First Organocatalytic Asymmetric Synthesis of 1-Benzamido-1,4-Dihydropyridine Derivatives



Laboratorio de Organocatálisis Asimétrica, Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) CSIC-Universidad de Zaragoza, C/Pedro Cerbuna 12, E-50009 Zaragoza, Spain
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(10), 2692; https://doi.org/10.3390/molecules23102692
Submission received: 30 September 2018
/
Revised: 16 October 2018
/
Accepted: 17 October 2018
/
Published: 19 October 2018
(This article belongs to the Special Issue Stereogenic Centers)
Abstract
:Preliminary results concerning the first asymmetric synthesis of highly functionalized 1-benzamido-1,4-dihydropyridine derivatives via the reaction of hydrazones with alkylidenemalononitriles in the presence of β-isocupreidine catalyst are reported. The moderate, but promising, enantioselectivity observed (40–54% ee), opens the door to a new area of research for the asymmetric construction of new chiral 1,4-dihydropyridine derivatives, whose enantioselective catalytic preparation are still very limited. Moreover, the use of hydrazones for the enantioselective construction of chiral 1,4-dihydropyridines has been overlooked in the literature so far. Therefore, our research represents a pivotal example in this field which remains still unexplored.
1. Introduction
1,4-Dihydropyridine (1,4-DHP) [1,2,3,4,5] ring is a challenging structural core in organic chemistry due to its biological properties [6,7,8,9], especially as calcium channel blockers (Figure 1) [10,11]. It is noteworthy that its range of application has been recently extended to other affections such as antioxidant, antidiabetic and antitumor agents [12].
Therefore, the potential of 1,4-dihydropyridines as valuable building blocks in organic synthesis has attracted the attention of many scientists [13,14]. Moreover, it has been found that the stereochemistry at C-4 can be related with both qualitative and quantitative differences in their biological activity. Thus, the control of the stereoselectivity in this chiral center has become an inspiring task of research and, therefore, there is a growing interest for the development of new enantioselective methods. However, there are only a few examples of organocatalytic enantioselective syntheses to obtain these compounds [15,16].
Recently, we have successfully contributed to this field with two pioneering works (Scheme 1) [16,17]. In the first example, we used Takemoto’s thiourea to synthesize chiral 2-oxospiro-[indole-3,4′-(1′,4′-dihydropyridine)] derivatives 1 with good reactivity and promising enantioselectivities (Scheme 1, route a) [17]. The most recent work has allowed us to obtain for the first time a family of substituted chiral 1,4-dihydropyridines 2 with very good results, using a bis-cinchona derivative as organocatalyst (Scheme 1, route b) [16].
Related to our work reported herein, we must remark that Yan and co-workers previously published two interesting racemic versions using benzohydrazides 3, instead of anilines, in two concomitant multicomponent processes to obtain 1-benzamidospiro[indoline-3,4′-pyridines] 4 [18] or benzamido-1,4-dihydropyridines 5 [19] (Scheme 2).
Based on this idea, we envisioned that the use of chiral amine-based catalysts could provide the first asymmetric version of a related reaction. Moreover, to the best of our knowledge, the use of hydrazones for the enantioselective construction of 1,4-dihydropyridines has been overlooked in the literature so far. It is worth noting that the development of new asymmetric strategies to obtain enantioenriched 1,4-DHPs is still desirable since there are only scarce examples in this field of research [15,16].
2. Results and Discussion
2.1. Synthesis of Starting Materials: Hydrazones 7 and Alkylidenemalononitriles 9
On the base of our recent works [16,17], we hypothesized that the enamine generated from hydrazones 7 could provide the same reactivity as reported in Scheme 1 and Scheme 2. With this aim in mind, we firstly synthesized six different hydrazones 7a–f as described below in Scheme 3.
