
Structure-Activity Relationships of Acyclic Selenopurine Nucleosides as Antiviral Agents
by
Pramod K. Sahu
†, 
Tamima Umme
†,
Jinha Yu
,
Gyudong Kim
,
Shuhao Qu
,
Siddhi D. Naik
and
Lak Shin Jeong
* Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea
*
Author to whom correspondence should be addressed.
†
The authors contributed equally to this work.
Molecules 2017, 22(7), 1167; https://doi.org/10.3390/molecules22071167
Submission received: 17 June 2017
/
Revised: 8 July 2017
/
Accepted: 10 July 2017
/
Published: 12 July 2017
(This article belongs to the Special Issue Nucleic Acid Chemistry and Nucleic Acid Drugs: A Themed Issue in Honor of Professor Lihe Zhang on the Occasion of His 80th Birthday)
Abstract
:A series of acyclic selenopurine nucleosides 3a–f and 4a–g were synthesized based on the bioisosteric rationale between oxygen and selenium, and then evaluated for antiviral activity. Among the compounds tested, seleno-acyclovir (4a) exhibited the most potent anti-herpes simplex virus (HSV)-1 (EC50 = 1.47 µM) and HSV-2 (EC50 = 6.34 µM) activities without cytotoxicity up to 100 µM, while 2,6-diaminopurine derivatives 4e–g exhibited significant anti-human cytomegalovirus (HCMV) activity, which is slightly more potent than the guanine derivative 4d, indicating that they might act as prodrugs of seleno-ganciclovir (4d).

1. Introduction
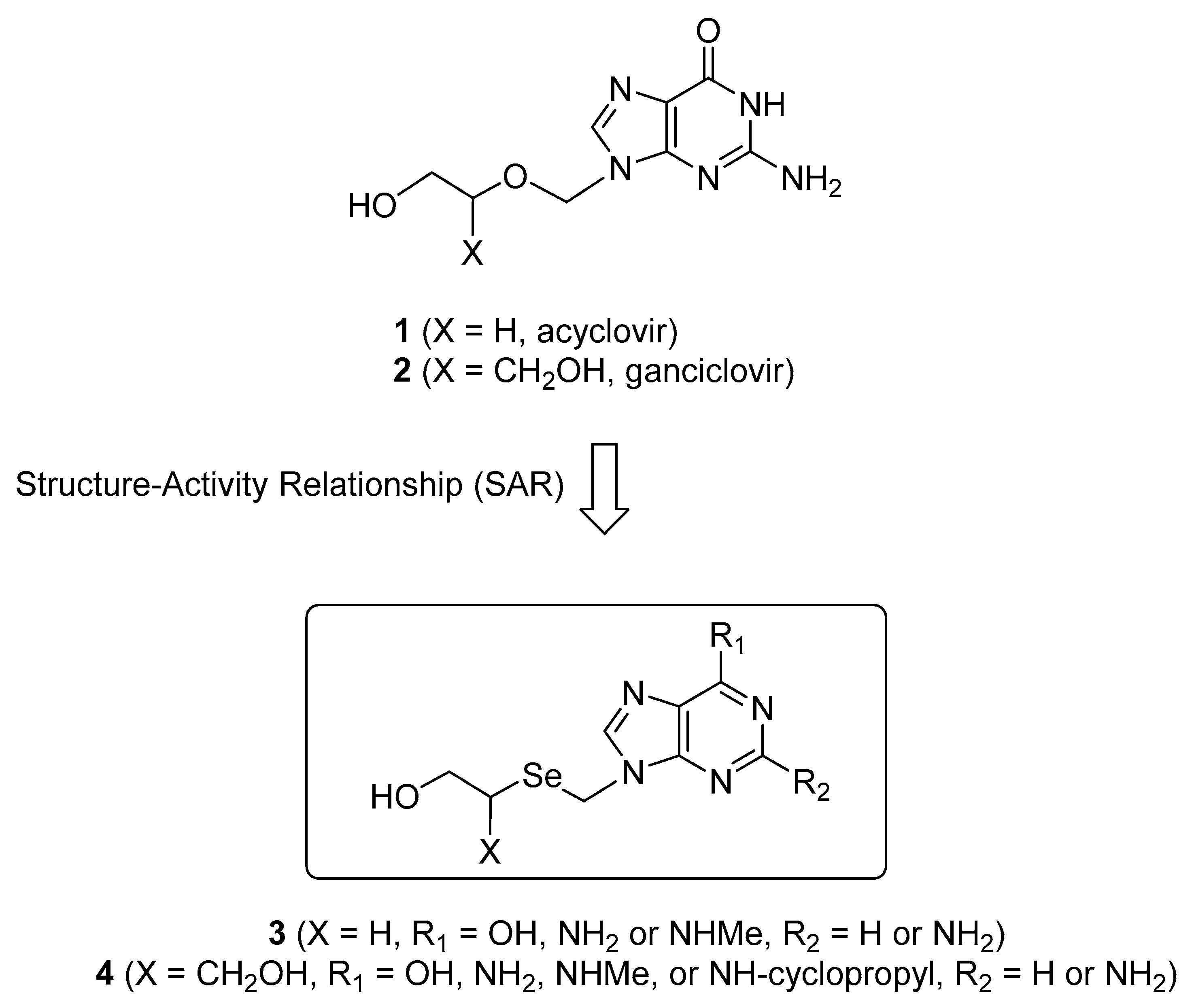
Modified nucleosides have continued to be fruitful resources for the development of antiviral agents [1]. Among these, acyclic nucleosides have been clinically used as drugs of choice for the treatment of herpetic viral infections such as herpes simplex virus (HSV), varicella-zoster virus (VZV), and human cytomegalovirus (HCMV) [2]. For example, acyclovir (1) [3,4] is widely used for the treatments of HSV- and VZV-infected patients, while ganciclovir (2) [5,6] is clinically used for the treatment of HCMV infections (Figure 1).
Compound 1 is intracellularly phosphorylated by viral-encoded thymidine kinase to its monophosphate, which is further phosphorylated to its triphosphate by cellular kinases. This triphosphate inhibits viral DNA polymerase reversibly by competing with the natural substrate, 2′-deoxyguanosine-5′-triphosphate (dGTP), and/or being incorporated into viral DNA chains, resulting in viral DNA chain termination [7,8]. On the other hand, compound 2 is also converted to its monophosphate by viral-encoded kinase (phosphotransferase), which is then subsequently converted into the triphosphate by cellular kinase [9]. This triphosphate inhibits HCMV DNA polymerase with a mechanism of action similar to 1 [9]. However, these drugs 1 and 2 have exhibited drawbacks such as hepatotoxicity [10], poor water solubility, and the appearance of resistant strains [11]. Although their poor water solubility has been solved by the amino acid ester prodrug approach, hepatotoxicity and resistance problems should still be solved. Thus, it has been highly desirable to develop new antiviral agents to tackle these problems.
