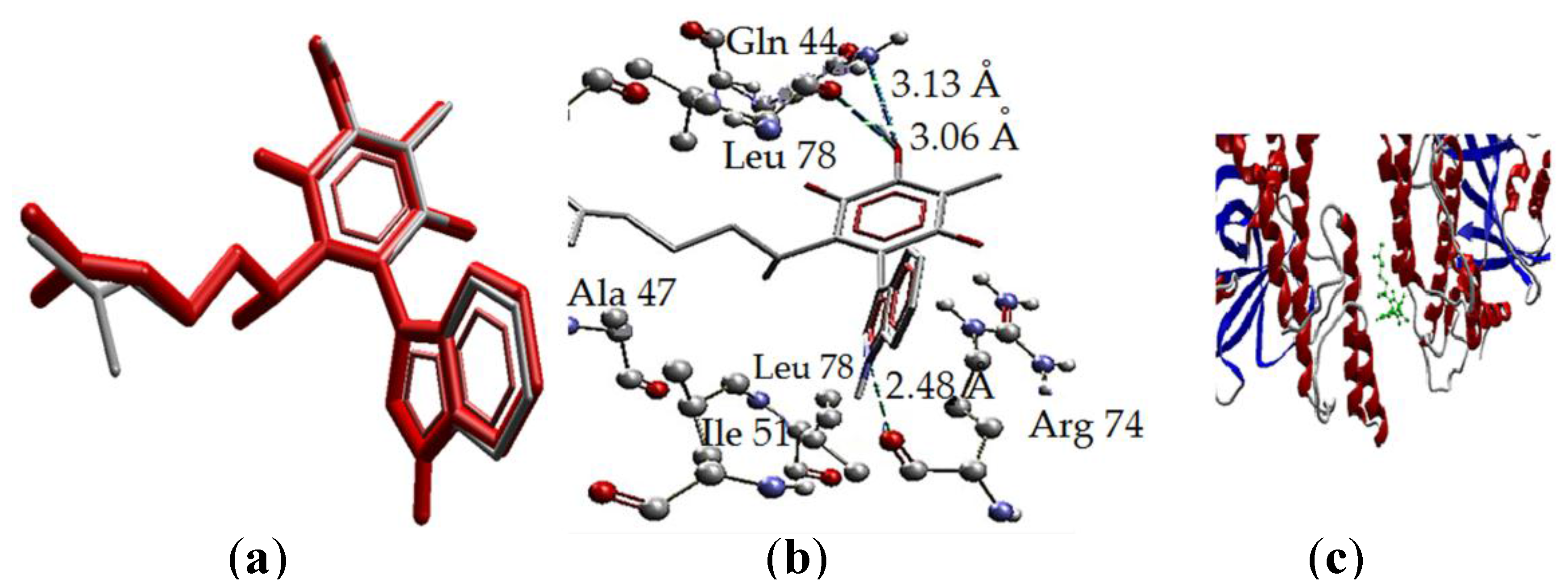
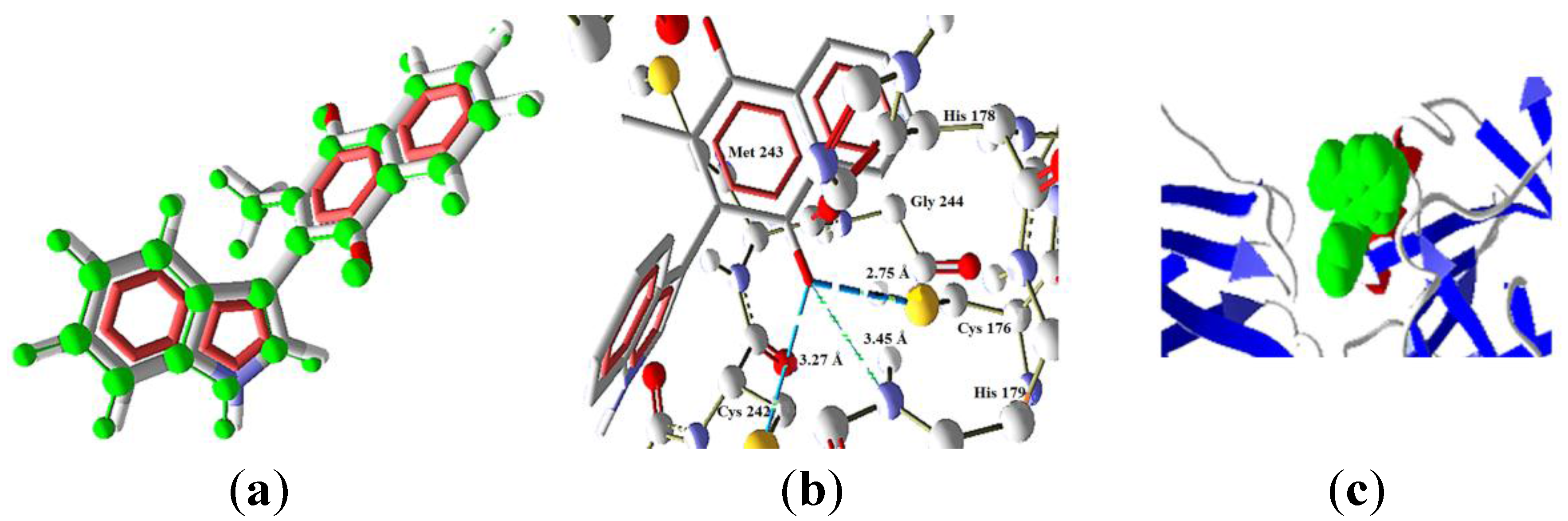
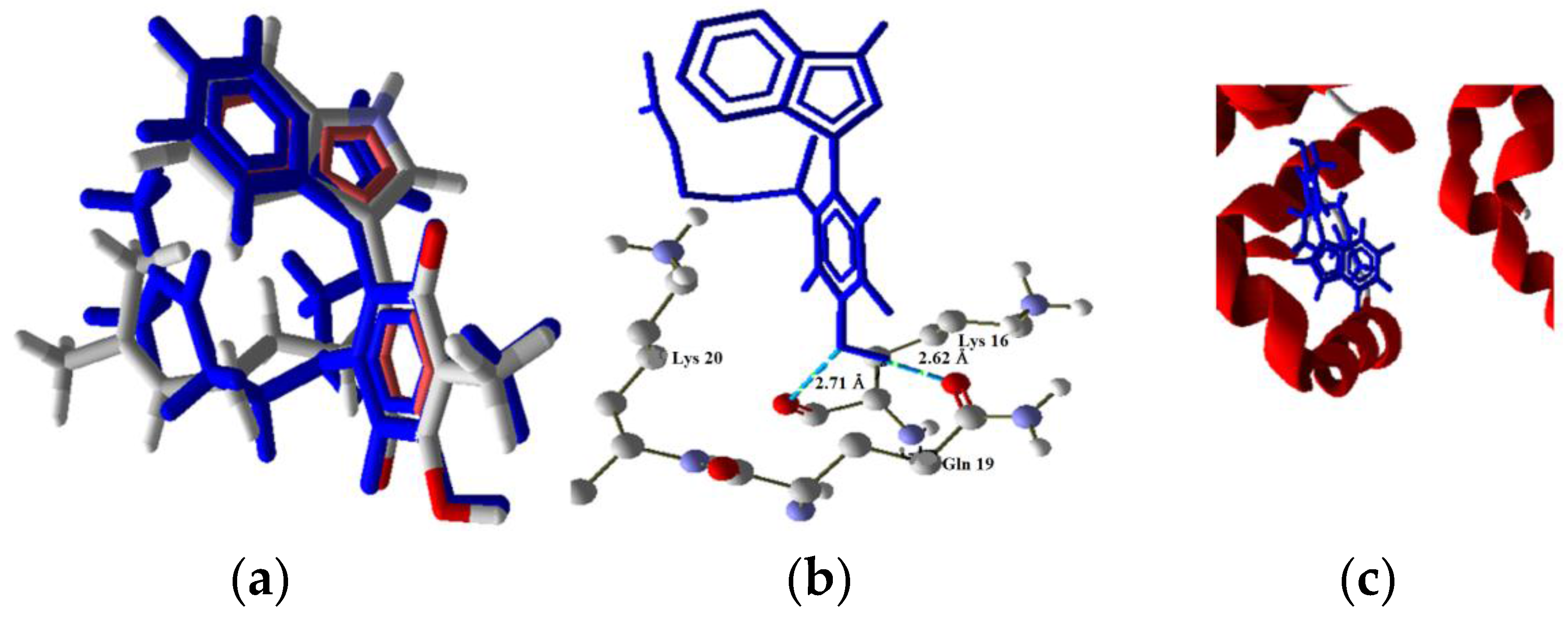
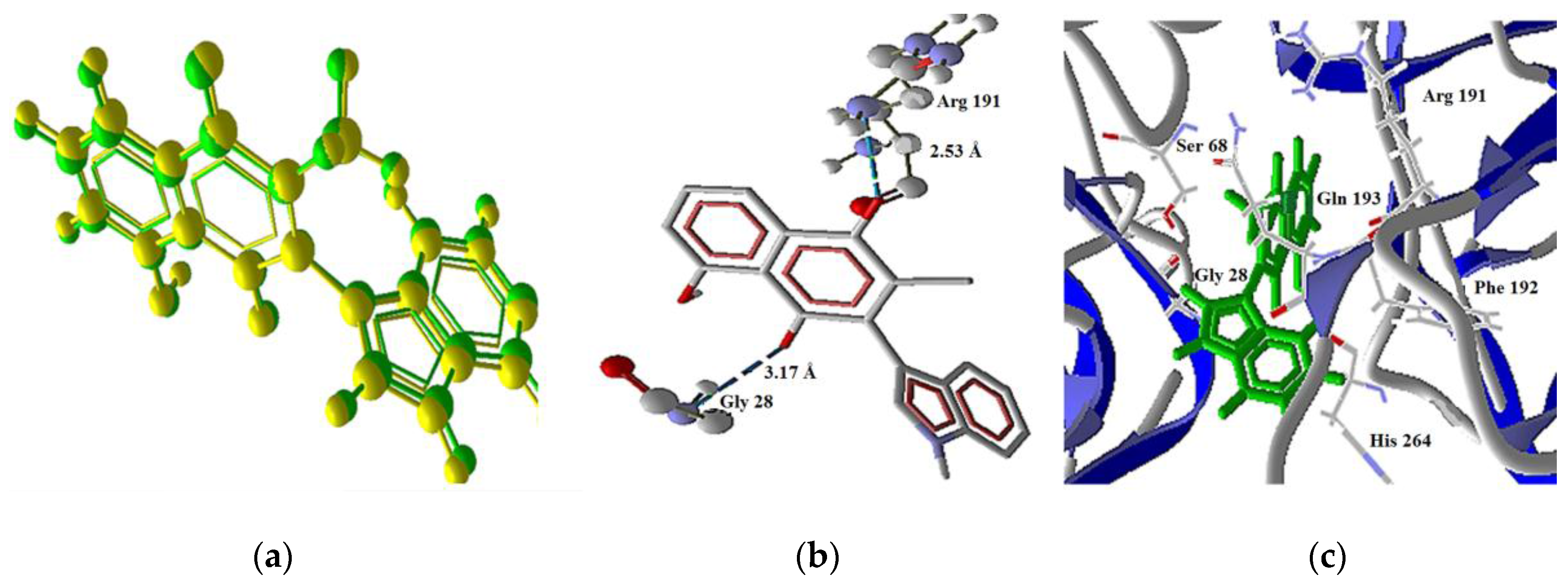
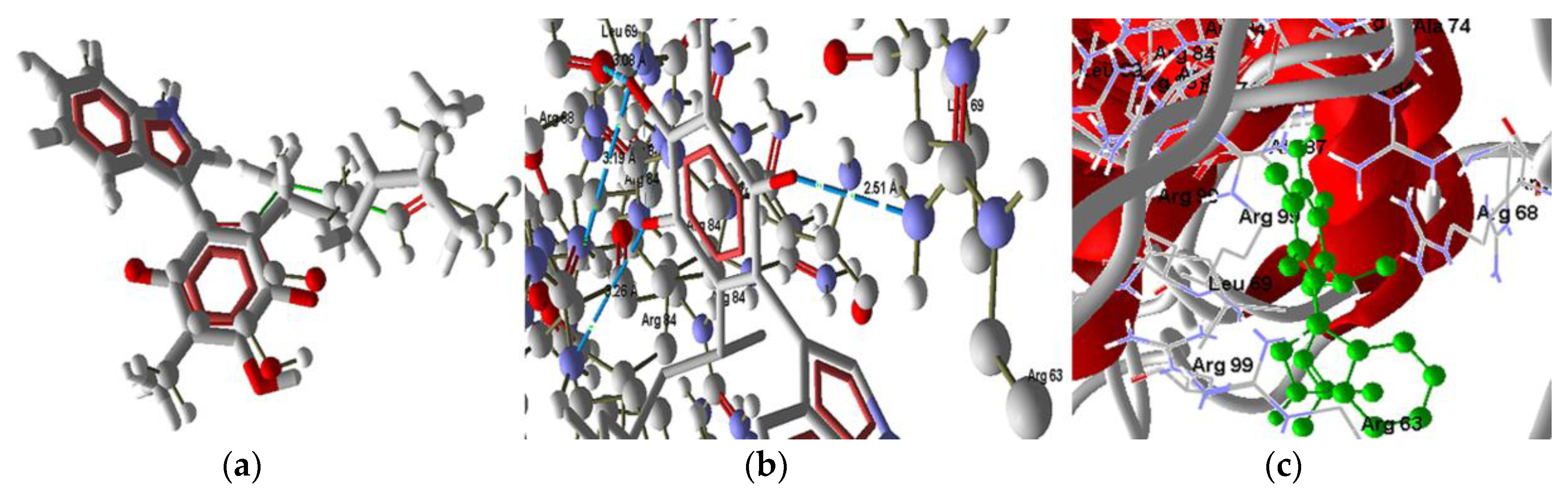
2.3 Docking Study in the Apoptosis Pathways
2.3.1. Intrinsic Pathway Considerations
