Comparative Transcriptome Analysis Reveals Adaptive Evolution of Notopterygium incisum and Notopterygium franchetii, Two High-Alpine Herbal Species Endemic to China

Abstract
:1. Introduction
2. Results
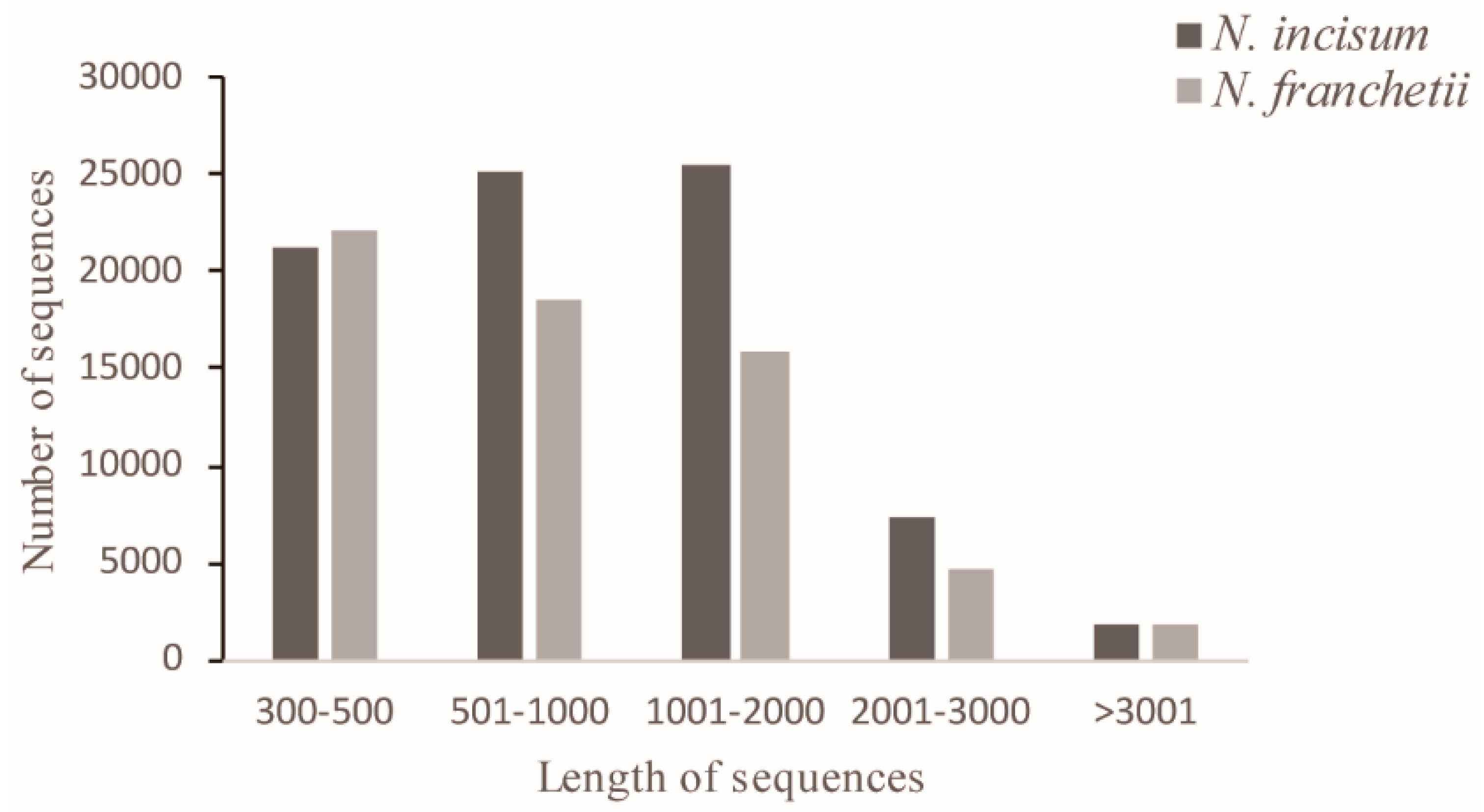
2.1. Sequencing and De Novo Assembly
2.2. Functional Annotation
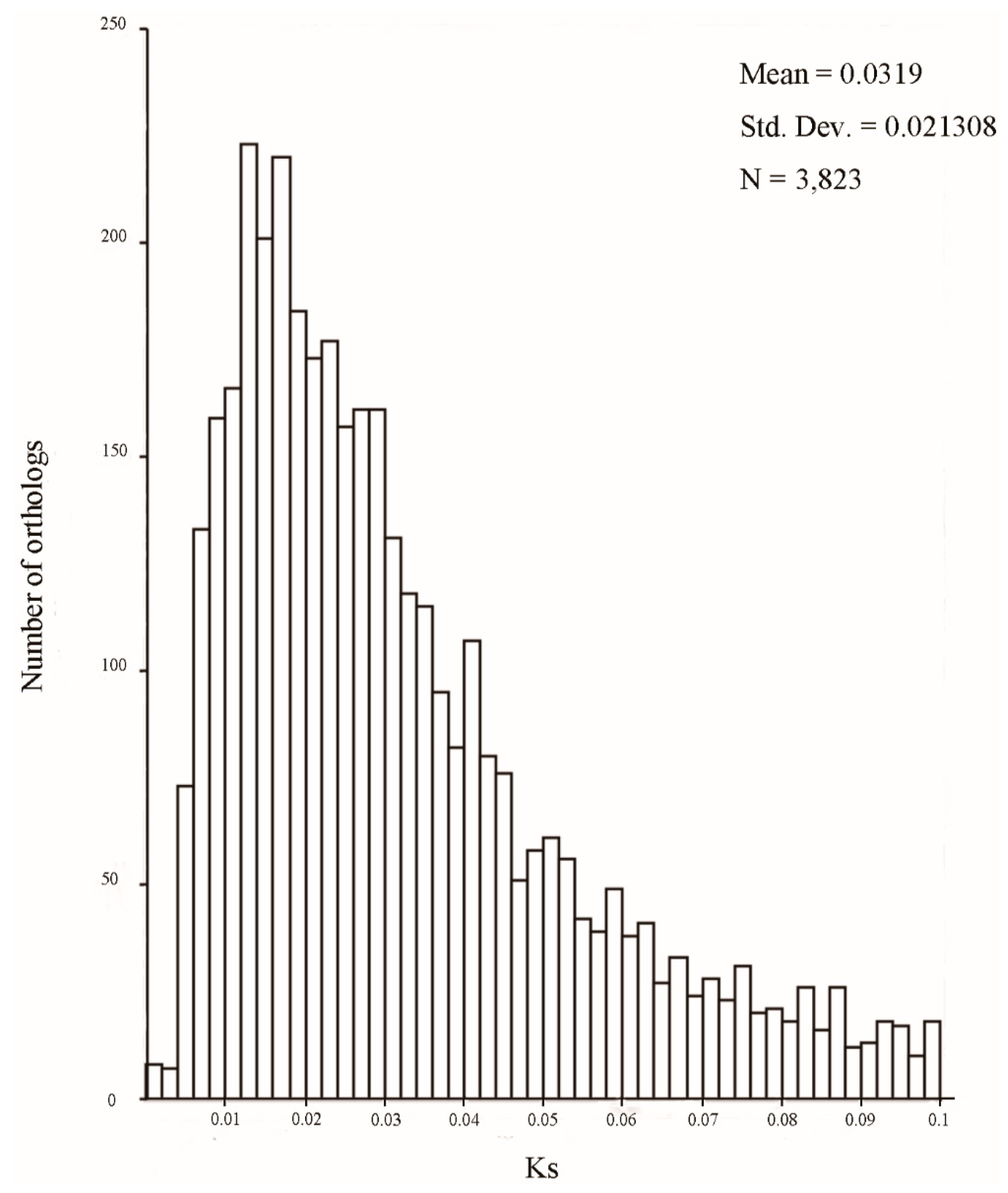
2.3. Putative Orthologs, Substitution Rates, and Species Divergence between N. incisum and N. franchetii
2.4. Functions and Metabolite Pathway Analysis under Positive Selection
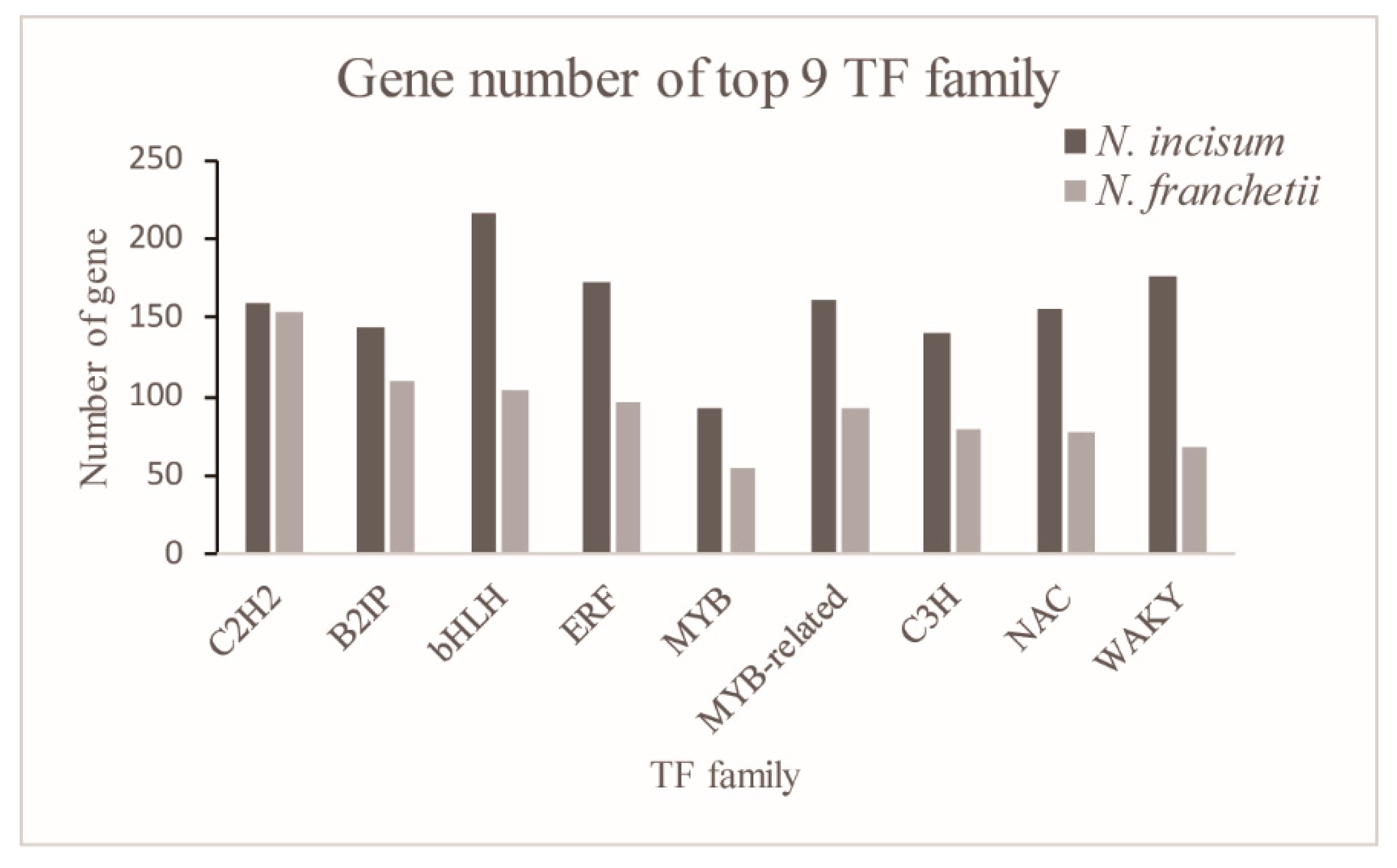
2.5. Transcription Factor Identification
2.6. SSR Identification
2.7. Genetic Diversity and Divergence of N. incisum and N. franchetii
3. Discussion
3.1. De Novo Assembly and Functional Annotation
3.2. Species Divergence between N. incisum and N. franchetii
3.3. Detecting Candidate Genes under Positive Selection
3.4. Polymorphic SSR Markers and Population Structure
4. Materials and Methods
4.1. Plant Material
4.2. RNA Preparation, Illumina Paired-End Sequencing
4.3. Transcriptome Assembly and Function Annotation
4.4. Ortholog Identification and Sequence Alignment
4.5. Substitution Rate Estimation and Selection Analyses
4.6. Transcription Factor Mining
4.7. Microsatellite Marker Detection
4.8. DNA Isolation and Primer Selection
4.9. Microsatellite DNA Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qiao, Q.; Wang, Q.; Han, X.; Guan, Y.; Sun, H.; Zhong, Y.; Huang, J.; Zhang, T. Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet Plateau. Sci. Rep. 2016, 6, 21729. [Google Scholar] [CrossRef] [PubMed]
- Wiener, G.; Han, J.L.; Long, R.J. The Yak; RAP Press: Bangkok, Thailand, 2003. [Google Scholar]
- Xin, G.S.; Long, R.J.; Guo, X.S.; Irvine, J.; Ding, L.M.; Ding, L.L.; Shang, Z.H. Blood mineral status of grazing Tibetan sheep in Northeast of the Qinghai-Tibetan Plateau. Livest. Sci. 2011, 136, 102–107. [Google Scholar] [CrossRef]
- Storz, J.F.; Scott, G.R.; Cheviron, Z.A. Phenotypic plasticity and genetic adaptation to high-altitude hypoxia in vertebrates. J. Exp. Biol. 2010, 213, 4125–4136. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Han, J.; Tang, X.; Ma, M.; Wang, K.; Zhang, X.; Ren, Q.; Chen, Q.; Qiu, Q. Comparative transcriptomic analysis revealed adaptation mechanism of Phrynocephalus erythrurus, the highest altitude Lizard living in the Qinghai-Tibet Plateau. BMC Evol. Boil. 2015, 15, 101. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Gao, J.; He, Y.; Guo, Y. Compositae plants differed in leaf cuticular waxes between high and low altitudes. Chem. Biodivers. 