Multinuclear NMR Measurements and DFT Calculations for Capecitabine Tautomeric Form Assignment in a Solution


Abstract
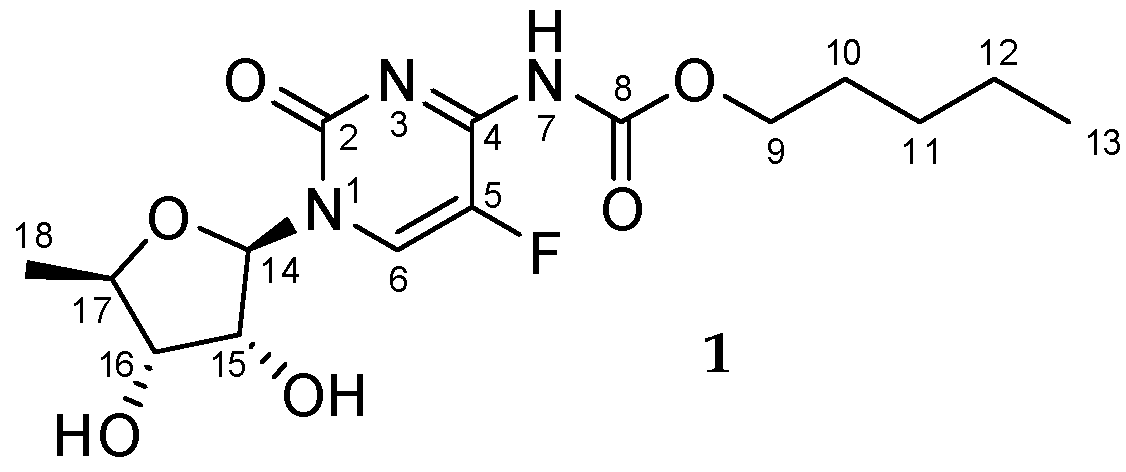
:
1. Introduction
2. Results and Discussion
2.1. Multinuclear MR Measurements in the DMSO and THF Solutions
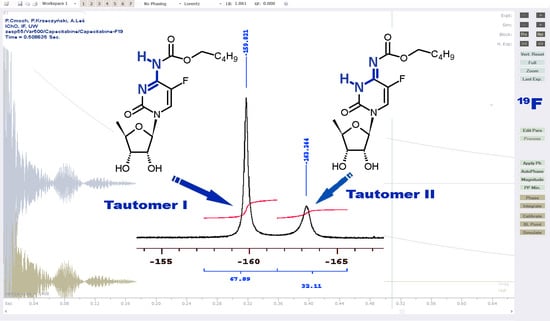
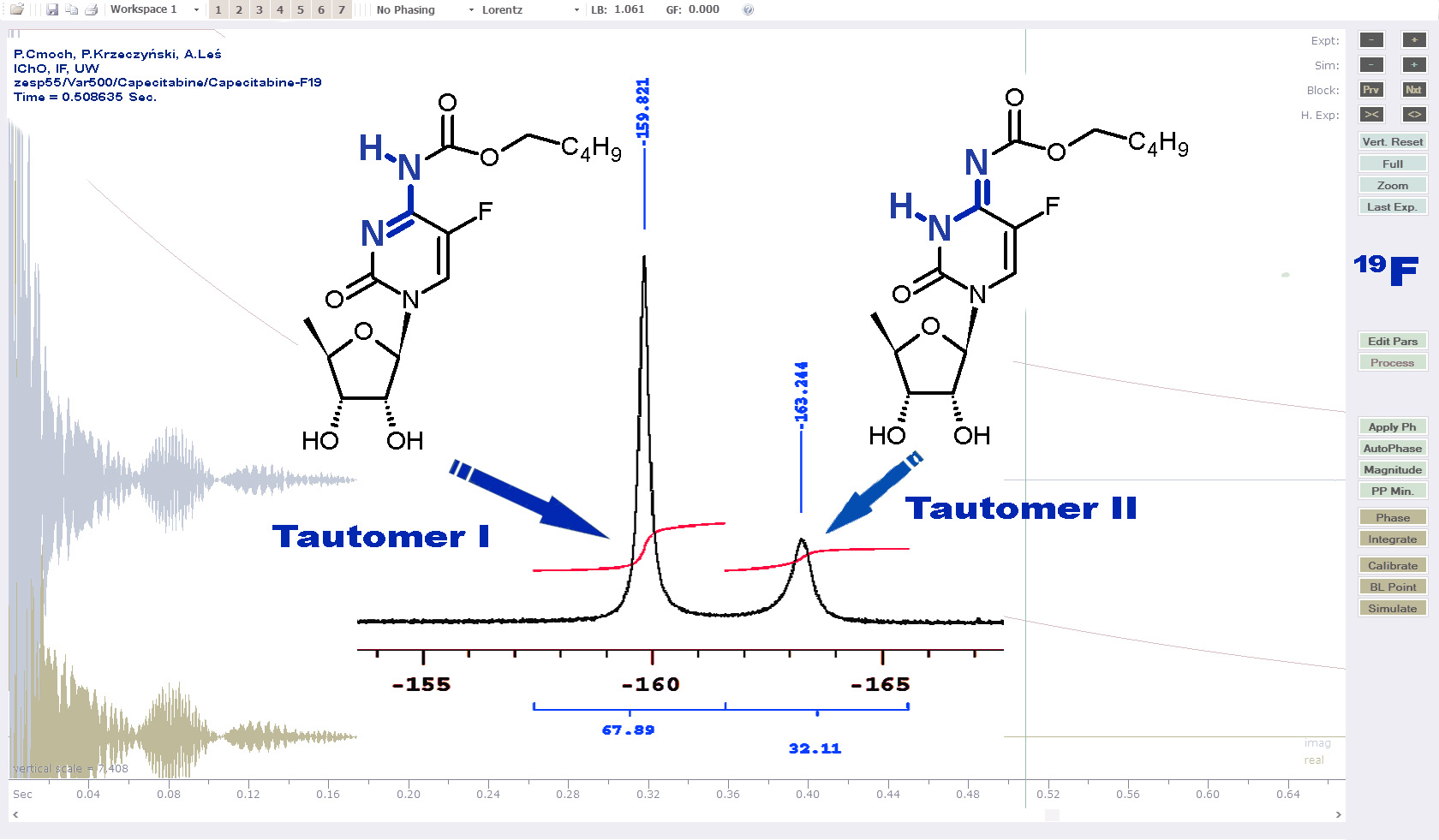
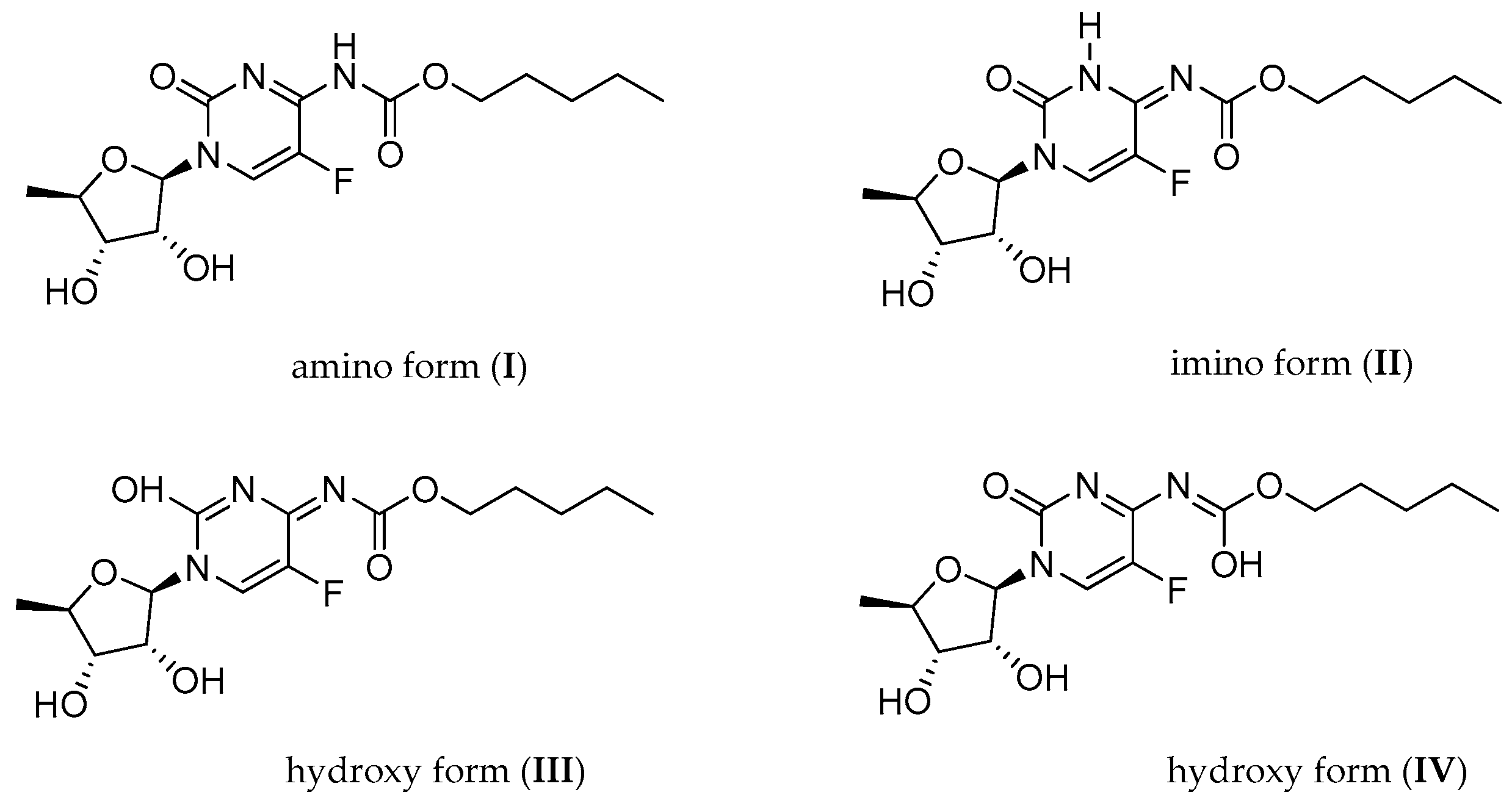
2.1.1. Temperature 298 K: Tautomers A and B Are Detected
2.1.2. Low-Temperature 1H, 19F Spectra: Tautomer A Becomes More Abundant
2.1.3. Methyl Derivatives and Identification of A/B Tautomers
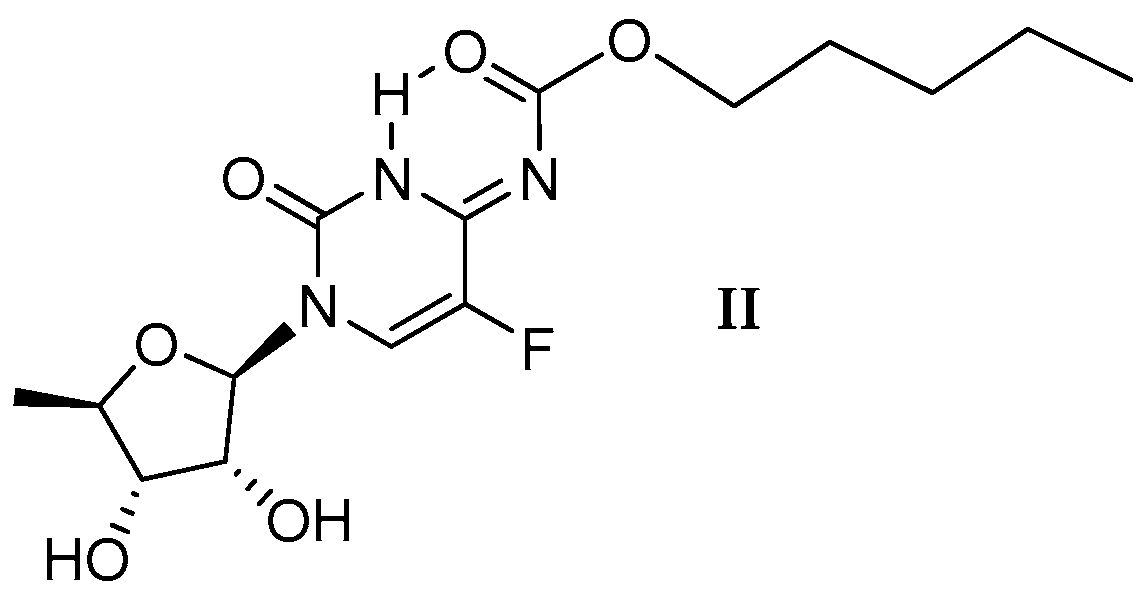
2.1.4. Capecitabine in THF: Form I Dominates over Form II
2.1.5. Capecitabine in the Aqueous Solution: Form I Is Exclusively Detected
2.1.6. Capecitabine in an Acidic Medium: Form I Is Protonated at the N3 Atom
2.2. Quantum Mechanical DFT Calculations
3. Materials and Methods
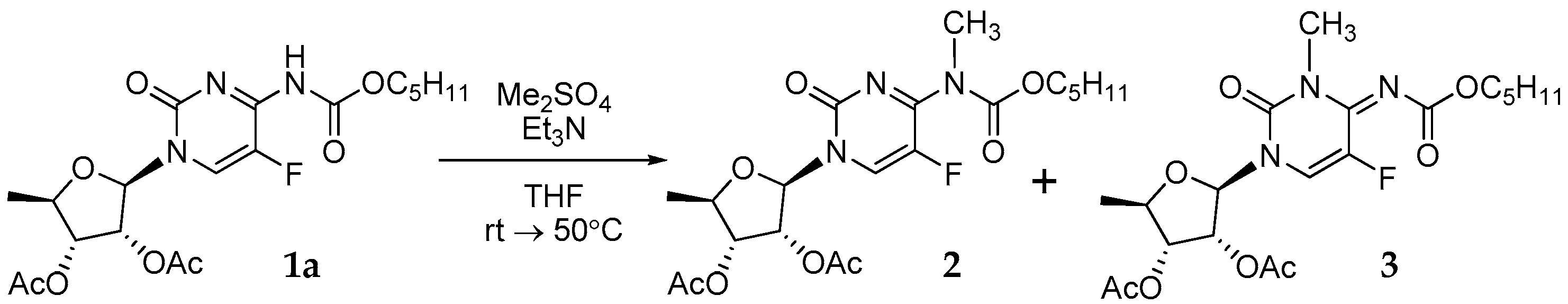
3.1. Synthesis of Capecitabine N3/N7-Methyl Derivatives 2 and 3 (Figure 4)
3.2. NMR Spectroscopy
3.3. DFT Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miwa, M.; Ura, M.; Nishida, M.; Sawada, N.; Ishikawa, T.; Mori, K.; Shimma, N.; Umeda, I.; Ishitsuka, H. Design of a Novel Oral Fluoropyrimidine Carbamate, Capecitabine, which Generates 5–Fluorouracil Selectively in Tumors by Enzymes Concentrated in Human Liver and Cancer Tissue. Eur. J. Cancer 1998, 34, 1274–1281. [Google Scholar] [CrossRef]
