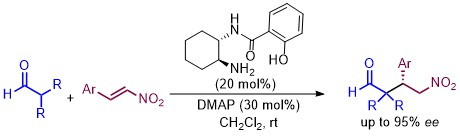
Asymmetric Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes Organocatalyzed by Chiral Monosalicylamides from trans-Cyclohexane-1,2-Diamines

 and
and 
Abstract

1. Introduction
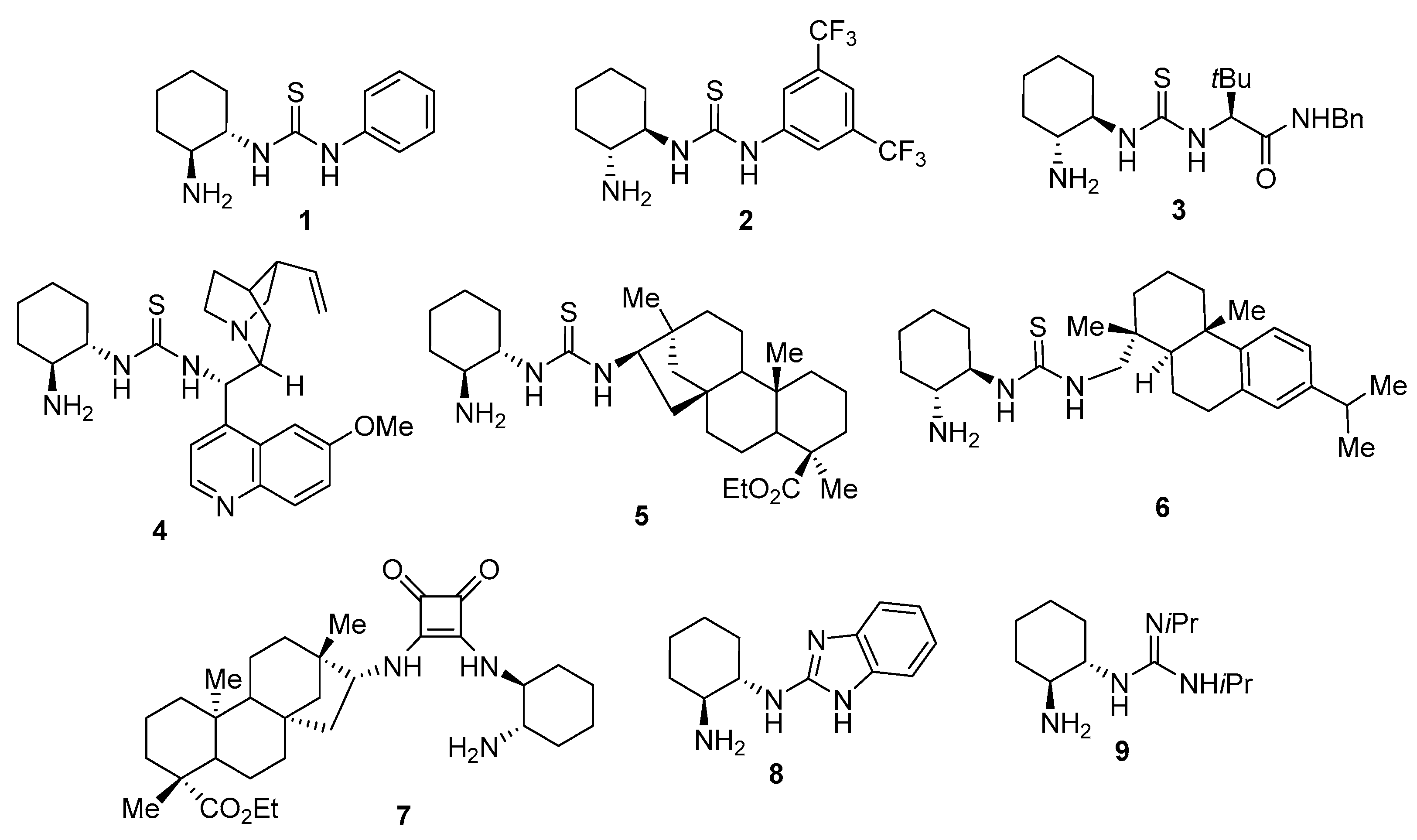
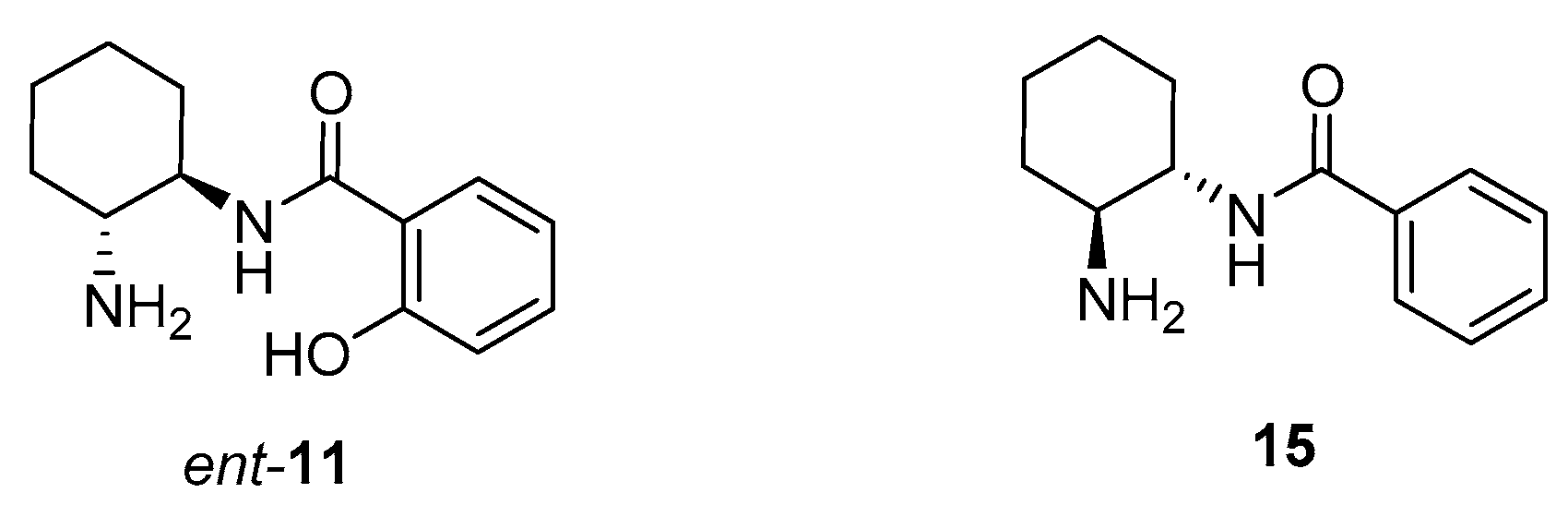
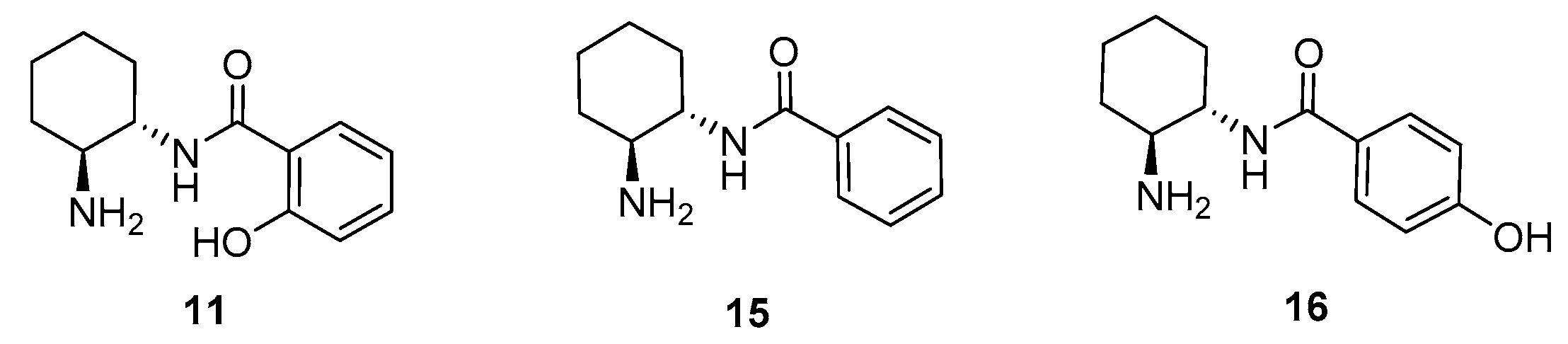
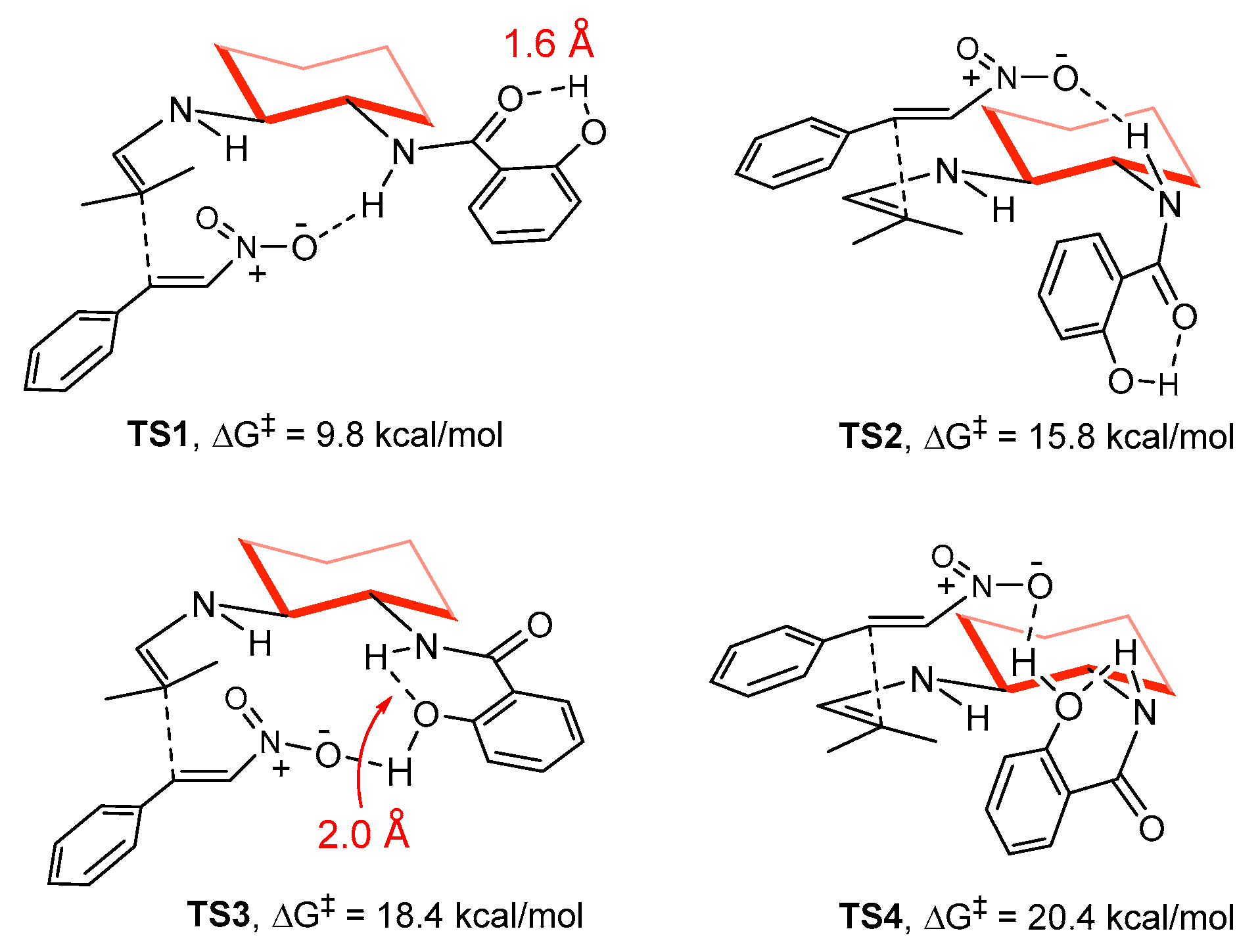
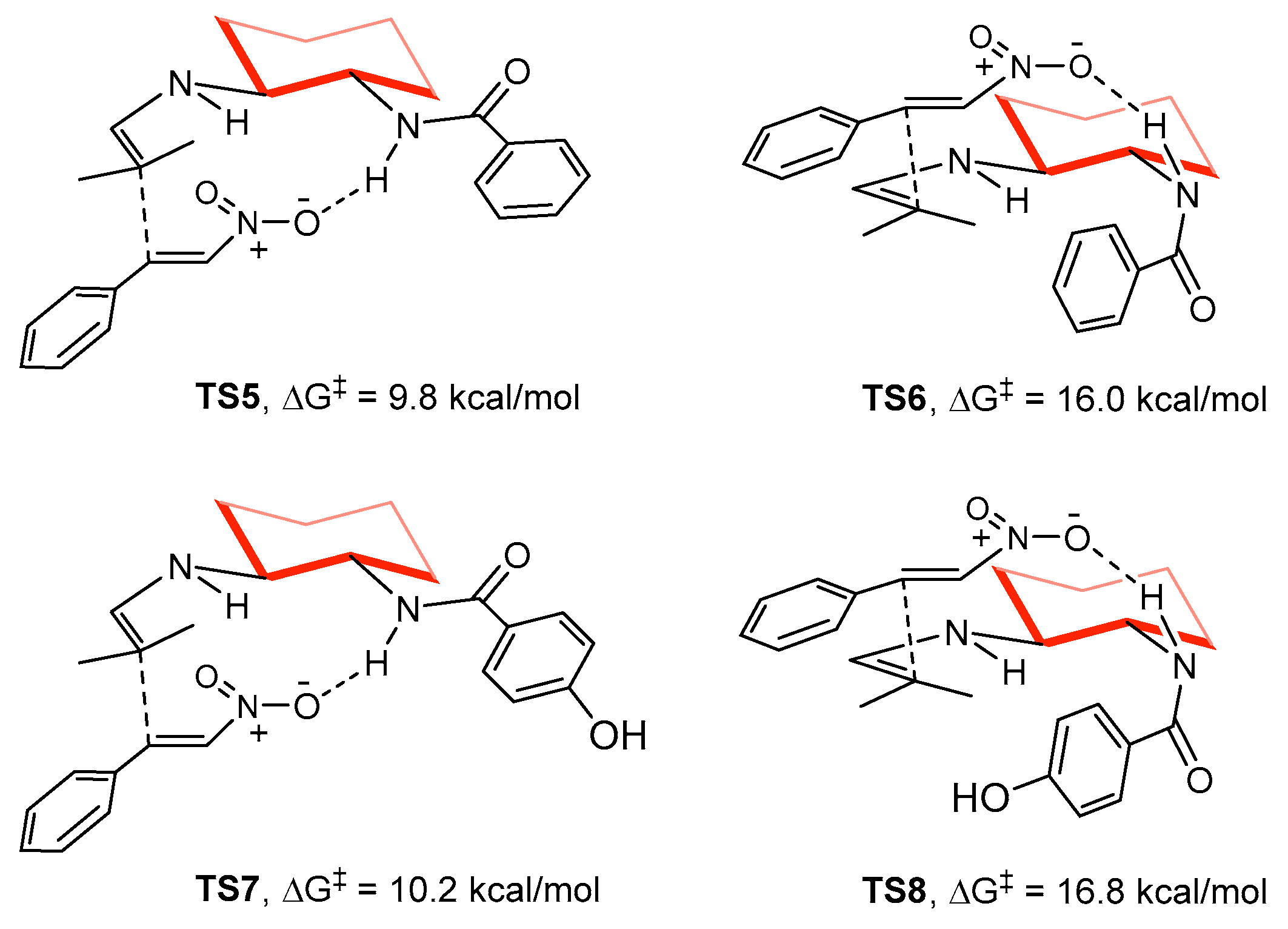
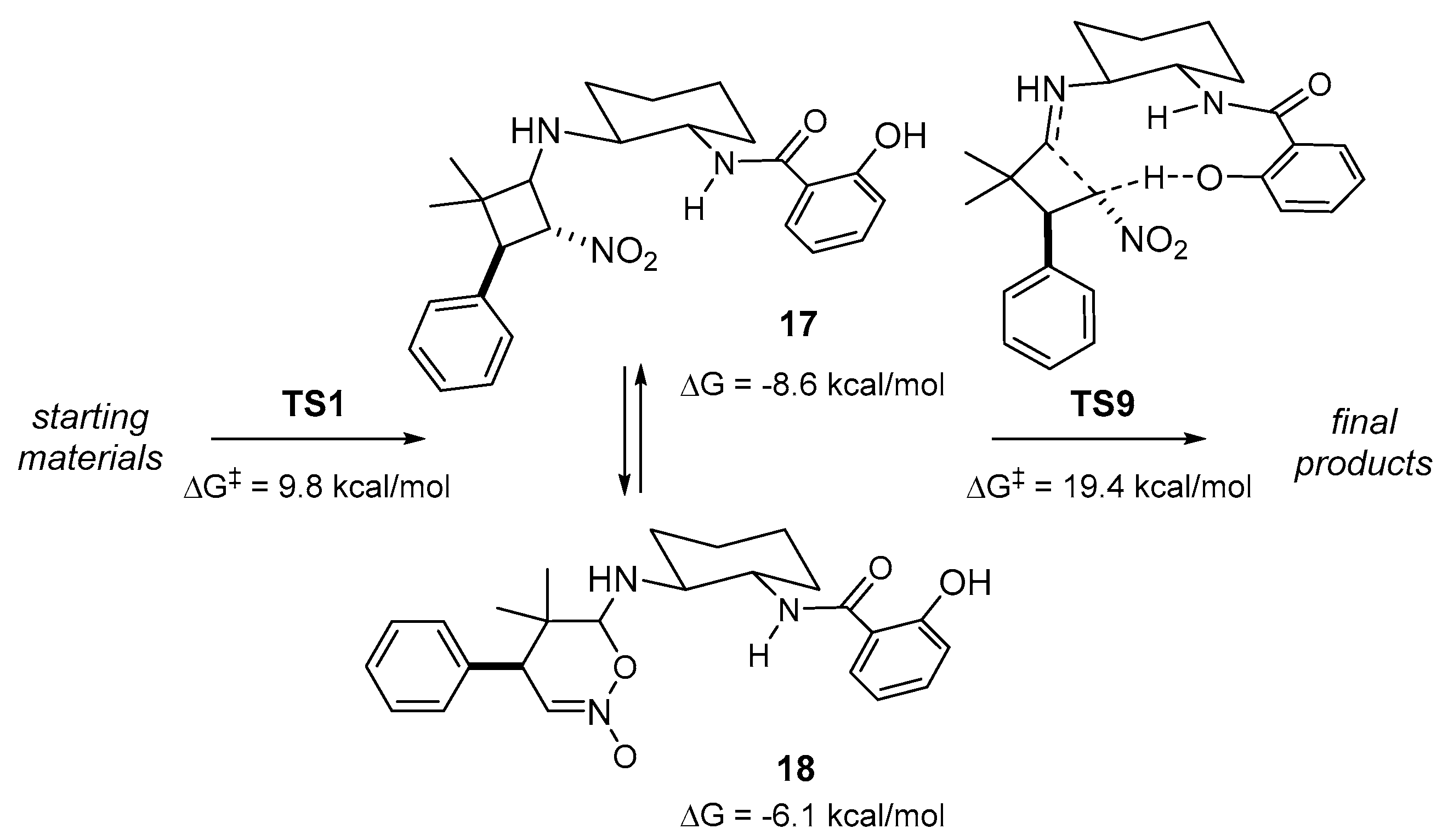
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Asymmetric Conjugate Addition Reaction
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aboul-Enein, M.N.; El-Azzouny, A.A.; Saleh, O.A.; Maklad, Y.A. On chemical structures with potent antiepileptic/anticonvulsant profile. Mini Rev. Med. Chem. 2012, 12, 671–700. [Google Scholar] [CrossRef] [PubMed]
- Andresen, H.; Aydin, B.E.; Mueller, A.; Iwersen-Bergmann, S. An overview of gamma-hydroxybutyric acid: Pharmacodynamics, pharmacokinetics, toxic effects, addiction, analytical methods, and interpretation of results. Drug Test. Anal. 2011, 3, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Gajcy, K.; Lochynski, S.; Librowski, T. A role of GABA analogues in the treatment of neurological diseases. Curr. Med. Chem. 2010, 17, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Ono, N. The Nitro Group in Organic Synthesis; John Wiley Sons, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Goksu, H.; Sert, H.; Kilbas, B.; Sen, F. Recent advances in the reduction of nitro compounds by heterogenous catalysts. Curr. Org. Chem. 2017, 21, 794–820. [Google Scholar] [CrossRef]
- Calderari, G.; Seebach, D. Asymmetric Michael additions. Stereoselective alkylation of chiral, non-racemic enolates by nitro olefins. Preparation of enantiomerically pure γ-aminobutyric and succinic acid derivatives. Helv. Chim. Acta 1985, 68, 1592–1604. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael additions to nitroalkenes. Eur. J. Org. Chem. 2002, 1877–1894. [Google Scholar] [CrossRef]
