Application of An Improved HPLC-FL Method to Screen Serine Palmitoyl Transferase Inhibitors

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Optimization of Analytical Procedure
2.2.1. Derivatization Conditions
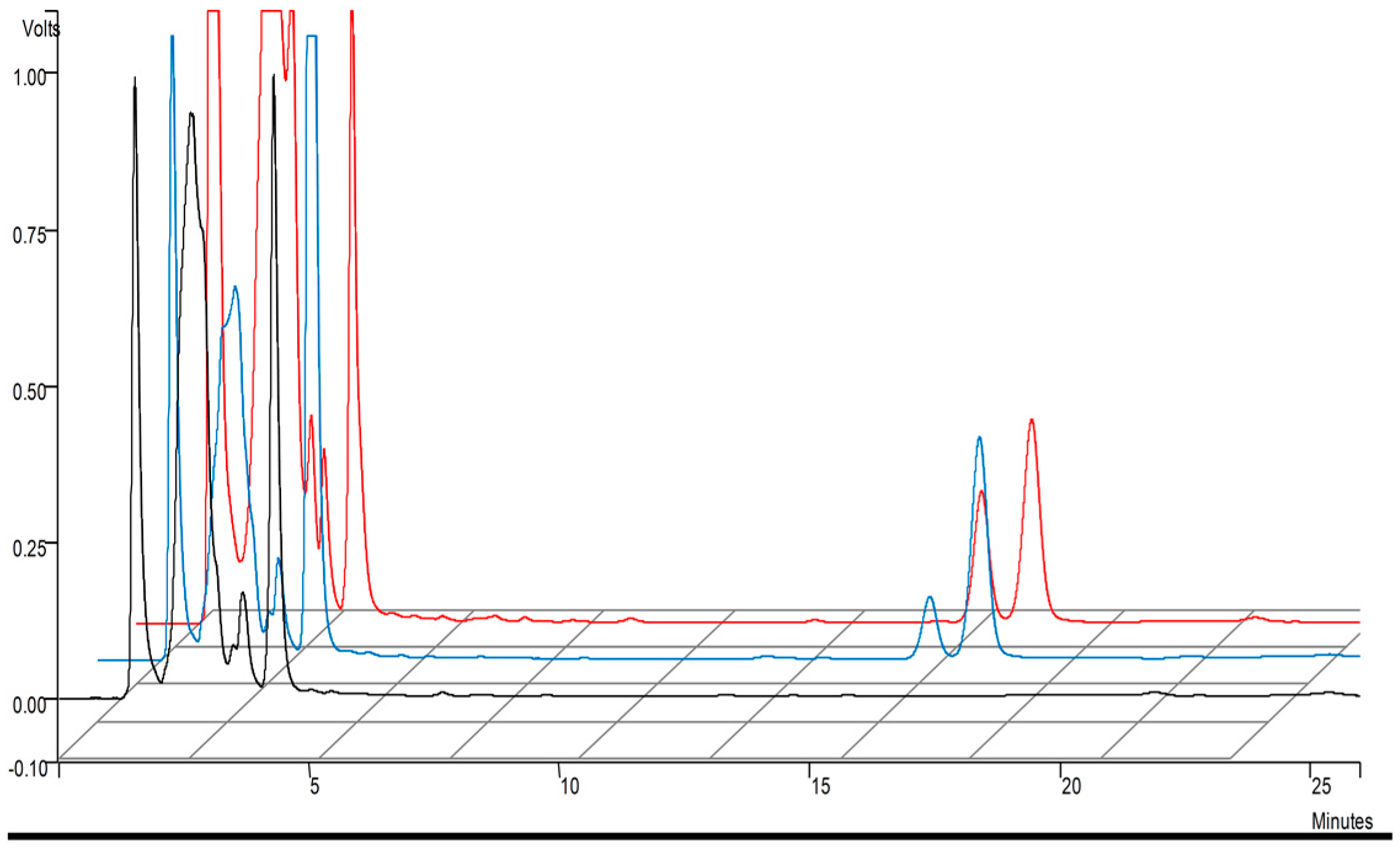
2.2.2. Chromatographic Conditions
2.2.3. Extraction Procedure
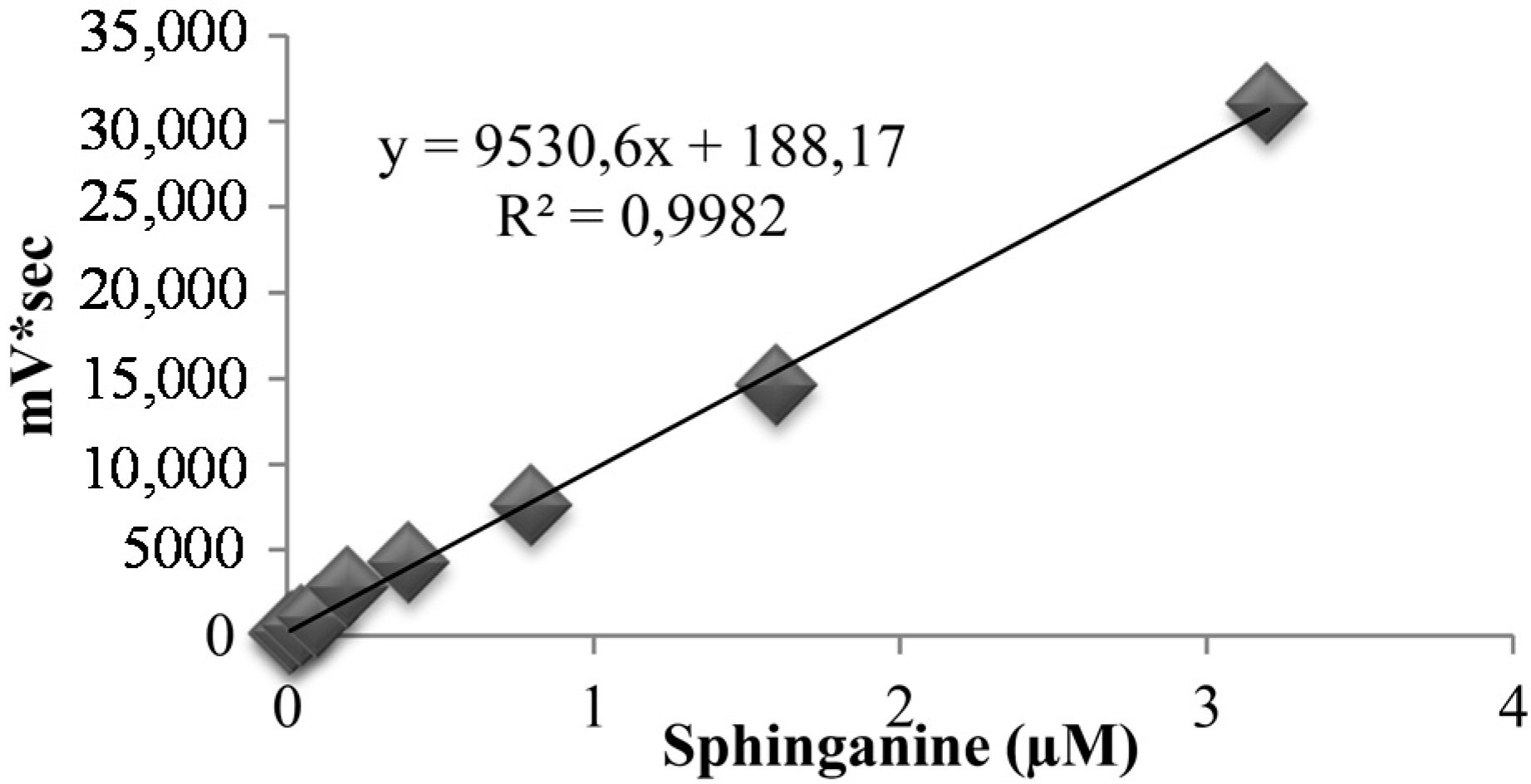
2.2.4. Method Validation
2.2.5. Incubation Period Evaluation
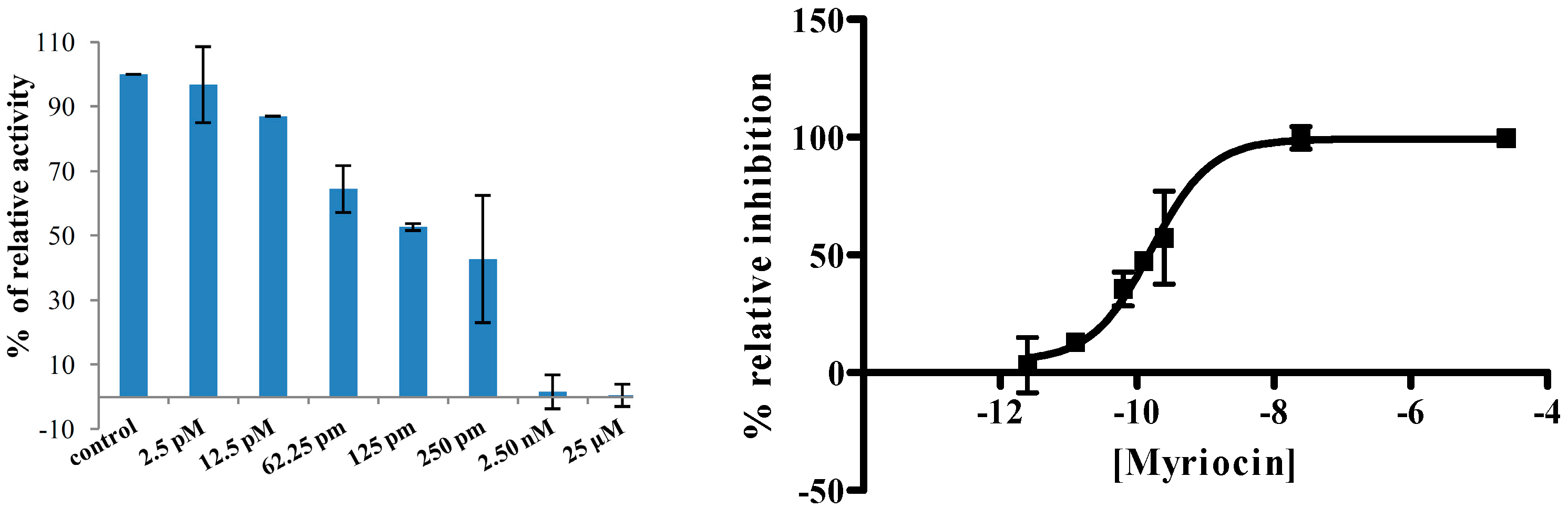
2.2.6. Screening of SPT Inhibitors
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. tert-Butyl 4-(2-nitro-4-cyanophenylamino)-piperidine-1-carboxylate (5)
3.1.2. tert-Butyl 4-(2-amino-4-cyanophenylamino)-1-piperidinecarboxylate (6)
3.1.3. tert-Butyl 4-(6-cyano-2-oxobenzimidazol-3-yl)piperidine-1-carboxylate (7)
3.1.4. 2,3-Dihydro-2-oxo-1-(piperidin-4-yl)-benzimidazole-5-carbonitrile (8)
3.1.5. {1-[2-(4-Chlorophenyl)-2-oxoethyl]piperidin-4-yl}-2,3-dihydro-2-oxo-benzimidazole-5-carbonitrile (2)
3.1.6. tert-Butyl-4-[2-oxo-5-(tetrazol-5-yl)-2,3-dihydro-benzimidazol-1-yl]piperidine-1-carboxylate (9)
3.1.7. 1-(Piperidin-4-yl)-5-(tetrazol-5-yl)-1H-benzimidazol-2-one (10)
3.1.8. {1-[2-(4-Chlorophenyl)-2-oxoethyl]piperidin-4-yl}-5-(2H-tetrazol-5-yl)-2,3-dihydro-benzimidazol-2-one (1)
3.2. Radiometric Inhibition Assay (RIA)
3.2.1. Materials
3.2.2. Radiometric Inhibition Assay (RIA) Procedure
3.3. HPLC-FL assay
3.3.1. Materials
3.3.2. HPLC-FL Assay Procedure
3.4. Cell Culture and SPT Cell Lysate Preparation
3.5. Incubation and Sample Extraction Procedure
3.6. Bioanalytical Method Validation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Biophys. Acta. 2003, 1632, 16–30. [Google Scholar] [CrossRef]