- Herr, R.J. Modern Drug Synthesis; Li, J.J., Johnson, D.S., Eds.; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Schellens, J.H.M. Capecitabine. Oncologist 2007, 12, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Walko, C.M.; Lindley, C. Capecitabine: A Review. Clin. Ther. 2005, 27, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Fluoropyrimidines in Cancer Therapy; Rustum, Y.M. (Ed.) Humana Press Inc.: Totowa, NJ, USA, 2003; pp. 261–284. ISBN 978-1-59259-337-8. [Google Scholar]
- Bautista, R.J.; Ravaschino, E.; Elhalem, E.; Coll Farma, S.L. Method for the Preparation of Capecitabine and Intermediates Used in Said Method. Eur. Pat. Appl. EP 2241 556 A1, 20 October 2010. [Google Scholar]
- Raghavendracharyulu, V.P.; Anant, M.M.; Srinivas, A.; Ramesh, B.; Rajasekhar, K.; Sekhar, M.; Anil, P.; Dr. Reddy’s Laboratories Ltd.; Dr. Reddy’s Laboratories, Inc. Process for Preparing Capecitabine. Patent Appl. WO 2008/131062 A2, 30 October 2008. [Google Scholar]
- Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H.; et al. The Design and Synthesis of a New Tumor-Selective Fluoropyrimidine Carbamate, Capecitabine. Bioorg. Med. Chem. 2000, 8, 1697–1706. [Google Scholar] [CrossRef]
- Hiriyanna, S.G.; Basavaiah, K. Impurity Profile Study of Capecitabine. Acta Chromatogr. 2008, 20, 609–624. [Google Scholar] [CrossRef]
- Hiriyanna, S.G. Identification, Separation, Isolation and Characterization of Major Impurities in Some Anticancer and Antipsychotic Drugs. Ph.D. Thesis, University of Mysore, Karnataka, India, February 2009. [Google Scholar]
