Abstract
Ion association is an important process in aqueous dissolution, precipitation, and crystallization of ionic inorganic, organic, and biological materials. Polyoxometalates (POMs) are good model compounds for understanding the complex relationships between lattice energy, ion-pairing in solution, and salt solubility. Here we perform calorimetric measurements to elucidate trends in cluster stability, lattice energy, and ion-pairing behavior studies of simple hexatantalate salts in neat water, parent hydroxide solutions, and molybdate melts, extending previous studies on the isostructural hexaniobates. High temperature calorimetry of alkali salts of hexatantalate reveals that the enthalpies of formation from oxides of the K, Rb, and Cs salts are more similar to each other than they are for their niobate analogues and that the tantalate cluster is energetically less stable than hexaniobate. Aqueous dissolution calorimetry reveals that the cesium salt of hexatantalate has a similar concentration dependence on its dissolution enthalpy to that of hexaniobate. However, unlike rubidium hexaniobate, rubidium hexatantalate also exhibits increased concentration dependence, indicating that hextantalate can undergo increased ion-pairing with alkali salts other than cesium, despite the dilute environments studied. Dissolution enthalpies of POM salts in the parent alkali hydroxides shows that protonation of clusters stabilizes lattices even more than the strongly associating heavy alkali cations do. Additionally, neither weak nor strong lattice ion associations necessarily correlates with respectively high or low aqueous solubility. These studies illuminate the importance of considering ion-pairing among the interrelated processes in the aqueous dissolution of ionic salts that can be extended to serving as a model of cation association to metal oxide surfaces.
Keywords:
polyoxometalates; hexatantalate; tantalum; countercations; ion-pairing; calorimetry; solubility 1. Introduction
Aqueous ion behavior is driven by many fundamental and interrelated physical processes. The solubility of ionic salts in water is predictable to some degree by the “hardness” or “softness” of the component cations and anions, which arises from their degrees of hydration upon dissolution, electron count in the frontier molecular orbitals, overall charge, and charge density [1,2,3,4]. Typically, close interactions between cations and anions (ion-pairing) in solutions predicates precipitation, with larger hydration spheres from more charge-dense species (i.e., Li+) maintaining solubility, according to the Hoffmeister series [5]. This factor competes with lattice energy, which is a sum of energies of all the bonds present in the lattice. For simple monoatomic ions such as alkali halides, similarly sized cations and anions stabilize each other to the greatest degree, decreasing solubility. The relationship between lattice energy and solubility is less well defined for alkali salts of oxoanions; in particular, the anomalous solubilities of some highly charged oxoanions including carbonate [6,7], which is further complicated by protonation in solution.
We define anomalous solubility as increased solubility with increased ion-pairing, while normal solubility is the opposite. As an initial approximation, oxoanions and POMs with low charge density exhibit the typical solubility trend (less soluble with increasing alkali countercation size), whereas those with high charge densities exhibit the reversed, atypical solubility trend. Some common POMs and simple oxoanions, their charge densities (described here as cluster charge divided by number of cluster atoms), and observed solubility trends as alkali salts are summarized in Table 1 and below we provide a qualitative explanation of these opposite solubility trends. We surmise that oxo species with higher charge densities exhibit this atypical trend due to their ability to disrupt the relatively weakly bound hydration spheres of larger alkali cations (i.e., Rb+ and Cs+), allowing for contact ion pairing. Because of the high POM/oxoanion charge, the alkali is strongly associated and therefore does not bridge to other oxoanions, suppressing aggregation and precipitation [8]. On the other hand, the high charge density of the same POMs/oxoanions does not completely satisfy bonding of the smaller and more charge-dense alkali cations (i.e., Li+ and Na+), therefore these alkalis bridge POM/oxoanions in solution, initiating precipitation. Considering the low-charge density POMs, they may also form contact ion-pairs with larger alkalis, but these alkalis are not as strongly associated to a single POM/oxoanion. Rather they bridged two or more of these anions in solution, initiating precipitation. Finally the low-charge density POMs likely cannot penetrate the large hydration sphere of the charge-dense alkalis (Li+, Na+), so there is no interaction in solution and solubility is retained.

Table 1.
Charge densities, defined as the total charge divided by the number of non-hydrogen atoms, and solubility trends with respect to alkali countercations of several prominent oxoanions and POMs.
