2. Results and Discussion
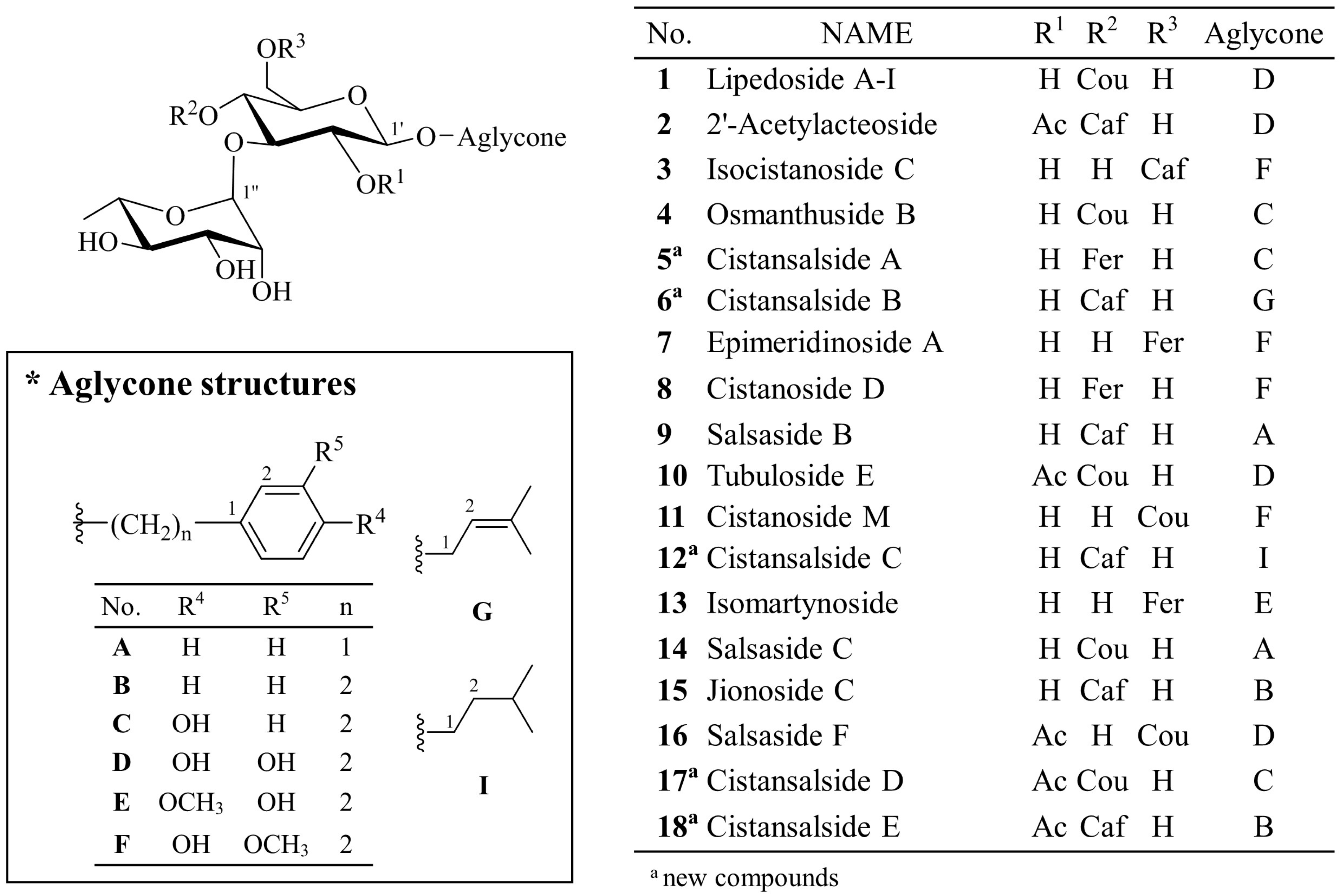
The phenylpropanoid-substituted diglycosides isolated from
C. salsa usually have structures based on disaccharide glycosides, which consist of a glucose and a rhamnose with a Rha (1→3) Glc linkage and one cinnamoyl substituent, such as coumaric acid (Cou), caffeic acid (Caf) and feruloyl acid (Fer), at the C-4 or C-6 position of glucose. The aglycone is commonly attached at the C-1 position of glucose. The structures of phenylpropanoid-substituted diglycosides with an acetyl group at the C-2 of glucose have frequently been reported [
6,
12,
16].
To perform the dereplication, MS fragmentation patterns of these compounds were analyzed by positive mode ESI-QTOF-MS. In MS spectra, all the phenylpropanoid-substituted diglycosides produced adduct ion peaks at [M + NH
4]
+, [M + K]
+ and [M + Na]
+, which provided the molecular weight and formula. The pattern of fragment ions could be found by successive losses of aglycone and glycoside residues ([M + H − Aglycone]
+, [M + H − Aglycone − Rha]
+ and [M + H − Aglycone – Rha − Glc (or Acetyl-Glc)]
+), which were useful for predicting the type of cinnamoyl substituent and sugars. The fragment ions at
m/z 163 of the caffeoyl group,
m/z 147 of the coumaroyl group or
m/z 177 of the feruloyl group give the characteristic signal of a cinnamoyl substituent in the phenylpropanoid-substituted diglycosides [
4,
15] (
Figure 2). The analysis of the fragment ions would provide useful information for the identification of the structures of phenylpropanoid-substituted diglycosides. However, their isomers could not be differentiated by MS spectrometry alone. For accurate identification of their complete structures, NMR spectra are required.