2016, 13, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Ma, L.; Sun, X.; Yang, S.; Kong, X.; Hu, X.; Yang, Y. Comparative proteomics analyses of Kobresia pygmaea adaptation to environment along an elevational gradient on the Central Tibetan Plateau. PLoS ONE 2014, 9, e98410. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G. Redox homeostasis and antioxidant signaling: A metabolic interface between stress perception and physiological responses. Plant Cell 2005, 17, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Vinocur, B.; Altman, A. Recent advances in engineering plant tolerance to abiotic stress: Achievements and limitations. Curr. Opin. Biotechnol. 2005, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Sun, X.; Kong, X.; Galvan, J.V.; Li, X.; Yang, S.; Yang, Y.; Yang, Y.; Hu, X. Physiological, biochemical and proteomics analysis reveals the adaptation strategies of the alpine plant Potentilla saundersiana at altitude gradient of the Northwestern Tibetan Plateau. J. Proteomics 2015, 112, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yang, L.; Zhao, J.; Wei, J.; Kong, X.; Wang, C.; Zhang, X.; Yang, Y.; Hu, X. Comparative proteomic analysis reveals the role of hydrogen sulfide in the adaptation of the alpine plant Lamiophlomis rotata to altitude gradient in the Northern Tibetan Plateau. Planta 2015, 241, 887–906. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.Y.; Chen, L.Y.; Muchuku, J.K.; Hu, G.W.; Wang, Q.F. Genetic adaptation of giant lobelias (Lobelia aberdarica and Lobelia telekii) to different altitudes in East African mountains. Front. Plant Sci. 2016, 7, 488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, H.F.; Wu, W.; Yu, H.; Ge, X.J. Comparative transcriptome analysis and marker development of two closely related Primrose species (Primula poissonii and Primula wilsonii). BMC Genom. 2013, 14, 329. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Yang, L.; Li, C.; Xu, W.; Chen, G. Genetic diversity in endangered Notopterygiumforbesii Boissieu based on intraspecies sequence variation of chloroplast DNA and implications for conservation. Biochem. Syst. Ecol. 2010, 38, 911–916. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Raven, P.H. Apiaceae through Ericaceae, In Flora of China; Science Press: Beijing, China, 2005; Volume 14, pp. 451–471. [Google Scholar]
- Wang, Y.P.; Pu, F.; Wang, P.; He, X.J. Studies on the systematics of the Chinese endemic genus Notopterygium. Acta Bot. Yunnanica 1996, 18, 424–430. [Google Scholar]
- She, M.; Pu, F. A new species of Notopterygium de Boiss. from China. J. Plant Resour. Environ. 1996, 6, 41–42. [Google Scholar]