- Rohliček, J.; Husak, M.; Gavenda, A.; Jegorov, A.; Kratochvil, B.; Fitch, A. Capecitabine from X–ray powder synchrotron data. Acta Cryst. 2009, E65, o1325–o1326. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Jamison, T.F. Rapid Continuous Synthesis of 5′–deoxyribonucleosides in flow via Brønsted Acid Catalyzed Glycosylation. Org. Lett. 2012, 14, 3348–3351. [Google Scholar] [CrossRef] [PubMed]
- Jhansi Rani, V.; Raghavendra, A.; Kishore, P.; Nanda Kumar, Y.; Hema Kumar, K.; Jagadeeswarareddy, K. Synthesis and biological activity evaluation of cytidine–5′–deoxy–5–fluoro–N–[(alkoxy/aryloxy)] carbonyl–cyclic 2′,3′–carbonates. Eur. J. Med. Chem. 2012, 54, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Malińska, M.; Krzeczyński, P.; Czerniec-Michalik, E.; Trzcińska, K.; Cmoch, P.; Kutner, A.; Woźniak, K. Crystal structure and tautomerism of capecitabine. J. Pharm. Sci. 2014, 103, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, S.; Ciuffreda, P.; Grisenti, P.; Ferraboschi, P. Synthesis of the antitumoral nucleoside capecitabine through a chemo-enzymatic approach. Tetrahedron Lett. 2015, 56, 5909–5913. [Google Scholar] [CrossRef]
- Rohliček, J.; Husak, M.; Gavenda, A.; Jegorov, A.; Kratochvil, B.; Fitch, A. Capecitabine from X–ray powder synchrotron data. Corrigendum. Acta Cryst. 2016, E72, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Tobias, S.; Günther, S. Tautomerism in OH–, SH–, and NH2–substituted pyrazines—A carbon–13 and nitrogen–15 NMR study. Tetrahedron Lett. 1982, 23, 4785–4788. [Google Scholar] [CrossRef]
