Synthesis and Evaluation of Novel Benzofuran Derivatives as Selective SIRT2 Inhibitors

 , ,
, ,
Abstract
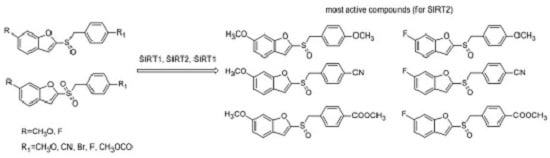
:
1. Introduction
2. Result and Discussion
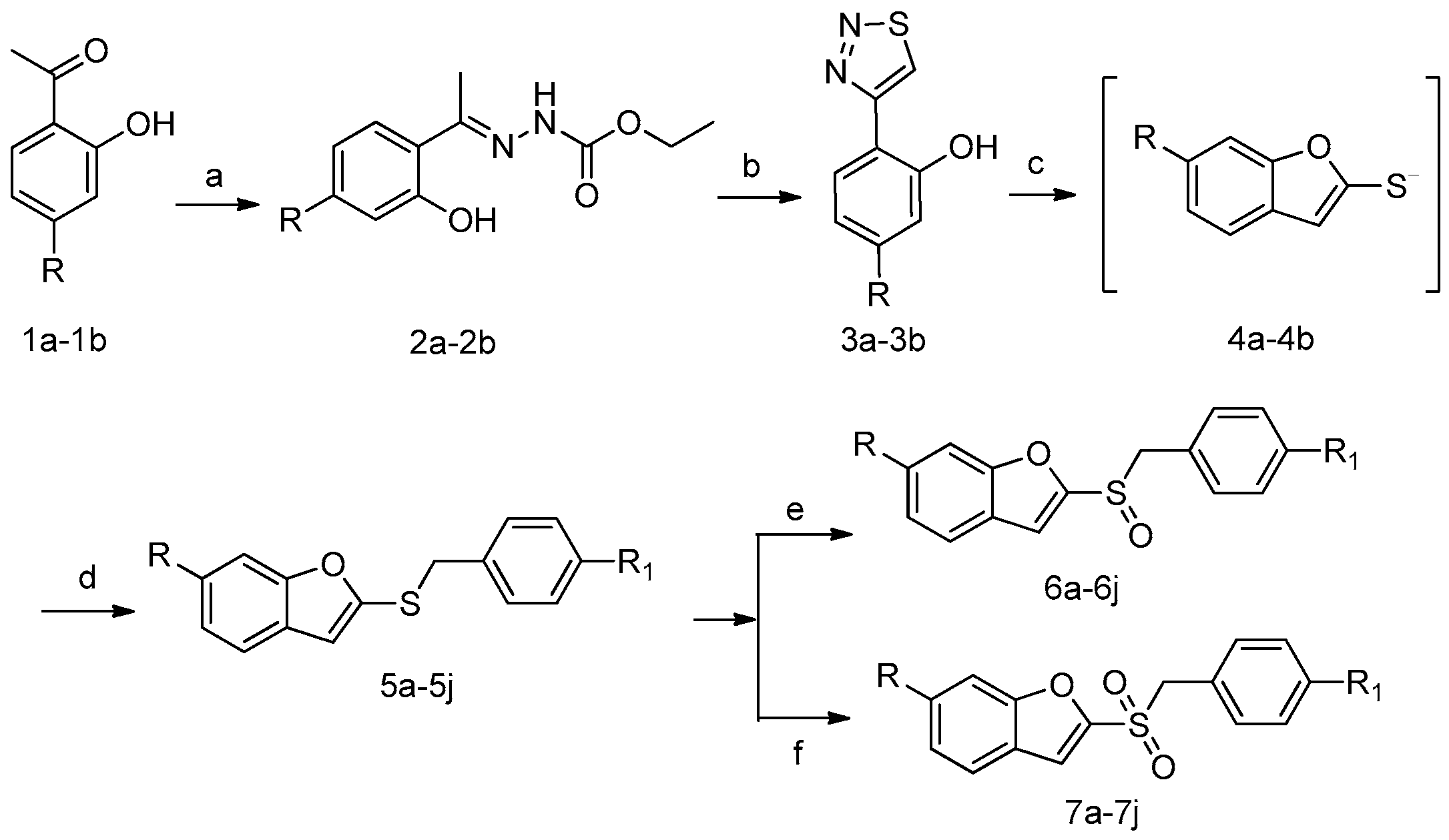
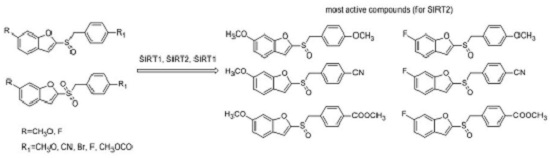
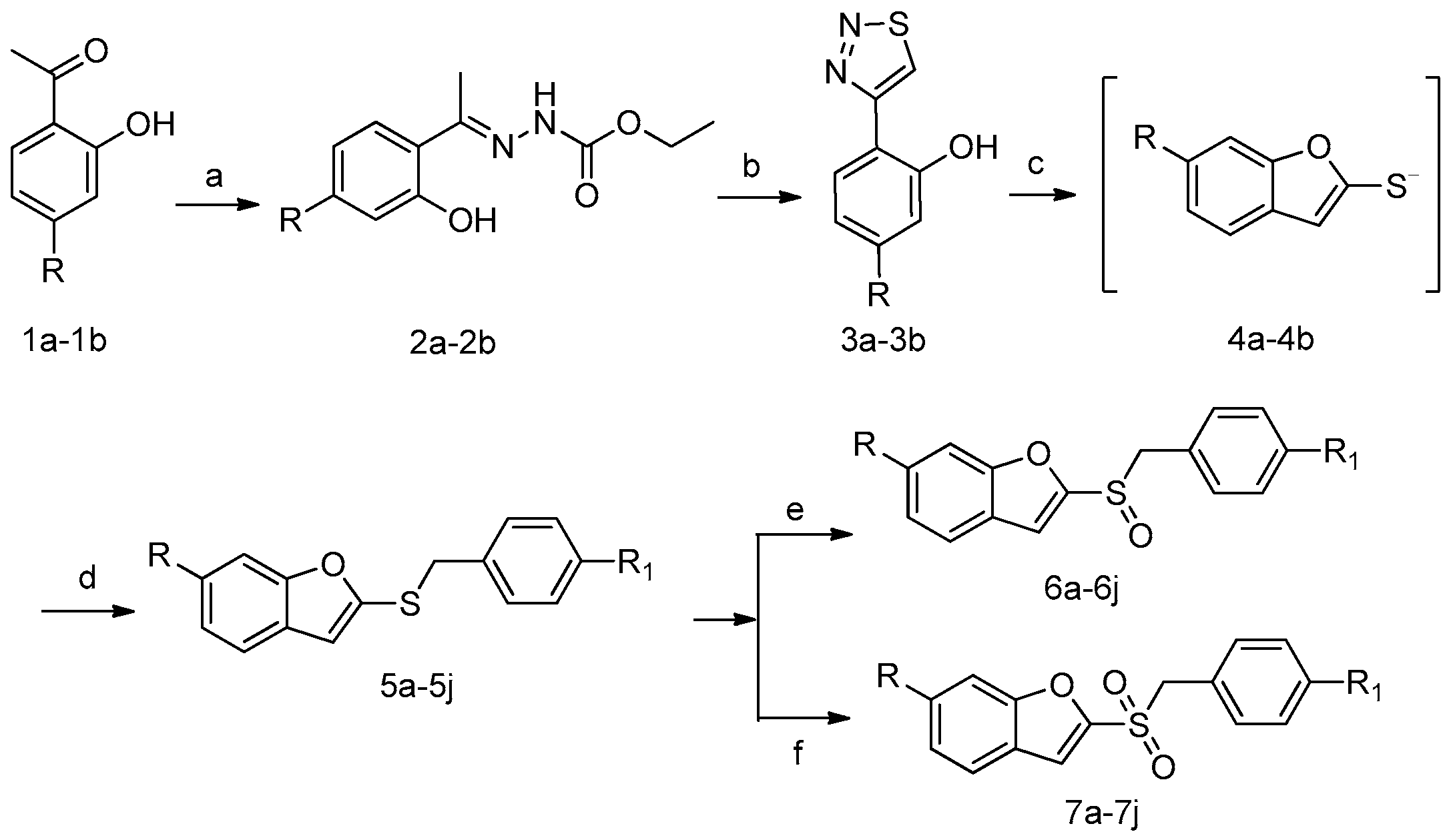
2.1. Chemistry
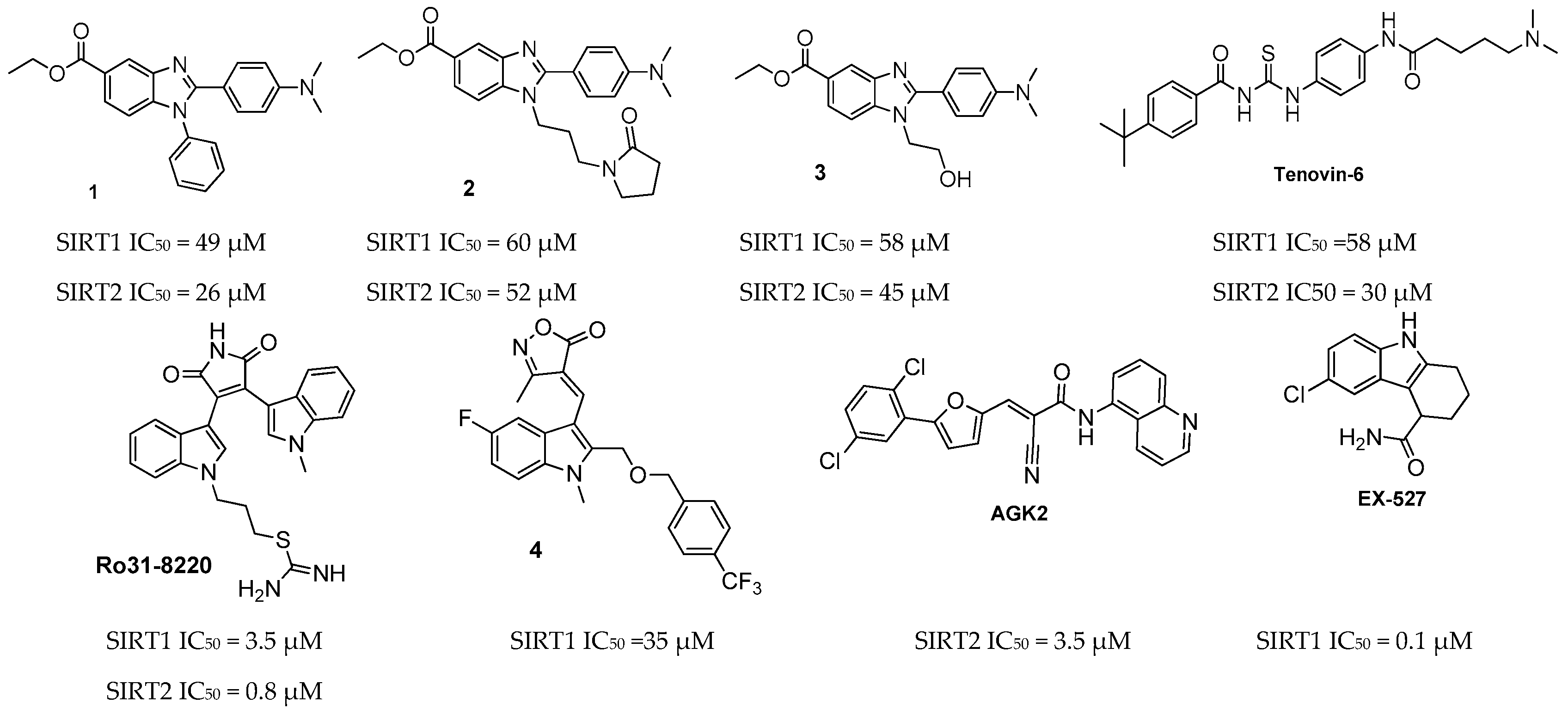
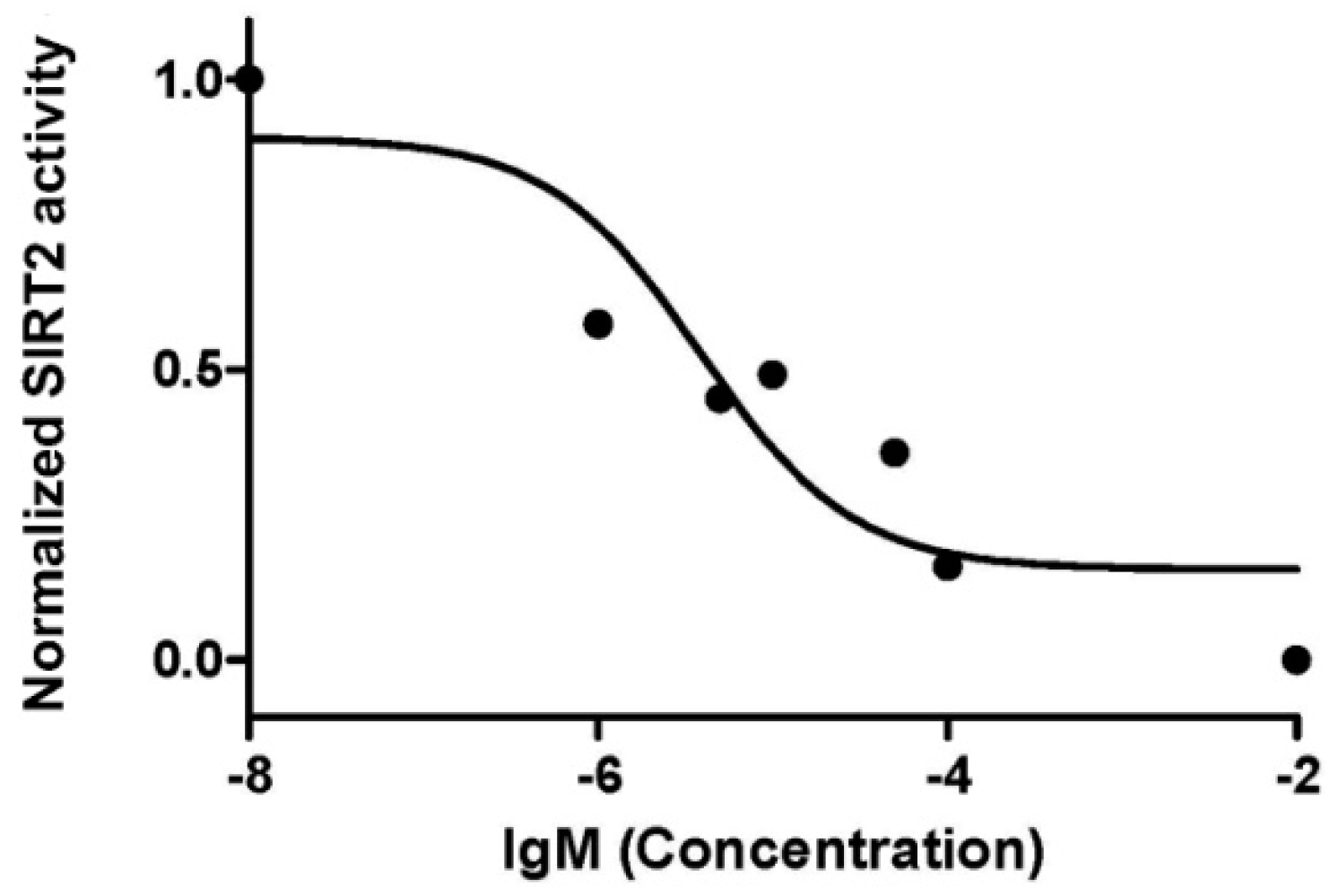
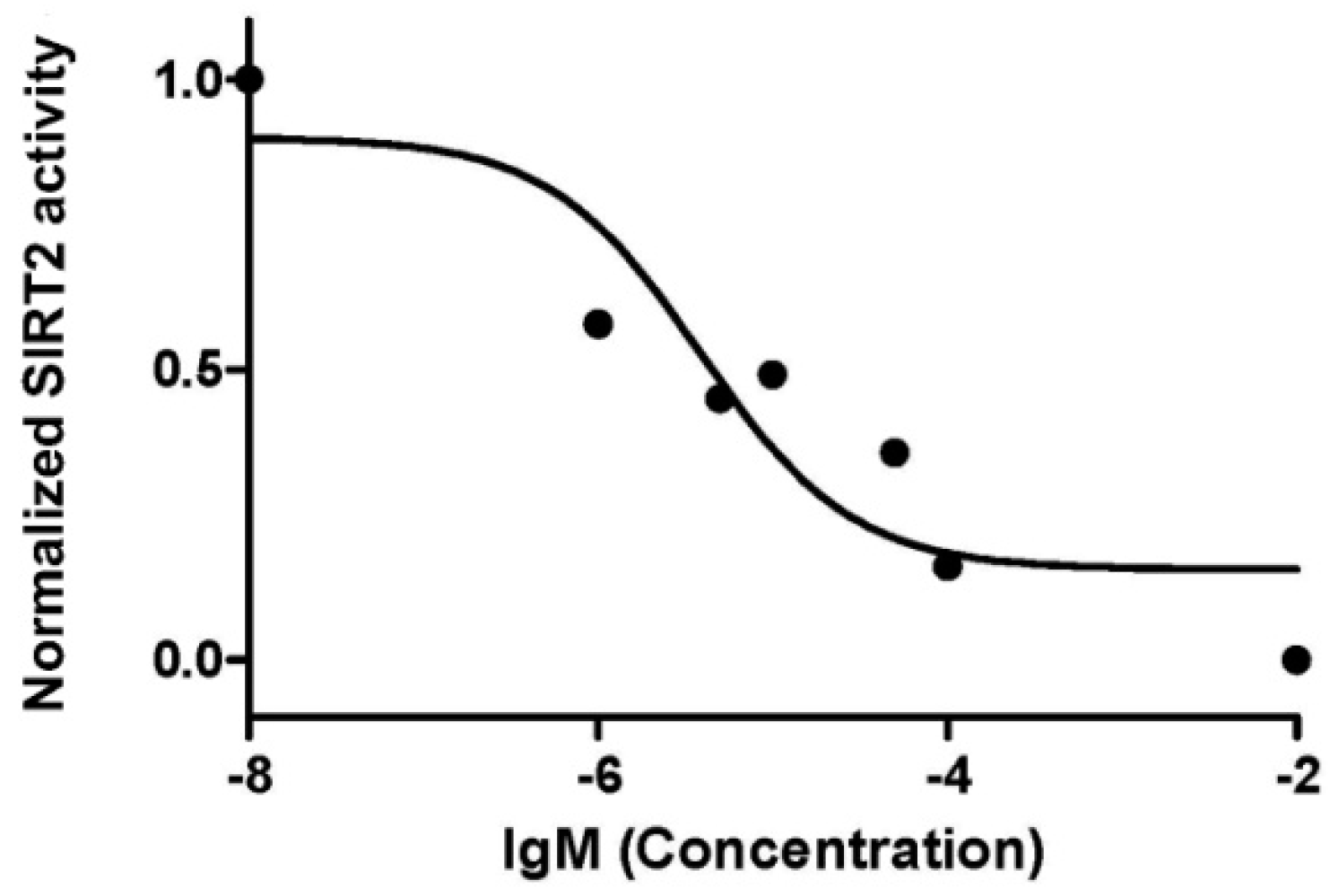
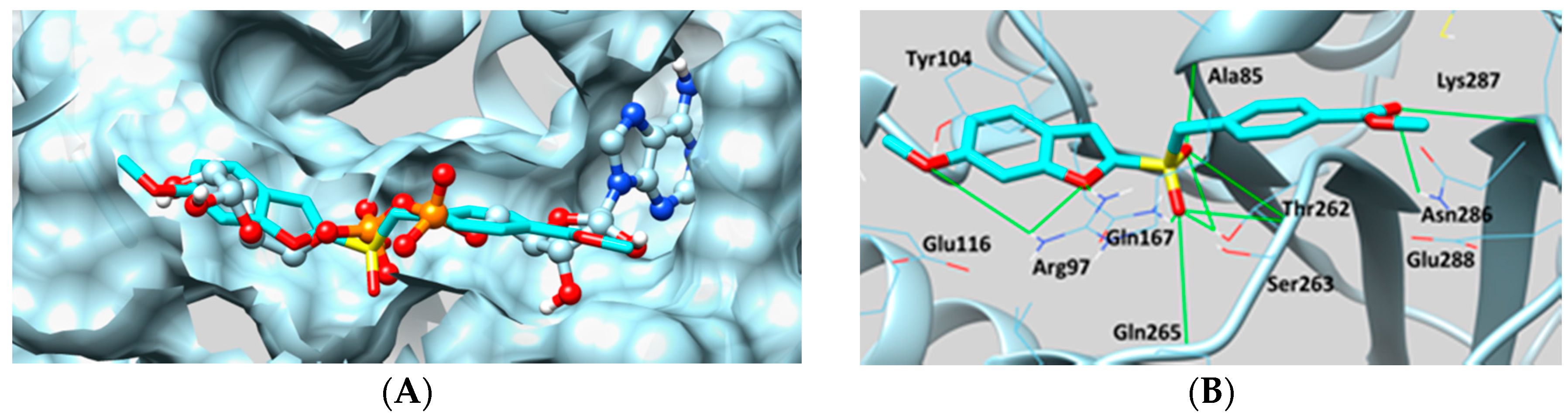
2.2. Sirtuin-Inhibitory Activity Evaluation
3. Experimental Section
3.1. Chemistry
3.2. Synthesis of Compounds 6a–6j
3.3. Synthesis of Compounds 7a–7j
3.4. Biological Evaluation
3.4.1. Expression and Purification of SIRT1−3
3.4.2. SIRT1-3 Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent histone deacetylase. Genome Biol. 2004, 5, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-likeproteins. Biochem. Biophys. Res. Commun. 