C. salsa was analyzed and the fingerprint of the EtOAc fraction was generated using the HPLC-DAD (diode array detector)-ESI-QTOF-MS method (
Figure 3). Each peak in the fingerprint of
C. salsa was predicted according to MS fragmentation features (
Table 1). Many phenylpropanoid-substituted diglycosides were screened out from this fraction, which was subjected to HPLC-QTOF-MS-guided isolation for the discovery of new phenylpropanoid-substituted diglycosides. Eighteen peaks including five new compounds were further identified and their structures were elucidated through extensive spectroscopic analysis.
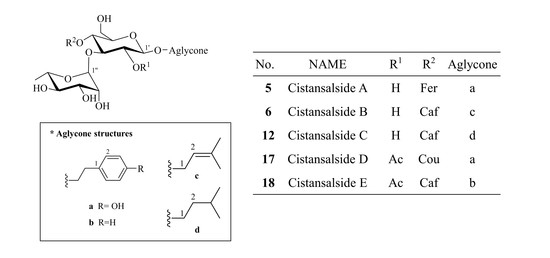
Cistansalside A (5) was obtained as a brown amorphous powder. Its molecular formula was determined to be C30H38O14 by positive mode high resolution (HR) ESI-QTOF-MS based on the adduct ion peak at m/z 645.2146 [M + Na]+ (calcd. for C30H38O14Na, 645.2154). A characteristic ion at m/z 177 suggested that a feruloyl substituent existed in its structure. The fragment ions at m/z 339 and m/z 485 suggested the presence of a rhamnose unit and a glucose unit as well.
The
13C-NMR spectrum showed 30 carbon atoms. Analysis of the
1H and HSQC spectra indicated the presence of two anomeric protons at δ
H 4.35 (1H, d,
J = 7.9 Hz, H-1′) and 5.02 (1H, s, H-1′′) and a methoxy proton at δ
H 3.80 (3H, s, 3′′′-OCH
3). The
1H-NMR spectrum showed an 1,3,4-trisubstituted benzene ring at δ
H 7.29 (1H, d,
J = 1.3 Hz, H-2′′′), 7.09 (1H, dd,
J = 8.3, 1.3 Hz, H-6′′′) and 6.79 (1H, d,
J = 8.3 Hz, H-5′′′), a
trans-olefin group at δ
H 7.53 (1H, d,
J = 15.9 Hz, H-7′′′) and 6.41 (1H, d,
J = 15.9 Hz, H-8′′′) and a
para-substituted benzene ring at δ
H 7.05 (2H, d,
J = 8.3 Hz, H-2, 6) and 6.67 (2H, d,
J = 8.3 Hz, H-3, 5) (
Table 2).
A 3,4-dihydroxyphenyl group was suggested by the HMBC correlations between H-2′′′ and a quaternary aromatic carbon at δC 149.4 (C-4′′′) and between H-5′′′ and C-1′′′ (δC 125.6) and C-3′′′ (δC 147.9). From the HMBC NMR spectrum, the correlations between a carbonyl carbon at δC 165.8 and H-8′′′ and between H-6′′′ and C-7′′′ (δC 145.5) suggested a 3,4-dihydroxylated cinnamoyl group. The HMBC correlation between the methoxy proton and C-3′′′ and the NOESY correlation between the methoxy proton and H-2′′′ confirmed the cinnamoyl substituent to be an (E)-feruloyl group.
The 4-hydroxyphenyl group was suggested by the HMBC correlations between H-3,5 and quaternary aromatic carbons at δC 155.7 (C-4) and 128.5 (C-1). A hydroxyethyl group was confirmed by the COSY correlations among H-7 (2H, δH 2.76, m), H-8a (1H, δH 3.90, m) and H-8b (1H, δH 3.61, m). From the HMBC NMR spectrum, the correlation between C-7 (δC 34.7) and H-2, 6 suggested a 4-hydroxyphenylethyl group, as an aglycone substituent.
Two sugar moieties, suggested by the MS fragment pattern, were double-checked by the NMR spectra and HPLC analysis of the acid hydrolysate. The absolute configurations of them were determined to be
d-glucose and
l-rhamnose using HPLC analysis of the acid hydrolysate [
17]. A β-glucose moiety and an
α-rhamnose moiety were established by coupling constants of the anomeric protons. The
1H-
1H COSY spectrum showed the sequential correlations from H-1′ to H-5′ and from H-1′′ to H-6′′ (
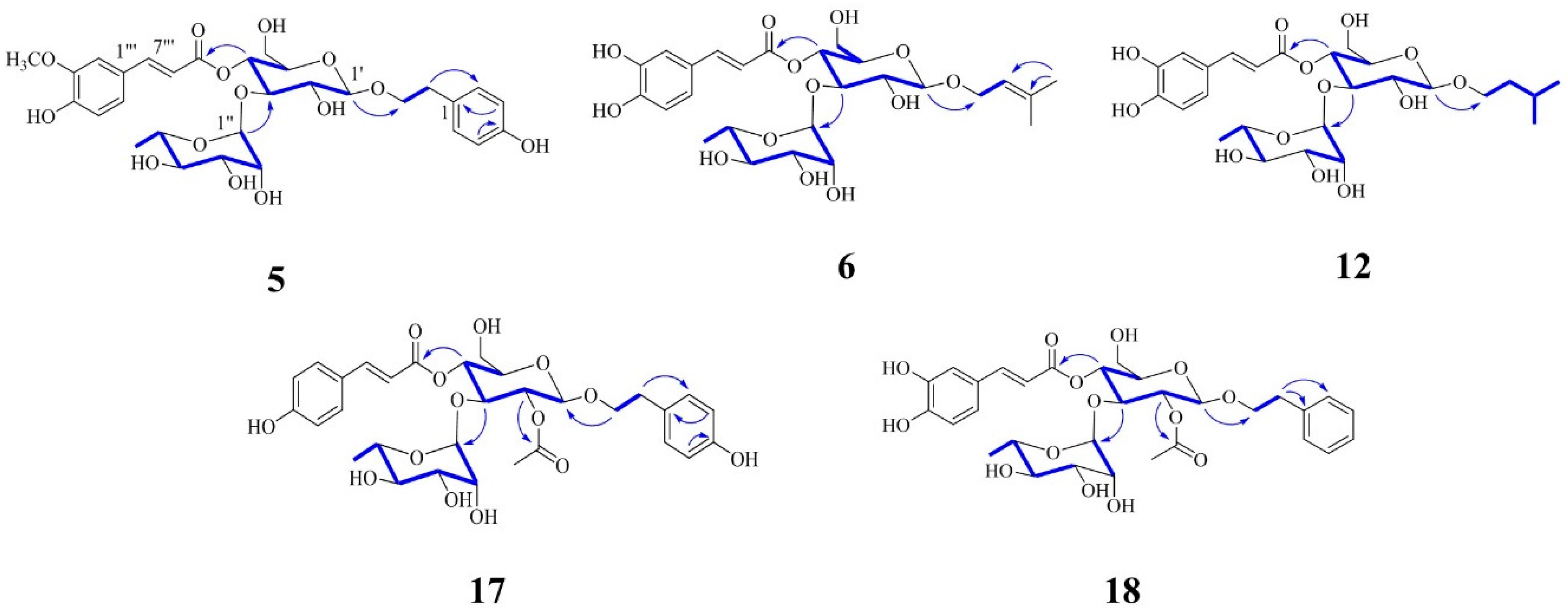
Figure 4).