- Yard, B.A.; Carter, L.G.; Johnson, K.A.; Overton, I.M.; Dorward, M.; Liu, H.; McMahon, S.A.; Oke, M.; Puech, D.; Barton, G.J.; et al. The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J. Mol. Biol. 2007, 370, 870–886. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.C.C.; Johnson, K.A.; Yard, B.A.; Lowther, J.; Carter, L.G.; Naismith, J.H.; Campopiano, D.J. The external aldimine form of serine palmitoyltransferase: Structural, kinetic, and spectroscopic analysis of the wild-type enzyme and HSAN1 mutant mimics. J. Biol. Chem. 2009, 284, 17328–17339. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, C.; Martinez–Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef] [PubMed]
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef] [PubMed]
- Gable, K.; Gupta, S.D.; Han, G.; Niranjanakumari, S.; Harmon, J.M.; Dunn, T.M. A Disease-causing Mutation in the Active Site of Serine Palmitoyltransferase Causes Catalytic Promiscuity. J. Biol. Chem. 2010, 285, 22846–22852. [Google Scholar] [CrossRef] [PubMed]
- Penno, A.; Reilly, M.M.; Houlden, H.; Laurá, M.; Rentsch, K.; von Eckardstein, A.; Hornemann, T. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J. Biol. Chem. 2011, 285, 11178–11187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippov, V.; Song, M.A.; Zhang, K.H.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Duerksen-Hughes, P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimers. Dis. 2012, 29, 537–547. [Google Scholar] [PubMed]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid B, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef] [PubMed]