2000, 273, 798. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulindeacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Beirowski, B.; Gustin, J.; Armour, S.M.; Yamamoto, H.; Viader, A.; North, B.J.; Michan, S.; Baloh, R.H.; Golden, J.P.; Schmidt, R.E.; et al. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.M.; Sarkander, J.; Kazantsev, A.G.; Outeiro, T.F. SIRT2 as a therapeutic target for age-relateddisorders. Front. Pharmacol. 2012, 3, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pais, T.F.; Szego, E.M.; Marques, O.; Miller-Fleming, L.; Antas, P.; Guerreiro, P.; de Oliveira, R.M.; Kasapoglu, B.; Outeiro, T.F. The NAD-dependent deacetylase sirtuin 2 is a suppressor ofmicroglial activation and brain inflammation. EMBO J. 2013, 32, 2603–2616. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Alam, H.B.; Liu, B.; Bronson, R.T.; Nikolian, V.C.; Wu, E.; Chong, W.; Li, Y. Selective inhibition of SIRT2 improves outcomes in a lethal septic model. Curr. Mol. Med. 2015, 15, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Eskandarian, H.A.; Impens, F.; Nahori, M.A.; Soubigou, G.; Coppee, J.Y.; Cossart, P.; Hamon, M.A. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 2013, 341, 1238858. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Zhu, Y.; Ozden, O.; Kim, H.S.; Jiang, H.; Deng, C.X.; Gius, D.; Vassilopoulos, A. SIRT2 is a tumor suppressor that connects aging, acetylome, cell cycle signaling, and carcinogenesis. Transl. Cancer Res. 2012, 1, 15–21. [Google Scholar] [PubMed]
- Yoon, Y.K.; Oon, C.E. Sirtuin inhibitors: An overview from medicinal chemistry perspective. Anticancer Agents Med. Chem. 2016, 16, 1–14. [Google Scholar]