2.3.2. Extrinsic Pathway Considerations
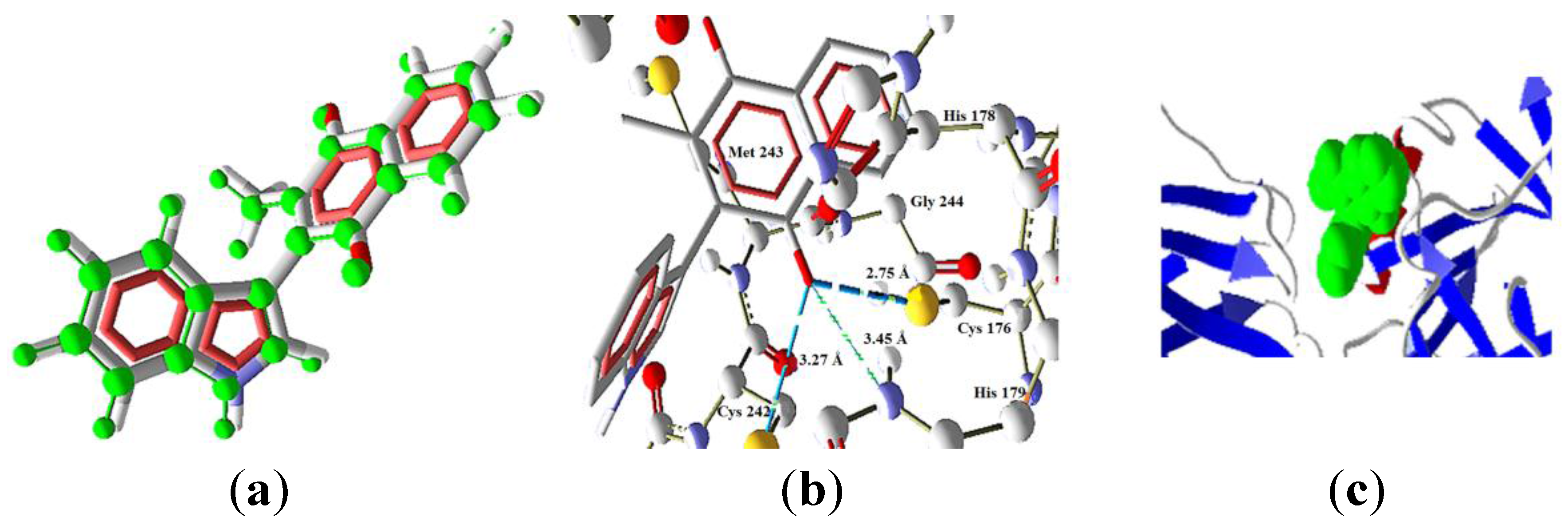
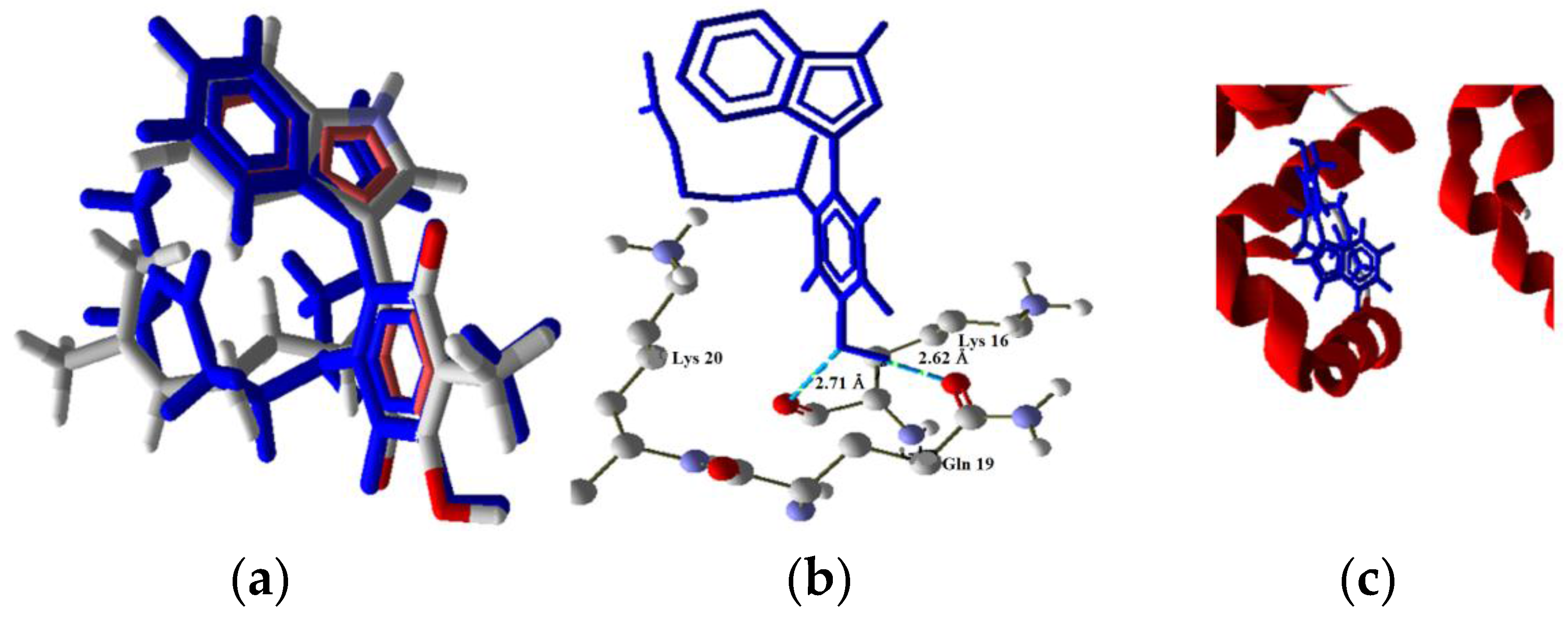
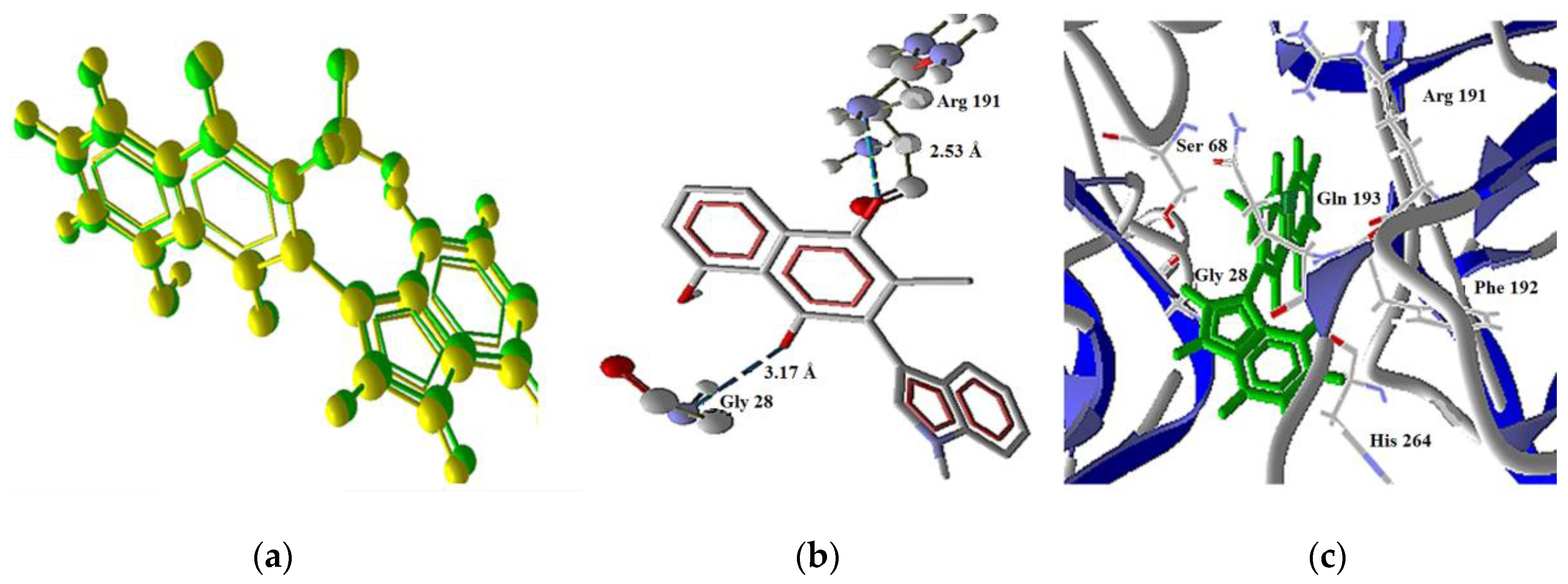
2.3.3. Intrinsic Pathway Docking Study Results
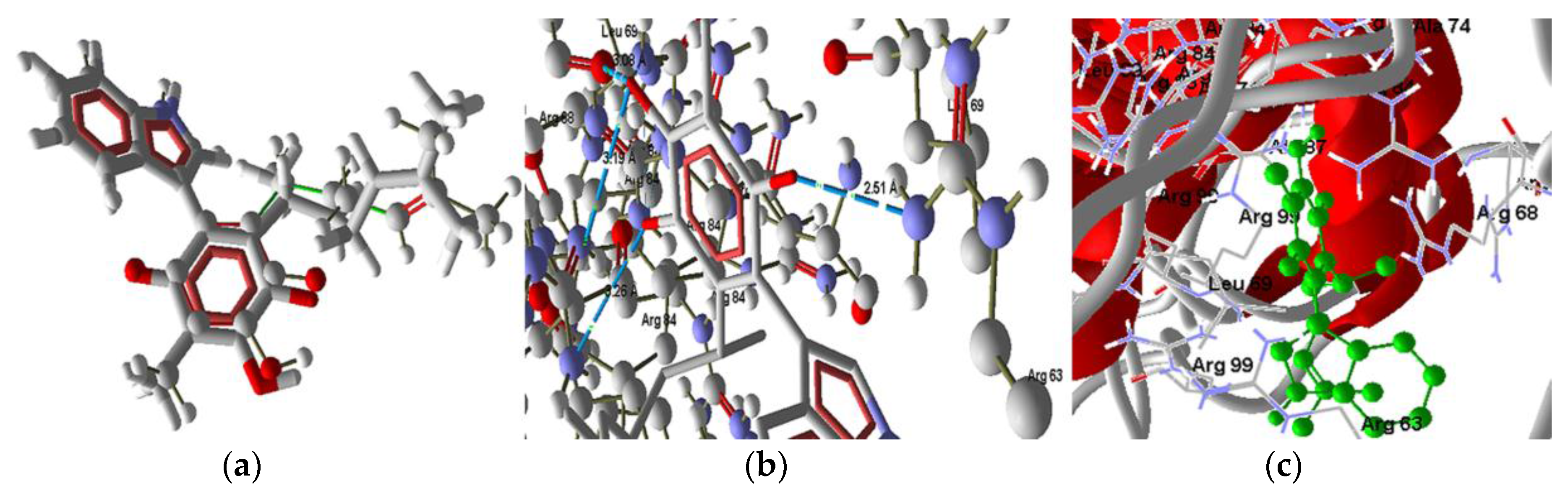
2.3.4. Extrinsic Pathway Docking Study