- Sulzer-Mosse, S.; Alexakis, A. Chiral amines as organocatalysts for asymmetric conjugate addition to nitroolefins and vinyl sulfones via enamine activation. Chem. Commun. 2007, 14, 3123–3135. [Google Scholar] [CrossRef] [PubMed]
- Roca-López, D.; Sadaba, D.; Delso, I.; Herrera, R.P.; Tejero, T.; Merino, P. Asymmetric organocatalytic synthesis of γ-nitrocarbonyl compounds through Michael and Domino reactions. Tetrahedron Asymmetry 2010, 21, 2561–2601. [Google Scholar] [CrossRef]
- Somanathan, R.; Chávez, D.; Servin, F.A.; Romero, J.A.; Navarrete, A.; Parra-Hake, M.; Aguirre, G.; Anaya, d.P.C.; González, J. Bifunctional organocatalysts in the asymmetric Michael additions of carbonylic compounds to nitroalkenes. Curr. Org. Chem. 2012, 16, 2440–2461. [Google Scholar] [CrossRef]
- Aitken, L.S.; Arezki, N.R.; Dell’Isola, A.; Cobb, A.J.A. Asymmetric organocatalysis and the nitro group functionality. Synthesis 2013, 45, 2627–2648. [Google Scholar]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Recent advances in asymmetric organocatalyzed conjugate additions to nitroalkenes. Molecules 2017, 22, 895. [Google Scholar] [CrossRef] [PubMed]
- Bennani, Y.L.; Hanessian, S. trans-1,2-Diaminocyclohexane derivatives as chiral reagents, scaffolds, and ligands for catalysis: Applications in asymmetric synthesis and molecular recognition. Chem. Rev. 1997, 97, 3161–3195. [Google Scholar] [CrossRef] [PubMed]
- Kouklovsky, C.; Langlois, Y.; Aguilar, E.; Fernández-García, J.M.; Sikervar, V. (1S,2S)-1,2-Diaminocyclohexane. In Encyclopedia of Reagents for Organic Synthesis; John Wiley Sons, Ltd.: New York, NY, USA, 2014. [Google Scholar]