- Yoon, Y.K.; Ali, M.A.; Wei, A.C.; Shirazi, A.N.; Parang, K.; Choon, T.S. Benzimidazoles as new scaffold of sirtuin inhibitors: Green synthesis, in vitro studies, molecular docking analysis and evaluation of their anti-cancer properties. Eur. J. Med. Chem. 2014, 83, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Panathur, N.; Gokhale, N.; Dalimba, U.; Koushik, P.V.; Yogeeswari, P.; Sriram, D. New indole-isoxazolone derivatives: Synthesis, characterization and in vitro SIRT1 inhibition studies. Bioorg. Med. Chem. Lett. 2015, 25, 2768–2772. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.K.; Ali, M.A.; Wei, A.C.; Choon, T.S.; Osman, H.; Parang, K.; Shirazi, A.N. Synthesis and evaluation of novel benzimidazole derivatives as sirtuin inhibitors with antitumor activities. Bioorg. Med. Chem. 2014, 22, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.; Hiremathad, A.; Singh, M.; Santos, M.A.; Keri, R.S. A review on antioxidant potential of bioactive heterocycle benzofuran: Natural and synthetic derivatives. Pharmacol. Rep. 2017, 69, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Petrov, M.L.; Teplyakov, F.S.; Androsov, D.A.; Yekhlef, M. New method of synthesis of benzo[b]furan-2-thiols from 4-(2-hydroxyaryl)-1,2,3-thiadiazoles. Russ. J. Org. Chem. 2009, 45, 1727–1728. [Google Scholar] [CrossRef]
- Petrov, M.L.; Iekhlev, M.; Teplyakov, F.S.; Androsova, D.A. Synthesis of Benzo[b]furan-2-thiolesfrom 4-(2-Hydroxyaryl)-1,2,3-thiadiazoles. Russ. J. Org. Chem. 2012, 48, 728–735. [Google Scholar] [CrossRef]
- Cui, H.Q.; Kamal, Z.; Ai, T.; Xu, Y.; More, S.S.; Chen, L. Discovery of potent and selective sirtuin 2 (SIRT2) inhibitors using a fragment-based approach. J. Med. Chem. 2014, 57, 8340–8357. [Google Scholar] [CrossRef] [PubMed]
- Moniot, S.; Schutkowski, M.; Steegborn, C. Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 2013, 182, 136–143. [Google Scholar] [CrossRef] [PubMed]