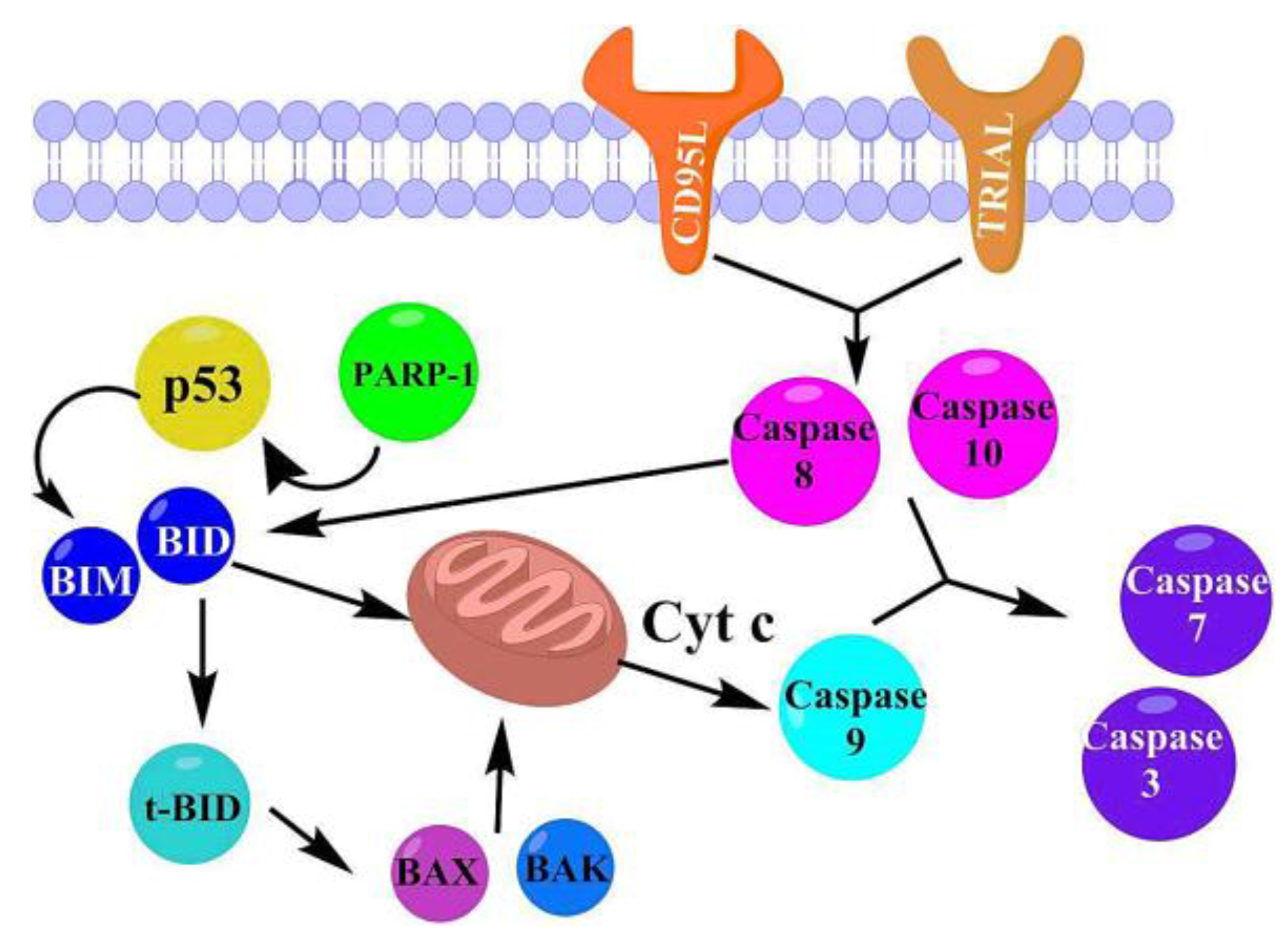
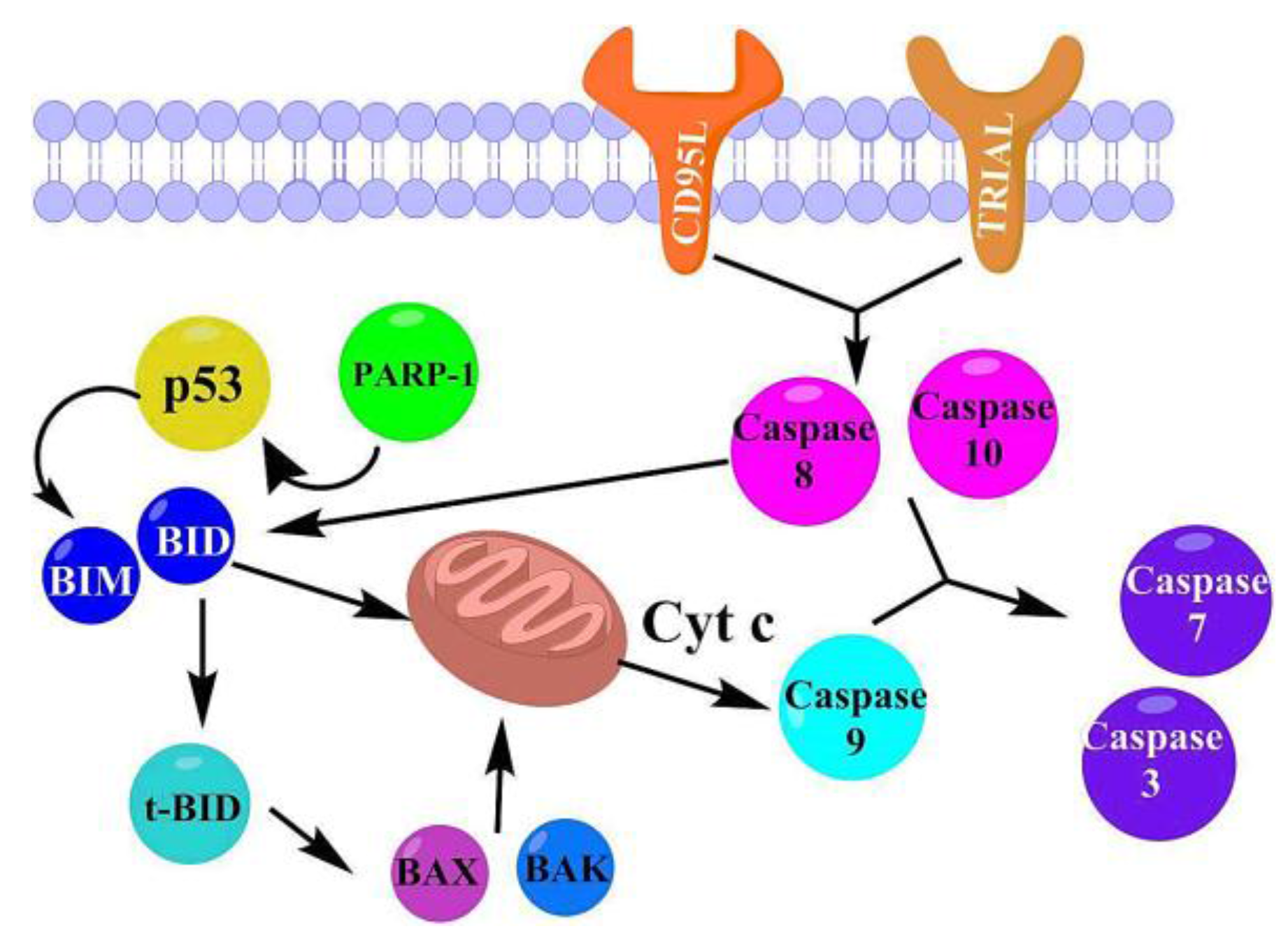
2.3.5. Proposed Apoptosis Pathways
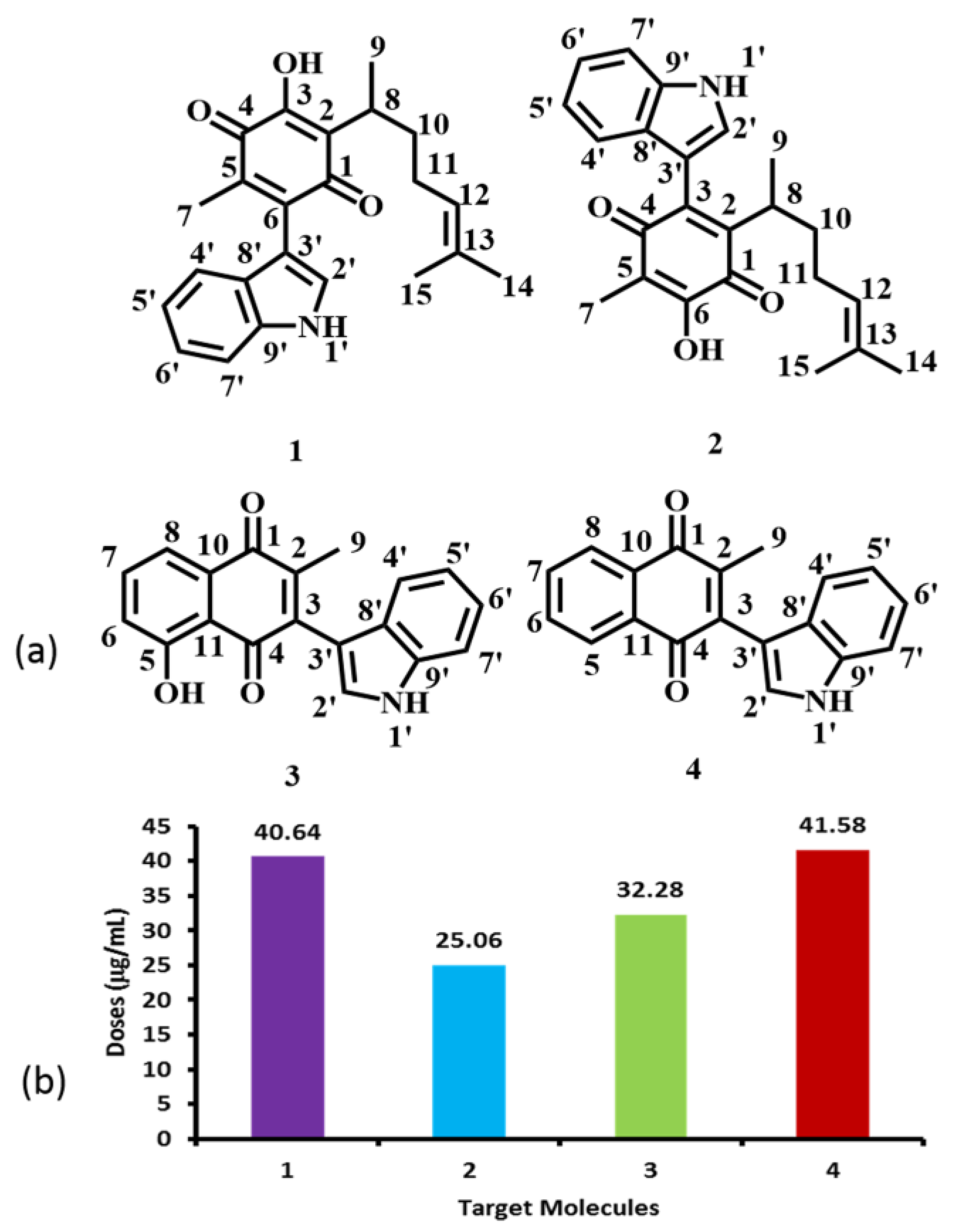
2.4. Pharmacological and Metabolic Properties
2.4.1. Target Molecules Absorption Predictions