- Zhang, X.-J.; Liu, S.-P.; Lao, J.-H.; Du, G.-J.; Yan, M.; Chan, A.S.C. Asymmetric conjugate addition of carbonyl compounds to nitroalkenes catalyzed by chiral bifunctional thioureas. Tetrahedron Asymmetry 2009, 20, 1451–1458. [Google Scholar] [CrossRef]
- Uehara, H.; Barbas, C.F., III. anti-Selective asymmetric Michael reactions of aldehydes and nitroolefins catalyzed by a primary amine/thiourea. Angew. Chem. Int. Ed. 2009, 48, 9848–9852. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, M.P.; Chen, Y.; Jacobsen, E.N. A chiral primary amine thiourea catalyst for the highly enantioselective direct conjugate addition of α,α-disubstituted aldehydes to nitroalkenes. Angew. Chem. Int. Ed. 2006, 45, 6366–6370. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Zou, Y.-Q.; Fu, L.; Ren, F.; Tan, F.; Xiao, W.-J. Highly enantioselective Michael addition of aldehydes to nitroolefins catalyzed by primary amine thiourea catalysts. Tetrahedron 2010, 66, 5367–5372. [Google Scholar] [CrossRef]
- Ma, Z.-W.; Liu, Y.-X.; Zhang, W.-J.; Tao, Y.; Zhu, Y.; Tao, J.-C.; Tang, M.-S. Highly enantioselective Michael additions of isobutyraldehyde to nitroalkenes promoted by amphiphilic bifunctional primary amine-thioureas in organic or aqueous medium. Eur. J. Org. Chem. 2011, 2011, 6747–6754. [Google Scholar] [CrossRef]
- Guo, X.-T.; Sha, F.; Wu, X.-Y. Highly enantioselective Michael addition of α,α-disubstituted aldehydes to nitroolefins. Res. Chem. Intermed. 2016, 42, 6373–6380. [Google Scholar] [CrossRef]
- Durmaz, M.; Sirit, A. Calixarene-based highly efficient primary amine-thiourea organocatalysts for asymmetric Michael addition of aldehydes to nitrostyrenes. Supramol. Chem. 2013, 25, 292–301. [Google Scholar] [CrossRef]