Hydrazones 7 were prepared after 12 h at room temperature with quantitative yields from differently substituted benzohydrazides 3, bearing different electronic properties in the aromatic ring (electron-withdrawing or electron-donating groups or none of them) (Scheme 3). In contrast to our previous works [16,17], the reaction between alkynyl 6 and hydrazide 3 leads to pure hydrazones 7 instead of the corresponding enamine (shown in Scheme 1), as previously obtained by us in the reaction between alkynyl 6 and an aniline.
2.2. Screening of the Reaction Conditions
To carry out the synthesis of the first chiral 1-benzamido-1,4-dihydropyridine derivatives 10 [16,17], we foresaw that a chiral organic base catalyst could promote this reaction, starting directly from the preformed intermediates: hydrazones 7 and malononitriles 9, giving rise to the desired final benzamido-1,4-dihydropyridines 10. To explore the viability of this hypothesis, the efficiency of different chiral organocatalysts I-X, with a base moiety in their structure, was initially studied in a model reaction between hydrazone 7a and malononitrile 9a (Scheme 5).
As reported in Scheme 5, the most promising value of enantioselectivity was achieved with β-isocupreidine X (32% ee), while the best reactivity was found with (DHQ)2Phal VI (81% yield). These results encouraged us to continue with both catalysts VI and X in the subsequent screening of different parameters to optimize this process (Table 1).
As shown in Table 1, parameters such as solvent, concentration of the reaction, amount of each reagent and catalyst were analyzed. In general, catalyst VI still provided better reactivity in some reactions media, such as MeCN, AcOEt, CH2Cl2 or MeOH (entries 1, 3, 5 and 13), in comparison with catalyst X (entries 2, 4, 6 and 14). However, better enantioselectivities were found with catalyst X in almost all solvents (except in CH2Cl2, entry 6). The best compromise in terms of reactivity and enantioselectivity was achieved in THF (entries 12, 15–20) using catalyst X. Concentration of the reaction medium led to a slight decrease in the enantioselectivity of the process with a smooth increase of the reactivity (entry 17). The opposite behavior is true when the reaction was diluted (entry 18). Variations in the amount of catalyst did not provide an appreciable improvement of the process (entries 19 and 20). Increasing the amount of reagents 7a or 9a gave rise to similar results in both cases (entries 15 and 16), although the use of 1.5 equivalent of 9a led to slightly better reactivity (entry 15). Therefore, we continue to the ensuing exploration of the scope of the reaction with these conditions (entry 15), as they are the best ones at this stage.
2.3. Scope of the Reaction
Once the reaction conditions were optimized, a series of 1-benzamido-1,4-dihydropyridine derivatives 10 were synthesized as shown in Scheme 6.
The final products 10 were isolated with high yields (up to 99%) and with moderate but promising enantioselectivities (up to 54% ee) since this work represents the first chiral version to obtain enantioenriched 1-benzamido-1,4-dihydropyridines. The results suggest a dependence of the reactivity of the process on the electronic properties of the aromatic ring in the alkylidenemalononitriles 9, since electron-withdrawing groups (4-NO2, 3-NO2, 3-Cl, 4-Cl, 4-Br or 4-CN) or heteroaromatic rings (2-furyl, 2-thienyl, 3-pyridyl) afforded better results in comparison with electrondonating substituents (4-Me and 4-MeO) or in the absence of substituents (1-naphthyl or phenyl). On the other hand, the enantioselectivity of the process does not have a clear correlation with the electronic properties of the aromatic ring in the alkylidenemalononitriles 9. Interestingly, we were able to obtain the desired 1-benzamido-1,4-dihydropyridines 10 changing the substituent in the aromatic ring of the hydrazone (7b–f), achieving more similar results in terms of enantioselectivity and reactivity than those obtained with hydrazone 7a.
Based on our previously reported works for the obtainment of chiral 1,4-dihydropyridine derivatives [16,17], in the field of cinchona alkaloids and in our own experience using this kind of organocatalysts [20,21,22], we think that the same mechanism could be operating in this case [16,17]. Thus, a first Michael addition reaction between the enamine generated in situ from hydrazones 7 and the alkylidenemalononitriles 9, followed by an intramolecular nucleophilic cyclization and a final tautomerization would yield 1-benzamido-1,4-dihydropyridines 10. However, in order to really know if β-isocupreidine X is acting as a bifunctional catalyst [23,24,25] and to better understand the role of this structure, more studies are ongoing in our lab.