Understanding the governing principles of solubility is important because it yields more complete models for thermodynamic calculations of aqueous speciation and ion association. Additionally, the solubility and association of ions in water affects their adsorption onto metal oxide surfaces [9], and knowledge of ion association can be used to fine-tune the electronic properties and chemical reactivities of catalysts, as well as design ion-specific sorbents [10,11,12].
Polyoxometalates (POMs), nanoscale molecular oxoanions of Group V and VI metals in their highest oxidation states, can generally be synthesized with any alkali cation, thus providing an excellent model for ion-pairing in water and at metal oxide surfaces [15,22]. These “molecular metal oxides” exhibit a wide range of aqueous behaviors that depend on their metal centers and, crucially, their countercations [23]. The isostructural and isovalent hexaniobate ([Nb6O19]8-, Nb) and hexatantalate ([Ta6O19]8-, Ta) POMs exhibit very similar pH stabilities, and aqueous solubilities with respect to alkali countercations [16,24,25]. In particular, Cs+ undergoes significant ion-pairing with these POMs, which increases their solubilities due to each Cs+ more favorably interacting with a single cluster rather than coordinating to multiple clusters, wherein binding to a single cluster inhibits aggregation and precipitation, even at high concentration [8]. However, subtle differences have been found in their degrees of ion association with Cs+. For example, Ta undergoes greater ion-pairing than Nb with Cs+ in dilute solutions in neat water, even at low concentration. This was attributed to the presence of relativistic effects in Ta, which results in the orbital interaction term having a larger contribution to the total bonding energy [8]. In other words, electrostatics alone are insufficient to describe cation association in the presence of Group V POMs, and covalent bonding between the alkalis and oxo ligands may also be considered, in order to arrive at a complete description of solution behavior.
2. Results & Discussion
2.1. Solid-State Calorimetry of Ta Salts
We previously conducted high-temperature oxide decomposition calorimetric studies on alkali (Li+, K+, Rb+, Cs+) salts of Nb [26]. Upon dropping pellets made from crystalline samples into molten sodium molybdate at 700 , we ascertained a trend of increasingly exothermic enthalpy of formation from oxides () with respect to alkali countercation size. To parallel this previous study, we performed measurements on the same alkali salts of Ta. Although the Na8[Ta6O19] salt can be synthesized, it was not studied because the analogous Nb salt is not readily obtained. Rather, Na7[HNb6O19] is the common form [27]. The measured is less exothermic for each alkali Ta6 analogue, compared to the Nb6 analogue (Figure 1). We rationalize this as mixing of Ta and O orbitals in Ta being poorer than the mixing between Nb and O in Nb [8,18], decreasing the thermodynamic stability of the Ta cluster in comparison to the Nb cluster. As with the observed trend for hexaniobate, the Li+ salt of Ta exhibits a far less exothermic (by more than a factor of two) than the larger alkali salts. This is explained by the structure of lithium hexaniobate [28,29]. The hexaniobate lattice contains adamantane-like Li-water clusters, which prevents extensive direct bonding between Li+ and Nb. A high-resolution single-crystal structure of Li–Ta has never been possible, due to poor crystal quality and perhaps disorder of the hydrated lithium in the lattice. We surmise that Li–Ta is not isostructural with the Li–Nb analogue (Li8Nb6O19·22H2O) due to the lack of a reported single-crystal structure for lithium hexatantalate [30,31]. However, Li is generally bonded to water in hydrated salt lattices, and this is reflected in its less exothermic formation enthalpy, compared to the salts of the heavier alkalis. Unlike Nb, the K+, Rb+, and Cs+ salts of Ta do not exhibit a strict trend of increasing (Figure 1). Instead, the three larger alkali salts are more similar in their . This suggests that Ta is slightly `less selective’ in its solid-state ion association than Nb.
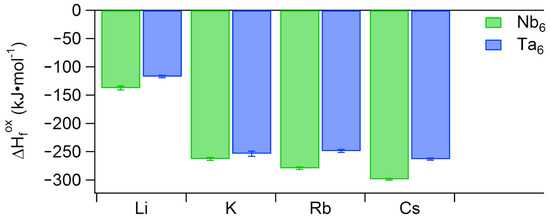
Figure 1.