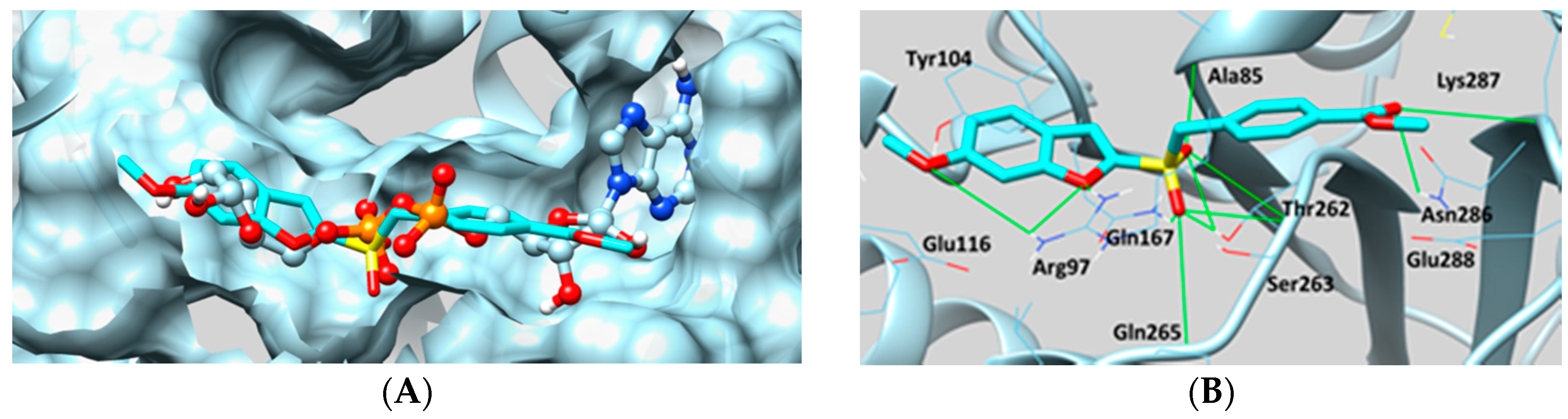
- Sakkiah, S.; Arooj, M.; Kumar, M.R.; Eom, S.H.; Lee, K.W. Identification of inhibitor binding site in human sirtuin 2 using molecular docking and dynamics simulations. PLoS ONE 2013, 8, e51429. [Google Scholar] [CrossRef] [PubMed]
- Tervo, A.J.; Kyrylenko, S.; Niskanen, P.; Salminen, A.; Leppänen, J.; Nyrönen, T.H.; Järvinen, T.; Poso, A. An insilico approach to discovering novel inhibitors of human sirtuin type 2. J. Med. Chem. 2004, 47, 6292–6298. [Google Scholar] [CrossRef] [PubMed]
- Sakkiah, S.; Thangapandian, S.; Park, C.; Son, M.; Lee, K.W. Molecular docking and dynamics simulation, receptor-based hypothesis: Application to identify novel sirtuin 2 inhibitors. Chem. Biol. Drug Des. 2012, 80, 315–327. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 6a–6j and 7a–7j are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | R | R1 | IC50 (µM) | ||