- Jiang, S.Y.; Sun, H.; Huang, X.J.; Zhou, Y.; Ma, X.J.; Yang, Z.R. Environmental pedology of Notopterygium incisum and N. forbesii. Chin. Tradit. Herb. Drugs 2005, 36, 918–921. [Google Scholar]
- Pu, F.; Wang, P.; Zheng, Z.; Wang, Y.P. A reclassification of Notopterygium boissieu (Umbelliferae). Acta Phytotaxon. Sin. 1999, 38, 430–436. [Google Scholar]
- Yang, J.; Yue, M.; Niu, C.; Ma, X.F.; Li, Z.H. Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium. Genes 2017, 8, E124. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.W.; Zhang, P.; Tao, H.Y.; Jiang, S.Y.; Zhou, Y. GC-MS analysis of essential oil constituents from rhizome and root of Notopterygium forbesii. J. Chin. Pharm. Sci. 2006, 15, 200–205. [Google Scholar]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.X.; Bai, G.Q.; Li, Z.H.; Kanwal, N.; Zhao, G.F. Comparative Transcriptome and Chloroplast Genome Analyses of Two Related Dipteronia Species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Boil. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef]
- Zheng, B.; Xu, Q.; Shen, Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai-Tibetan Plateau: Review and speculation. Quat. Int. 2002, 97, 93–101. [Google Scholar] [CrossRef]
- Sun, Y.; Li, L.; Li, L.; Zou, J.; Liu, J. Distributional dynamics and interspecific gene flow in Picea likiangensis and P. wilsonii triggered by climate change on the Qinghai-Tibet Plateau. J. Biogeogr. 2015, 42, 475–484. [Google Scholar] [CrossRef]
- Noctor, G.; Mhamdi, A.; Chaouch, S.; Han, Y.; Neukermans, J.; Marquez Garcia, B.; Queval, G.; Foyer, C.H. Glutathione in plants: An integrated overview. Plant Cell Environ. 2012, 35, 454–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Zhang, S.Z.; Whitworth, R.J.; Stuart, J.J.; Chen, M.S. Unbalanced activation of glutathione metabolic pathways suggests potential involvement in plant defense against the gall midge Mayetiola destructor in wheat. Sci. Rep. 2015, 5, 8092. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Bonthala, V.S.; Mayes, K.; Moreton, J.; Blythe, M.; Wright, V.; May, S.T.; Massawe, F.; Mayes, S.; Twycross, J. Identification of gene modules associated with low temperatures response in Bambara groundnut by network-based analysis. PLoS ONE 2016, 11, e0148771. [Google Scholar] [CrossRef] [PubMed]
- Birol, I.; Behsaz, B.; Hammond, S.A.; Kucuk, E.; Veldhoen, N.; Helbing, C.C. De novo transcriptome assemblies of Rana (Lithobates) catesbeiana and Xenopus laevis tadpole livers for comparative genomics without reference genomes. PLoS ONE 2015, 10, e0130720. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Yang, L.; Wang, D.; Zhu, J.K.; Lang, Z.B. De novo assembly and analysis of the transcriptome of Ocimumamericanum var. pilosum under cold stress. BMC Genom. 2016, 17, 209. [Google Scholar]
