The Positional Isomeric Effect on the Structural Diversity of Cd(II) Coordination Polymers, Using Flexible Positional Isomeric Ligands Containing Pyridyl, Triazole, and Carboxylate Fragments

 ,
,
Abstract
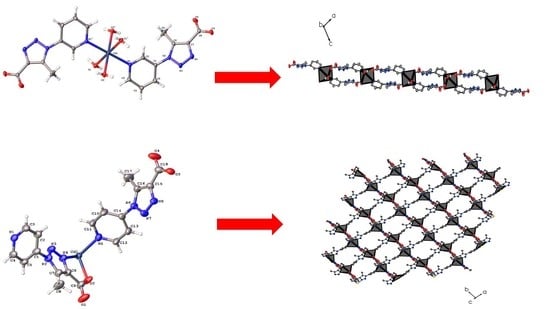
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Instruments
2.2. Single-Crystal X-ray Diffraction
2.3. Synthetic Procedures
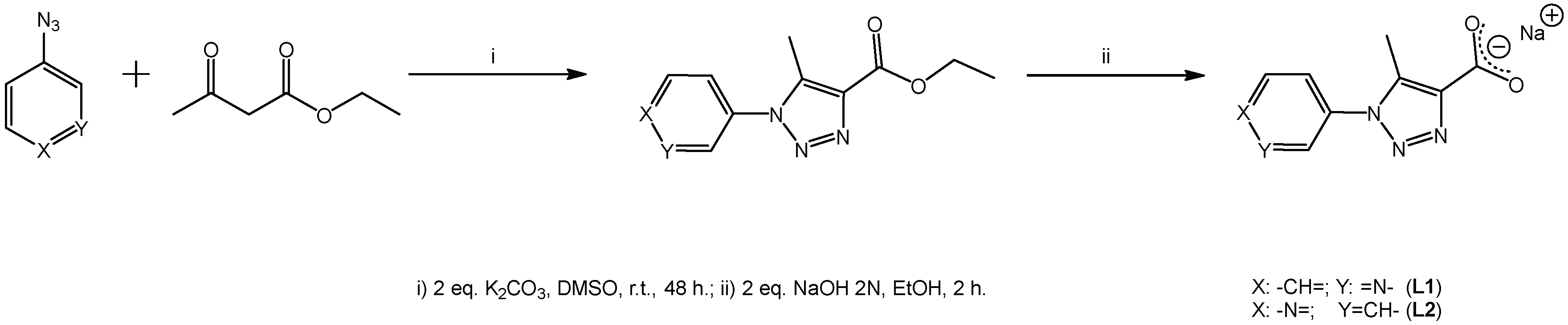
2.3.1. General Procedure for the Syntheses of Ligands L1 and L2
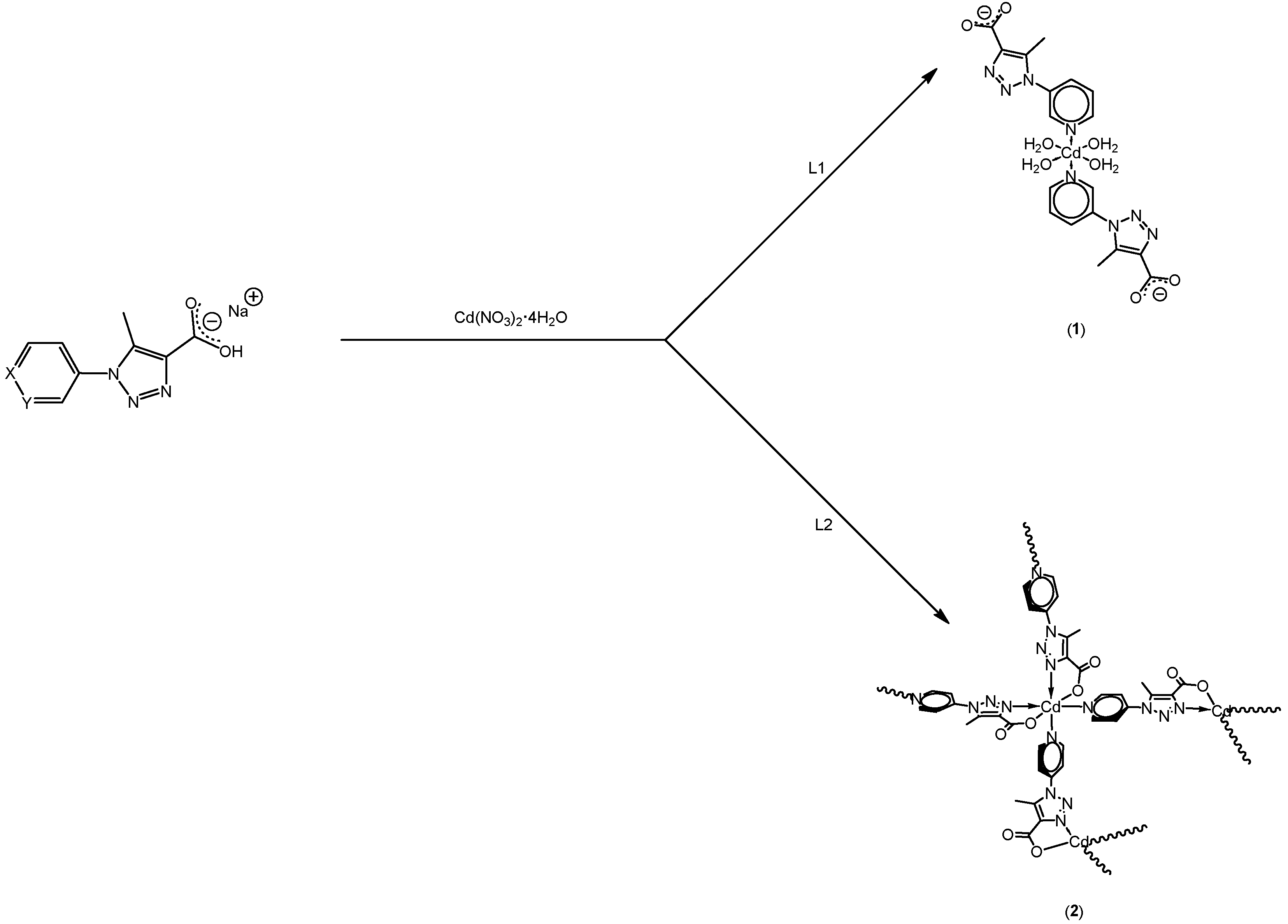
2.3.2. Synthesis of Tetraaqua–bis(5-methyl-1-(3-pyridin)-1H-1,2,3-triazole-carboxilate) Cadmium(II) [Cd(L1)2·4H2O] (1)
2.3.3. Synthesis of Catena-tetra(5-methyl-1-(4-pyridin)-1H-1,2,3-triazole-carboxilate) Cadmium(II) Pentahydrate [Cd(L2)4]n (2)
3. Results
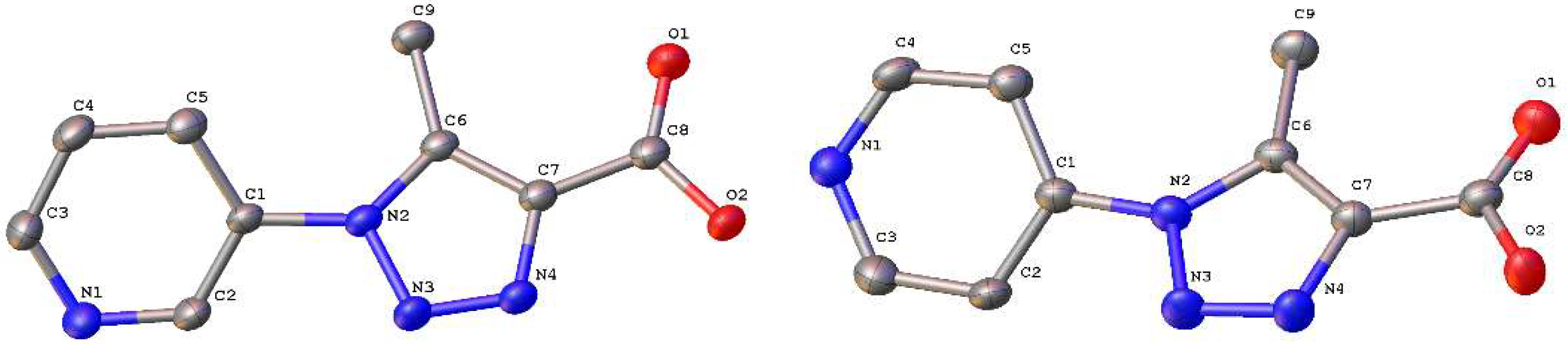
3.1. Syntheses of L1 and L2
3.2. Syntheses of 1 and 2
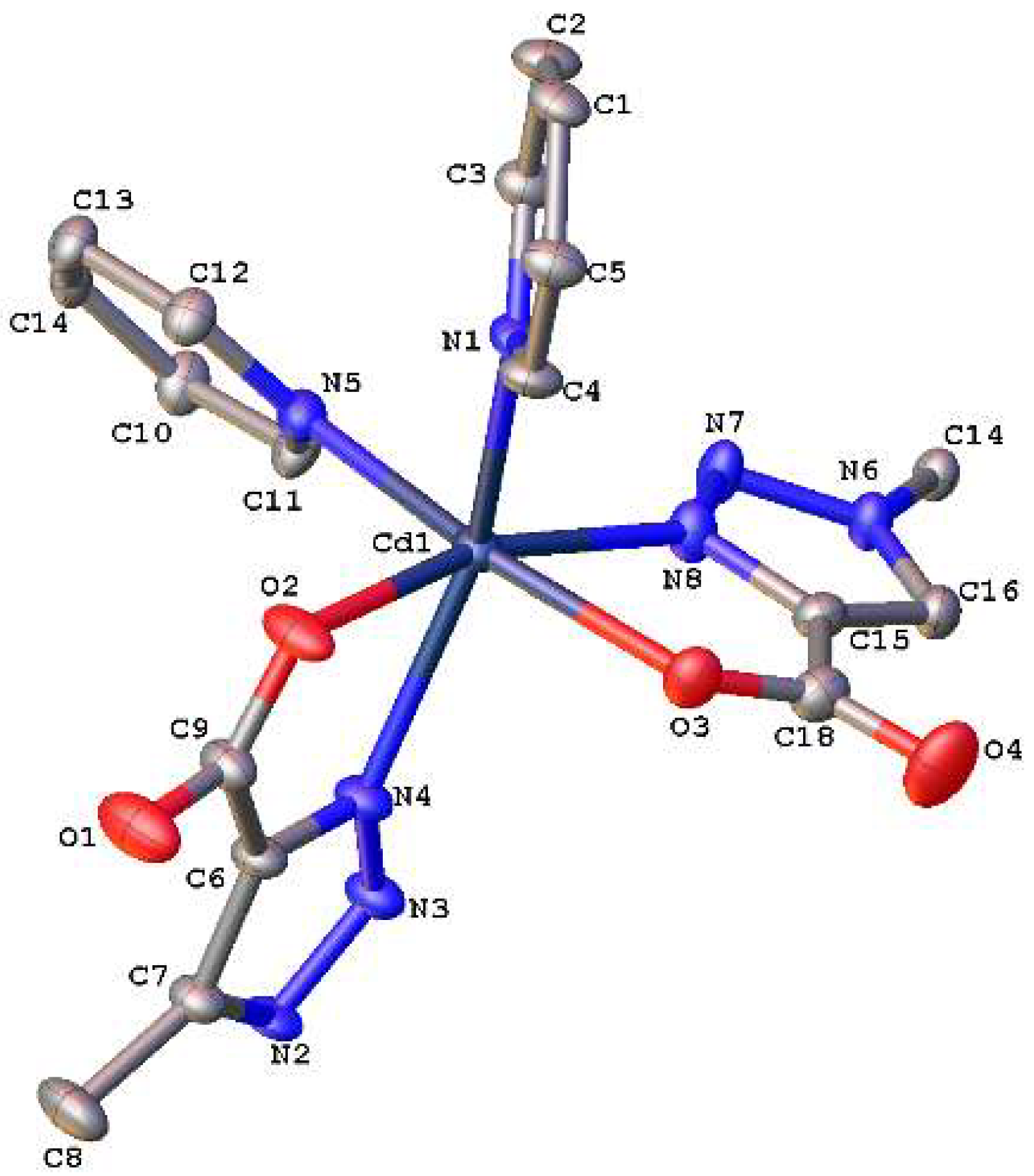
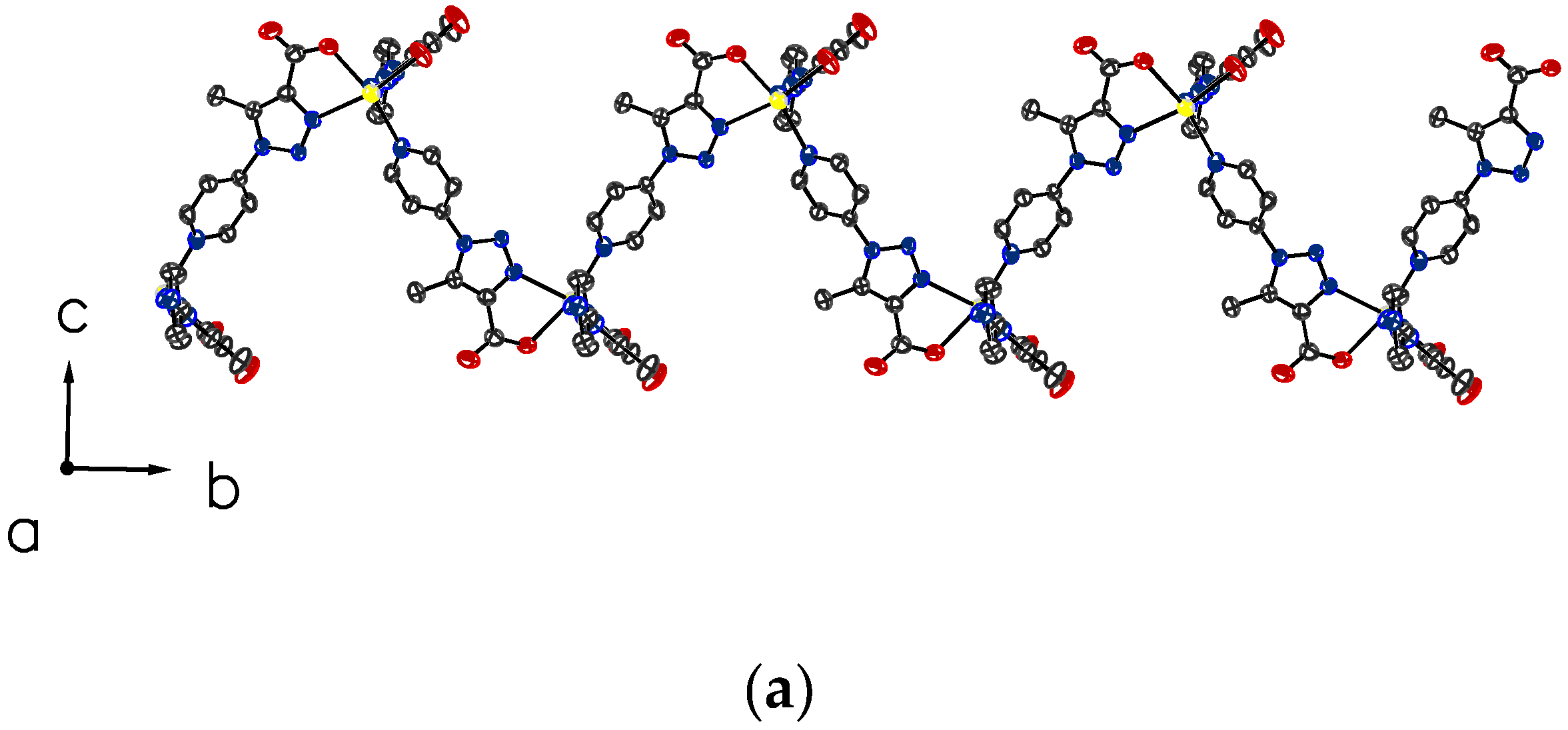
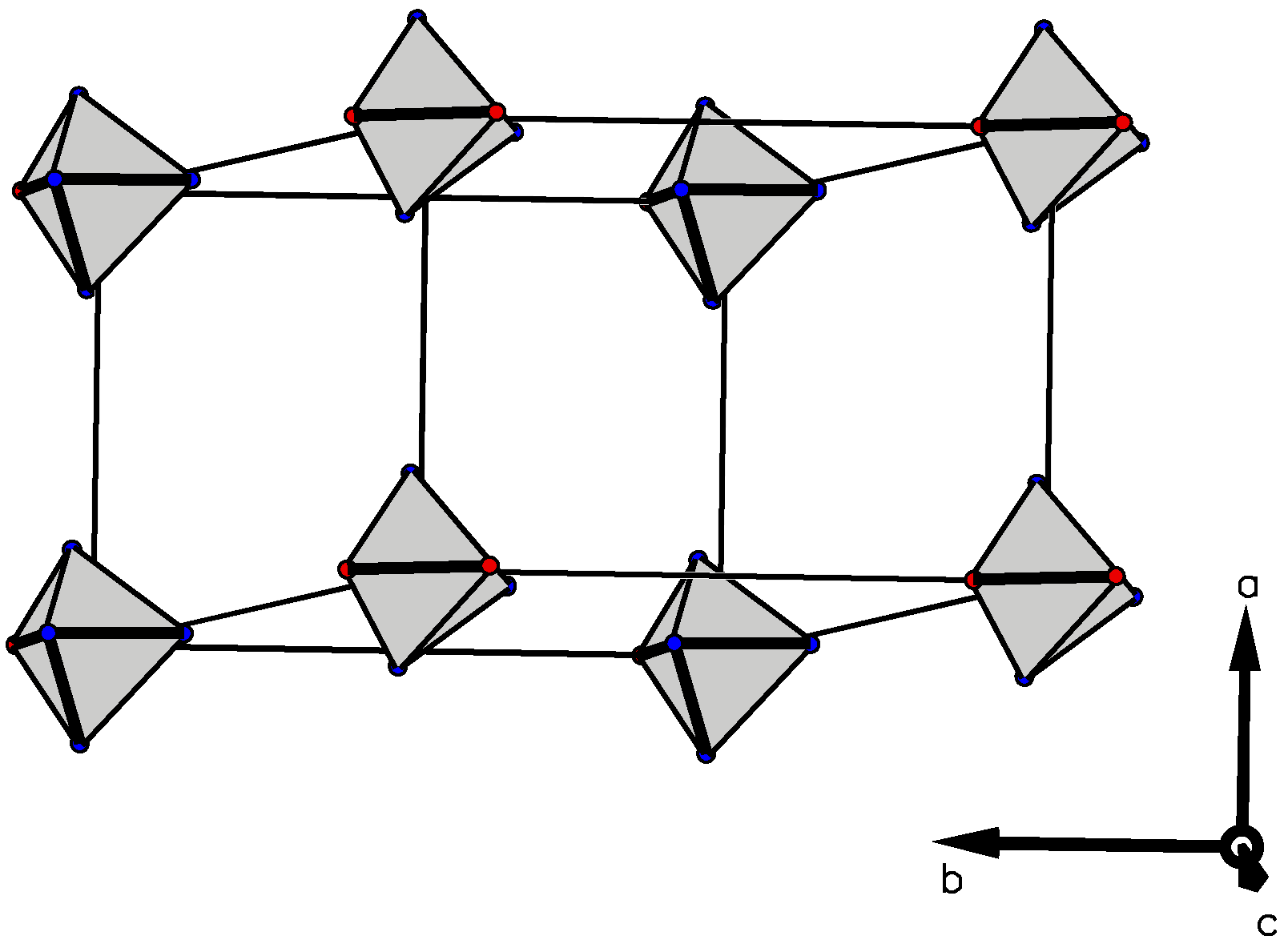
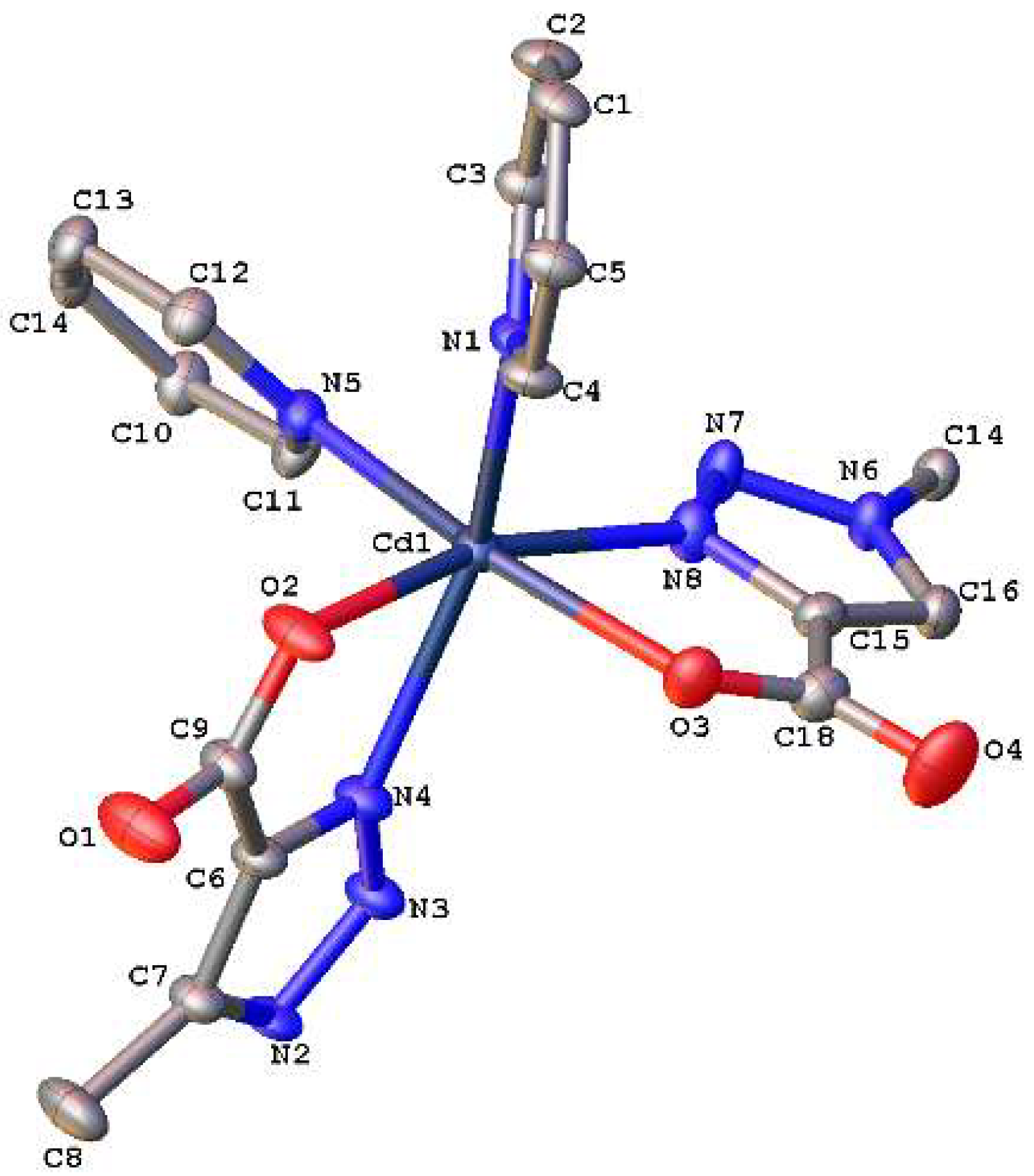
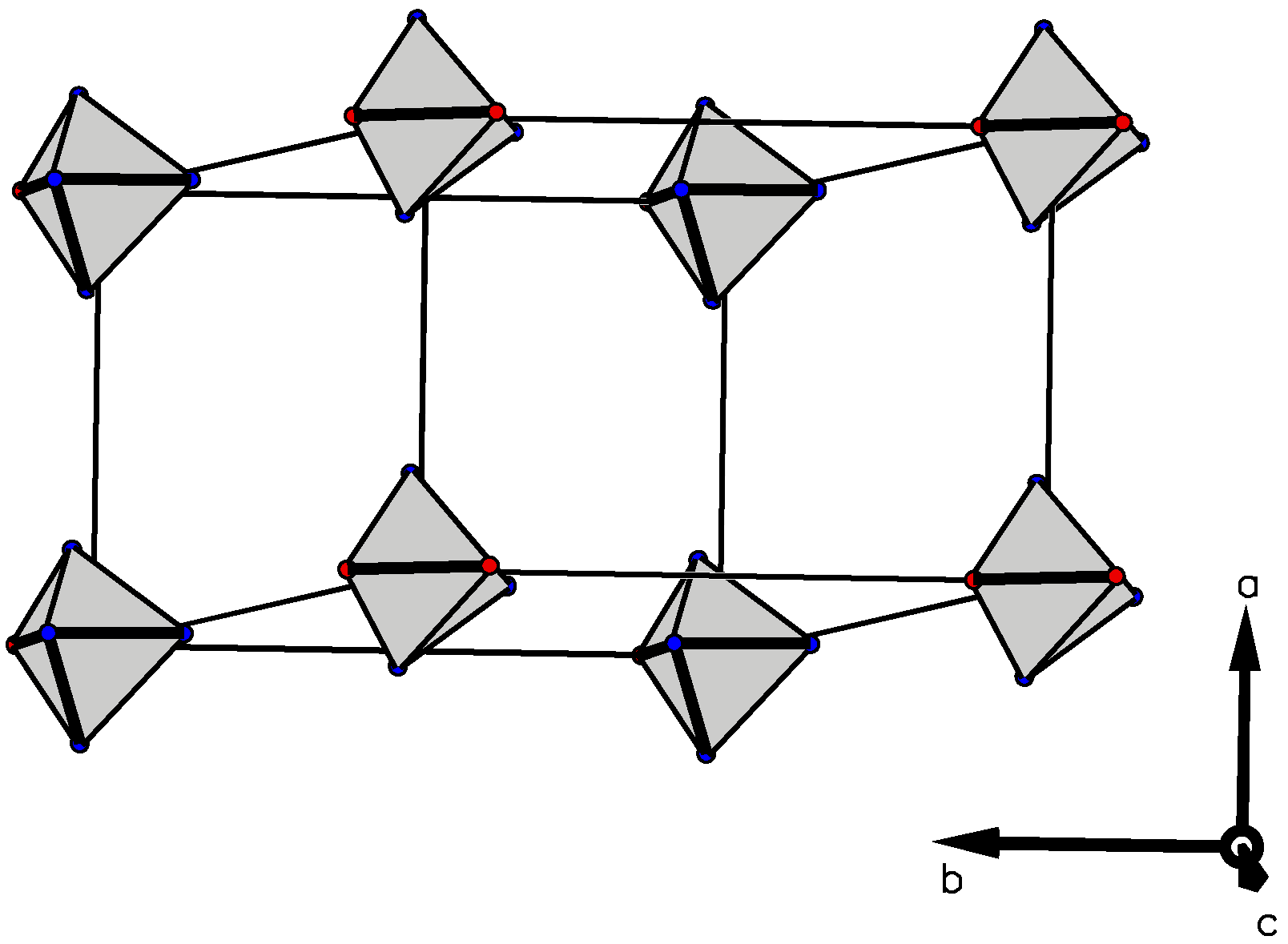
3.3. Crystallographic Studies
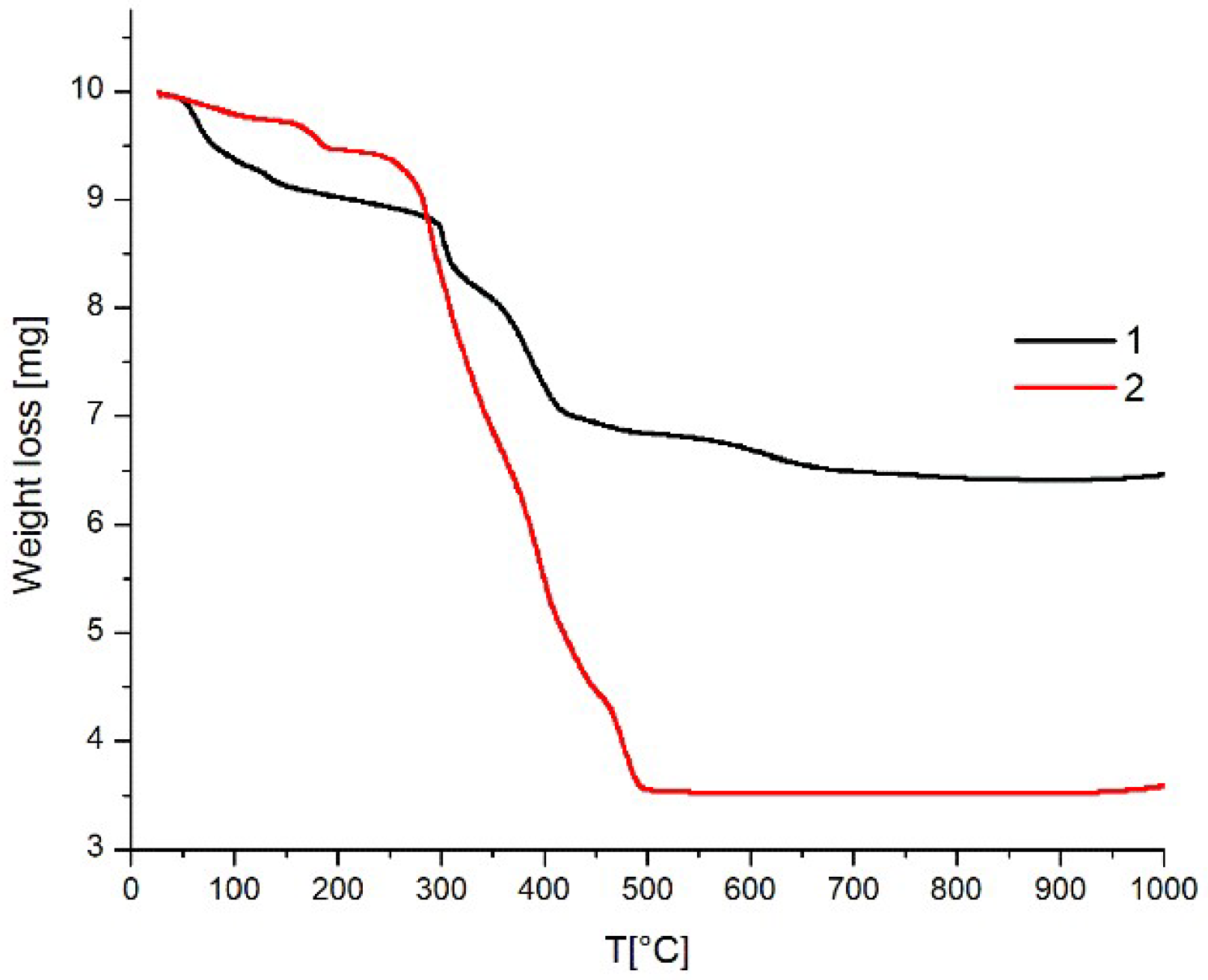
3.4. Thermal Stability Studies
3.5. Emission Spectra Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Robin, A.Y.; Fromm, K.M. Coordination polymer networks with O- and N-donors: What they are, why and how they are made. Coord. Chem. Rev. 2006, 250, 2127–2157. [Google Scholar] [CrossRef]
- Pettinari, C.; Tăbăcaru, A.; Galli, S. Coordination polymers and metal-organic frameworks based on poly(pyrazole)-containing ligands. Coord. Chem. Rev. 2016, 307, 1–31. [Google Scholar] [CrossRef]