In preliminary accounts, we synthesized the selenium analogues 3 (X = H, R1 = OH, R2 = NH2, seleno-acyclovir) and 4 (X = CH2OH, R1 = OH, R2 = NH2, seleno-ganciclovir) of 1 and 2 because they are in bioisosteric relationships; we also evaluated them for antiviral activity [12]. As expected, seleno-acyclovir exhibited potent anti-HSV activity, while seleno-ganciclovir exerted significant anti-HCMV activity [11]. Thus, based on these findings, it was very interesting to carry out the structure-activity relationship (SAR) study modifying the C2 and/or C6 positions of the guanine base of seleno-acyclovir and seleno-ganciclovir, which might overcome the drawbacks such as high cytotoxicity and poor water solubility caused by the guanine base. Herein, we report the full accounts of acyclic selenopurine nucleosides 3 and 4 modified at the C2 and/or C6 position as antiviral agents.
2. Results and Discussion
2.1. Chemistry
Scheme 1 illustrates the synthesis of 6-chloro- and 2-amino-6-chloropurine derivatives 11 and 12, which serve as the versatile intermediates for the synthesis of various seleno-acyclovir analogues [11]. 2-Bromoethanol (5) was converted to the glycosyl donor 8 according to our previously reported procedure [12]. 2-Bromoethanol (5) was treated with selenium dianion prepared in situ by heating with selenium powder and hydrazine hydrate in aqueous KOH solution to afford the diselenide 6, which was protected with the tert-butyldiphenylsilyl (TBDPS) group to give 7. Treatment of 7 with NaBH4 followed by trapping with methylene bromide yielded the glycosyl donor 8. Condensation of 8 with 6-chloropurine and 2-amino-6-chloropurine in the presence of K2CO3 produced the desired N9-6-chloropurine derivative 9 and N9-2-amino-6-chloropurine derivative 10, respectively, along with concomitant formations of the corresponding N7-isomers in negligible amounts. The N9-isomers were confirmed by the chemical shift of the C5 signal of 13C-NMR. The pronounced difference in N7- and N9-isomers reside in the C5 signals of 13C-NMR [13]. In general, the C5 signal (~132 ppm) in the N9-isomer is shifted downfield by ~10 ppm relative to the corresponding shift in the N7-isomer [13]. The chemical shift of the C5 in 9 was 131.69 ppm, indicating that it is a N9-isomer. The N9-isomer 9 was further confirmed after being converted to the final 4a. Removals of the TBDPS group in 9 and 10 with n-Bu4NF afforded the key intermediates 11 and 12, respectively.
Conversion of the key intermediates 11 and 12 into the seleno-acyclovir analogues 3a–c and 4a–c is shown in Scheme 2. Treatment of 11 and 12 with 2-mercaptoethanol and NaOMe in MeOH yielded seleno-inosine derivative 3a (80%) and seleno-acyclovir 4a [12] (94%), respectively. The N9-isomer 4a was further confirmed by comparing the C5 signal (δ 116.73 ppm) of 4a with that (δ 116.73 ppm) of acyclovir (1) [12]. Compounds 11 and 12 were also converted to the N6-methyladenine derivative 3b and the 2-amino-N6-methyladenine derivative 4b, respectively, by heating with 40% aqueous methylamine solution in MeOH. To overcome the low water solubility of the hypoxanthine or guanine base, 11 and 12 were transformed to the adenine derivative 3c and 2,6-diamino derivative 4c, respectively, by heating with t-butanolic ammonia at 85 °C.
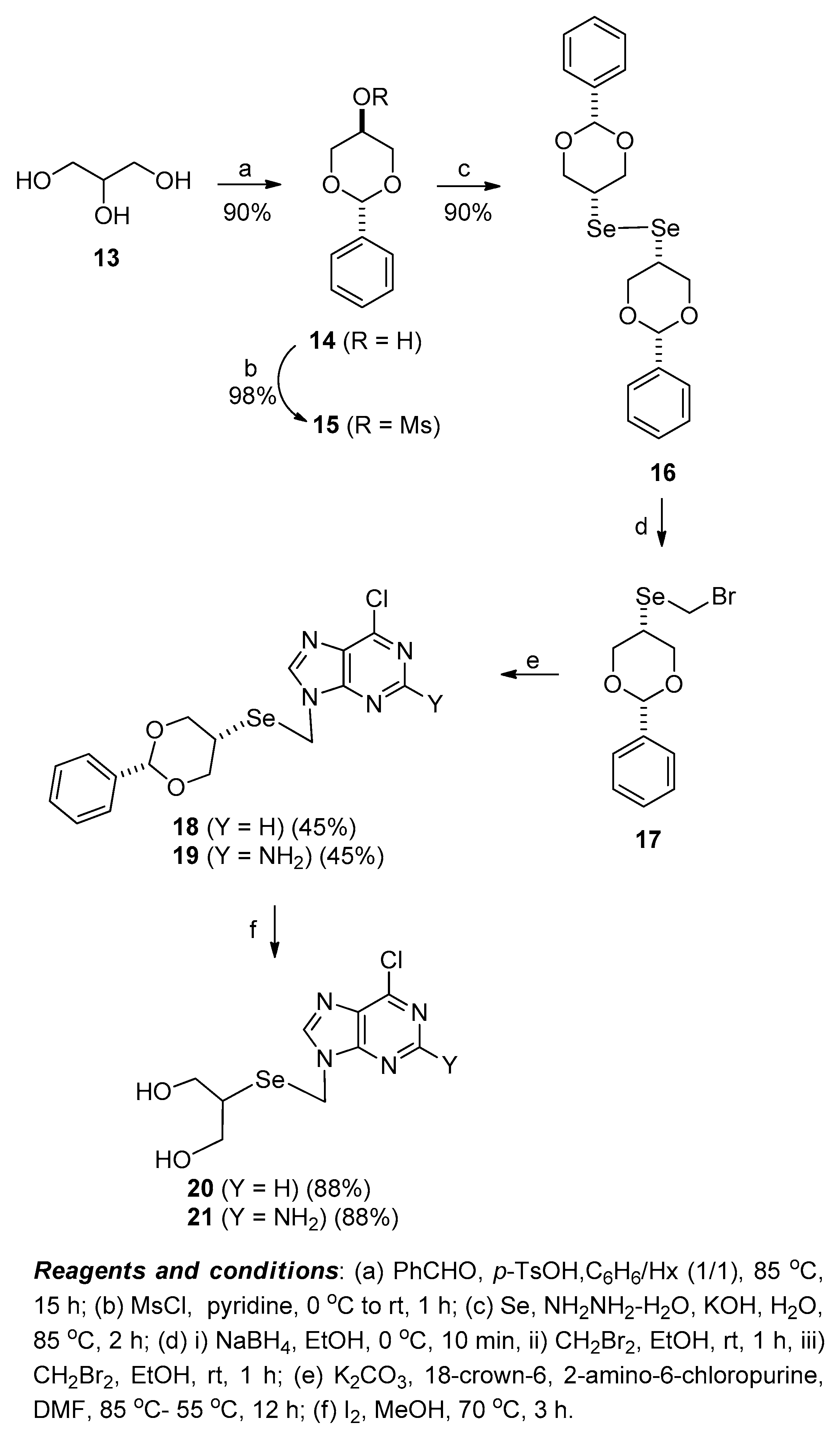
For the synthesis of seleno-ganciclovir analogues, the key intermediates 6-chloropurine derivative 20 and 2-amino-6-chloropurine derivative 21 were first synthesized, starting from glycerol (13), as depicted in Scheme 3 [12]. Glycerol (13) was converted to the glycosyl donor 17 according to our previously reported procedure [12]. Glycerol (13) was protected with the 1,3-benzylidene group by treating with benzaldehyde in the presence of p-TsOH to give 14, which was treated with MsCl to give the mesylate 15. Using similar conditions (Se, NH2NH2-H2O, and KOH) employed in the preparation of 6, compound 15 was smoothly converted to the diselenide 16. The reduction of diselenide 16 with NaBH4 followed by the treatment with CH2Br2 yielded the glycosyl donor 17. Without purification, compound 17 was condensed with 6-chloropurine and 2-amino-6-chloropurine using the same procedure employed in the preparation of 9 and 10 to afford 18 and 19, respectively. The removal of 1,3-benzylidene in 18 and 19 was achieved by treating with iodine in MeOH to give 20 and 21, respectively.