From the 1H-NMR spectrum, the downfield shift of H-4′ (δH 4.71) suggested an acyl-substituent on glucose. The HMBC correlation between H-4′ and C-9′′′ (δC 165.8) confirmed that a feruloyl substituent was located at the C-4′ position. The aglycone was located at C-1′ according to the HMBC correlation between H-1′ and C-8 (δc 70.2). The HMBC correlation between H-1′′ and C-3′ (δC 79.2) gave us the position of rhamnose in this structure. Thus, the structure of 5 was established to be 4-hydroxyphenylethyl-O-α-l-rhamnopyranosyl-(1→3)-4-O-(E)-feruloyl-β-d-glucopyranoside and the compound was named cistansalside A.
Cistansalside B (6) was obtained as a brown amorphous powder. Its molecular formula was determined to be C26H36O13 based on the 13C-NMR data and a (+)-HR-ESI-QTOF-MS peak at m/z 579.2054 [M + Na]+ (calcd. for C26H36O13Na, 579.2048). Fragment ions including a [M + H − Aglycone − Rha − Glc]+ ion at m/z 163, a [M + H − Aglycone − Rha]+ ion at m/z 325 and a [M + H − Aglycone]+ ion at m/z 471 were also detected. A characteristic ion at m/z 163 suggested that a caffeoyl group existed in the structure.
The
1H-NMR spectrum showed an 1,3,4-trisubstituted benzene ring at δ
H 7.02 (1H, s, H-2′′′), 6.97 (1H, d,
J = 8.3 Hz, H-6′′′) and 6.75 (1H, d,
J = 8.3 Hz, H-5′′′), a
trans-olefin at δ
H 7.47 (1H, d,
J = 15.8 Hz, H-7′′′) and 6.19 (1H, d,
J = 15.8 Hz, H-8′′′). The
1H and HSQC NMR spectra showed two anomeric protons at δ
H 4.29 (1H, d,
J = 7.9 Hz, H-1′) and 5.01 (1H, s, H-1′′), an olefinic proton at δ
H 5.13 (1H, dd,
J = 7.6, 6.5 Hz, H-2), two methylene protons at δ
H 4.23 (1H, dd,
J = 12.2, 6.5 Hz, H-1a) and 4.13 (1H, dd,
J = 12.2, 7.6 Hz, H-1b) and germinal methyl groups at δ
H 1.71 (3H, s, H-4) and 1.63 (3H, s, H-5) (
Table 2).
From the HMBC spectrum, the 3,4-dihydroxyphenyl group was suggested by the correlations between H-2′′′ and a quaternary aromatic carbon at δC 149.0 (C-4′′′) and between H-5′′′ and another quaternary carbons at δC 125.5 (C-1′′′) and 148.5 (C-3′′′). From the HMBC NMR spectrum, the correlations between H-6′′′ and C-7′′′ (δC 145.6) and between the carbonyl carbon at δc 165.7 (C-9′′′) and H-8′′′ suggested an (E)-caffeoyl group.
A 3-methylbutenyl group, an aglycone substructure, was suggested by the COSY correlations of H-2 with H-1a and H-1b and the HMBC correlations between H-4 and H-5 and the olefinic carbons, at δC 120.7 (C-2) and 136.4 (C-3).
Two sugar moieties were established by the NMR spectra analysis and HPLC spectra analysis of the acid hydrolysate, with MS fragment pattern. The absolute configuration of them were determined to be
d-glucose and
l-rhamnose using HPLC analysis of the acid hydrolysate [
17]. These sugar moieties were defined as a β-glucose and an
α-rhamnose by coupling constants of the anomeric protons. The
1H-
1H COSY spectrum showed the sequential correlations from H-1′ to H-6′ and from H-1′′ to H-6′′ (
Figure 4).
A downshifted glucose proton at δH 4.70 (H-4′) suggested an acyl-substituent on glucose. From the HMBC spectrum, the correlation between H-4′ and C-9′′′ confirmed the position of caffeoyl substituent. The HMBC correlations between H-1′ and C-1 (δC 64.5) and between H-3′ (δH 3.68) and C-1′′ (δC 101.2) suggested the positions of each substituents. Accordingly, the structure of 6 was determined as 3-methylbutenyl-O-α-l-rhamnopyranosyl-(1→3)-4-O-(E)-caffeoyl-β-d-gluco-pyranoside and named cistansalside B.