- Ma, Z.-W.; Liu, X.-F.; Sun, B.; Huang, X.-H.; Tao, J.-C. Chiral primary amine-squaramide catalyzed highly enantioselective Michael addition of isobutyraldehyde to nitroolefins. Synthesis 2017, 49, 1307–1314. [Google Scholar] [CrossRef]
- Fernandes, T.d.A.; Vizcaíno-Milla, P.; Ravasco, J.M.J.M.; Ortega-Martinez, A.; Sansano, J.M.; Nájera, C.; Costa, P.R.R.; Fiser, B.; Gómez-Bengoa, E. Bifunctional primary amine 2-aminobenzimidazole organocatalyst anchored to trans-cyclohexane-1,2-diamine in enantioselective conjugate additions of aldehydes. Tetrahedron Asymmetry 2016, 27, 118–122. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Fiser, B.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Michael addition of isobutyraldehyde to nitroalkenes organocatalyzed by chiral primary amine-guanidines. Tetrahedron Asymmetry 2014, 25, 462–467. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Chinchilla, R. Solvent-dependent enantioswitching in the Michael addition of α,α-disubstituted aldehydes to maleimides organocatalyzed by mono-N-Boc-protected cyclohexa-1,2-diamines. Tetrahedron Asymmetry 2014, 25, 1091–1094. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Fiser, B.; Gómez-Bengoa, E.; Chinchilla, R. Solvent-induced reversal of enantioselectivity in the synthesis of succinimides by the addition of aldehydes to maleimides catalysed by carbamate-monoprotected 1,2-diamines. Eur. J. Org. Chem. 2015, 1218–1225. [Google Scholar] [CrossRef]
- Flores-Ferrandiz, J.; Stiven, A.; Sotorrios, L.; Gómez-Bengoa, E.; Chinchilla, R. Enantioselective addition of aryl ketones and acetone to nitroalkenes organocatalyzed by carbamate-monoprotected cyclohexa-1,2-diamines. Tetrahedron Asymmetry 2015, 26, 970–979. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Chinchilla, R. Organocatalytic enantioselective conjugate addition of aldehydes to maleimides in deep eutectic solvents. Tetrahedron Asymmetry 2017, 28, 302–306. [Google Scholar] [CrossRef]