The key intermediates 20 and 21 were converted to the seleno-ganciclovir analogues 3d–f and 4d–g, as shown in Scheme 4. Treatment of 20 and 21 with 2-mercaptoethanol and NaOMe afforded the seleno-inosine derivative 3d and the seleno-ganciclovir 4d [12], respectively. The N9 regiochemistry of 4d was further confirmed by comparing the C5 signal (δ 116.56 ppm) of 4d with that (δ 116.40 ppm) of ganciclovir (2) [12]. The 6-chlororpurine derivative 20 was converted to the N6-methyladenine derivative 3e and the adenine derivative 3f by treating with methylamine and ammonia, respectively. The 2-amino-6-chloropurine derivative 21 was treated with methylamine, ammonia, and cyclopropylamine to afford the 2-amino-N6-methylaminopurine derivative 4e, the 2,6-diaminopurine derivative 4f, and the 2-amino-N6-cyclopropylaminopurine derivative 4g, respectively.
2.2. Antiviral Activity
All the final compounds 3a–f and 4a–g were assayed for their antiviral activity against several herpes viruses such as HSV-1 (strain F, VR-733), HSV-2 (strain MS, VR-540), VZV (Ellen, VR-1367), and HCMV (Davis, VR-807) [12,14]. Cytotoxicity data were measured in HEL299 (CCL-137) cells as described previously [12,14]. As shown in Table 1, compound 4a exhibited the most potent anti-HSV-1 and HSV-2 activities, whereas compound 4e exerted the most potent antiviral activity against HCMV, although it is moderate. It is interesting to note that 2,6-disubstituted nucleosides exhibited antiviral activity, but 6-substituted nucleosides were totally inactive up to 100 µM, regardless of the substitution at the X. Compounds 4b and 4c also exhibited significant antiviral activities against HSV-1 and HSV-2, indicating that they might be deaminated by the cellular nucleoside deaminase to serve as prodrugs of seleno-acyclovir (4a), although this should be confirmed by an adenosine deaminase test. Similarly, compounds 4e–g, which seem to act as prodrugs of seleno-ganciclovir (4d), also exhibited significant anti-HCMV activity and they were more potent than the parent compound 4d. However, all compounds were neither active nor toxic up to 100 µM against VZV. The finding that all synthesized compounds were less potent than the reference compounds 1 and 2 might be explained by the difficulty in phosphorylation induced by steric effects of the bulky selenium atom, although it was much relieved by the freely rotatable acyclic single bond [15].
3. Materials and Methods
Proton (1H) and carbon (13C)-NMR spectra were recorded on a Bruker AV 400 (Bruker Corporation, Rheinstettern, Germany) (400/100 MHz) spectrometer. Chemical shifts are reported in ppm (δ) with residual solvents as the internal standard. UV spectra were recorded on a PerkinElmer Lambda25 in methanol. Melting points were determined on a Barnstead Electrothermal 9100 instrument. Mass spectra were recorded on a fast atom bombardment (FAB). All reactions involving air- or moisture-sensitive conditions were routinely carried out under an inert atmosphere of dry nitrogen. Reactions were checked by thin layer chromatography (Kieselgel 60 F254, Merck, Darmstadt, Germany). Spots were detected by viewing under UV light, and by colorizing with charring after dipping in a p-anisaldehyde solution. The crude compounds were purified by column chromatography on silica gel (Kieselgel 60, 230–400 mesh, Merck, Darmstadt, Germany). All solvents were purified and dried by standard techniques just before use.
3.1. General Procedure for Base Condensation
To a stirring solution of 7 [12] or 16 [12] (5.52 mmol) in ethanol (25 mL), NaBH4 (46.88 mmol) was added at 0 °C, followed by the addition of a solution of CH2Br2 (683.47 mmol) in ethanol (100 mL). The reaction mixture was stirred at room temperature for 1 h and evaporated. The residue was treated with aqueous NaHCO3 (50 mL) and extracted with ethyl acetate (3 × 150 mL). The organic layer was dried (MgSO4), filtered, and evaporated to give crude bromide 8. A suspension of K2CO3 (27.68 mmol), 18-crown-6 (10.36 mmol), and 6-chloropurine (13.81 mmol) or 2-amino-6-chloropurine (13.81 mmol) in N,N-dimethylformamide (100 mL) was stirred under N2 at 85 °C for 3 h. To this mixture, a solution of 8 in N,N-dimethylformamide (DMF) (10 mL) was added and the reaction mixture was stirred at 55 °C for 7 h, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexanes:ethyl acetate = 1:1) to give the base condensed product 9, 10, 18, or 19.