Cistansalside C (12), a brown amorphous powder, was determined to have a molecular formula of C26H38O13 by (+)-HR-ESI-QTOF-MS, which showed a peak at m/z 581.2213 [M + Na]+ (calcd. for C26H38O13Na, 581.2205). A characteristic ion at m/z 163 suggested that a caffeoyl substituent existed in the structure. The fragment ions at m/z 325 and m/z 471 suggested the existence of a rhamnose unit and a glucose unit.
Comparison of the NMR spectra of 12 with those of 6 showed that they were similar except for the aglycone structure. In the NMR spectra of 12, two paraffinic carbons at δC 38.0 (C-2) and 24.4 (C-3) were observed instead of two olefinic carbons at δC 120.7 (C-2) and 136.4 (C-3) in the aglycone of 6. The germinal methyl groups (δH 0.88) in the aglycone of 12 were shifted upfield relative to H-4 and H-5 (δH 1.71 and 1.63) in the aglycone of 6 (Table 2). The aglycone of 12 was suggested to be a 3-methylbutyl group, which was confirmed by the 1H and COSY NMR spectra. Peaks of 3-methylbutyl group were observed at δH 3.81 (1H, m, H-1a), 3.48 (1H, m, H-1b), 1.71 (1H, m, H-3), 1.44 (2H, m, H-2) and 0.88 (H-4, 5).
Two sugar moieties were reaffirmed by the HPLC analysis of the acid hydrolysate and the NMR spectra analysis as well as MS fragment pattern. The absolute configurations of the sugars were identified as
d-glucose and
l-rhamnose using HPLC analysis of the acid hydrolysate [
17]. A β-glucose moiety and an
α-rhamnose moiety were confirmed by coupling constants of the anomeric protons. The
1H-
1H COSY spectrum showed the sequential correlations from H-1′ to H-5′, from H-1′′ to H-3′′ and from H-6′′ to H-4′′ (
Figure 4).
The position of substituents were confirmed by means of the HMBC analysis. In the HMBC spectrum, the correlations between H-1′ and C-1 (δC 67.2), between H-3′ (δH 3.68) and C-1′′ (δC 101.2) and between H-4′ (δH 4.70) and C-9′′′ (δC 165.7) were detected. Consequently, the structure of 12 was established to be 3-methylbutyl-O-α-l-rhamnopyranosyl-(1→3)-4-O-(E)-caffeoyl-β-d-gluco-pyranoside and named cistansalside C.
Cistansalside D (17), an amorphous brown powder, was determined to have a molecular formula of C31H38O14 by the positive mode high-resolution ESI-QTOF-MS, which showed an adduct ion peak at m/z 657.2147 [M + Na]+ (calcd. for C31H38O14Na, 657.2154). Fragment ions including a [M + H − Aglycone − Rha − Acetyl Glc]+ ion at m/z 147, a [M + H − Aglycone − Rha]+ ion at m/z 351 and a [M + H − Aglycone]+ ion at m/z 497 were also detected. A characteristic ion at m/z 147 suggested that a coumaroyl substituent was present in its structure. The fragment ions at m/z 351 and m/z 497 suggested the existence of a rhamnose unit and an acetyl-substituted glucose unit.
The
13C-NMR spectrum revealed the presence of 31 carbon atoms. Two anomeric protons at δ
H 4.61 (1H, m, H-1′) and 4.60 (1H, s, H-1′′) were observed in the
1H and HSQC spectra. The
1H-NMR spectrum indicated the presence of a methyl group at δ
H 1.97 (3H, s, acetyl-CH
3), a
trans-olefin at δ
H 7.55 (1H, d,
J = 15.9 Hz, H-7′′′) and 6.34 (1H, d,
J = 15.9 Hz, H-8′′′) and two
para-substituted benzene rings at δ
H 7.53 (2H, d,
J = 6.9 Hz, H-3′′′, 5′′′) and 6.79 (2H, d,
J = 6.9 Hz, H-2′′′, 6′′′)/ δ
H 6.98 (2H, d,
J = 8.0 Hz, H-2, 6) and 6.65 (2H, d,
J = 8.0 Hz, H-3, 5) (
Table 2). From the HMBC NMR spectrum, the correlations between a carbonyl carbon at δ
c 165.5 (C-9′′′) and H-8′′′ and between H-2′′′, 6′′′ and C-7′′′ (δ
c 145.4) suggested an (
E)-coumaroyl group. The HMBC correlation between a methyl proton peak and a carbonyl carbon at δ
c 169.0 confirmed the presence of an acetyl group.
A 4-hydroxyphenyl group was suggested based on the HMBC correlations between H-3, 5 and quaternary aromatic carbons at δC 155.6 (C-4) and 128.6 (C-1). A hydroxylated ethyl group was confirmed by the COSY NMR signals at δH 2.66 (2H, t, J = 6.1 Hz, H-7), 3.90 (1H, m, H-8a) and 3.54 (1H, m, H-8b). The HMBC correlations between H-7 and C-1 (δC 128.6) and C-2, 6 (δC 129.7) suggested a 4-hydroxyphenylethyl group, as an aglycone substructure.
Two sugar moieties, suggested from MS fragment pattern, were reconfirmed by the HPLC analysis of the acid hydrolysate and NMR spectra.
d-glucose and
l-rhamnose were elucidated using HPLC analysis of the acid hydrolysate [
17]. A β-glucose moiety and an
α-rhamnose moiety were established by coupling constants of the anomeric protons. The
1H-
1H COSY spectrum showed the sequential correlations from H-1′ to H-5′ and from H-1′′ to H-6′′ (
Figure 4).