- Costes, J.-P.; Duhayon, C.; Mallet-Ladeira, S.; Shova, S.; Vendier, L. Does the sign of the Cu-Gd magnetic interaction depend on the number of atoms in the bridge? Chem. Eur. J. 2016, 22, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D.; Golinski, J. Synthesis of open chain 2,3-disubstituted 4-nitroketone by diastereoselective Michael addition of (E)-enamines to (E)-nitroolefins. A topological rule for C,C-bond forming processes between prochiral centers. Helv. Chim. Acta 1981, 64, 1413–1423. [Google Scholar] [CrossRef]
- Seebach, D.; Beck, A.K.; Golinski, J.; Hay, J.N.; Laube, T. Über den sterischen Verlauf von Enaminen aus offenkettigen Aldehyden und Ketonen mit Nitroolefinen zu 2,3-disubstituierten 4-Nitroketonen. Helv. Chim. Acta 1985, 68, 162–172. [Google Scholar] [CrossRef]
- Földes, T.; Madarász, A.; Révész, Á.; Dobi, Z.; Varga, S.; Hamza, A.; Nagy, P.R.; Pihko, P.M.; Pápai, I. Stereocontrol in diphenylprolinol silyl ether catalyzed Michael additions: Steric shielding or Curtin-Hammett scenario? J. Am. Chem. Soc. 2017, 139, 17052–17063. [Google Scholar] [CrossRef] [PubMed]
- Burés, J.; Armstrong, A.; Blackmond, D. Explaining anomalies in enamine catalysis: “Downstream species” as a new paradigm for stereocontrol. Acc. Chem. Res. 2016, 49, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, G.; Rahaman, H.; Madarász, A.; Pápai, I.; Melarto, M.; Valkonen, A.; Pihko, P.M. Dihydrooxazine oxides as key Intermediates in organocatalytic Michael additions of aldehydes to nitroalkenes. Angew. Chem. Int. Ed. 2012, 51, 13144–13148. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Pujol, M.D. Straightforward synthesis of nitroolefins by microwave- or ultrasound-assisted Henry reaction. Tetrahedron Lett. 2011, 52, 2629–2632. [Google Scholar] [CrossRef]