- Strettoi, E.; Gargini, C.; Novelli, E.; Sala, G.; Piano, I.; Gasco, P.; Ghidoni, R. Inhibition of ceramide biosynthesis preserves photoreceptor structure and function in a mouse model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2010, 107, 18706–18711. [Google Scholar] [CrossRef] [PubMed]
- Piano, I.; Novelli, E.; Gasco, P.; Ghidoni, R.; Strettoi, E.; Gargini, C. Cone survival and preservation of visual acuity in an animal model of retinal degeneration. Eur. J. Neurosci. 2013, 37, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Reforgiato, M.R.; Milano, G.; Fabriàs, G.; Casas, J.; Gasco, P.; Paroni, R.; Samaja, M.; Ghidoni, R.; Caretti, A.; Signorelli, P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res. Cardiol. 2016, 111, 12. [Google Scholar] [PubMed]
- Caretti, A.; Torelli, R.; Perdoni, F.; Falleni, M.; Tosi, D.; Zulueta, A.; Casas, J.; Sanguinetti, M.; Ghidoni, R.; Borghi, E.; et al. Inhibition of ceramide de novo synthesis by myriocin produces the double effect of reducing pathological inflammation and exerting antifungal activity against A. fumigatus airways infection. Biochim. Biophys. Acta 2016, 1860, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Caretti, A.; Bragonzi, A.; Facchini, M.; De Fino, I.; Riva, C.; Gasco, P.; Musicanti, C.; Casas, J.; Fabriàs, G.; Ghidoni, R.; et al. Anti-inflammatory action of lipid nanocarrier-delivered myriocin: Therapeutic potential in cystic fibrosis. Biochim. Biophys. Acta 2014, 