From the 1H-NMR spectrum, the downfield shift of H-2′ (δH 4.69) and H-4′ (δH 4.80) suggested the acyl-substituted position on glucose. The connections between glucose and two acyl groups were confirmed by the HMBC correlations between H-4′ (δH 4.80) and C-9′′′ and between H-2′ and a carbonyl carbon of an acetyl group (δC 169.0). The positions of an aglycone and a rhamnose were given by the HMBC correlations between H-3′ (δH 3.95) and C-1′′′ and between H-8 and C-1′. Accordingly, the structure of 17 was assigned as 4-hydroxyphenylethyl-2-O-acetyl-O-α-l-rhamnopyranosyl-(1→3)-4-O-(E)-coumaroyl-β-d-glucopyranoside and named cistansalside D.
Cistansalside E (18) was isolated as an amorphous brown powder. Its molecular formula was determined to be C31H38O14 by 13C-NMR data and the positive mode high-resolution ESI-QTOF-MS peak at m/z 657.2166 [M + Na]+ (calcd. for C31H38O14Na, 657.2154). Additionally, fragment ions including a caffeoyl ion at m/z 163, an [M + H − Aglycone − Rha]+ ion at m/z 367 and an [M + H − Aglycone]+ ion at m/z 513 were also detected. The fragment ions suggested the existence of a rhamnose unit and an acetyl-substituted glucose unit.
The
1H-NMR spectrum suggested the presence of two anomeric protons at δ
H 4.63 (1H, d,
J = 8.2 Hz, H-1′) and 4.60 (1H, s, H-1′′) and two acyl-substituted glucose protons at δ
H 4.71 (1H, dd,
J = 8.9, 8.2 Hz, H-2′) and 4.81 (1H, dd,
J = 9.7, 9.5 Hz, H-4′). The
1H and HMBC spectra suggested the existence of an acetyl group at δ
H 1.94 (3H, s, acetyl-CH
3), an (
E)-caffeoyl moiety at δ
H 7.48 (1H, d,
J = 15.9 Hz, H-7′′′), 7.02 (1H, d,
J = 1.6 Hz, H-2′′′), 6.98 (1H, dd,
J = 8.1, 1.6 Hz, H-6′′′), 6.76 (1H, d,
J = 8.1 Hz, H-5′′′) and 6.21 (1H, d,
J = 15.9 Hz, H-8′′′) and a mono-substituted benzene ring at δ
H 7.29 (2H, m, H-3, 5), 7.21 (2H, m, H-2, 6) and 7.20 (1H, m, H-4) (
Table 2). A phenylethyl group, an aglycone substructure, was suggested by the HMBC correlations between H-7 (2H, δ
H 2.80, m) and C-1 (δ
C 138.8) and between H-7 and C-2, 6 (δ
C 128.9) and the COSY NMR signals of H-7 with H-8a (1H, δ
H 3.99, m) and H-8b (1H, δ
H 3.63, m).
Two sugar moieties were reaffirmed by the HPLC analysis of the acid hydrolysate and NMR spectra analysis. The absolute configurations of the sugars were elucidated using HPLC analysis of the acid hydrolysate, which were confirmed to be
d-glucose and
l-rhamnose [
17]. A β-glucose moiety and an α-rhamnose moiety were established by coupling constants of the anomeric protons. The
1H-
1H COSY spectrum showed the sequential correlations from H-1′ to H-5′, from H-1′′ to H-2′′ and from H-6′′ to H-3′′ (
Figure 4).
From the HMBC spectrum, the correlations between H-1′ and C-8 (δC 69.4), between H-2′ and a carbonyl carbon of an acetyl group (δC 169.1), between H-3′ (δH 3.95) and C-1′′ (δC 102.0) and between H-4′ (δH 4.81) and C-9′′′ (δC 165.6) confirmed the position of substituents in the structure. Therefore, the structure of 18 was determined to be phenylethyl-2-O-acetyl-O-α-l-rhamnopyranosyl-(1→3)-4-O-(E)-caffeoyl-β-d-glucopyranoside and named cistansalside E.
Most of the
trans-cinnamoyl substituents were isomerized to the
cis-isoform in vitro. Light has been reported to convert
trans-cinnamic acid derivatives into
cis-isoforms [
18,
19]. The equilibrium of the
trans-
cis conversion of the cinnamoyl substituents was observed to maintain approximately 70% of the isolates in the
trans-isoform. For the olefin protons of the
cis form, peaks at approximately 6.90 ppm (d,
J = 12~13 Hz, H-7′′′) and 5.80 ppm (d,
J = 12~13 Hz, H-8′′′) were assignable in the
1H-NMR spectra, whereas peaks at approximately 7.55 ppm (d,
J = 15.8 Hz, H-7′″) and 6.40 ppm (d,
J = 15.8 Hz, H-8′′′) were observed for
trans form [
20]. In the
1H-NMR spectrum of
trans-
cis mixtures, peaks for two olefinic protons (H-7′′′ and H-8′′′) were observed in the ratio of 7:3 (
trans:
cis).
13C-NMR peaks of the
cis form were similar to those of the
trans form.