- Kuster, G.J.T.; Steeghs, R.H.J.; Scheeren, H.W. Novel five/five- and six/five-membered bicyclic nitroso acetals from high-pressure-promoted cyclization reactions of p-methoxybenzyl vinyl ether, 1-nitro-2-heteroaryl ethenes, and mono- and di-substituted olefins. Eur. J. Org. Chem. 2001, 553–560. [Google Scholar] [CrossRef]
- Bai, J.-F.; Xu, X.-Y.; Huang, Q.-C.; Peng, L.; Wang, L.-X. Highly asymmetric Michael additions of α,α-disubstituted aldehydes to β-nitroalkenes promoted by chiral pyrrolidine-thiourea bifunctional catalysts. Tetrahedron Lett. 2010, 51, 2803–2805. [Google Scholar] [CrossRef]
- Porta, R.; Coccia, F.; Annunziata, R.; Puglisi, A. Comparison of different polymer- and silica-supported 9-amino-9-deoxy-epi-quinines as recyclable organocatalysts. ChemCatChem 2015, 7, 1490–1499. [Google Scholar] [CrossRef]
- Rasappan, R.; Reiser, O. Cyclohexane-1,2-diamines: Efficient catalysts for the enantioselective conjugate addition of ketones to nitro olefins. Eur. J. Org. Chem. 2009, 1305–1308. [Google Scholar] [CrossRef]
- Ting, Y.-F.; Chang, C.; Reddy, R.J.; Magar, D.R.; Chen, K. Pyrrolidinyl-camphor derivatives as a new class of organocatalyst for direct asymmetric Michael addition of aldehydes and ketones to β-nitroalkenes. Chem. Eur. J. 2010, 16, 7030–7038. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functional and systematic testing of four M06-class functional and 12 other functional. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3047. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Mennuci, B.; Tomasi, J. Ab initio study of ionic solutions by a polarizable continuum model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF versión of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. (Theochem) 1999, 464, 211–226. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 11 and ent-11 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst (mol %) | Additive (mol %) a | Solvent | t (d) | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|---|