2.4.2. Target Molecule Metabolism
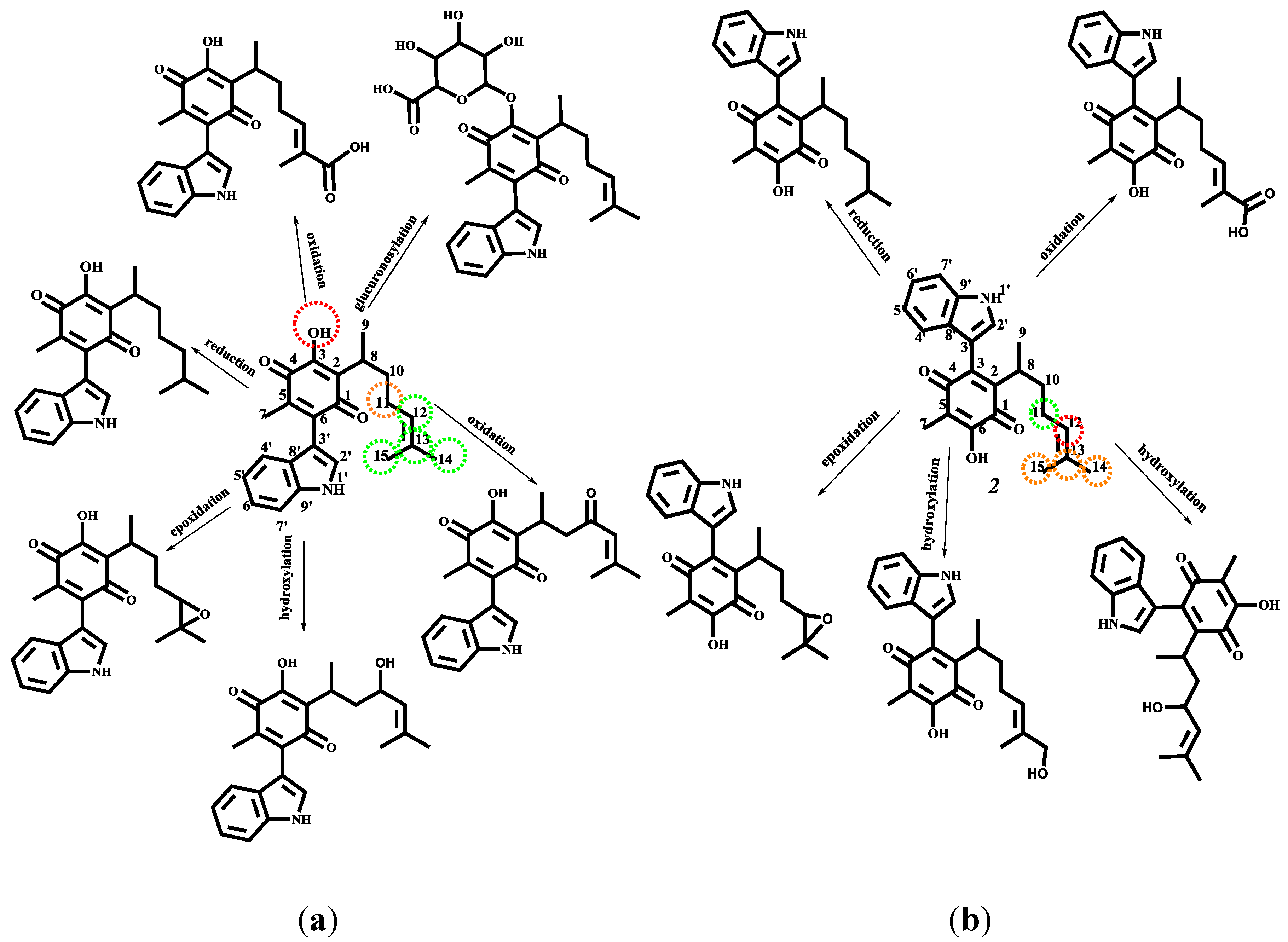
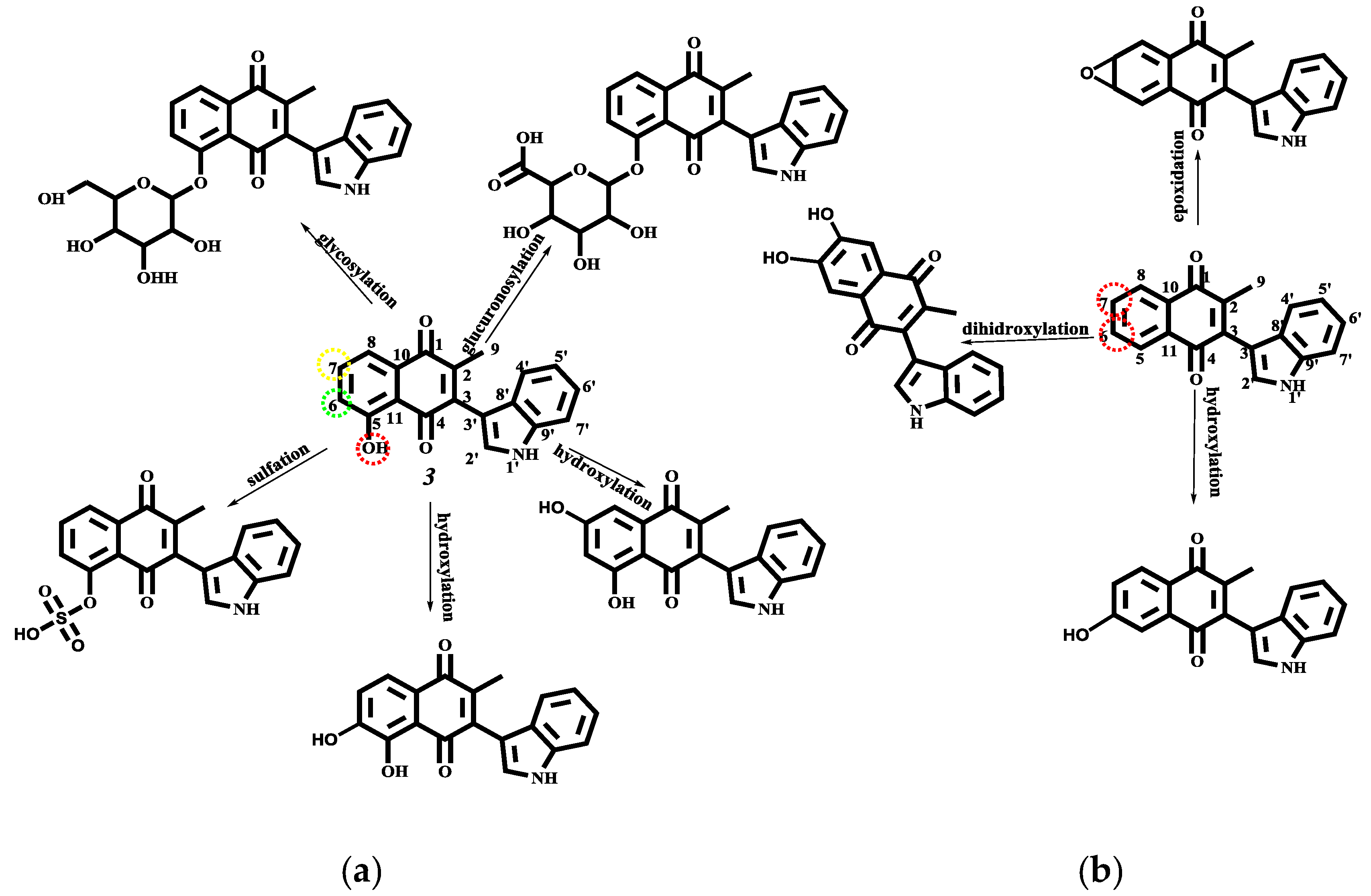
2.4.3. Phase I and II Metabolic Pathway
3. Methods
3.1. Reactivity Parameters
3.2 Toxicological and Physicochemical Properties Prediction
3.3 Docking Study
3.3.1 Docking Details
3.3.2. Molecular Docking Simulation
3.4. Pharmacological and Metabolic Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nomenclature of Quinones with Isoprenoid Side-Chains. Available online: http://www.chem.qmul.ac.uk/iupac/misc/quinone.html (accessed on 10 February 2016).