9-((2-O-tert-Butyldiphenylsilyloxyethylselanylmethyl)-6-chloro-9H-purin (9). Yield: 46%; colorless syrup; UV (MeOH) λmax 264 nm; 1H-NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.17 (s, 2H), 7.68–7.65 (m, 4H), 7.46–7.37 (m, 6H), 5.43 (s, 2H), 3.93 (t, J = 6 Hz, 2H), 2.88 (t, J = 6.4 Hz, 2H), 1.05 (s, 9H); 13C-NMR (100 MHz, CDCl3) δ 152.13, 151.51, 151.24, 144.86, 135.54, 133.11, 131.69, 129.89, 127.79, 64.07, 34.20, 27.93, 26.81, 19.17; MS (ESI) m/z 531.0888 (M + H)+; Anal. Calcd. for C24H27ClN4OSeSi: C, 54.39; H, 5.14; N, 10.57. Found: C, 54.89; H, 4.99; N, 10.43.
9-((2-O-tert-Butyldiphenylsilyloxyethylselanyl)methyl)-6-chloro-9H-purin-2-amine (10). Yield: 45%; white solid; m.p. 115–120 °C; UV (MeOH) λmax 310 nm; 1H-NMR (400 MHz, CDCl3) δ 7.81 (s, 1H), 7.6–7.65 (m, 4H), 7.46–7.37 (m, 6H), 5.22 (s, 2H), 3.92 (t, J = 6.8 Hz, 2H), 2.86 (t, J = 6.4 Hz, 2H), 1.05 (s, 9H); MS (ESI) m/z 546.0995 (M + H)+; Anal. Calcd. for C24H28ClN5OSeSi: C, 52.89; H, 5.18; N, 12.85. Found: C, 52.99; H, 4.98; N, 12.45.
9-((2-Phenyl-1, 3-dioxan-5-ylselanyl)methyl)-6-chloro-9H-purin (18). Yield: 45%; colorless syrup; UV (MeOH) λmax 264 nm; 1H-NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 8.27 (s, 1H), 7.41–7.39 (m, 2H), 7.33–7.31 (m, 3H), 5.46 (s, 2H), 5.43 (s, 1H), 4.29 (dd, J = 4.6 Hz, 11.4 Hz, 2H), 3.78 (t, J = 11.8 Hz, 2H), 3.65–3.57 (m, 1H); MS (ESI) m/z 411.0113 (M + H)+; Anal. Calcd. for C16H15ClN4O2Se: C, 46.90; H, 3.69; N, 13.67. Found: C, 46.56; H, 3.39; N, 13.89.
9-((2-Phenyl-1,3-dioxan-5-ylselanyl)methyl)-6-chloro-9H-purin-2-amine (19). Yield: 45%; white solid; m.p. 164–166 °C; UV (MeOH) λmax 310 nm; 1H-NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.43–7.40 (m, 2H), 7.36–7.32 (m, 3H), 5.44 (s, 1H), 5.27 (s, 2H), 5.24 (s, 2H), 4.32 (dd, J = 4.5 and 11.4 Hz, 2H), 3.82–3.76 (m, 2H), 3.69–3.63 (s, 1H); 13C-NMR (125 MHz, CD3OD) δ 159.3, 153.4, 151.8, 141.4, 137.6, 129.1, 128.3, 125.9, 125.2, 101.4, 71.4. 35.4, 33.4; MS (FAB) m/z 426.0117 (M + H)+; Anal. Calcd. for C16H16ClN5O2Se: C, 45.24; H, 3.80; N, 16.49. Found: C, 45.33; H, 4.10; N, 16.55.
3.2. General Procedure for TBDPS Removals
To a solution of 9 or 10 (4.036 mmol) in tetrahydrofuran (THF) (40 mL), tetra-n-butylammonium fluoride (2.06 mmol, 1 M solution in THF) was added under N2, and the reaction mixture stirred at room temperature for 1 h and evaporated. The residue was purified by column chromatography (hexanes:ethyl acetate = 1:9) to give 11 or 12, respectively.
2-((6-Chloro-9H-purin-9-yl)methylselanyl) ethanol (11). Yield: 92%; white solid; m.p. 98–100 °C; UV (MeOH) λmax 264 nm; 1H-NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 8.32 (s, 1H), 5.61 (s, 2H), 4.02 (q, J = 5.6 Hz, 2H), 2.88 (t, J = 5.6 Hz, 2H), 2.65 (t, J = 5.6 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ 152.30, 151.72, 151.69, 145.47, 132.03, 63.41, 34.54, 28.28; MS (ESI) m/z 290.9713 (M + H)+.
2-((2-Amino-6-chloro-9H-purin-9-yl)methylselanyl) ethanol (12). Yield: 45%; white solid; m.p. 150–152 °C; UV (MeOH) λmax 310 nm; 1H-NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 6.95 (br s, 2H, exchangeable), 5.35 (s, 2H), 3.59 (t, J = 6.4 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 159.8, 153.6, 149.5, 142.7, 123.4, 61.4, 33.8, 27.7; MS (ESI) m/z 307.9807 (M + H)+.
3.3. General Procedure for the 1,3-Benzylidene Removals
To a solution of 18 or 19 (7.06 mmol) in MeOH (20 mL), iodine (0.2 mL, 0.1 M solution in MeOH) was added and the reaction mixture was heated at 60 °C for 4 h. Then, the reaction mixture was quenched with few drops of aqueous sodium thiosulfate and evaporated. The residue was purified by silica gel column chromatography (CH2Cl2:MeOH = 24:1) to give 20 or 21, respectively.
2-((6-Chloro-9H-purin-9-yl)methylselanyl) propane-1, 3-diol (20). Yield: 88%; white solid; m.p. 140–143 °C; UV (MeOH) λmax 263 nm; 1H-NMR (400 MHz, CD3OD) δ 8.75 (s, 1H), 8.69 (s, 1H), 5.68 (s, 2H), 3.79 (s, 2H), 3.77 (s, 2H), 3.27 (s, 1H); 13C-NMR (100 MHz, CD3OD) δ 153.89, 153.78, 152.11, 148.97, 133.23, 64.54, 49.63, 35.80; MS (FAB) m/z 326.1516 (M + H)+.