- Férey, G. Hybrid porous solids: Past, present, future. Chem. Soc. Rev. 2008, 37, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Almeida Paz, F.A.; Klinowski, J.; Vilela, S.M.F.; Tomé, J.P.C.; Cavaleiro, J.A.S.; Rocha, J. Ligand design for functional metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 1088–1110. [Google Scholar] [CrossRef] [PubMed]
- Bellussi, G.; Carati, A.; Rizzo, C.; Millini, R. New trends in the synthesis of crystalline microporous materials. Catal. Sci. Technol. 2013, 3, 833–857. [Google Scholar] [CrossRef]
- Lalonde, M.; Bury, W.; Karagiaridi, O.; Brown, Z.; Hupp, J.T.; Farha, O.K. Transmetalation: Routes to metal exchange within metal-organic frameworks. J. Mater. Chem. A 2013, 1, 5453–5468. [Google Scholar] [CrossRef]
- Falcaro, P.; Ricco, R.; Doherty, C.M.; Liang, K.; Hill, A.J.; Styles, M.J. MOF positioning technology and device fabrication. Chem. Soc. Rev. 2014, 43, 5513–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, A.U.; Trukhan, N.; Müller, U. Industrial applications of metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houk, R.J.T. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Heine, J.; Müller-Buschbaum, K. Engineering metal-based luminescence in coordination polymers and metal–organic frameworks. Chem. Soc. Rev. 2013, 42, 9232. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yue, Y.; Qian, G.; Chen, B. Luminescent functional metal-organic frameworks. Chem. Rev. 2012, 112, 1126–1162. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Lanthanide-Based Luminescent Hybrid Materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar] [CrossRef] [PubMed]