- De la Loza, R.; Leopoldo. Discurso pronunciado por el catedrático de química medica de la Escuela de Medicin. In Escritos de Leopoldo Río de la Loza; Noriega, J.M., Ed.; Imprenta de Ignacio Escalante: México City, México, 1911; Volume 1, pp. 94–100. [Google Scholar]
- Joseph-Nathan, P.; Santillan, R.L. The chemistry of perezone and its consequences. Stud. Nat. Prod. Chem. 1989, 5, 763–813. [Google Scholar]
- Sánchez, I.H.; Yanez, R.; Enríquez, R.; Joseph-Nathan, P. Reaction mechanism change in the Lewis acid catalyzed perezone-pipitzol transformation. J. Org. Chem. 1981, 46, 2818–2819. [Google Scholar] [CrossRef]
- Sánchez, I.H.; Basurto, F.; Joseph-Nathan, P. The stereocontrol of the perezone to pipitzol transformation. J. Nat. Prod. 1984, 47, 382–383. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, A.; Barrio, H.; Collera, O.; Enríquez, R.G.; Ortiz, B.; Sánchez-Obregón, R.; Yu, M. Isomerization of perezone into isoperezone and preparation of dihydroisoperezinone. Nat. Prod. Lett. 1994, 4, 133–139. [Google Scholar] [CrossRef]
- Burgueño-Tapia, E.; Joseph-Nathan, P. 13C-NMR substituent chemical shifts in hydroxyl-p-benzoquinones. Magn. Reson. Chem. 2000, 38, 390–393. [Google Scholar] [CrossRef]
- Burgueño-Tapia, E.; Joseph-Nathan, P. Detailed studies of perezone rearrangements. Monatsh. Chem./Chem. Mon. 1997, 128, 651–658. [Google Scholar] [CrossRef]
- Martínez, J.; Velasco-Bejarano, B.; Delgado-Reyes, J.F.; Pozas, R.; Torres Domínguez, H.M.; Trujillo-Ferrara, J.G.; Miranda, R. Eco-contribution to the chemistry of perezone, a comparative study, using different modes of activation and solventless conditions. Nat. Prod. Commun. 2008, 3, 1465–1468. [Google Scholar]
- Enriquez, R.; Ortega, J.; Lozoya, X. Active components in Perezia roots. J. Ethnopharmacol. 1980, 2, 389–393. [Google Scholar] [CrossRef]
- Alarcon-Aguilar, F.J.; Roman-Ramos, R.; Jimenez-Estrada, M.; Reyes-Chilpa, R.; Gonzalez-Paredes, B.; Flores-Saenz, J.L. Effects of three Mexican medicinal plants (Asteraceae) on blood glucose levels in healthy mice and rabbits. J. Ethnopharmacol. 1997, 55, 171–177. [Google Scholar] [CrossRef]
- De La de Peña, A.; Izaguirre, R.; Baños, G.; Viveros, M.; Enríquez, R.G.; Fernández-G, J.M. Effect of perezone, aminoperezone and their corresponding isomers isoperezone and isoaminoperezone upon in vitro platelet aggregation. Phytomedicine 2001, 8, 465–468. [Google Scholar]
- Sanchez-Torres, L.E.; Torres-Martínez, J.A.; Godinez-Victoria, M.; Omar, J.M.; Velasco-Bejarano, B. Perezone and its isomer isoperezone induce caspase-dependent and caspase-independent cell death. Phytomedicine 2010, 17, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Lozada, M.C.; Soria-Arteche, O.; Apan, M.T.R.; Nieto-Camacho, A.; Enríquez, R.G.; Izquierdo, T.; Jiménez-Corona, A. Synthesis, cytotoxic and antioxidant evaluations of amino derivatives from perezone. Bioorg. Med. Chem. 2012, 20, 5077–5084. [Google Scholar] [CrossRef] [PubMed]
- Koulouri, S.; Malamidou-Xenikaki, E.; Spyroudis, S. Acid-catalyzed addition of indoles to hydroxyquinones. Tetrahedron 2005, 61, 10894–10902. [Google Scholar] [CrossRef]
- Zhang, H.B.; Liu, L.; Chen, Y.J.; Wang, D.; Li, C.J. “On Water”-Promoted Direct Coupling of Indoles with 1, 4-Benzoquinones without Catalyst. Eur. J. Org. Chem. 2006, 2006, 869–873. [Google Scholar] [CrossRef]
- Pliego, R.G.; Juan, E.R.; Miranda, R.; Villalobos-Molina, R.; Delgado, F.; Osnaya, R.; Ferrara, J.T. Vasodilator effects of bis-dihydropyridines structurally related to nifedipine. Med. Chem. 2006, 2, 527–534. [Google Scholar]
- Velasco, B.; Trujillo-Ferrara, J.G.; Castillo, L.H.F.; Miranda, R.; Sánchez-Torres, L.E. In vitro apoptotic activity of 2,2-diphenyl-1,3, 2-oxazaborolidin-5-ones in L5178Y cells. Life Sci. 2007, 80, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Noguez, M.O.; García, A.; Ibarra, C.; Cabrera, A.; Aceves, J.M.; Nicolas, M.I.; Miranda, R. Green synthesis of bis-Biginelli esters, with vasodilatory effects, their mass spectrometric and physical studies. Catalysts 2009, 4, 6–10. [Google Scholar]
- Reyes, L.; Corona, S.; Arroyo, G.; Delgado, F.; Miranda, R. Eco-Contribution for the production of N-arylnitrones: Solvent-Free and assisted by microwaves. Int. J. Mol. Sci. 2010, 11, 2576–2583. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Romero-Vega, S.; Abeja-Cruz, R.; Álvarez-Toledano, C.; Miranda, R. Green Approach-Multicomponent Production of Boron-Containing Hantzsch and Biginelli Esters. Int. J. Mol. Sci. 2013, 14, 2903–2915. [Google Scholar] [CrossRef] [PubMed]
- Zarco Juarez, M.; Martínez, J.O.; Noguez Cordova, O.; Nicolás Vazquez, M.I.; Ramírez-Apan, T.; Pérez Flores, J.; Arroyo Razo, G.A. A green approach to the production of hybrid diindolylmethane-phenylboronic acids via a 3MCR: Promising antineoplasic molecules. J. Chem. 2013, 2013, 531208. [Google Scholar] [CrossRef]
- Escobedo-González, R.G.; Pérez Martínez, H.; Nicolás-Vázquez, M.I.; Martínez, J.; Gómez, G.; Serrano, J.N.; Miranda Ruvalcaba, R. Green Production of Indolylquinones, Derivatives of Perezone, and Related Molecules, Promising Antineoplastic Compounds. J. Chem. 2016, 2016, 3870529. [Google Scholar] [CrossRef]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1044. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Xu, J.; Hagler, A. Chemoinformatics and drug discovery. Molecules 2002, 7, 566–600. [Google Scholar] [CrossRef]
- Kumar, A.; Mohan, C.G.; Mishra, P.C. Molecular electrostatic potential and field as descriptors of hydrogen bonding and molecular activity. Effects of hybridization displacement charge. J. Mol. Struct. Theochem. 1996, 361, 135–144. [Google Scholar] [CrossRef]
- Politzer, P.; Laurence, P.R.; Jayasuriya, K. Molecular Electrostatic Potentials: An Effective Tool for the Elucidation of Biochemical Phenomena. Environ. Health Persp. 1985, 61, 191–202. [Google Scholar] [CrossRef]
- OSIRIS Property Explorer. Actelion Pharmaceuticals Ltd.: Allschwil, Switzerland, 2010. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 2 February 2016).