Solid-state (enthalpy of formation from oxides, kJ (mol-Nb)) values for alkali salts of Ta6 (this study) and Nb6 (prior study) [26].
2.2. Dissolution Energy of Ta Salts
While high-temperature calorimetry provides information about the stability of the clusters and their association with lattice species, solution calorimetry elucidates hydration enthalpies of the ions upon dissolution, as well as their solid-state lattice interactions. Enthalpy of dissolution () at room temperature of the Nb salts in neat water showed that the Cs-salt has greater concentration dependence in its dissolution enthalpy compared to the other alkali salts. By extension, the degree of structural change became less extensive at higher concentrations, indicating that the Cs+ underwent maximum ion-pairing with Nb6, even at the low concentrations (<2 mM). Analogous measurements of Ta6 salts also showed concentration dependence for each alkali salt (Figure 2). Li+ again cannot be easily compared to the larger alkalis, due to its far more extensive coordination to water molecules in the solid state. This is reflected in its overall lower values of compared to the other three alkali salts, meaning it is not necessary to disrupt extensive bonding between the clusters and alkalis to dissolve them, which is an endothermic process. However, the steeper slope of the Cs–Ta trend was matched by the Rb+ salt, with K+ having a smaller slope. This indicates that rubidium and cesium undergo similar degrees of ion-pairing with Ta in solution, which is different from the observed Nb trend, in which the cesium salt clearly had the greatest dependence on concentration. Like the high-temperature calorimetry, this too indicates that Ta is `less selective’ in its ion association than Nb (in both solution and solid state). On the other hand, it has been noted that Nb is more basic than Ta, meaning it protonates upon dissolution in water, at the bridging oxos sites [32,33]. Since protonation is an endothermic process that is not equal between the Ta and Nb, it is difficult to compare the dissolution enthalpy with exact certainty.
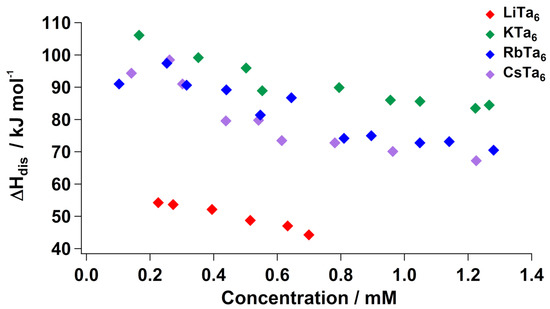
Figure 2.
Enthalpy of aqueous dissolution () of Li–Ta6, K–Ta6, Rb–Ta6 and Cs–Ta6 in water, normalized for lattice water (i.e., representing dissolution of the dehydrated forms Li8[Ta6O19], K8[Ta6O19], Rb8[Ta6O19] and Cs8[Ta6O19]; the hydrated enthalpies are provided in Tables S5–S9).
We dissolved the K, Rb and Cs Ta salts in 1 molar solutions of each alkali cation’s parent hydroxide (Figure 3). The Li+ salt was omitted due to its insolubility in LiOH. Like Nb, alkali salts of Ta do not have a concentration dependence when dissolved in base, due to a lack of cluster protonation and the excess cations in solution forcing the “maximally associated” state at all concentrations. The generally more exothermic dissolution enthalpies can be related to the lack of protonation of the clusters with high concentration of base. There is a distinct trend in with respect to alkali cation size; with Cs–Ta being most exothermic, followed by Rb-Ta6 and then K–Ta. This trend suggested that less energy is required to dissociate the Cs–Ta lattice adequately to achieve dissolution, compared to the K and Rb analogues. This is exactly consistent with ion-pairing persisting in solution: increasing K–Ta< Rb–Ta< Cs–Ta. The solutions remain monodisperse and no appreciable amount of impurities form with added solid Ta, accounted for by previous small-angle X-ray scattering studies of hexatantalate in 1 molar parent hydroxide solution [24]. We also do not expect any impurities or cluster alteration upon dissolution in hydroxide solutions because the hexatantalate form is most stable in conditions of high concentration hydroxide, confirming that the flatlining of is strictly from maximal A–Ta ion-pairing.