- Yogo, M.; Hirota, K.; Senda, S. Pyrimidine Derivatives and Related Compounds. Part 42. J. Chem. Soc. Perkin Trans. 1982, 1, 473. [Google Scholar] [CrossRef]
- Markowski, V.; Glenn, R.; Sullivan, G.R.; John, D.; Roberts, J.D. Nitrogen–15 Nuclear Magnetic Resonance Spectroscopy of Some Nucleosides and Nucleotides. J. Am. Chem. Soc. 1977, 99, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Kozerski, L.; Sierzputowska-Gracz, H.; Krzyżosiak, W.; Bratek-Wiewiórowska, M.; Jaskólski, M.; Wiewiórowski, M. Comparative structural analysis of cytidine, ethenocytidine and their protonated salts III. 1H, 13C and 15N NMR studies at natural isotope abundance. Nucl. Acids Res. 1984, 12, 2205–2223. [Google Scholar] [CrossRef] [PubMed]
- Martino, R.; Gilard, V.; Desmoulin, F.; Malet-Martino, M. Fluorine–19 or phosphorus–31 NMR spectroscopy: A suitable analytical technique for quantitative in vitro metabolic studies of fluorinated or phosphorylated drugs. J. Pharm. Biomed. Anal. 2005, 38, 871–891. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-L.; Troy, H.; Judson, I.; Leek, R.; Leach, M.; Stubbs, M.; Harris, A.; Griffiths, J.R. Noninvasive Measurements of Capecitabine Metabolism in Bladder Tumors Overexpressing Thymidine Phosphorylase by Fluorine-19 Magnetic Resonance Spectroscopy. Clin. Cancer Res. 2004, 10, 3863–3870. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yan, J.; Sun, P.; Xu, K.; Li, S.; Yang, H.; Li, H. Comparative analysis of the interaction of capecitabine and gefitinib with human serum albumin using 19F nuclear magnetic resonance-based approach. J. Pharm. Biomed. Anal. 2016, 129, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5651. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. The Basis Set Convergence of Spin-Spin Coupling Constants Calculated by Density Functional Methods. J. Chem. Theory Comput. 2006, 2, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Helgaker, T.; Watson, M.; Handy, N.C. Analytical calculation of nuclear magnetic resonance indirect spin-spin coupling constants at the generalized gradient approximation and hybrid levels of density–functional theory. J. Chem. Phys. 2000, 113, 9402–9409. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound 1 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | Chemical Shift (ppm) | Ratio of the Integrals c | Δν1/2 (Hz) | |||
| DMSO-d6 | THF-d8 | DMSO-d6 | THF-d8 | DMSO-d6 | THF-d8 | |
| 1H | 10.52 (A) | 9.61 (A) | 66.5 | 48.6 | 62 | 185 |
| 298 K | 11.68 (B) | 11.63 (B) | 33.5 | 52.4 | 168 | 141 |
| 1H | 10.21 (A) | 71.7 | 5 | |||
| 218 K | 11.88 (B) | 28.3 | 11 | |||
| 19F | −163.3 (B) | −164.7 (B) | 32.1 | 51.8 | 250 | 176 |
| 298 K | −159.8 (A) | −162.6 (A) | 67.9 | 48.2 | 115 | 192 |
| 19F | −163.3 (B) | 30.0 | 17 | |||
| 218 K | −161.5 (A) | 70.0 | 64 | |||
| b | 13C-NMR Chemical Shifts (ppm) DMSO-d6 at 298 K | |||||
| Form A | Form B | |||||
| C6 | 129.7 | 124.7 | ||||
| C5 | 137.0 [244 Hz] d | Not found, (140.6) [224.3 Hz] f | ||||
| C2, C4, C8 | 151.2, 152.5, 153.7 e | 147.7, 161.0, not found e | ||||