- Stavila, V.; Talin, A.A.; Allendorf, M.D. MOF-based electronic and opto-electronic devices. Chem. Soc. Rev. 2014, 43, 5994–6010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yam, V.W.-W.; Lo, K.K.-W. Luminescent polynuclear d10 metal complexes. Chem. Soc. Rev. 1999, 28, 323–334. [Google Scholar] [CrossRef]
- Huang, R.W.; Wei, Y.S.; Dong, X.Y.; Wu, X.H.; Du, C.X.; Zang, S.Q.; Mak, T.C. Hypersensitive dual-function luminescence switching of a silver-chalcogenolate cluster-based metal-organic framework. Nat. Chem. 2017, 9, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Su, H.F.; Tan, Y.Z.; Schein, S.; Lin, S.C.; Liu, W.; Wang, S.A.; Wang, W.G.; Tung, C.H.; Sun, D.; et al. Assembly of silver Trigons into a buckyball-like Ag180 nanocage. Proc. Natl. Acad. Sci. USA 2017, 114, 12132–12137. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Feng, L.; Su, H.F.; Wang, Z.; Zhao, Q.Q.; Wang, X.P.; Tung, C.H.; Sun, D.; Zheng, L.S. Anisotropic Assembly of Ag52 and Ag76 Nanoclusters. J. Am. Chem. Soc. 2018, 140, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Du, X.S.; Li, B.; Wang, J.Y.; Li, G.P.; Gao, G.G.; Zang, S.Q. Atom-Precise Modification of Silver(I) Thiolate Cluster by Shell Ligand Substitution: A New Approach to Generation of Cluster Functionality and Chirality. J. Am. Chem. Soc. 2018, 140, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Carlucci, L.; Ciani, G.; Proserpio, D.M. Polycatenation, polythreading and polyknotting in coordination network chemistry. Coord. Chem. Rev. 2003, 246, 247–289. [Google Scholar] [CrossRef]