- Rispe, C.; Fabrice, L.; Daciana, P.; Anthony, B.; Sylvie, H.; Gaël, L.T.; Denis, T.; Julie, J.; François, D. De novo transcriptome assembly of the grapevine phylloxera allows identification of genes differentially expressed between leaf-and root-feeding forms. BMC Genom. 2016, 17, 219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, P.; Lascoux, M.; Meagher, T.R.; Liu, J. Rapidly evolving genes and stress adaptation of two desert poplars, Populuseu phratica and P. pruinosa. PLoS ONE 2013, 8, e66370. [Google Scholar]
- Wang, Y.; Hey, J. Estimating divergence parameters with small samples from a large number of loci. Genetics 2010, 184, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Lee, K.; Kang, H. Emerging roles of RNA-binding proteins in plant growth, development, and stress responses. Mol. Cells 2016, 39, 179. [Google Scholar] [PubMed]
- Califice, S.; Baurain, D.; Hanikenne, M.; Motte, P.A. Single Ancient Origin for Prototypical Serine/Arginine-Rich Splicing Factors1. Plant Physiol. 2012, 158, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, R.; Tuteja, N. Analysis of DNA repair helicase UvrD from Arabidopsis thaliana and Oryza sativa. Plant Physiol. Biochem. 2013, 71, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Ghanta, S.; Chattopadhyay, S. Glutathione as a signaling molecule: Another challenge to pathogens. Plant Signal Behav. 2011, 6, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Pociecha, E.; Plazek, A.; Janowiak, F.; Waligorski, P.; Zwierzykowski, Z. Changes in abscisic acid, salicylic acid and phenylpropanoid concentrations during cold acclimation of androgenic forms of Festulolium (Festuca pratensis×Lolium multiflorum) in relation to resistance to pink snow mould (Microdochium nivale). Plant Breed. 2009, 128, 397–403. [Google Scholar] [CrossRef]
- He, W.; Zhuang, H.; Fu, Y.; Guo, L.; Guo, B.; Guo, L.; Zhang, Y.; Wei, Y. De novo transcriptome assembly of a Chinese locoweed (Oxytropis ochrocephala) species provides insights into genes associated with drought, salinity, and cold tolerance. Front. Plant Sci. 2015, 6, 1086. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.R.; Joshi, G.; Kukreja, B.; Malik, V.; Arora, P.; Pandey, R.; Shukla, R.N.; Bankar, K.G.; Katiyar-Agarwal, S.; Goel, S.; et al. Global insights into high temperature and drought stress regulated genes by RNA-Seq in economically important oilseed crop Brassica juncea. BMC Plant Biol. 2015, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.K.; Chen, G.; Zhang, J.L.; Zhang, Y.J.; Xie, X.L.; Zhao, Z.P.; Pan, Y.; Hu, Z.L. The abiotic stress-responsive NAC-type transcription factor SlNAC4 regulates salt and drought tolerance and stress-related genes in tomato (Solanum lycopersicum). Plant Cell Rep. 2014, 33, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Puranik, S.; Sahu, P.P.; Srivastava, P.S.; Prasad, M. NAC proteins: Regulation and role in stress tolerance. Trends Plant Sci. 2012, 17, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, D.; Yu, M.; Baldwin, D.; Gruber, M.; Sharpe, A.; Parkin, I.; Whitwill, S.; Lydiate, D. Molecular characterization of Brassicanapus NAC domain transcriptional activators induced in response to biotic and abiotic stress. Plant Mol. Boil. 2003, 53, 383–397. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Zhen, W.; Jun, T.; Rong, H.; Peng, W.; Hou, X.L.; Song, X.M.; Xiong, A.S. Genome-wide analysis of the R2R3-MYB transcription factor genes in Chinese cabbage (Brassica rapa ssp. pekinensis) reveals their stress and hormone responsive patterns. BMC Genom. 2015, 16, 17. [Google Scholar]
- Muthamilarasan, M.; Khandelwal, R.; Yadav, C.B.; Bonthala, V.S.; Khan, Y.; Prasad, M. Identification and molecular characterization of MYB transcription factor superfamily in C4 model plant foxtail millet (Setariaitalica L.). PLoS ONE 2014, 9, e109920. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Huang, W.; Guan, J. Transcriptomic analysis provides insight into high-altitude acclimation in domestic goats. Gene 2015, 567, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Kavas, M.; Baloğlu, M.C.; Atabay, E.S.; Ziplar, U.T.; Daşgan, H.Y.; Ünver, T. Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol. Genet. Genom. 2016, 291, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Y.; Chiou, S.J.; Ko, C.Y.; Lin, T.Y. Functional characterization of three ethylene response factor genes from Bupleurumkaoi indicates that BkERFs mediate resistance to Botrytis cinerea. J. Plant Physiol. 2011, 168, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Chikagawa, Y.; Kato, M.; Maeda, K.; Ozeki, Y. Upregulation of the promoter activity of the carrot (Daucuscarota) phenylalanine ammonia-lyase gene (DcPAL3) is caused by new members of the transcriptional regulatory proteins, DcERF1 and DcERF2, which bind to the GCC-box homolog and act as an activator to the DcPAL3 promoter. J. Plant Res. 2008, 121, 499. [Google Scholar] [PubMed]
- Sun, Y.Z.; Niu, Y.Y.; Li, Y.; Zhu, Y.J.; Luo, H.M.; Chen, S.L. Cloning and bioinformatic analysis of PqERF1 gene in Panax quinquefolius. Acta Pharm. Sin. 2011, 46, 1008–1014. [Google Scholar]