| C14 | 91.1 | 89.6 | ||||
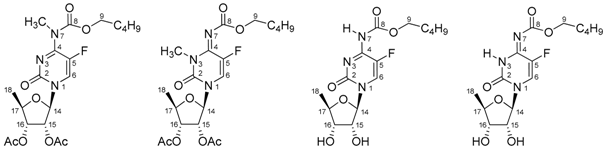
| 2 | 3 | A = I | B = II | |
|---|---|---|---|---|
| N1 | −226.2 | −255.9 | −221.7 | −244.6 |
| C2 | 153.0 | 149.6 | 154.0 1 | 147.4 |
| N3 | −123.9 | −245.6 | −139.3 | −236.5 |
| C4 | 159.2 [12.1 Hz] 2 | 145.6 [26.0 Hz] 2 | 154.4 1 [11.8 Hz] 2 | 154.0 1,3 |
| C5 | 140.5 [243.9 Hz] 2 | 139.3 [229.6 Hz] 2 | 137.9 [243.6 Hz] 2 | 140.3 [232.4 Hz] 2 |
| F | −154.5 | −161.0 | −161.5 | −163.3 |
| H6/C6 | 7.91/130.3 [36.2 Hz] 2 | 7.52/121.3 [36.0 Hz] 2 | 7.96/129.7 [34.0 Hz] 2 | 7.86/126.5 [34.5 Hz] 2 |
| N7 | −276.2 | −151.7 | −268.9 | (−197.8) 4 |
| H7/H3 | - | - | 10.08 | 11.88 |
| CH3 | 3.29/34.7 | 3.27/30.4 | - | - |
| C8 | 154.5 | 160.4 | 151.7 | 164.7 |
| H9/C9 | 4.17/67.8 | 4.05/66.4 | 4.11/66.5 | 4.05/66.3 |
| H10/C10 | 1.65/29.2 | 1.63/29.4 | 1.65/29.4 | 1.65/29.4 |
| H11/C11 | 1.35/28.8 | 1.35/29.0 | 1.35/28.9 | 1.35/29.2 |
| H12/C12 | 1.35/23.2 | 1.35/23.2 | 1.35/23.5 | 1.35/23.6 |
| H13/C13 | 0.91/14.3 | 0.90/14.3 | 0.91/14.7 | 0.91/14.7 |
| H14/C14 | 5.87/91.2 | 5.88/90.1 | 5.62/93.3 | 5.76/91.0 |
| H15/C15 | 5.46/74.6 | 5.40/73.7 | 4.17/75.5|6.01 5 | 4.28/74.6|5.69 5 |
| H16/C16 | 5.12/74.9 | 5.08/74.8 | 3.63/75.5|4.61 5 | 3.74/75.5|4.87 5 |
| H17/C17 | 4.19/78.9 | 4.11/78.7 | 3.99/80.2 | 3.92/80.5 |
| H18/C18 | 1.43/18.2 | 1.39/18.4 | 1.42/18.2 | 1.36/18.7 |
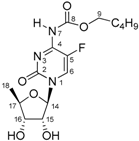
| D2O | THF + HClO4 | |
|---|---|---|
| N1 | −225.0 | −221.0 |
| C2 | 157.0 | 145.6 |
| N3 | (−158.4) 1 | −225.8 |
| C4 | 157.1 [12.6 Hz] 2 | 152.9 [20.2 Hz] 2 |