- Oshio, H.; Saito, Y.; Ito, T. Cluster Assembly by Hydrogen Bonds: Channel Structure of Cu4L4 Cubanes. Angew. Chem. Int. Ed. Engl. 1997, 36, 2673–2675. [Google Scholar] [CrossRef]
- Du, M.; Bu, X.H.; Guo, Y.M.; Liu, H.; Batten, S.R.; Ribas, J.; Mak, T.C. First CuIIdiamondoid net with 2-fold interpenetrating frameworks. The role of anions in the construction of the supramolecular arrays. Inorg. Chem. 2002, 41, 4904–4908. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Noveron, J.C.; Resendiz, M.J.E.; Liu, J.; Yamamoto, T.; Parker, D.; Cinke, M.; Nguyen, C.V.; Arif, A.M.; Stang, P.J. Self-assembly of flexible supramolecular metallacyclic ensembles: Structures and adsorption properties of their nanoporous crystalline frameworks. J. Am. Chem. Soc. 2004, 126, 10645–10656. [Google Scholar] [CrossRef] [PubMed]
- Song, L.C.; Zhang, W.X.; Hu, Q.M. Syntheses and Characterizations of Novel One-dimensional Coordination Polymers Self-assembled from Co(NCS)2 and Flexible Diester-bridged Pyridine-based Ligands. Chin. J. Chem. 2010, 20, 1421–1429. [Google Scholar] [CrossRef]
- Huang, F.P.; Tian, J.L.; Gu, W.; Liu, X.; Yan, S.P.; Liao, D.Z.; Cheng, P. Co(II) coordination polymers: Positional isomeric effect, structural and magnetic diversification. Cryst. Growth Des. 2010, 10, 1145–1154. [Google Scholar] [CrossRef]