2-((2-Amino-6-chloro-9H-purin-9-yl)methylselanyl) propane-1,3-diol (21). Yield: 88%; white solid; m.p. 160–162 °C; UV (MeOH) λmax 310 nm; 1H-NMR (400 MHz, CD3OD) δ 8.18 (s, 1H), 5.45 (s, 2H), 3.85–3.76 (m, 4H), 3.3–3.33 (m, 1H); 13C-NMR (100 MHz, CD3OD + CDCl3) δ 162.1, 155.5, 152.3, 144.7, 125.6, 71.8, 64.4, 48.8, 35.0; MS (ESI) m/z 337.9915 (M + H)+; Anal. Calcd. for C9H12ClN5O2Se: C, 32.11; H, 3.59; N, 20.80. Found: C, 32.01; H, 3.91; N, 20.56.
3.4. Conversion of 6-Chloro Derivatives to 6-Keto Derivatives
A solution of 6-chloro derivative 11, 12, 20, or 21 (0.358 mmol), 2-mercaptoethanol (1.297 mmol) and NaOMe (1.797 mmol) in methanol (15 mL) was refluxed for 4 days at 75 °C. After completion of the reaction, the reaction mixture was cooled and neutralized with acetic acid. The solvent was removed under reduced pressure and the residue was chromatographed (CH2Cl2:MeOH = 7:1) to give the 6-keto derivative 3a, 4a, 3d, or 4d.
9-((2-Hydroxyethylselanyl)methyl)-1H-purin-6-(9H)-one (3a). Yield: 80%; white solid; m.p. 225–227 °C; UV (MeOH) λmax, 248 nm; 1H-NMR (400 MHz, DMSO-d6) δ 8.16 (s, 1H), 8.06 (s, 1H), 5.44 (s, 2H), 4.89 (br s, 1H, exchangeable), 3.58–3.56 (m, 2H), 2.81 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 156.64, 148.03, 145.87, 139.96, 124.13, 61.29, 33.95, 27.55; MS (ESI) m/z 275.0041 (M + H)+; Anal. Calcd. for C8H10N4O2Se: C, 35.18; H, 3.69; N, 20.51. Found: C, 35.19; H, 3.56; N, 20.11.
9-((2-Hydroxyethylselanyl)methyl)-2-amino-1H purin-6(9H)-one (4a) [12]. Yield: 94%; white solid: m.p. 266–268 °C; UV (MeOH) λmax 259 nm; 1H-NMR (400 MHz, DMSO-d6) δ 10.60 (br s, 1H, exchangeable with D2O), 7.78 (s, 1H), 6.49 (br s, 2H), 5.23 (s, 2H), 4.86 (t, J = 5.3 Hz, 1H), 3.58 (q, J = 6.5 Hz, 2H), 2.79 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 156.7, 153.7, 150.9, 137.1, 116.6, 61.4, 33.4, 27.3; MS (ESI) m/z 290.0152 (M + H)+; Anal. Calcd. for C8H11N5O2Se: C, 33.34; H, 3.85; N, 24.30. Found: C, 33.14; H, 4.15; N, 24.01.
9-((1,3-Dihydroxypropan-2-ylselanyl)methyl)-1H purin-6(9H)-one (3d). Yield: 85%; white solid; m.p. 189–182 °C; UV (MeOH) λmax 247 nm; 1H-NMR (400 MHz, DMSO-d6) δ 8.17 (s, 1H), 8.06 (s, 1H), 5.46 (s, 2H), 4.84 (br s, 2H, exchangeable), 3.65–3.56 (m, 4H), 3.21–3.17 (m, 1H); 13C-NMR (100 MHz, DMSO-d6) δ 156.68, 148.01, 145.84, 139.93, 124.06, 61.49, 47.30, 33.23; MS (ESI) m/z 305.0142 (M + H)+; Anal. Calcd. for C9H12N4O3Se: C, 35.66; H, 3.99; N, 18.48. Found: C, 35.26; H, 4.13; N, 18.08.
9-((1,3-Dihydroxypropan-2-ylselanyl)methyl)-2-amino-1H-purin-6(9H)-one (4d) [12]. Yield: 70%; white solid; m.p. 204–207 °C; UV (MeOH) λmax 258 nm; 1H-NMR (400 MHz, DMSO-d6) δ 10.69 (br s, 1H, exchangeable with D2O), 7.78 (s, 1H), 6.59 (s, 2H, exchangeable with D2O), 5.5 (s, 2H), 4.84 (t, J = 5.3 Hz, 2H, exchangeable with D2O), 3.69–3.64 (m, 2H), 3.62-3.56 (m, 2H, exchangeable with D2O), 3.21–3.16 (m, 1H); 13C-NMR (100 MHz, DMSO-d6) δ 156.8, 153.8, 150.9, 137.0, 116.6, 61.6, 47.0, 32.8; MS (ESI) m/z 320.0254 (M + H)+; Anal. Calcd. for C9H13N5O3Se: C, 33.97; H, 4.12; N, 22.01. Found: C, 34.12; H, 4.42; N, 22.37.
3.5. Conversion of 6-Chloro Derivatives to N6-Methylamino Derivatives
A solution of 6-chloro derivative 11, 12, 20, or 21 (0.342 mmol) and methylamine (10 mL, 40% aqueous solution) in methanol (10 mL) was heated at 85 °C for 48 h in a steel bomb. After completion of the reaction, the solvent was removed and the residue was purified by silica gel column chromatography (CH2Cl2:MeOH = 20:1) to give the N6-methylamino derivative 3b, 4b, 3e, or 4e.
2-((6-(Methylamino)-9H-purin-9-yl)methylselanyl) ethanol (3b). Yield: 70%; white solid; m.p. 171–173 °C; UV (MeOH) λmax 266 nm; 1H-NMR (400 MHz, CD3OD) δ 8.26 (s, 1H), 8.17 (s, 1H), 5.49 (s, 2H), 3.73 (t, J = 6.6 Hz, 2H), 3.10 (broad s, 3H), 2.84 (t, J = 6.6 Hz, 2H); 13C-NMR (100 MHz, CD3OD) δ 157.58, 154.69, 150.18, 142.64, 121.46, 64.11, 35.66, 28.88; MS (ESI) m/z 288.0363 (M + H)+; Anal. Calcd. for C9H13N5OSe: C, 37.77; H, 4.58; N, 24.47. Found: C, 37.78; H, 4.55; N, 24.31.