- Escobedo-González, R.; Méndez-Albores, A.; Villarreal-Barajas, T.; Aceves-Hernández, J.M.; Miranda-Ruvalcaba, R.; Nicolás-Vázquez, M.I. Theoretical Study of 8-Chloro-9-Hydroxy-Aflatoxin B1, the Conversion Product of Aflatoxin B1 by Neutral Electrolyzed Water. Toxins 2016, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Polkam, N.; Ramaswamy, V.R.; Rayam, P.; Allaka, T.R.; Anantaraju, H.S.; Dharmarajan, S.; Perumal, Y.; Gandamalla, D.; Yellu, N.R.; Balasubramanian, S.; et al. Synthesis, molecular properties prediction and anticancer, antioxidant evaluation of new edaravone derivatives. Bioorg. Med. Chem. Lett. 2016, 10, 2562–2568. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration cheminformatics. Available online: http://www.molinspiration.com/ (accessed on 2 February 2016).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 4–25. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Sirajuddin, M.; Ali, S.; Khalid, N.; Tahir, M.N.; Khan, H.; Ansari, T.M. Pharmacological investigations and Petra/Osiris/Molinspiration (POM) analyses of newly synthesized potentially bioactive organotin (IV) carboxylates. J. Photochem. Photobiol. B 2000, 158, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Thasni, K.A.; Sivakumar, K.C.; Nair, R.S.; Banerji, A.; Somasundaram, V.; Srinivas, P. Structure activity relationship of plumbagin in BRCA1 related cancer cells. Mol. Carcinogen. 2003, 52, 392–403. [Google Scholar]
- Yan, W.; Tu, B.; Liu, Y.Y.; Wang, T.Y.; Qiao, H.; Zhai, Z.J.; Tang, T.T. Suppressive effects of plumbagin on invasion and migration of breast cancer cells via the inhibition of STAT3 signaling and down-regulation of inflammatory cytokine expressions. Bone Res. 2013, 1, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Kaji, A.; Saito, R.; Nomura, M.; Miyamoto, K.I.; Kiriyama, N. Mechanism of the cytotoxicity of asterriquinone, a metabolite of Aspergillus terreus. Anticancer Res. 1996, 17, 3675–3679. [Google Scholar]
- Li, X.; Zheng, S.L.; Li, X.; Li, J.L.; Qiang, O.; Liu, R.; He, L. Synthesis and anti-breast cancer activity of new indolylquinone derivatives. Eur. J. Med. Chem. 2012, 54, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Hou, X.; Jacob, C. 1,4-Naphthoquinones: From oxidative damage to cellular and inter-cellular signaling. Molecules 2014, 19, 14902–14918. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.S.; Parveen, S.; Beg, M.A.; Suhail, M.; Chaudhary, A.G.; Damanhouri, G.A.; Rehan, M. Anticancer compound plumbagin and its molecular targets: A structural insight into the inhibitory mechanisms using computational approaches. PLoS ONE 2014, 9, e87309. [Google Scholar] [CrossRef] [PubMed]
- Escobedo-González, R.G.; Bahena, L.; Arias Tellez, J.L.; Hinojosa Torrés, J.; Ruvalcaba, R.M.; Aceves Hernández, J.M. Characterization and comparison of perezone with some analogues. Experimental and theoretical study. J. Mol. Struct. 2015, 1097, 98–105. [Google Scholar] [CrossRef]
- Wang, X.D.; Li, C.Y.; Jiang, M.M.; Dong, Li.; Wen, P.; Song, X.; Zhang, J. Induction of apoptosis in human leukemia cells through an intrinsic pathway by cathachunine, a unique alkaloid isolated from Catharanthus roseus. Phytomedicine 2016, 23, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar]
- Hongmei, Z. Extrinsic and Intrinsic Apoptosis Signal Pathway Review. In Apoptosis and Medicine; Ntuli, T.M., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Liu, C.; Zhou, M.; Li, Q.; Wang, R.; Kang, J. Screening of Small-Molecule Inhibitors of Protein-Protein Interaction with Capillary Electrophoresis Frontal Analysis. Anal. Chem. 2016, 88, 8050–8057. [Google Scholar] [CrossRef] [PubMed]
- Isabelle, M.; Moreel, X.; Gagné, J.P.; Rouleau, M.; Ethier, C.; Gagné, P.; Poirier, G.G. Investigation of PARP-1, PARP-2, and PARG interactomes by affinity-purification mass spectrometry. Proteome Sci. 2010, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- East China University of Science and Technology, School of Pharmacy, ADMETSAR, Shanghai Key Laboratory of New Drug Design, Laboratory of Molecular Modeling and Design. Available online: http://lmmd.ecust.edu.cn:8000 (accessed on 10 February 2016).
- Pham The, H.; González-Álvarez, I.; Bermejo, M.; Mangas Sanjuan, V.; Centelles, I.; Garrigues, T.M.; Cabrera-Pérez, M.Á. In Silico Prediction of Caco-2 Cell Permeability by a Classification QSAR Approach. Mol. Inf. 2011, 30, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Y.; Liang, H.; Bender, A.; Glen, R.C.; Yan, A. P-glycoprotein substrate models using support vector machines based on a comprehensive data set. J. Chem. Inf. Model. 2011, 51, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Broccatelli, F.; Carosati, E.; Neri, A.; Frosini, M.; Goracci, L.; Oprea, T.I.; Cruciani, G.A. Novel approach for predicting P-glycoprotein (ABCB1) inhibition using molecular interaction fields. J. Med. Chem. 2011, 54, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.; Zhao, Q.; Peng, H.; Hou, T. ADME evaluation in drug discovery. 10. Predictions of P-glycoprotein inhibitors using recursive partitioning and naive Bayesian classification techniques. Mol. Pharm. 2011, 8, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Didziapetris, R.; Japertas, P.; Avdeef, A.; Petrauskas, A. Classification analysis of P-glycoprotein substrate specificity. J. Drug Targ. 2003, 11, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.K.; Agarwal, S.; Raghava, G.P. Prediction of cytochrome P450 isoform responsible for metabolizing a drug molecule. BMC Pharmacol. 2010, 10, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Reisfeld, B.; Mayeno, A.N. Cytochromes P450: A structure-based summary of biotransformations using representative substrates. Drug Metab. Rev. 2008, 40, 1–100. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V. Guide to Cytochromes P450: Structure and Function; Taylor & Francis: London, UK, 2001. [Google Scholar]
- Cheng, F.; Yu, Y.; Zhou, Y.; Shen, Z.; Xiao, W.; Liu, G.; Tang, Y. Insights into molecular basis of cytochrome p450 inhibitory promiscuity of compounds. J. Chem. Inf. Model. 2011, 51, 2482–2495. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Lahari, B.L.; Rajkumar, T.; Reddy, L.S.S.; Reddy, Y.S.R.; Sivudu, G.; Krishna, P.N. In-silico Design, Synthesis and Biological Evaluation of Some Michael Adducts from Chalcones. Inventi Impact Med. Chem. 2015, 2015, 119–132. [Google Scholar]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Frisch, M.J.; Trucks, W.G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D.J. Density-Functional thermochemistry. III. The role of exact exchange. Chem. Phys. 1993, 98, 5648–5656. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Gill, P.M.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. The performance of the Becke-Lee-Yang-Parr (B-LYP) density functional theory with various basis sets. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef]
- London, F. Théorie quantique des courants interatomiques dans les combinaisons aromatiques. J. Phys. Radium 1937, 8, 397–409. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21 + G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Erdogdu, Y.; Tahir Güllüo-lu, M.; Kurt, M. DFT, FT-Raman, FT-IR and NMR studies of 2-fluorophenylboronic acid. J. Raman. Spectrosc. 2009, 40, 1615–1623. [Google Scholar] [CrossRef]
- Gangadharan, R.P.; Krishnan, S.S. First Order Hyperpolarizabilities, NPA and Fukui Functions of Cyclohexanone by Density Functional Theory Method. Acta. Phys. Pol. A 2015, 127, 748–752. [Google Scholar] [CrossRef]
- Hunter, C.A. Quantifying intermolecular interactions: Guidelines for the molecular recognition toolbox. Angew. Chem. Int. Edit. 2004, 43, 5310–5324. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Nakanishi, I.; Warizaya, M.; Iwashita, A.; Kido, Y.; Hattori, K.; Fujii, T. Inhibitor-induced structural change of the active site of human poly (ADP-ribose) polymerase. FEBS Lett. 2004, 556, 43–46. [Google Scholar] [CrossRef]
- Wang, Y.; Rosengarth, A.; Luecke, H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr. D 2007, 63, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.J.; Li, H.; Salvesen, G.S.; Yuan, J.; Wagner, G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 1999, 96, 615–624. [Google Scholar] [CrossRef]
- Ha, N.C.; Kim, J.S. Complex structure of BCL-XL and mutated BIM BH3 domain. Available online: http://www.rcsb.org/pdb/explore/explore.do?structureId=4YK9 (accesed on 10 February 2016).
- Liu, W.; Ramagopalm, U.A.; Zhan, C.; Bonanno, J.B.; Bhosle, R.C.; Nathenson, S.G.; Almo, S.C. Crystal structure of FASL and DcR3 complex. Structure 2016, 24, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Nemoviová, I.; Benedict, C.A.; Zajonc, D.M. Structure of human cytomegalovirus UL141 binding to TRAIL-R2 reveals novel, non-canonical death receptor interactions. PLoS Pathog. 2013, 9, e1003224. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Kordich, J.J.; Huang, T.H.; Piasecki, J.; Bush, T.L.; Sullivan, T.; O’Neill, J.W. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell 2014, 26, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.K.; Serrano, P.; Geralt, M.; Wuthrich, K. NMR structure of the SH3 domain of human RAS p21 protein activator (GTPase activating protein) 1. Available online: http://www.rcsb.org/pdb/explore/explore.do?structureId=2M51 (accesed on 10 February 2016).