- Du, M.; Jiang, X.J.; Zhao, X.J. Molecular tectonics of mixed-ligand metal-organic frameworks: Positional isomeric effect, metal-directed assembly, and structural diversification. Inorg. Chem. 2007, 46, 3984–3995. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.P.; Tian, J.L.; Chen, G.J.; Li, D.D.; Gu, W.; Liu, X.; Yan, S.P.; Liao, D.Z.; Cheng, P. A case study of the ZnII-BDC/bpt mixed-ligand system: Positional isomeric effect, structural diversification and luminescent properties. Cryst. Eng. Comm. 2010, 12, 1269–1279. [Google Scholar] [CrossRef]
- Liu, T.; Wang, S.; Lu, J.; Dou, J.; Niu, M.; Li, D.; Bai, J. Positional isomeric and substituent effect on the assemblies of a series of d10 coordination polymers based upon unsymmetric tricarboxylate acids and nitrogen-containing ligands. Cryst. Eng. Comm. 2013, 15, 5476–5489. [Google Scholar] [CrossRef]
- Li, N.Y.; Ge, Y.; Wang, T.; Wang, S.J.; Ji, X.Y.; Liu, D. Construction of a series of coordination polymers based on 1,4-naphthalenedicarboxylic and flexible dipyridyl ligands. Cryst. Eng. Comm. 2014, 16, 2168. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEX3 Package. APEX3, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Brito, I.; Kesternich, V.; Pérez-Fehrmann, M.; Araneda, C.; Cárdenas, A. Crystal structure of ethyl 5-methyl-1-(pyridin-3-yl)-1H-1,2,3-triazole-4-carboxylate, C11H12N4O2. Zeitschrift Für Krist—New Cryst. Struct. 2017, 232, 1011–1012. [Google Scholar] [CrossRef]
- Senthil, S.; Kamalraj, V.R.; Wu, S.L. Synthesis and characterization of ferroelectric liquid crystal dimers containing thioester and carboxylate linking groups in the inner side of the molecule. J. Mol. Struct. 2008, 886, 175–182. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths Determined by X-ray and Neutron-Diffraction. 