|---|---|---|---|---|---|
| SIRT1 | SIRT2 | SIRT3 | |||
| 6a | CH3O | CH3O | >100 | 13.52 | >100 |
| 6b | CH3O | CN | >100 | 15.14 | >100 |
| 6c | CH3O | Br | >100 | 85.14 | >100 |
| 6d | CH3O | F | >100 | 95.21 | >100 |
| 6e | CH3O | CH3OCO | >100 | 15.68 | >100 |
| 6f | F | CH3O | >100 | 27.89 | >100 |
| 6g | F | CN | >100 | 32.75 | >100 |
| 6h | F | Br | >100 | 74.93 | >100 |
| 6i | F | F | >100 | 88.07 | >100 |
| 6j | F | CH3OCO | >100 | 19.70 | >100 |
| 7a | CH3O | CH3O | >100 | 7.66 | >100 |
| 7b | CH3O | CN | >100 | 8.09 | >100 |
| 7c | CH3O | Br | >100 | 17.76 | >100 |
| 7d | CH3O | F | >100 | 43.93 | >100 |
| 7e | CH3O | CH3OCO | >100 | 3.81 | >100 |
| 7f | F | CH3O | >100 | 7.92 | >100 |
| 7g | F | CN | >100 | 8.85 | >100 |
| 7h | F | Br | >100 | 20.14 | >100 |
| 7i | F | F | >100 | 51.42 | >100 |
| 7j | F | CH3OCO | >100 | 6.16 | >100 |
| Tenovin6 | - | - | 37.50 | 15.32 | 89.31 |
| AGK2 | - | - | >100 | 1.56 | 52.80 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Cui, H.; Yu, X.; Peng, T.; Wang, G.; Wen, X.; Sun, Y.; Liu, S.; Zhang, S.; Hu, L.; et al. Synthesis and Evaluation of Novel Benzofuran Derivatives as Selective SIRT2 Inhibitors. Molecules 2017, 22, 1348. https://doi.org/10.3390/molecules22081348
Zhou Y, Cui H, Yu X, Peng T, Wang G, Wen X, Sun Y, Liu S, Zhang S, Hu L, et al. Synthesis and Evaluation of Novel Benzofuran Derivatives as Selective SIRT2 Inhibitors. Molecules. 2017; 22(8):1348. https://doi.org/10.3390/molecules22081348
Chicago/Turabian StyleZhou, Yumei, Huaqing Cui, Xiaoming Yu, Tao Peng, Gang Wang, Xiaoxue Wen, Yunbo Sun, Shuchen Liu, Shouguo Zhang, Liming Hu, and et al. 2017. "Synthesis and Evaluation of Novel Benzofuran Derivatives as Selective SIRT2 Inhibitors" Molecules 22, no. 8: 1348. https://doi.org/10.3390/molecules22081348