1840, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, J.M.; Clarke, D.J.; McMahon, S.A.; Lowther, J.P.; Beattie, A.E.; Langridge-Smith, P.R.R.; Broughton, H.B.; Dunn, T.M.; Naismith, J.H.; Campopiano, D.J. The chemical basis of serine palmitoyltransferase inhibition by Myriocin. J. Am. Chem. Soc. 2013, 135, 14276–14285. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.D.; Wang, E.; Merrill, A.H. Enzymology of long-chain base synthesis by liver: Characterization of serine palmitoyltransferase in rat liver microsomes. Arch. Biochem. Biophys. 1984, 228, 282–291. [Google Scholar] [CrossRef]
- Rütti, M.F.; Richard, S.; Penno, A.; von Eckardstein, A.; Hornemann, T. An improved method to determine serine palmitoyltransferase activity. J. Lipid Res. 2009, 5, 1238–1244. [Google Scholar]
- Bolton, G.M.; Hutchings, R.H.; Kohrt, J.T.; Park, W.K.C.; Van Huis, C.A. Inhibitors of Serine Palmitoyltransferase. Int. Pat. EP2121656 A1, 25 November 2007. [Google Scholar]
- Amantini, D.; Beleggia, R.; Fringuelli, R.; Pizzo, F.; Vaccaro, L. TBAF-Catalyzed synthesis of 5-substituted 1H-tetrazoles under solventless conditions. J. Org. Chem. 2004, 69, 2896–2898. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H.; Wang, E.; Mullins, R.E.; Jamison, W.C.; Nimkar, S.; Liotta, D.C. Quantitation of free sphingosine in liver by high-performance liquid chromatography. Anal. Biochem. 1988, 171, 373–381. [Google Scholar] [CrossRef]
- European Medicines Agency; Committee for Medicinal Products for Human Use. Guideline on Bioanalytical Method Validation; European Medicines Agency: London, UK, 2011; EMEA/CHMP/EWP/192217/2009. [Google Scholar]