2-((2-Amino-6-(methylamino)-9H-purin-9-yl)methylselanyl) ethanol (4b). Yield: 85%; white solid; m.p. 183–185 °C; UV (MeOH) λmax 282 nm; 1H-NMR (400 MHz, CD3OD) δ 7.81 (s, 1H), 5.35 (s, 2H), 3.75 (t, J = 6.7 Hz, 2H), 3.04 (broad s, 3H), 2.85 (t, J = 6.6 Hz, 2H); 13C-NMR (75 MHz, CD3OD) δ 162.91, 158.07, 139.54, 115.59, 64.28, 64.28, 35.28, 31.54, 28.58; MS (ESI) m/z 303.0472 (M + H)+; Anal. Calcd. for C9H14N6OSe: C, 35.89; H, 4.69; N, 27.90. Found: C, 36.18; H, 4.32; N, 28.01.
2-((6-(Methylamino)-9H-purin-9-yl)methylselanyl)propane-1,3-diol (3e). Yield: 50%; white solid; m.p. 180–183 °C; UV (MeOH) λmax 266 nm; 1H-NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.22 (s, 1H), 7.69 (br s, 1H, exchangeable), 5.48 (s, 2H), 4.84 (t, J = 5.1 Hz, 2H, exchangeable), 3.67–3.56 (m, 4H), 3.25–3.21 (m, 1H), 2.96 (br s, 3H); 13C-NMR (75 MHz, DMSO-d6) δ 154.90, 152.56, 148.14, 140.19, 119.20, 61.55, 47.26, 33.03, 27.01; MS (ESI) m/z 318.0469 (M + H)+; Anal. Calcd. for C10H15N5O2Se: C, 37.98; H, 4.78; N, 22.15. Found: C, 37.99; H, 4.36; N, 22.01.
2-((2-Amino-6-(methylamio)-9H-purin-9-yl)methylselanyl) propane-1,3-diol (4e). Yield: 50%; white solid; m.p. 140–143 °C; UV (MeOH) λmax 282 nm; 1H-NMR (400 MHz, DMSO-d6) δ 7.77 (s, 1H), 7.16 (br s, 1H, exchangeable), 5.88 (s, 2H, exchangeable), 5.28 (s, 2H), 4.82 (t, J = 5.2 Hz, 2H, exchangeable), 3.70–3.65 (m, 2H), 3.63-3.57 (m, 2H), 3.21–3.16 (m, 1H), 2.89 (br s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 160.28, 155.38, 136.57, 113.43, 105.24, 61.67, 46.88, 32.61, 26.90, 26.85; MS (ESI) m/z 333.0573 (M + H)+; Anal. Calcd. for C10H16N6O2Se: C, 36.26; H, 4.87; N, 25.37. Found: C, 36.56; H, 443; N, 25.08.
3.6. Conversion of 6-Chloro Derivatives to N6-Amino Derivatives
6-Chloro derivative 11, 12, 20, or 21 (0.342 mmol) and NH3/t-butanol (10 mL) were taken in a steel bomb and heated at 85 °C for 12 h. The solvent was removed and the residue was purified by silica gel column chromatography (CH2Cl2:MeOH = 24:1) to give N6-amino derivative 3c, 4c, 3f, 4f, or 4g.
2-((6-Amino-9H-purin-9-yl)methylselanyl)ethanol (3c). Yield: 95%; white solid; m.p. 175–177 °C; UV (MeOH) λmax 260 nm; 1H-NMR (400 MHz, CD3OD) δ 8.25 (s, 1H), 8.23 (s, 1H), 5.52 (s, 2H), 3.74 (t, J = 6.4 Hz, 2H), 2.86 (t, J = 6.4 Hz, 2H); 13C-NMR (100 MHz, CD3OD) δ 157.41, 153.94, 150.52, 142.52, 120.17, 63.35, 34.95, 28.16; MS (ESI) m/z 274.0202 (M + H)+; Anal. Calcd. for C8H11N5OSe: C, 35.30; H, 4.07; N, 25.73. Found: C, 35.20; H, 4.47; N, 25.43.
2-((2,6-Diamino-9H-purin-9-yl)methylselanyl) ethanol (4c). Yield: 70%; white solid; m.p. 84–86 °C; UV (MeOH) λmax 281 nm; 1H-NMR (400 MHz, DMSO-d6) δ 7.80 (s, 1H), 6.73 (s, 2H, exchangeable), 5.86 (s, 2H, exchangeable), 5.28 (s, 2H), 4.89 (t, J = 5.3 Hz, 1H, exchangeable), 3.60 (q, J = 6.3 Hz, 2H), 2.81 (t, J = 6.7 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6) δ 160.32, 156.07, 151.42, 136.99, 113.11, 61.46, 33.28, 27.22; MS (ESI) m/z 289.0314 (M + H)+; Anal. Calcd. for C8H12N6OSe: C, 33.46; H, 4.21; N, 29.26. Found: C, 33.20; H, 4.41; N, 29.43.
2-((6-Amino-9H-purin-9-yl)methylselanyl)propane-1,3-diol (3f). Yield: 80%; white solid; m.p. 186–188 °C; UV (MeOH) λmax 260 nm; 1H-NMR (400 MHz, DMSO-d6) δ 8.23 (s, 1H), 8.16 (s, 1H), 7.23 (s, 2H, exchangeable), 5.47 (s, 2H), 4.85 (t, J = 5.1 Hz, 2H, exchangeable), 3.67–3.57 (m, 4H), 3.24–3.21 (m, 1H); 13C-NMR (100 MHz, DMSO-d6) δ 155.93, 152.53, 149.11, 140.47, 118.72, 61.57, 47.24, 32.96; MS (ESI) m/z 304.0292 (M + H)+; Anal. Calcd. for C9H13N5O2Se: C, 35.77; H, 4.34; N, 23.18. Found: C, 35.89; H, 4.12; N, 23.02.