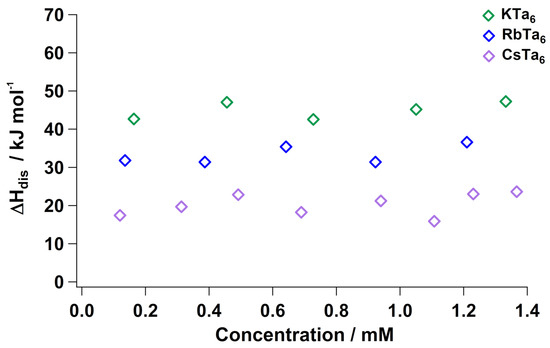
Figure 3.
Enthalpy of aqueous dissolution () of Li–Ta6, K–Ta6, Rb–Ta6 and Cs-Ta6 in their parent hydroxide (1 M) solutions, normalized for lattice water. The hydrated enthalpies are provided in Tables S10–S13.
Finally, we compared the of the tetramethylammonium salt of Ta ([(CH3)4N]6H2O19·21H2O, TMA–Ta) in water and in 1 molar TMAOH to the previously measured Nb analogue (Figure 4). As with the analogous TMA–Nb study, does not have a concentration dependence in either solution environment due to the lack of ion association between TMA+ and Ta. However, is less exothermic for TMA–Ta (∼ ) than it is for TMA–Nb (∼ ) in water. As described above, this is because Ta is a weaker base than Nb [25], resulting in a lesser degree of deprotonation, consistent with fewer exothermic events occurring in solution. The difference in between TMA–Ta and TMA–Nb is greatly enhanced in 1M TMAOH (∼ and (∼ , respectively). This is due to the tendency for TMA–Nb to form oligomeric chains in neat water, whereas TMA–Ta typically forms dimers [24]. Thus, the vast difference in exothermic dissolution is further explained by the degree of hydrolysis of the respective assemblies—a far greater number of exothermic events occur in dissociating the Nb6 chains than in dissociating Ta dimers. The is nonetheless more exothermic for TMA–Ta in 1M TMAOH than it is in neat water due to the disassembly of these dimers.
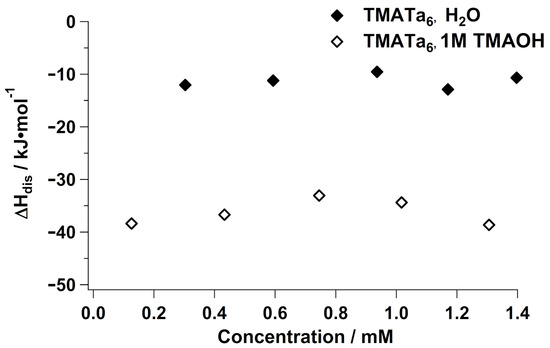
Figure 4.
Enthalpy of dissolution of TMA–Ta6 in neat water and in 1M TMAOH.
3. Materials and Methods
3.1. Determination of Drop Solution Enthalpies
was measured in a custom-made isoperibol Tian-Calvet twin microcalorimeter. Pellets of about 5 mg were loosely pressed, weighed, and dropped from room temperature into 3Na2O4MoO3 molten solvent at 702 . The calorimeter assembly was washed with oxygen at 43 . Oxygen was bubbled through the solvent at to aid dissolution, evolve water vapor, and to maintain oxidizing conditions. The calorimeter was calibrated against the heat content of 5 mg pellets of high-purity Al2O3 (99.997%, Alfa Aesar) dropped into an empty crucible.
3.2. Determination of Room Temperature Dissolution Enthalpies
was measured using a CSC 4400 isothermal microcalorimeter operated at 25 . About 10–25 of each sample was hand pressed into a pellet and dropped one at a time into of H2O. Each experiment was repeated in 1 M AOH (A = K, Rb, Cs, TMA) for the respective clusters. The lithium salt was omitted due to its insolubility.
The calorimeter was calibrated by dissolving 15 mg pellets of KCl in water with stirring at 25 . Hydrous and anhydrous cluster dissolution enthalpy values in water are reported in Tables S5–S9 and parent hydroxide values are reported in Tables S10–S13.
Enthalpies of anhydrous clusters were found by subtracting the enthalpy of dissolution of lattice water (0.4 ) [34] and then adjusting for the relative molar weights of the hydrated and anhydrous clusters, namely:
3.3. Thermogravimetric Analysis
Thermogravimetry was used to determine number of lattice waters per cluster. These experiments were performed on a Setaram LabSys Evo. The sample was heated in an alumina crucible from 25 to 600 in an inert atmosphere at 10 . The total weight loss was determined at the temperature where weight % reached a minimum (Figures S5–S8).