- Miyake, Y.; Kosutsumi, Y.; Nakamura, S.; Fujita, T.; Kawasaki, T. Serine palmitoyl transferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem. Biophys. Res. Commun. 1995, 211, 396–403. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 2 and 5–10 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QC Sample | Accuracy | Precision | |
|---|---|---|---|
| Within-Run | Between-Run | ||
| LOW (100 nM) | 9.92% | 9.1% | 13.3% |
| MEDIUM (800 nM) | 5.73% | 14.8% | 11.0% |
| HIGH (1600 nM) | 8.57% | 6.3% | 7.4% |
| Compound | % of Residual | SPT Activity |
|---|---|---|
| RIA | HPLC-FL | |
| 1 | 1.8 ± 0.8 | 1.3 ± 0.5 |
| 2 | 5.7 ± 2.1 | 4.1 ± 1.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertini, S.; Saccomanni, G.; Carlo, S.D.; Digiacomo, M.; Gargini, C.; Piano, I.; Campisi, G.M.; Ghidoni, R.; Macchia, M.; Manera, C. Application of An Improved HPLC-FL Method to Screen Serine Palmitoyl Transferase Inhibitors. Molecules 2017, 22, 1198. https://doi.org/10.3390/molecules22071198
Bertini S, Saccomanni G, Carlo SD, Digiacomo M, Gargini C, Piano I, Campisi GM, Ghidoni R, Macchia M, Manera C. Application of An Improved HPLC-FL Method to Screen Serine Palmitoyl Transferase Inhibitors. Molecules. 2017; 22(7):1198. https://doi.org/10.3390/molecules22071198
Chicago/Turabian StyleBertini, Simone, Giuseppe Saccomanni, Sara Del Carlo, Maria Digiacomo, Claudia Gargini, Ilaria Piano, Giuseppe Matteo Campisi, Riccardo Ghidoni, Marco Macchia, and Clementina Manera. 2017. "Application of An Improved HPLC-FL Method to Screen Serine Palmitoyl Transferase Inhibitors" Molecules 22, no. 7: 1198. https://doi.org/10.3390/molecules22071198