- Chen, W.; Liu, Y.X.; Jiang, G.F. De novo assembly and characterization of the testis transcriptome and development of EST-SSR markers in the cockroach Periplaneta americana. Sci. Rep. 2015, 5, 11144. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, H.W.; Fu, X.N.; Li, X.H.; Gao, H.W. Development of simple sequence repeat markers and diversity analysis inalfalfa (Medicago sativa L.). Mol. Biol. Rep. 2013, 40, 3291–3298. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cai, C.F.; Cheng, F.Y.; Cui, H.L.; Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Li, Y.D.; Hui, M.; Cui, Z.; Liu, Y.; Song, C.W.; Shi, G.H. Comparative transcriptomic analysis provides insights into the molecular basis of the metamorphosis and nutrition metabolism change from zoeae to megalopae in Eriocheirsinensis. Genom. Proteom. 2015, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. Solexa QA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson., J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genom. Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Wu, Y.; Zhang, L.; Li, X.W.; Zhao, X.Y.; Wang, W.H.; Gao, Z.S.; Xia, Y.P. De novo assembled transcriptome analysis and SSR marker development of a mixture of six tissues from Lilium oriental hybrid ‘Sorbonne’. Plant Mol. Biol. Rep. 2015, 33, 281–293. [Google Scholar] [CrossRef]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. ISMB 1999, 99, 138–148. [Google Scholar]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genom. Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Vatansever, R.; Koc, I.; Ozyigit, I.I.; Sen, U.; Uras, M.E.; Anjum, N.A.; Eduarda, P.; Filiz, E. Genome-wide identification and expression analysis of sulfate transporter (SULTR) genes in potato (Solanum tuberosum L.). Planta 2016, 244, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Puniya, B.L.; Kulshreshtha, D.; Kumar, S.; Kaur, J.; Ramachandran, S. De novo transcriptome sequencing reveals important molecular networks and metabolic pathways of the plant, Chlorophytum borivilianum. PLoS ONE 2013, 8, e83336. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Wu, Y.; Li, D.Q.; Wang, G.Q.; Li, X.; Xia, Y.P. Transcriptomic analysis of the underground renewal buds during dormancy transition and release in ‘Hangbaishao’ peony (Paeonia lactiflora). PLoS ONE 2015, 10, e0119118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Xie, D.; Liu, W.; Peng, Q.; He, X. De novo assembly and characterization of the transcriptome, and development of SSR markers in wax gourd (Benicasa hispida). PLoS ONE 2013, 8, e71054. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.W.; Hoffman, J.R.; Hewitt, T.L.; Beasley, R.R.; Lance, S.L.; Jones, K.L.; Morris, T.J.; Zanatta, D.T. Development and characterization of 29 microsatellite markers for Ligumianasuta (Bivalvia: Unionidae) using an Illumina sequencing approach. Biochem. Syst. Ecol. 2016, 66, 239–242. [Google Scholar] [CrossRef]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Gao, T.; Han, Z.; Zhang, X.; Luo, J.; Yanagimoto, T.; Zhang, H. Population genetic differentiation of the black rockfish Sebastes schlegelii revealed by microsatellites. Biochem. Syst. Ecol. 2016, 68, 170–177. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N. incisum | N. franchetii | |
|---|---|---|
| Sequencing data | ||
| Total number of reads | 55,833,880 | 60,052,340 |
| Total length of reads (bp) | 6,979,235,000 | 9,007,851,000 |
| GC (%) | 40.57 | 43.91 |
| Q20 (%) | 97.09 | 97.31 |
| Final assembly | ||
| Total number of unigenes | 81,446 | 63,153 |
| Total length of unigenes | 86,924,837 | 62,615,693 |
| N50 length of assembly | 1435 | 1405 |
| Mean of length of assembly | 1067 | 991 |
| Database | N. incisum | N. franchetii | |||
|---|---|---|---|---|---|
| Number | Percentage (%) | Number | Percentage (%) | ||
| Annotated | NR | 69,141 | 84.89 | 45,267 | 71.68 |
| Swiss-Prot | 53,805 | 66.06 | 39,518 | 62.58 | |
| KOG | 45,705 | 56.12 | 33,217 | 52.6 | |
| KEGG | 31,488 | 38.66 | 23,288 | 36.88 | |
| GO | 48,515 | 59.57 | 29,474 | 46.67 | |
| Total | 69,237 | 85 | 47,774 | 75.65 | |
| Unannotated | 12,209 | 14.99 | 15,379 | 24.35 | |
| Total | 81,446 | 100 | 63,153 | 100 | |
| Orthologous Unigenes | GO Terms | BLASTx to NR Database | Ka/Ks |
|---|---|---|---|
| ORTHOMCL12869 | RNA splicing | serine/arginine-rich SC35 | 1.45 |
| ORTHOMCL14798 | RNA splicing | RNA-binding protein with multiple splicing | 1.04 |
| ORTHOMCL15032 | RNA splicing | Serine/arginine-rich splicing factor 12 | 1.66 |
| ORTHOMCL17505 | RNA splicing | U1 small nuclear ribonucleoprotein (U1 snRNP) | 1.22 |
| ORTHOMCL18047 | RNA splicing | tRNA 2′-phosphotransferase 1 | 2.93 |
| ORTHOMCL16216 | mismatch repair | DNA mismatch repair protein MSH4 | 1.15 |
| ORTHOMCL16135 | acetate metabolic process | acetyl-coenzyme A synthetase | 1.98 |
| ORTHOMCL16124 | heat shock protein binding | cysteine and histidine-rich domain-containing protein RAR1 | 1.2 |
| ORTHOMCL17922 | catalytic-type peptidase activity | NEDD8-specific protease 1 | 2.4 |
| KEGG pathway | |||
| ORTHOMCL11569 | Plant–pathogen interaction | RPM1 interacting protein 4 transcript 2 | 1.3 |