All the isolates were tested for their inhibitory effects on LPS-induced NO production in RAW 264.7 cells. Dexamethasone was used as a positive control and its IC
50 was 7.0 μM. Of the tested compounds, compounds
5 (IC
50 42.7 ± 6.6 μM),
11 (IC
50 37.3 ± 2.2 μM),
13 (IC
50 40.0 ± 4.0 μM) and
18 (IC
50 27.9 ± 0.8 μM) showed moderate inhibitory activities on inducible NO synthase, while the other compounds were inactive in this assay (IC
50 values > 100 μM). To verify whether these compounds had cytotoxicity, cell viability was measured employing MTT assay. As a result, none of them displayed significant cytotoxicity (
Supplemental Figure S6-1). These four compounds were selected to evaluate for their inhibitory activity against NF-κB pathway in LPS-stimulated RAW 264.7 cells. Stimulation of RAW 264.7 cells with LPS induced the phosphorylation of IκBα and NF-κB (p65) after 0.5 h of incubation. The phosphorylation of NF-κB (p65) was significantly reduced by pretreatment with compounds
11,
13 and
18 as shown by western blot analysis (
Figure 5). Therefore, compounds
11,
13 and
18 might exert anti-inflammatory effects via the inhibition of NF-κB in macrophages.
3. Materials and Methods
3.1. General Experiment Procedure
Optical rotations were measured with a Jasco P-2000 digital polarimeter (Jasco, Tokyo, Japan). UV spectra were recorded on a Chirascan plus Circular Dichroism spectrometer (Chirascan, APL, UK). IR spectra were recorded using Jasco FT/IR-4200 spectrophotometer. High-resolution electrospray ionization quadrupole-time-of-flight mass spectrometry (HR-ESI-qTOF-MS) was performed on an Agilent 6530 Accurate-Mass Q-TOF LC/MS equipped with Agilent 1260 Infinity series (Agilent Technologies, Inc., Palo Alto, CA, USA), and the column used was a Jasco SFCpak Crest C18T-5 column (i.d. 150 × 4.6 mm, 5 μm) . MassHunter Workstation Software was used for data acquisition.
1D (1H and 13C) and 2D (1H-1H COSY, HSQC, HMBC, NOESY) NMR spectra were obtained with a Jeol LA 300 (Jeol, Tokyo, Japan), Bruker AVANCE-400, Bruker AVANCE-500, Bruker AVANCE-600 and Bruker AVANCE 800 HD spectrometers coupled with cryoprobe (Bruker, Ettlingen, Germany). DMSO-d6 (Cambridge Isotope Laboratories, Inc. Andover, MA, USA) was used as NMR solvent and reference peaks (δH 2.50 and δC 39.5). Column chromatography (CC) was performed using Sephadex LH-20 (25–100 μm; Pharmacia, Uppsala, Sweden) or Kieselgel 60 silica gel (40–63 μm, 230–400 mesh, Art. 9385; Merck, Darmstadt, Germany). Thin-layer chromatography (TLC) was conducted on pre-coated Kieselgel 60 silica gel F254 plates (Art. 5715; Merck). Spots on TLC were detected using UV lamp at 254 nm and 365 nm (VL-4.LC, 365/254; Vilber Lourmat, Torcy, France). Medium pressure liquid chromatography (MPLC) was performed on a RediSep 120 g silica flash column (Isco, Lincoln, NE, USA) and Kiesegel 60 silica gel (40–63 μm, 230–400 mesh, Art. 9385; Merck) using a Combiflash companion (Isco). The high pressure liquid chromatography (HPLC) system was a Gilson HPLC equipped with a Gilson 321 pump and UV.VIS 151 detector (Gilson, Middleton, WI, USA), using semi-preparative ODS columns (Luna 5 μm C18 (2) 100 Å, i.d. 250 × 10 mm, 5 μm, Phenomenex Inc., Torrance, CA, USA; Hypersil GOLD™ aQ 175 Å, i.d. 250 × 10 mm, 5 μm, Thermo Scientific™, Hennigsdorf, Germany; Inno C18 column 120 Å, i.d. 250 × 10 mm, 5 μm, Young Jin Biochrom Co., Ltd., Seongnam, Korea). The analytical RP-HPLC system was a Waters 2695 alliance system with a 996 Photodiode Array (PDA) detector (Waters Corp, Milford, MA, USA), and the column used was a Hypersil™ BDS C18 column (130 Å, i.d. 150 × 4.6 mm, 5 μm, Thermo Scientific™). Formic acid was purchased from Daejung Chemicals & Metals Co., Ltd. (Seoul, Korea). HPLC grade solvents were purchased from Fisher Scientific Korea Ltd. (Seoul, Korea). H2SO4, Na2CO3 and first grade solvents for extraction, fractionation and isolation were purchased from Daejung Chemical & Metals Co. Ltd. (Seoul, Korea). l- and d-cysteine methyl ester hydrochloride and o-tolylisothiocyanate were purchased from Tokyo Chemical Industry (Tokyo, Japan).
3.2. Plant Material
The whole plants of C. salsa, which were collected from the Shinjang Uyghur, were imported through Daerim Pharmaceutical Wholesale Company (Cheongju, Korea). They were identified by Prof. Dr. Jehyun Lee (Dongguk University, Seoul, Korea). The voucher specimen (SNUPH2016-03) was deposited in the Herbarium of Medicinal Plant Garden, College of Pharmacy, Seoul National University.
3.3. Extraction and Isolation
The dried whole plants of C. salsa (5.7 kg) were chopped and extracted three times with MeOH (20 L) at room temperature with sonication for 99 min. After removal of the solvent in vacuo, The crude extract (1.35 kg) was suspended in H2O (5 L), then partitioned with EtOAc (5 L). The EtOAc residue (55.3 g) was separated into 16 fractions (E01-16) on silica gel chromatography eluting with CHCl3/MeOH (50:1–0:1, step-gradient system).
E08 (611.7 mg) was subjected to silica gel medium-pressure liquid chromatography (25 g) and eluted with CHCl3/MeOH (18:1–0:1, step-gradient system) and gave 11 fractions (E08a-k). From E08g, compounds 13 (0.7 mg) and 18 (0.6 mg) were purified using a Luna 5 μm HPLC column and isocratic elution with 28% aq. MeCN. HPLC purification (Hypersil GOLD, 25% aq. MeCN) of E08h (138.5 mg) furnished compounds 7 (10.6 mg), 8 (17.2 mg), 14 (13.4 mg) and 17 (4.9 mg).