1. Bond Lengths in Organic-Compounds. J. Chem. Soc.; Perkin Trans 2. 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Cisterna, J.; Cárdenas, A.; Brito, I. Crystal structure of 4-aminopyridinium 4-acetyl-(pyridin-4-yl)-1H-1,2,3-triazol-5-olate monohydrate, C14H16N6O3. Zeitschrift Für Krist—New Cryst. Struct. 2018, 233, 571–573. [Google Scholar] [CrossRef]
- Macksasitorn, S.; Hu, Y.; Stork, J.R. Homoconjugated 4-aminopyridine salts: Influence of anions on network topology. Cryst. Eng. Comm. 2013, 15, 1698–1705. [Google Scholar] [CrossRef]
- Lu, M.; Wang, L.Y.; Hu, J.L.; Wang, Y.Y.; Yang, G.P. Syntheses, structures, and photoluminescence of a series of d10 coordination polymers with R-isophthalate (R. = -OH, -CH3, and -C.(CH3)3). Cryst. Growth Des. 2009, 9, 5334–5342. [Google Scholar] [CrossRef]
- Meng, G.X.; Feng, Y.M.; Wang, Y.; Gao, Y.L.; Wan, W.W.; Huang, X.T. Effect of pH on the self-assembly of three Cd(II) coordination compounds containing 5-(4-pyridyl)tetrazole: Syntheses, structures, and properties. J. Coord. Chem. 2015, 68, 1705–1718. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, G.P.; Zhao, Y.; Wu, W.P.; Liu, B.; Wang, Y.Y. Three new solvent-directed Cd(ii)-based MOFs with unique luminescent properties and highly selective sensors for Cu2+cations and nitrobenzene. Dalt. Trans. 2015, 44, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Dobretsov, G.E.; Syrejschikova, T.I.; Smolina, N.V. On mechanisms of fluorescence quenching by water. Biophysics (Oxf) 2014, 59, 183–188. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | L1H | L2H | 1 | 2 |
|---|---|---|---|---|