| 1 | 11 (20) | - | PhMe | 2 | 10 d | 79 (S) |
| 2 | 11 (20) | - | DMF | 2 | 26 | 9 (S) |
| 3 | 11 (20) | - | CH2Cl2 | 2 | 10 d | 84 (S) |
| 4 | 11 (20) | DMAP (20) | CH2Cl2 | 2 | 81 | 92 (S) |
| 5 | 11 (20) | Imidazole (20) | CH2Cl2 | 2 | 10 d | 76 (S) |
| 6 | 11 (20) | Pyridine (20) | CH2Cl2 | 2 | 10 d | 81 (S) |
| 7 | 11 (20) | TMG (20) | CH2Cl2 | 2 | 43 | 91 (S) |
| 8 | 11 (20) | DBU (20) | CH2Cl2 | 2 | 31 | 38 (S) |
| 9 | 11 (20) | DABCO (20) | CH2Cl2 | 2 | 47 | 93 (S) |
| 10 | 11 (20) | PhCO2H (20) | CH2Cl2 | 2 | 10 d | 71 (S) |
| 11 | 11 (20) | 4-O2NC6H4CO2H (20) | CH2Cl2 | 2 | 10 d | 78 (S) |
| 12 | 11 (20) | 3,4-(MeO)2C6H3CO2H (20) | CH2Cl2 | 2 | 10 d | 75 (S) |
| 13 | 11 (10) | DMAP (20) | CH2Cl2 | 2 | 17 | 94 (S) |
| 14 | 11 (20) | DMAP (10) | CH2Cl2 | 2 | 41 | 94 (S) |
| 15 | 11 (20) | DMAP (30) | CH2Cl2 | 2 | 72 | 95 (S) |
| 16 | ent-11 (20) | DMAP (30) | CH2Cl2 | 2 | 74 | 95 (R) |
| 17 | 15 (20) | DMAP (30) | CH2Cl2 | 3 | 40 | 79 (S) |
| 18 | 10 (20) | DMAP (30) | CH2Cl2 | 3 | 30 | 65 (S) |

| Entry | Aldehyde | β-Nitroalkene | t (d) | γ-Nitroaldehyde | ||||
|---|---|---|---|---|---|---|---|---|
| R1,R2 | No. | R3 | No. | No. | Yield (%) a | eeb (%) b | ||
| 1 | Me,Me | 12a | Ph | 13a | 2 | (S)-14aa | 72 | 95 |
| 2 | Me,Me | 12a | 4-MeC6H4 | 13b | 2 | (S)-14ab | 67 | 92 |
| 3 | Me,Me | 12a | 4-MeOC6H4 | 13c | 2 | (S)-14ac | 91 | 92 |
| 4 | Me,Me | 12a | 3,4-(OCH2O)C6H3 | 13d | 2 | (S)-14ad | 64 | 85 |
| 5 | Me,Me | 12a | 3,4,5-(MeO)3C6H2 | 13e | 2 | (S)-14ae | 85 | 94 |
| 6 | Me,Me | 12a | 4-FC6H4 | 13f | 2 | (S)-14af | 62 | 92 |