E09 (1.15 g) was further purified on a silica-MPLC column (20 g) eluting with CHCl3/MeOH (10:1–0:1, step-gradient system) to give 11 subfractions (E09a-k). E09e (398.7 mg) was subjected to Sephadex LH-20 (MeOH) and yielded eight subfractions (E09e1-8). E09e6 was subsequently purified using a Hypersil GOLD HPLC column (22% aq. MeCN) to afford compound 11 (0.4 mg). From E09e7, compound 5 (0.6 mg) was purified by isocratic eluction from a Luna 5 μm HPLC column with 40% aq. MeOH.
E10 (1.20 g) was separated into nine fractions (E10a-i) on Sephadex LH-20 column eluting with MeOH. From E10g (147.5 mg), compounds 4 (13.4 mg), 9 (8.0 mg) and 15 (5.0 mg) were isolated by HPLC separation (Inno, 23% aq. MeCN). Fraction E10h (470.0 mg) was purified on a Hypersil GOLD column by isocratic elution (40% aq. MeOH) to yield compounds 1 (3.8 mg), 10 (42.1 mg) and 16 (3.0 mg).
E11 (2.96 g) was subjected to Sephadex LH-20 eluting with MeOH to give nine fractions (E11a-i). E11e was separated by Sep-Pak C18 cartridge eluting stepwise with 10%, 20%, 30%, 50% and 100% aq. MeOH to yield seven fractions (E11e1-7), followed by Luna 5 μm HPLC (28% aq. MeCN) to give compounds 3 (3.4 mg), 6 (1.4 mg) and 12 (1.2 mg).
E12 (19.0 g) was subjected to silica MPLC (120 g) using a CHCl3/MeOH step-gradient system to give six fractions (18:1–0:1, E12a-f). E12f was chromatographed on a Sephadex LH-20 column (MeOH), yielding seven fractions (E12f1-7). HPLC purification (Hypersil GOLD, 25% aq. MeCN) of E12f7 furnished compound 2 (3.3 mg). All solvents used for HPLC were 0.05% formic acid buffer. The common flow rate for HPLC and MPLC chromatography was 3 and 40 mL/min, respectively.
3.4 Characterization
Cistansalside A (
5): brown amorphous powder;
−33.7 (
c 0.1, MeOH); UV(MeOH) λ
max nm (log ε) 332 (3.18); IR (neat) ν
max 3359, 1748, 1705, 1602, 1516 cm
−1;
1H-NMR (800 MHz) and
13C-NMR (200 MHz) data, see
Table 2; HRMS (ESI-TOF)
m/z [M + Na]
+ 645.2146 (calcd. for C
30H
38O
14Na, 645.2154).
Cistansalside B (
6): brown amorphous powder;
−72.9 (
c 0.1, MeOH); UV(MeOH) λ
max nm (log ε) 333 (3.32); IR (neat) ν
max 3400, 1705, 1603, 1516 cm
−1;
1H-NMR (500 MHz) and
13C-NMR (125 MHz) data, see
Table 2; HRMS (ESI-TOF)
m/z [M + Na]
+ 579.2054 (calcd. for C
26H
36O
13Na, 579.2048).
Cistansalside C (
12): brown amorphous powder;
−61.6 (
c 0.1, MeOH); UV(MeOH) λ
max nm (log ε) 337 (3.25); IR (neat) ν
max 3359, 1704, 1602, 1508 cm
−1;
1H-NMR (800 MHz) and
13C-NMR (200 MHz) data, see
Table 2; HRMS (ESI-TOF)
m/z [M + Na]
+ 581.2213 (calcd. for C
26H
38O
13Na, 581.2205).
Cistansalside D (
17): brown amorphous powder;
−51.1 (
c 0.1, MeOH); UV(MeOH) λ
max nm (log ε) 221 (3.53), 315 (3.52); IR (neat) ν
max 3358, 1746, 1722, 1603, 1516, 1232, 1157, 1039 cm
−1;
1H-NMR (400 MHz) and
13C-NMR (75 MHz) data, see
Table 2; HRMS (ESI-TOF)
m/z [M + Na]
+ 657.2147 (calcd. for C
31H
38O
14Na, 657.2154).
Cistansalside E (
18): brown amorphous powder;
−59.2 (
c 0.1, MeOH); UV(MeOH) λ
max nm (log ε) 336 (3.22); IR (neat) ν
max 3370, 1741, 1712, 1602, 1231, 1157 cm
−1;
1H-NMR (800 MHz) and
13C-NMR (200 MHz) data, see
Table 2; HRMS (ESI-TOF)
m/z [M + Na]
+ 657.2166 (calcd. for C
31H
38O
14Na, 657.2154).
3.5. HPLC-QTOF-MS Analysis
Chromatographic-mass spectrometry analysis was performed on an Agilent 1260 Infinity series LC system (Agilent Technologies, Inc., USA). The analytical column was a SFCpak Crest C18T-5 column (i.d. 150 × 4.6 mm, 5 μm, Jasco, Japan). The mobile phase consisted of 0.1% (v/v) formic acid in MeCN (A) and water (B) using a gradient elution of 0–35 min (23% A), 35–45 min (23–28% A), 45–75 min (28% A) and 75–80 min (90% A). The flow rate was kept at 0.3 mL/min. The absorbance was measured at 320 nm. The conditions of the ESI source were as follows: drying gas (N2) flow rate, 10 L/min; drying gas temperature, 350 °C; nebulizer, 30 psig; sheath gas flow rate, 12.0 L/min; sheath gas temperature, 350 °C; capillary, 4000 V; skimmer, 60 V; octapole RF, 750 V; fragmentor voltage, 180 V; positive mode. The system was operated under Masshunter workstation software. The mass range was set at m/z 50–1000.