3.4. Syntheses
3.4.1. Alkali Salts
The larger alkali salts of [Ta6O19]8- were synthesized by refluxing a ≈40 mM solution of their peroxotantalate analogues (A3Ta(O2)4; A = K, Rb, Cs) in concentrated parent alkali hydroxide solution for 4 h, except for the lithium salt, which was formed by metathesis of the potassium salt in 1 M LiOH solution. The peroxotantalates were formed by adding TaCl5 to 40 mL of cold 30% (w/w) H2O2 solution, adding concentrated AOH solutions (A = K, Rb, Cs), and precipitating with ethanol. Full syntheses are described in a prior publication [27].
3.4.2. Tetramethylammonium Salt
Tetramethylammonium hexatanatalate ([(CH3)4N]6H2Ta6O19) was synthesized by adding 1.32 g of (NH4)3Ta(O2)4 to 8.25 mL of 1.4 M tetramethylammonium hydroxide and refluxing for 5 h. The solution was then microfiltered, 40 mL isopropyl alcohol was added, and then the resulting solution was centrifuged to yield a small denser layer containing the product. The supernatant was discarded and further addition of 30 mL isopropyl alcohol yielded a white precipitate, which was washed with more isopropyl alcohol and oven-dried under vacuum at 60 [24].
4. Conclusions
This study represents a rare quantitative evaluation of combined high temperature and aqueous dissociation energy of water-soluble metal-oxo clusters, elucidating the correlation between ion-association and acid-base behavior in dissolution of cluster salts, as well as the stability of the clusters. Experimentally determined enthalpy of formation of the Nb and Ta hexametalate POMs showed lower thermodynamic stability of the Ta-analogues compared to the Nb-analogues, regardless of the alkali counterion. This is consistent with the slightly larger ionic radius of Ta5+ than Nb5+, and corresponding weaker M–O bonds within the POM. While all cluster salts exhibit negative enthalpy of formation from oxides, there is a periodic trend of increasingly exothermic enthalpy with increasing alkali size. Notably fewer negative enthalpies are measured for the Li-salts compared to the heavier congeners, due to minimal cluster-cation association within the lattice (lower lattice energy). Aqueous dissolution enthalpy is the net sum of both endothermic and exothermic processes including protonation and/or deprotonation of the oxoanion POMs, dissociation of ions, and hydration of ions. With the various solutions (neat water and parent hydroxides) and various countercations (alkalis and tetramethylammounium, TMA) for the Nb and Ta hexametalates, we can conclude the following about cation-oxoanion lattice energetics. First, protonation of oxoanions such as POMs stabilize lattices even more than the strongly associated heavier alkali cations. Second, solubility of oxoanions cannot be described as inversely correlated with their lattice energy: we observed both the most negative and the most positive lattice energies for the two most soluble salts, respectively with TMA+ and with Cs+ counterions. This means that lattice energy, as classically described by the Born-Haber cycle, does not necessarily predict solubility of oxoanion salts, or lattices containing aqua species.
Supplementary Materials
The following are available online. Thermochemical Cycles (with oxide drop solution enthalpies taken from previous studies) [35], values for hydrated (non-normalized for water) enthalpies of dissolution, thermogravimetric analyses, and energy dispersive X-ray spectra are available as Supplementary Information.
Author Contributions
Conceptualization, D.J.S., A.N. and M.N.; Formal analysis, D.J.S., G.P.N., A.N. and M.N.; Funding acquisition, A.N. and M.N.; Investigation, D.J.S., G.P.N., A.N. and M.N.; Methodology, D.J.S., G.P.N., A.N. and M.N.; Project administration, A.N. and M.N.; Resources, A.N. and M.N.; Supervision, G.P.N. and A.N.; Visualization, D.S., A.N. and M.N.; Writing—original draft, D.J.S.; Writing—review & editing, D.J.S., G.P.N., A.N. and M.N.