2-((2,6-Diamino-9H-purin-9-yl)methylselanyl)propane-1,3-diol (4f). Yield: 75%; white solid; m.p. 193–196 °C; UV (MeOH) λmax 282 nm; 1H-NMR (400 MHz, DMSO-d6) δ 7.79 (s, 1H), 6.67 (s, 2H, exchangeable), 5.81 (d, J = 2.8 Hz, 2H, exchangeable), 5.28 (s, 2H), 4.84 (t, J = 5.1 Hz, 1H, exchangeable), 3.71–3.66 (m, 2H), 3.63–3.57 (m, 2H), 3.29 (s, 1H, exchangeable), 3.23–3.16 (m, 1H); 13C-NMR (100 MHz, DMSO-d6) δ 160.20, 156.03, 151.36, 136.98, 113.09, 61.66, 61.55, 46.84, 32.61; MS (ESI) m/z 319.0414 (M + H)+; Anal. Calcd. for C9H14N6O2Se: C, 34.08; H, 4.45; N, 26.49. Found: C, 34.48; H, 4.17; N, 26.83.
2-((2-Amino-6-(cyclopropylamino)-9H-purin-9-yl)methylselanyl)propane-1,3-diol (4g). To a solution of 44 (100 mg, 0.296 mmol) in ethanol (10 mL) in a steel bomb, cyclopropylamine (0.103 mL, 1.486 mmol) and triethylamine (0.272 mL, 1.950 mmol) were added and the mixture was heated at 100 °C for 48 h. After completion of the reaction, the solvent was removed and the residue was purified by silica gel chromatography (CH2Cl2:MeOH = 20:1 to give 4d (52 mg, 55%) as a white solid: m.p. 140–143 °C; UV (MeOH) λmax 285 nm; 1H-NMR (400 MHz, DMSO-d6) δ 7.79 (s, 1H), 7.29 (br s, 1H, exchangeable), 5.88 (s, 2H, exchangeable), 5.29 (s, 2H), 4.84 (t, J = 5.2 Hz, 2H, exchangeable), 3.71–3.66 (m, 2H), 3.63–3.57 (m, 2H), 3.22–3.19 (m, 1H), 3.03 (br s, 1H), 0.68-0.62 (m, 2H), 0.59–0.58 (m, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 160.19, 155.81, 150.94, 136.73, 113.35, 69.68, 61.63, 46.91, 32.66, 23.86, 6.44; MS (ESI) m/z 359.0731 (M + H)+; Anal. Calcd. for C12H18N6O2Se: C, 40.34; H, 5.08; N, 23.52. Found: C, 40.14; H, 5.17; N, 23.12.
3.7. Antiviral Activity and Cytotoxicity Assays
Antiviral activity was measured using a standard cytopathic (CPE) inhibition assay as described before [13]. Briefly, Vero cells in stationary phase were infected with the virus at a multiplicity of infection of 2–4 CCID50 (50% cell culture inhibitory dose) per each well of 96-well plates. After 2 h of adsorption at 37 °C, the liquid was aspirated and 100 μL of Dulbeco’s modified eagle’s media (DMEM)/2% fetal bovine serum (FBS) containing a compound was applied to each well in duplicate for each concentration and further incubated for 3 days. Antiviral activity was measured by MTT assay and expressed as the EC50. Cytocidal assay was performed as a control experiment for the antiviral assay. It was carried out simultaneously with the antiviral assay described above using mock instead of virus for infection, and cell viability was measured by MTT assay. The concentration of the compound responsible for 50% reduction of cell growth was calculated and expressed as CC50.
4. Conclusions
We have synthesized various acyclic seleno-purine nucleosides 3a–f and 4a–g, and evaluated them for anti-herpetic activity. The key diselenides 6 and 16 were synthesized by treating bromide 5 and mesylate 15 with selenium powder and hydrazine hydrate in aqueous KOH solution, respectively. The glycosyl donors 8 and 17 were synthesized by treating diselenides 7 and 16 with NaBH4 followed by trapping with methylene bromide, and then condensed with 6-chloropurine or 2-amino-6-chloropurine anion in a SN2 manner. Among the compounds tested, seleno-acyclovir (4a) showed the most potent anti-HSV-1 and HSV-2 activities, while the seleno-ganciclovir analogue 4e exhibited the most potent anti-HCMV activity. It seems that 2,6-diaminopurine nucleosides might be converted to seleno-acyclovir (4a) and seleno-ganciclovir (4d) by cellular nucleoside deaminases, serving as prodrugs of 4a and 4d. Although we could not discover more potent compounds than the reference compounds, acyclovir (1) or ganciclovir (2), it is expected that these synthesized nucleosides can be a new template for the design of novel acyclic nucleoside analogues.
Acknowledgments
This work was supported by the grants from Mid-Career Research Program (370C-20160046) of National Research Foundation (NRF), Korea. Antiviral assay by Chong-Kyo Lee (KRICT, Korea) is greatly appreciated.
Author Contributions
L.S.J. conceived and designed the experiments; P.K.S. and T.U. performed the experiments; P.K.S and L.S.J. analyzed the data; J.Y., G.K., S.Q., and, S.D.N. contributed reagents/materials/analysis tools; L.S.J. wrote the paper. All the authors contributed in writing and proofreading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Herdewijn, P. (Ed.) Modified Nucleosides in Biochemistry, Biotechnology and Medicine; Wiley-VCH: Weinheim, Germany, 2008; pp. 1–658. [Google Scholar]
- Freeman, S.; Gardiner, J.M. Acyclic nucleosides as antiviral compounds. Mol. Biotechnol. 1996, 5, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Elion, G.B.; Furman, P.A.; Fyfe, J.A.; de Miranda, P.; Beauchamp, L.; Schaeffer, H.J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl)guanine. Proc. Natl. Acad. Sci. USA 1977, 74, 5716–5720. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, H.J.; Beauchamp, L.; de Miranda, P.; Elion, G.B.; Bauer, D.J.; Collins, P. 9-(2-Hydroxyethoxymethyl)guanine activity against viruses of the herpes group. Nature 1978, 272, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Field, A.K.; Davies, M.E.; DeWitt, C.; Perry, V.; Liou, R.; Germershausen, J.; Karkas, J.D.; Ashton, W.T.; Johnson, D.B.R.; Tolman, R.L. 9-{[2-Hydroxy-l-(hydroxymethyl)ethoxy]methyl}guanine: A selective inhibitor of herpes group virus replication. Proc. Natl. Acad. Sci. USA 1983, 80, 4139–4143. [Google Scholar] [CrossRef] [PubMed]