| 7 | Me,Me | 12a | 2-ClC6H4 | 13g | 2 | (R)-14ag c | 50 | 87 |
| 8 | Me,Me | 12a | 4-ClC6H4 | 13h | 2 | (S)-14ah | 70 | 88 |
| 9 | Me,Me | 12a | 4-BrC6H4 | 13i | 2 | (S)-14ai | 50 | 94 |
| 10 | Me,Me | 12a | 4-F3CC6H4 | 13j | 2 | (S)-14aj | 51 | 93 |
| 11 | Me,Me | 12a | 2-Naphthyl | 13k | 2 | (S)-14ak | 68 | 91 |
| 12 | Me,Me | 12a | 3-Pyridinyl | 13l | 2 | (S)-14al | 43 | 91 |
| 13 | Me,Me | 12a | 2-Furanyl | 13m | 2 | (S)-14am | 82 | 92 |
| 14 | -(CH2)4- | 12b | Ph | 13a | 3 | (S)-14ba | 60 | 94 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Guillén, J.R.; Flores-Ferrándiz, J.; Gómez, C.; Gómez-Bengoa, E.; Chinchilla, R. Asymmetric Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes Organocatalyzed by Chiral Monosalicylamides from trans-Cyclohexane-1,2-Diamines. Molecules 2018, 23, 141. https://doi.org/10.3390/molecules23010141
Martínez-Guillén JR, Flores-Ferrándiz J, Gómez C, Gómez-Bengoa E, Chinchilla R. Asymmetric Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes Organocatalyzed by Chiral Monosalicylamides from trans-Cyclohexane-1,2-Diamines. Molecules. 2018; 23(1):141. https://doi.org/10.3390/molecules23010141
Chicago/Turabian StyleMartínez-Guillén, José R., Jesús Flores-Ferrándiz, Cecilia Gómez, Enrique Gómez-Bengoa, and Rafael Chinchilla. 2018. "Asymmetric Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes Organocatalyzed by Chiral Monosalicylamides from trans-Cyclohexane-1,2-Diamines" Molecules 23, no. 1: 141. https://doi.org/10.3390/molecules23010141
APA StyleMartínez-Guillén, J. R., Flores-Ferrándiz, J., Gómez, C., Gómez-Bengoa, E., & Chinchilla, R. (2018). Asymmetric Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes Organocatalyzed by Chiral Monosalicylamides from trans-Cyclohexane-1,2-Diamines. Molecules, 23(1), 141. https://doi.org/10.3390/molecules23010141