Funding
The studies performed at OSU and UCD by DS and MN were supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award # DE-SC0010802. GPN and AN acknowledge support by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical and Geosciences, under award FG02-97ER14749.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| POM | Polyoxometalate |
| Nb | Hexaniobate |
| Ta | Hexatantalate |
| TMA | Tetramethylammonium |
References
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Ayers, P.W. An elementary derivation of the hard/soft-acid/base principle. J. Chem. Phys. 2005, 122, 141102. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Evans, J.N. The Fukui function: a key concept linking frontier molecular orbital theory and the hard-soft-acid-base principle. J. Am. Chem. Soc. 1995, 117, 7756–7759. [Google Scholar] [CrossRef]
- Ayers, P.W.; Parr, R.G.; Pearson, R.G. Elucidating the hard/soft acid/base principle: A perspective based on half-reactions. J. Chem. Phys. 2006, 124, 194107. [Google Scholar] [CrossRef] [PubMed]
- Oncsik, T.; Trefalt, G.; Borkovec, M.; Szilagyi, I. Specific ion effects on particle aggregation induced by monovalent salts within the Hofmeister series. Langmuir 2015, 31, 3799–3807. [Google Scholar] [CrossRef] [PubMed]
- Szwarc, M. Ions and Ion Pairs in Organic Reactions; John Wiley & Sons: Hoboken, NJ, USA, 1974; Volume 2. [Google Scholar]
- Linke, W.F. Solubilities, Inorganic and Metal Organic Compounds: A Compilation of Solubility Data from the Periodical Literature; Van Nostrand: Syracuse, NY, USA, 1965; Volume 2. [Google Scholar]
- Sures, D.J.; Serapian, S.A.; Kozma, K.; Molina, P.I.; Bo, C.; Nyman, M. Electronic and relativistic contributions to ion-pairing in polyoxometalate model systems. Phys. Chem. Chem. Phys. 2017, 19, 8715–8725. [Google Scholar] [CrossRef] [PubMed]
- Boehm, H. Acidic and basic properties of hydroxylated metal oxide surfaces. Discuss. Faraday Soc. 1971, 52, 264–275. [Google Scholar] [CrossRef]
- Connor, G.P.; Holland, P.L. Coordination chemistry insights into the role of alkali metal promoters in dinitrogen reduction. Catal. Today 2017, 286, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Bonzel, H. Alkali-promoted gas adsorption and surface reactions on metals. J. Vac. Sci. Technol. A Vac. Surf. Films 1984, 2, 866–872. [Google Scholar] [CrossRef]
- Mross, W. Alkali doping in heterogeneous catalysis. Catal. Rev. Sci. Eng. 1983, 25, 591–637. [Google Scholar] [CrossRef]
- Seidell, A. Solubilities of Inorganic and Organic Compounds C. 2; D. Van Nostrand Company: New York, NY, USA, 1919. [Google Scholar]
- Perry, D.L. Handbook of Inorganic Compounds; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Nyman, M.; Alam, T.M.; Bonhomme, F.; Rodriguez, M.A.; Frazer, C.S.; Welk, M.E. Solid-state structures and solution behavior of alkali salts of the [Nb6O19]8- Lindqvist ion. J. Clust. Sci. 2006, 17, 197–219. [Google Scholar] [CrossRef]
- Nyman, M. Polyoxoniobate chemistry in the 21st century. Dalton Trans. 2011, 40, 8049–8058. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Nyman, M.; Rodriguez, M.A. Soluble heteropolyniobates from the bottom of group IA. Angew. Chem. Int. Ed. 2011, 50, 12514–12517. [Google Scholar] [CrossRef] [PubMed]
- Sures, D.J.; Molina, P.I.; Miró, P.; Zakharov, L.N.; Nyman, M. Cesium salts of niobo-tungstate isopolyanions with intermediate group V–group VI character. New J. Chem. 2016, 40, 928–936. [Google Scholar] [CrossRef]
- Krtil, J. Exchange properties of ammonium salts of 12-heteropolyacids—II: Separation of rubidium and caesium on ammonium phosphotungstate. J. Inorg. Nucl. Chem. 1961, 19, 298–303. [Google Scholar] [CrossRef]