- Laskin, O.L.; Stahl-Bayliss, C.M.; Kalman, C.M.; Rosecan, L.R. Use of ganciclovir to treat serious cytomegalovirus infections in patients with AIDS. J. Infect. Dis. 1987, 155, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.E.; Spector, T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J. Biol. Chem. 1989, 264, 7405–7411. [Google Scholar]
- Reardon, J.E. Herpes simplex virus type 1 and human DNA polymerase interactions with 2′-deoxyguanosine 5′-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition. J. Biol. Chem. 1989, 264, 19039–19044. [Google Scholar] [PubMed]
- De Clercq, E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004, 2, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Mde, F.; Dos Santos, O.F.; Boim, M.A.; Razvickas, C.V.; de Moura, L.A.; Ajzen, H.; Schor, N. Nephrotoxicity of acyclovir and ganciclovir in rats: Evaluation of glomerular hemodynamics. J. Am. Soc. Nephrol. 1997, 8, 361–367. [Google Scholar] [PubMed]
- Starafeld, L.; Chou, S. Antiviral drug resistance: Mechanisms and clinical implications. Infect. Dis. Clin. N. Am. 2010, 24, 413–437. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.K.; Umme, T.; Yu, J.; Nayak, A.; Kim, G.; Noh, M.; Lee, J.-Y.; Kim, D.-D.; Jeong, L.S. Selenoacyclovir and selenoganciclovir: Discovery of a new template for antiviral agents. J. Med. Chem. 2015, 58, 8734–8738. [Google Scholar] [CrossRef] [PubMed]
- Paquette, L.A.; Dong, S. Stereoselective synthesis of β-anomeric 4′-thiaspirocyclic ribonucleosides carrying the full complement of RNA-level hydroxyl substitution. J. Organ. Chem. 2005, 70, 5655–5664. [Google Scholar] [CrossRef] [PubMed]
- Jeong, L.S.; Kim, H.O.; Moon, H.R.; Hong, J.H.; Yoo, S.J.; Choi, W.J.; Chun, M.W.; Lee, C.-K. Syntheses and structure-activity relationships of novel apio and thioapio dideoxydidehydronucleosides as anti-HCMV agents. J. Med. Chem. 2001, 44, 806–813. [Google Scholar] [CrossRef]
- Yu, J.; Sahu, P.K.; Kim, G.; Qu, S.; Choi, Y.; Song, J.; Lee, S.K.; Noh, M.; Park, S.; Jeong, L.S. Design, synthesis and cellular metabolism study of 4′-selenonucleosides. Future Med. Chem. 2015, 7, 1643–1655. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
The rationale for the design of acyclic selenopurine nucleosides 3 and 4 based on the potent antiviral activity of 1 and 2.
Figure 1.
The rationale for the design of acyclic selenopurine nucleosides 3 and 4 based on the potent antiviral activity of 1 and 2.

Scheme 1.
Synthesis of 6-chloro- and 2-amino-6-chloropurine derivatives 11 and 12.

Scheme 2.
Synthesis of seleno-acyclovir analogues 3a–c and 4a–c.

Scheme 3.
Synthesis of key intermediates 6-chloropurine derivative 20 and 2-amino-6-chloropurine derivative 21 starting from glycerol (13).
Scheme 3.
Synthesis of key intermediates 6-chloropurine derivative 20 and 2-amino-6-chloropurine derivative 21 starting from glycerol (13).

Scheme 4.
Synthesis of seleno-ganciclovir analogues 3d–f and 4d–f.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Antiviral activities of all the final nucleosides 3a-f and 4a-g.
| Compound (X, R1, R2) | EC50 (µM) a | CC50 (µM) b | |||
|---|---|---|---|---|---|
| HSV-1 | HSV-2 | VZV | HCMV | HEL299 | |
| 3a (X = H, R1 = OH, R2 = H) | >100 | >100 | >100 | >100 | >100 |
| 3b (X = H, R1 = NHMe, R2 = H) | >100 | >100 | >100 | >100 | >100 |
| 3c (X = H, R1 = NH2, R2 = H) | >100 | >100 | >100 | >100 | >100 |
| 3d (X = CH2OH, R1 = OH, R2 = H) | >100 | >100 | >100 | >100 | >100 |
| 3e (X = CH2OH, R1 = NHMe, R2 = H) | >100 | >100 | >100 | >100 | >100 |
| 3f (X = CH2OH, R1 = NH2, R2 = H) | >100 | >100 | >100 | 65.2 | >100 |
| 4a (X = H, R1 = OH, R2 = NH2) c | 1.47 | 6.34 | >100 | >100 | >100 |
| 4b (X = H, R1 = NHMe, R2 = NH2) | 14.3 | 17.6 | >100 | >100 | >100 |
| 4c (X = H, R1 = NH2, R2 = NH2) | 15.4 | 23.2 | >100 | >100 | >100 |
| 4d (X = CH2OH, R1 = OH, R2 = NH2) c | >100 | >100 | >100 | 53.1 | >100 |
| 4e (X = CH2OH, R1 = NHMe, R2 = NH2) | >100 | >100 | >100 | 32.1 | >100 |
| 4f (X = CH2OH, R1 = NH2, R2 = NH2) | >100 | >100 | >100 | 34.3 | >100 |
| 4g (X = CH2OH, R1 = NH-cyclopropyl, R2 = NH2) | >100 | >100 | >100 | 41.1 | >100 |
| 1 (Acyclovir) | 0.66 | 1.02 | 6.4 | 18.9 | >100 |
| 2 (Ganciclovir) | 0.90 | 1.40 | 11.1 | 2.14 | >100 |
a The effective concentration required to inhibit virus-induced cytopathic effect by 50%; b The cytotoxic concentration of the compound responsible for 50% reduction of cell viability; c Reference [11].
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sahu, P.K.; Umme, T.; Yu, J.; Kim, G.; Qu, S.; Naik, S.D.; Jeong, L.S. Structure-Activity Relationships of Acyclic Selenopurine Nucleosides as Antiviral Agents. Molecules 2017, 22, 1167. https://doi.org/10.3390/molecules22071167
AMA Style
Sahu PK, Umme T, Yu J, Kim G, Qu S, Naik SD, Jeong LS. Structure-Activity Relationships of Acyclic Selenopurine Nucleosides as Antiviral Agents. Molecules. 2017; 22(7):1167. https://doi.org/10.3390/molecules22071167
Chicago/Turabian StyleSahu, Pramod K., Tamima Umme, Jinha Yu, Gyudong Kim, Shuhao Qu, Siddhi D. Naik, and Lak Shin Jeong. 2017. "Structure-Activity Relationships of Acyclic Selenopurine Nucleosides as Antiviral Agents" Molecules 22, no. 7: 1167. https://doi.org/10.3390/molecules22071167