| ORTHOMCL16124 | Plant–pathogen interaction | cysteine and histidine-rich domain-containing protein RAR1 | 1.2 |
| ORTHOMCL13297 | Plant–pathogen interaction | probable calcium-binding protein CML45 | 1.61 |
| ORTHOMCL17232 | Plant–pathogen interaction | pathogenesis-related protein PR-1 type | 4.32 |
| ORTHOMCL18122 | Plant–pathogen interaction | probable cyclic nucleotide-gated ion channel 17 | 2.93 |
| ORTHOMCL17558 | Glutathione metabolism | glutathione peroxidase | 1.21 |
| ORTHOMCL17864 | Glutathione metabolism | glutathione S-transferase | 1.54 |
| ORTHOMCL15235 | Ribosome biogenesis in eukaryotes | nucleolar protein 56 | 5.42 |
| ORTHOMCL11943 | Ribosome biogenesis in eukaryotes | probable mediator of RNA polymerase II transcription subunit 36b | 1.09 |
| ORTHOMCL20729 | Ribosome biogenesis in eukaryotes | nucleolar protein 56-like | 1.37 |
| Transcription factors | |||
| ORTHOMCL11146 | WRKY | WRKY transcription factor 48 | 1.02 |
| ORTHOMCL13242 | WRKY | WRKY transcription factor 22 | 2.35 |
| ORTHOMCL17007 | NAC | NAC transcription factor | 1.05 |
| ORTHOMCL20774 | NAC | NAC domain-containing protein 21/22 | 1.16 |
| ORTHOMCL16173 | MYB | MYB-like protein X | 3.22 |
| ORTHOMCL17851 | MYB | transcription factor MYB48 isoform X1 | 1.74 |
| ORTHOMCL11581 | bHLH | transcription factor bHLH61 isoform 1 | 1.05 |
| ORTHOMCL14612 | bHLH | transcription factor bHLH81 | 1.16 |
| ORTHOMCL12079 | bHLH | transcription factor bHLH100 | 1.37 |
| ORTHOMCL11075 | bHLH | transcription factor bHLH130 | 2.61 |
| ORTHOMCL18712 | ERF | transcription factor DcERF1 | 2.29 |
| ORTHOMCL11417 | ERF | ethylene response factor 2.1 | 1.14 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Liu, M.-L.; Yue, M.; Zhao, Z.; Zhao, G.-F.; Li, Z.-H. Comparative Transcriptome Analysis Reveals Adaptive Evolution of Notopterygium incisum and Notopterygium franchetii, Two High-Alpine Herbal Species Endemic to China. Molecules 2017, 22, 1158. https://doi.org/10.3390/molecules22071158
Jia Y, Liu M-L, Yue M, Zhao Z, Zhao G-F, Li Z-H. Comparative Transcriptome Analysis Reveals Adaptive Evolution of Notopterygium incisum and Notopterygium franchetii, Two High-Alpine Herbal Species Endemic to China. Molecules. 2017; 22(7):1158. https://doi.org/10.3390/molecules22071158
Chicago/Turabian StyleJia, Yun, Mi-Li Liu, Ming Yue, Zhe Zhao, Gui-Fang Zhao, and Zhong-Hu Li. 2017. "Comparative Transcriptome Analysis Reveals Adaptive Evolution of Notopterygium incisum and Notopterygium franchetii, Two High-Alpine Herbal Species Endemic to China" Molecules 22, no. 7: 1158. https://doi.org/10.3390/molecules22071158