- Briand, L.E.; Baronetti, G.T.; Thomas, H.J. The state of the art on Wells–Dawson heteropoly-compounds: A review of their properties and applications. Appl. Catal. A Gen. 2003, 256, 37–50. [Google Scholar] [CrossRef]
- Ginsberg, A.P. Inorganic Syntheses; John Wiley & Sons: Hoboken, NJ, USA, 1990. [Google Scholar]
- Fullmer, L.B.; Mansergh, R.H.; Zakharov, L.N.; Keszler, D.A.; Nyman, M. Nb2O5 and Ta2O5 thin films from polyoxometalate precursors: A single proton makes a difference. Cryst. Growth Des. 2015, 15, 3885–3892. [Google Scholar] [CrossRef]
- Nyman, M.; Burns, P.C. A comprehensive comparison of transition-metal and actinyl polyoxometalates. Chem. Soc. Rev. 2012, 41, 7354–7367. [Google Scholar] [CrossRef] [PubMed]
- Fullmer, L.B.; Molina, P.I.; Antonio, M.R.; Nyman, M. Contrasting ion-association behaviour of Ta and Nb polyoxometalates. Dalton Trans. 2014, 43, 15295–15299. [Google Scholar] [CrossRef] [PubMed]
- Deblonde, G.J.P.; Moncomble, A.; Cote, G.; Bélair, S.; Chagnes, A. Experimental and computational exploration of the UV-visible properties of hexaniobate and hexatantalate ions. RSC Adv. 2015, 5, 7619–7627. [Google Scholar] [CrossRef]
- Sures, D.J.; Sahu, S.K.; Molina, P.I.; Navrotsky, A.; Nyman, M. Distinctive interactions of cesium and hexaniobate in water. ChemistrySelect 2016, 1, 1858–1862. [Google Scholar] [CrossRef]
- Anderson, T.M.; Rodriguez, M.A.; Bonhomme, F.; Bixler, J.N.; Alam, T.M.; Nyman, M. An aqueous route to [Ta6O19]8- and solid-state studies of isostructural niobium and tantalum oxide complexes. Dalton Trans. 2007, 4517–4522. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.M.; Thoma, S.G.; Bonhomme, F.; Rodriguez, M.A.; Park, H.; Parise, J.B.; Alam, T.M.; Larentzos, J.P.; Nyman, M. Lithium Polyniobates. A Lindqvist-supported lithium- water adamantane cluster and conversion of hexaniobate to a discrete Keggin complex. Cryst. Growth Des. 2007, 7, 719–723. [Google Scholar] [CrossRef]
- Hou, Y.; Fast, D.B.; Ruther, R.E.; Amador, J.M.; Fullmer, L.B.; Decker, S.R.; Zakharov, L.N.; Dolgos, M.R.; Nyman, M. The atomic level journey from aqueous polyoxometalate to metal oxide. J. Solid State Chem. 2015, 221, 418–425. [Google Scholar] [CrossRef]
- Nyman, M.; Anderson, T.M.; Provencio, P.P. Comparison of aqueous and non-aqueous soft-chemical syntheses of lithium niobate and lithium tantalate powders. Cryst. Growth Des. 2008, 9, 1036–1040. [Google Scholar] [CrossRef]
- Deblonde, G.J.P.; Coelho-Diogo, C.; Chagnes, A.; Cote, G.; Smith, M.E.; Hanna, J.V.; Iuga, D.; Bonhomme, C. Multinuclear solid-state NMR investigation of hexaniobate and hexatantalate compounds. Inorg. Chem. 2016, 55, 5946–5956. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Ozawa, Y.; Yagasaki, A. Which is the most basic oxygen in [Ta6O19]8-?—Synthesis and structural characterization of [H2Ta6O19]6-. Inorg. Chem. Commun. 2011, 14, 115–117. [Google Scholar] [CrossRef]
- Matsumoto, M.; Ozawa, Y.; Yagasaki, A. Long hydrogen-bonded rod of molecular oxide: A hexatantalate tetramer. Inorg. Chem. 2012, 51, 5991–5993. [Google Scholar] [CrossRef] [PubMed]
- Robie, R.A.; Hemmingway, B.; Fisher, J.R. Thermodynamic Properties of Minerals and Related Substances at 298. 15 K and 1 Bar (105 Pascals) Pressure and at Higher Temperatures; U.S. Geological Survey: Reston, VA, USA, 1978.
- Zlotnik, S.; Sahu, S.K.; Navrotsky, A.; Vilarinho, P.M. Pyrochlore and perovskite potassium tantalate: Enthalpies of formation and phase transformation. Chem. Eur. J. 2015, 21, 5231–5237. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).