| C5 | 140.5 [245.2 Hz] 2 | 136.4 [233.4 Hz] 2 |
| F | −163.2 | −165.8 |
| H6/C6 | 8.06/131.3 [33.4 Hz] 2 | 8.36 [6.0 Hz]/135.4 [34.6 Hz] 2 |
| N7 | (−271.6) 1 | −259.4 |
| C8 | 155.6 | 154.3 |
| H9/C9 | 4.23/70.0 | 4.32/69.2 |
| H10/C10 | 1.70/30.3 | 1.73/28.9 |
| H11/C11 | 1.35/30.0 | 1.38/28.6 |
| H12/C12 | 1.34/24.4 | 1.36/23.1 |
| H13/C13 | 0.88/16.0 | 0.90/14.3 |
| H14/C14 | 5.78/94.6 | 5.69/94.5 |
| H15/C15 | 4.30/77.4 | 4.26/75.5 |
| H16/C16 | 3.88/76.9 | 3.81/75.1 |
| H17/C17 | 4.21/82.5 | 4.05/81.3 |
| H18/C18 | 1.47/20.1 | 1.42/18.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cmoch, P.; Krzeczyński, P.; Leś, A. Multinuclear NMR Measurements and DFT Calculations for Capecitabine Tautomeric Form Assignment in a Solution. Molecules 2018, 23, 161. https://doi.org/10.3390/molecules23010161
Cmoch P, Krzeczyński P, Leś A. Multinuclear NMR Measurements and DFT Calculations for Capecitabine Tautomeric Form Assignment in a Solution. Molecules. 2018; 23(1):161. https://doi.org/10.3390/molecules23010161
Chicago/Turabian StyleCmoch, Piotr, Piotr Krzeczyński, and Andrzej Leś. 2018. "Multinuclear NMR Measurements and DFT Calculations for Capecitabine Tautomeric Form Assignment in a Solution" Molecules 23, no. 1: 161. https://doi.org/10.3390/molecules23010161
APA StyleCmoch, P., Krzeczyński, P., & Leś, A. (2018). Multinuclear NMR Measurements and DFT Calculations for Capecitabine Tautomeric Form Assignment in a Solution. Molecules, 23(1), 161. https://doi.org/10.3390/molecules23010161