| Empirical Formula | C9H8N4O2 | C9H8N4O2 | C18H22CdN8O8 | [C18H14CdN8O4]n |
| Formula mass, g·mol−1 | 204.19 | 204.19 | 590.84 | 518.77 |
| Collection T, K | 295.21 | 296.27 | 295.51 | 297.22 |
| crystal system | orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| space group | Pna21 | Cc | P21/c | P21/n |
| a (Å) | 13.4617(12) | 18.5248(10) | 9.9864(5) | 10.7368(8) |
| b (Å) | 3.7613(3) | 3.7425(2) | 7.3650(4) | 14.8147(14) |
| c (Å) | 17.9448(16) | 13.0186(7) | 15.7758(9) | 15.7859(14) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 92.154(4) | 104.846(3) | 99.017(3) |
| γ (°) | 90 | 90 | 90 | 90 |
| V (Å3) | 908.61(14) | 901.93(8) | 1121.57(11) | 2479.9(4) |
| Z | 4 | 4 | 2 | 4 |
| ρcalcd (g·cm−3) | 14.926 | 1.504 | 1.750 | 1.389 |
| Crystal size (mm) | 0.402 × 0.24 × 0.108 | 0.414 × 0.129 × 0.114 | 0.13 × 0.102 × 0.073 | 0.459 × 0.433 × 0.258 |
| F(000) | 425.5 | 424.0 | 596.0 | 1032.0 |
| abs coeff (mm−1 ) | 0.931 | 0.938 | 8.372 | 0.917 |
| θ range (°) | 9.86 to 122.94 | 9.556 to 117.866 | 9.16 to 118.36 | 5.5 to 56.75 |
| range h,k,l | −14/15, −4/4, −19/19 | −20/20, −4/4, −13/14 | −11/11, −8/8, −17/17 | −12/14, −1 9/19, −21/21 |
| No. total refl. | 13089 | 8282 | 24642 | 52471 |
| No. unique refl. | 1328 | 1242 | 1598 | 6169 |
| Comp. θmax (%) | 94.0 | 97.0 | 98.3 | 99.4 |
| Max/min transmission | 0.765/0.904 | 0.865/0.899 | 0.413/0.543 | 0.663/0.78 |
| Data/Restraints/Parameters | 1328/5/138 | 1242/2/139 | 1598/0/164 | 6169/0/282 |
| Final R [I > 2σ(I)] | R1 = 0.0490, wR2 = 0.1363 | R1 = 0.0445, wR2 = 0.1004 | R1 = 0.0392, wR2 = 0.0779 | R1 = 0.0415, wR2 = 0.0922 |