3.6. Acid Hydrolysis
Compounds were hydrolyzed using 1 N H2SO4 (100 μL) heated with a water bath at 90 °C for 2 h, then neutralized with saturated aqueous Na2CO3 solution. After the solutions were dried under a stream of N2, the products and standard sugars (d-Glc, l-Glc, l-Rha) were dissolved in pyridine (100 μL) containing l-cysteine methyl ester hydrochloride (0.5 mg). An l-rhamnose sample was dissolved in pyridine (100 μL) containing d-cysteine methyl ester hydrochloride (0.5 mg). After that, they were heated at 60 °C for 1 h. The solutions were treated with 1 μL (1.11 mg) of o-tolylisothiocyanate, which were heated again at 60 °C for 1 h. Each final mixture was directly analyzed by analytical RP-HPLC (Hypersil™ BDS C18 column, 17% aq. MeCN, 0.8 mL/min, 40 min, 35 °C). The tR of the peak at 21.9 and 40.4 min coincided with that of the thiocarbamoyl thiazolidine derivative of d-glucose and l-rhamnose, respectively.
3.7. Cell Culture
Murine macrophages, RAW 264.7, were obtained from the Korean Research Institute of Bioscience and Biotechnology (Daejeon, Korea), and grown in RPMI medium containing 10% fetal bovine serum and 100 U/mL penicillin/streptomycin sulfate. Cells were incubated in a humidified 5% CO2 atmosphere at 37 °C.
3.8. Drugs and Chemicals
RPMI, penicillin and streptomycin were purchased from HyClone (Logan, UT, USA). Bovine serum albumin and LPS were purchased from Sigma (St. Louis, MO, USA).
3.9. Measurement of NO Production
The nitrite concentration in the culture medium was measured as an indicator of NO production according to the Griess reaction. The cells were seeded at 2 × 105 cells/well in 96-well culture plates. After pre-incubation of the RAW 264.7 cells for 18 h, the cells were pretreated with compounds (50 µM, 10 µM, 5 µM or 1 µM) and stimulated with LPS (500 ng/mL) for 24 h. Test compounds dissolved in DMSO. Cells were also treated with 0.05% DMSO as a vehicle control. RAW 264.7 cells (2 × 105 cells/well) were cultured in 96-well plates using RPMI without phenol red, and pretreated with samples for 0.5 h. Cellular NO production was induced by the addition of 500 ng/mL final concentration LPS and a 24 h incubation. Following incubation, 100 µL of conditioned media was mixed with the same volume of Griess reagent and incubated for 15 min. The absorbance of the mixture at 540 nm was measured with an ELISA microplate reader (Benchmark, Bio-Rad Laboratories, Richmond, CA, USA). The values obtained were compared with those of standard concentrations of sodium nitrite dissolved in RPMI, and the concentrations of nitrite in the conditioned media of sample-treated cells were calculated.
3.10. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay for Cell Viability
Cells were seeded into 96-well plates at a density of 5 × 104 cells/well and incubated with serum-free media in the presence of samples. Test compounds dissolved in DMSO. Cells were also treated with 0.05% DMSO as a vehicle control. Following incubation for 24 h, 10 µL MTT (5 mg/mL in saline) was added and incubation was continued for further 4 h. Mitochondrial succinate dehydrogenase in live cells converts MTT into visible formazan crystals during incubation. The formazan crystals were then solubilized in dimethyl sulfoxide and the absorbance was measured at 540 nm using an enzyme-linked immunosorbent assay (ELISA) microplate reader (Benchmark, Bio-Rad Laboratories). Relative cell viability was calculated compared with the absorbance of the untreated control group. All experiments were performed in triplicate.
3.11. Immunoblot Analysis
Protein expression was assessed by western blotting according to standard procedures. Briefly, RAW264.7 cells were cultured in 60 mm culture dishes (2 × 106/mL), following by pretreatment 50 μM of compounds. Cells were washed twice in ice cold PBS (pH 7.4), the cell pellets were resuspended in lysis buffer on ice for 15 min, and the cell debris was then removed by centrifugation. Protein concentration was determined using Bio-Rad protein assay reagent according to the manufacturer’s instructions. Protein (20–30 μg) was mixed 1:1 with 2× sample buffer (20% glycerol, 4% SDS, 10% 2-ME, 0.05% bromophenol blue, and 1.25 M Tris [pH 6.8]), loaded onto 8 or 15% SDS-PAGE gels, and run at 150 V for 90 min. Cellular proteins were transferred onto ImmunoBlot polyvinylidene difluoride membranes (Bio-Rad) using a Bio-Rad semi-dry transfer system according to the manufacturer’s instructions. The membranes were then incubated overnight with the resprective p-NF-κB, NF-κB, p-IκBα and β-actin primary antibodies (Abcam, Cambridge, UK) in Tris-buffered saline containing 5% skimmed milk and 0.1% Tween 20. The following day, the blots were washed three times with Tris-buffered saline (0.1% Tween 20) and incubated for 1 h with an HRP-conjugated secondary anti-IgG antibody (diluted 1:2000–1:20,000). The blots were washed again three times with Tris-buffered saline (0.1% Tween 20), and immunoreactive bands were developed using the chemiluminescent substrate ECL Plus (Amersham Biosciences, Piscataway, NJ, USA).
3.12. Statistical Analysis
Experimental data are presented as the mean ± SEM. The level of statistical significance was determined by analysis of variance (ANOVA) followed by Dunnett’s t-test for multiple comparisons. p Values less than 0.05 were considered significant.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}