- Suzuki, M.; Youle, R.J.; Tjandra, N. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell 2000, 103, 645–654. [Google Scholar] [CrossRef]
- Moldoveanu, T.; Liu, Q.; Tocilj, A.; Watson, M.; Shore, G.; Gehring, K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol. Cell 2006, 24, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Molegro, A.P.S. MVD Molegro Virtual Docker 5.0; Molegro: Aarhus C, Denmark, 2011. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 1.3; Schrçdinger LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Feixiong, C.; Weihua, L.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for evaluating chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar]
- Metaprint2D-React metabolic product predictor. Available online: http://www-metaprint2d.ch.cam.ac.uk/metaprint2d-react (accessed on 25 February 2016).
- Correa-Basurto, J.; Bello, M.; Rosales-Hernandez, M.C.; Hernández-Rodríguez, M.; Nicolás-Vázquez, I.; Rojo-Domínguez, A.; Flores-Sandoval, C.A. QSAR, docking, dynamic simulation and quantum mechanics studies to explore the recognition properties of cholinesterase binding sites. Chem. Biol. Interact. 2014, 209, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.; Arnby, C.H.; Carlsson, L.; Smith, J.; Stein, V.; Glen, R.C. Reaction site mapping of xenobiotic biotransformations. J. Chem. Inf. Model. 2007, 47, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Piechota, P.; Cronin, M.T.; Hewitt, M.; Madden, J.C. Pragmatic approaches to using computational methods to predict xenobiotic metabolism. J. Chem. Inf. Model. 2013, 53, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.; Zamora, I. New methods in predictive metabolism. J. Comput.-Aided Mol. Des. 2002, 16, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.C.; Alves, V.M.; Fraga, C.A.; Barreiro, E.J.; de Oliveira, V.; Andrade, C.H. Combination of docking, molecular dynamics and quantum mechanical calculations for metabolism prediction of 3,4-methylenedioxybenzoyl-2-thienylhydrazone. J. Mol. Model. 2012, 18, 2065–2078. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Energy (eV) | |
|---|---|---|
| HOMO | LUMO | |
| 1 | −5986 | −3374 |
| 2 | −5959 | −3428 |
| 3 | −6013 | −3347 |
| 4 | −5905 | −3184 |
| Property | Compound | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| Toxicity Risks | Mutagenic | N | N | N | N |
| Tumorigenic | N | N | N | N | |
| Irritant | H | H | N | N | |
| Reproductive effect | N | N | N | N | |
| Physicochemical Properties | cLogP | 4.6 | 4.6 | 2.83 | 3.17 |
| Solubility (Log S) | −4.07 | −4.07 | −4.15 | −4.46 | |
| Mol. weight | 363 | 363 | 303 | 287 | |
| TPSA | 70.16 | 70.16 | 70.16 | 49.93 | |
| Druglikeness | −0.87 | −0.87 | 2.29 | −0.57 | |
| Drug Score | 0.24 | 0.24 | 0.74 | 0.5 | |
| Protein (PDB) | Target Molecules | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||
| Score | RMSD | Score | RMSD | Score | RMSD | Score | RMSD | |
| PARP-1 (1UKO) | −144.263 | 22.569 | −136.380 | 0.702 | −103.384 | 0.117 | −102.680 | 0.254 |
| p53 (2OCJ) | −115.059 | 5.92 | −102.806 | 2.274 | −107.622 | 1.975 | −97.960 | 0.212 |
| BID (2BID) | −157.069 | 0.654 | −133.750 | 52.699 | −141.290 | 0.082 | −137.550 | 0.16 |
| BIM (4YK9) | −126.480 | 6.99 | −141.566 | 0.837 | −99.742 | 2.744 | −100.855 | 0.142 |
| CD95L (4MSV) | −99.340 | 0.6789 | −132.437 | 0.457 | −102.592 | 0.17 | −105.411 | 0.127 |
| TRAIL-R2 (4I9X) (4N90) | −126.081 | 1.342 | −137.349 | 16.801 | −101.123 | 6.465 | −101.083 | 7.82 |
| −140.412 | 1.6765 | −132.437 | 136.013 | −121.757 | 0.136 | −122.564 | 31.917 | |
| t-BiID (2M51) | No docking | No docking | −1790.830 | 0.454 | −1552.800 | 0.073 | −1556.770 | 0.389 |
| BAX (1FI6) | −140.360 | 9.819 | −138.366 | 0.884 | −130.975 | 0.08 | −126.530 | 0.072 |
| BAK (2IMT) | −116.520 | 0.3111 | −135.800 | 0.868 | −121.235 | 0.146 | −95.060 | 2.179 |
| Absorption Model | Molecules | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||
| Result | P | Result | P | Result | P | Result | P | |
| Blood-Brain Barrier | BBB+ | 0.650 | BBB+ | 0.556 | BBB− | 0.570 | BBB+ | 0.872 |
| Human Intestinal Absorption | HIA+ | 1 | HIA+ | 1 | HIA+ | 1 | HIA+ | 1 |
| Caco-2Permeability | Caco2+ | 0.500 | Caco2+ | 0.507 | Caco2+ | 0.527 | Caco2+ | 0.646 |
| P-glycoprotein Substrate | S | 0.722 | S | 0.692 | S | 0.620 | NS | 0.511 |
| P-glycoprotein Inhibitor | I | 0.536 | I | 0.604 | NI | 0.818 | I | 0.742 |
| I | 0.946 | I | 0.946 | NI | 0.897 | NI | 0.711 | |
| Renal Organic Cation Transporter | NI | 0.769 | NI | 0.793 | NI | 0.834 | NI | 0.746 |
| Metabolism Model | Molecules | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||
| Result | P | Result | P | Result | P | Result | P | |
| CYP450 2C9 Substrate | NS | 0.764 | NS | 0.794 | NS | 0.740 | NS | 0.734 |
| CYP450 2D6 Substrate | NS | 0.810 | NS | 0.807 | NS | 0.818 | NS | 0.814 |
| CYP450 3A4 Substrate | S | 0.678 | S | 0.646 | NS | 0.569 | NS | 0.500 |
| CYP450 1A2 Inhibitor | I | 0.637 | I | 0.664 | I | 0.912 | I | 0.914 |
| CYP450 2C9 Inhibitor | I | 0.510 | NI | 0.511 | I | 0.907 | I | 0.894 |
| CYP450 2D6 Inhibitor | NI | 0.812 | NI | 0.812 | NI | 0.687 | NI | 0.530 |
| CYP450 2C19 Inhibitor | NI | 0.589 | NI | 0.619 | I | 0.719 | I | 0.840 |
| CYP450 3A4 Inhibitor | NI | 0.809 | NI | 0.808 | NI | 0.830 | NI | 0.707 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escobedo-González, R.; Vargas-Requena, C.L.; Moyers-Montoya, E.; Aceves-Hernández, J.M.; Nicolás-Vázquez, M.I.; Miranda-Ruvalcaba, R. In silico Study of the Pharmacologic Properties and Cytotoxicity Pathways in Cancer Cells of Various Indolylquinone Analogues of Perezone. Molecules 2017, 22, 1060. https://doi.org/10.3390/molecules22071060
Escobedo-González R, Vargas-Requena CL, Moyers-Montoya E, Aceves-Hernández JM, Nicolás-Vázquez MI, Miranda-Ruvalcaba R. In silico Study of the Pharmacologic Properties and Cytotoxicity Pathways in Cancer Cells of Various Indolylquinone Analogues of Perezone. Molecules. 2017; 22(7):1060. https://doi.org/10.3390/molecules22071060
Chicago/Turabian StyleEscobedo-González, René, Claudia Lucia Vargas-Requena, Edgar Moyers-Montoya, Juan Manuel Aceves-Hernández, María Inés Nicolás-Vázquez, and René Miranda-Ruvalcaba. 2017. "In silico Study of the Pharmacologic Properties and Cytotoxicity Pathways in Cancer Cells of Various Indolylquinone Analogues of Perezone" Molecules 22, no. 7: 1060. https://doi.org/10.3390/molecules22071060
APA StyleEscobedo-González, R., Vargas-Requena, C. L., Moyers-Montoya, E., Aceves-Hernández, J. M., Nicolás-Vázquez, M. I., & Miranda-Ruvalcaba, R. (2017). In silico Study of the Pharmacologic Properties and Cytotoxicity Pathways in Cancer Cells of Various Indolylquinone Analogues of Perezone. Molecules, 22(7), 1060. https://doi.org/10.3390/molecules22071060