| R indices (all data) | R1 = 0.0618, wR2 = 0.1415 | R1 = 0.0557, wR2 = 0.1077 | R1 = 0.0614, wR2 = 0.0849 | R1 = 0.0631, wR2 = 0.1041 |
| Goodness of fit/F2 | 1.094 | 1.124 | 1.065 | 1.036 |
| Largest diff. Peak/hole(eÅ−3) | 0.33/−0.35 | 0.18/−0.15 | 0.72/−0.58 | 1.93/−0.63 |
| Flack Parameter | 0.0(5) | 0.4(2) | --- | --- |
| Compound | λmax Excitation (nm) | λmax Emission (nm) |
|---|---|---|
| L1 | 366 | 448 |
| L2 | 360 | 413 |
| 1 | 366 | 446 |
| 2 | 366 | 412 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cisterna, J.; Araneda, C.; Narea, P.; Cárdenas, A.; Llanos, J.; Brito, I. The Positional Isomeric Effect on the Structural Diversity of Cd(II) Coordination Polymers, Using Flexible Positional Isomeric Ligands Containing Pyridyl, Triazole, and Carboxylate Fragments. Molecules 2018, 23, 2634. https://doi.org/10.3390/molecules23102634
Cisterna J, Araneda C, Narea P, Cárdenas A, Llanos J, Brito I. The Positional Isomeric Effect on the Structural Diversity of Cd(II) Coordination Polymers, Using Flexible Positional Isomeric Ligands Containing Pyridyl, Triazole, and Carboxylate Fragments. Molecules. 2018; 23(10):2634. https://doi.org/10.3390/molecules23102634
Chicago/Turabian StyleCisterna, Jonathan, Catherine Araneda, Pilar Narea, Alejandro Cárdenas, Jaime Llanos, and Iván Brito. 2018. "The Positional Isomeric Effect on the Structural Diversity of Cd(II) Coordination Polymers, Using Flexible Positional Isomeric Ligands Containing Pyridyl, Triazole, and Carboxylate Fragments" Molecules 23, no. 10: 2634. https://doi.org/10.3390/molecules23102634