3. Materials and Methods
3.1. General Experimental Methods
Purification of reaction products was carried out by column chromatography using silical-gel (0.063–0.200 mm). Analytical thin layer chromatography was performed on 0.25 mm silical gel 60-F plates. ESI (Zaragoza, Spain) and MicroTof-Q Bruker mass analyzer (Zaragoza, Spain) were used for HRMS measurements. HPLC was performed on analytical HPLC Waters (Delta 600 Separation Module, 2996 Photodiode Array Detector, Zaragoza, Spain). IR spectra have been registered in a PerkinElmer Spectrum 100 FT-IR Spectrometer (Zaragoza, Spain). The optical rotation measurements were taken on a JASCO Digital polarimeter DIP-370 (Zaragoza, Spain). NMR spectroscopy was conducted using a Bruker AVANCE–II spectrometer (Zaragoza, Spain). 1H-NMR spectra were recorded at 300 and 400 MHz; 13C-APT-NMR spectra were recorded at 75 and 100 MHz; (min: minor isomer); DMSO-d6 was used as the deuterated solvent. Chemical shifts were reported in the δ scale relative to residual DMSO (2.50 ppm) for 1H-NMR and to the central line of DMSO-d6 (39.52 ppm) for 13C-APT-NMR. Spectral data for many of the synthesized compounds are consistent with values previously reported in the literature: hydrazone 7a [26]; alkylidenemalononitriles 9a [27], 9b [27], 9c [28], 9d [28], 9e [27], 9f [29], 9g [30], 9i [27], 9j [27], 9k [28], 9l [31] and 9m [31]; and 1,4-dihydropyridines 10ac [19], 10ae [19], 10af [19] and 10ai [19]. Melting points were recorded on a Gallenkamp MDP350 Variable Heater melting point apparatus without correction. Catalysts I [32], II [32], VI [33], VII [34], VIII [33], IX [35] and X [36] were commercially available and III [37], IV [38] and V [38] were synthesized as reported.
3.2. General Procedure for the Synthesis of Hydrazones 7
To a solution of the corresponding hydrazide 3 (5 mmol) in ethanol (15 mL), dimethyl acetylenedicarboxylate 6 (5 mmol, 615 μL) was slowly added due to the exothermic characteristics of the reaction. The reaction mixture was stirred for 12 h at room temperature. Then, the solvent was evaporated under vacuum, and the reaction crude was purified by filtration, washing the white precipitate with small portions of ethanol (3 × 3 mL) giving rise to the corresponding final adduct 7 (Scheme 3).
(E)-Dimethyl 2-(2-(benzoyl)hydrazono)succinate (7a) [26]: Following the general procedure, compound 7a was obtained as a white solid in a 98% yield.
(E)-Dimethyl 2-(2-(4-nitrobenzoyl)hydrazono)succinate (7b): Following the general procedure, compound 7b was obtained as a white solid in a 98% yield. mp 146–148 °C. 1H-NMR (300 MHz, DMSO-d6) δ 3.65 (s, 3H), 3.76 (s, 3H), 3.90 (s, 2H), 8.06 (d, J = 8.1 Hz, 2H), 8.35 (d, J = 8.5 Hz, 2H), 11.70 (s, 1H). 13C-APT-NMR (75 MHz, DMSO-d6) δ 32.6 (1C), 52.2 (1C), 52.6 (1C), 123.3 (2C), 130.3 (2C), 138.8 (1C), 149.3 (1C), 164.4 (1C), 168.2 (3C). IR (neat) (cm–1) ν 3451, 3244, 3113, 1729, 1690, 1671, 1520, 1349, 1241, 1124, 1005, 852, 719. HRMS (ESI+) calcd for C13H13N3NaO7 346.0646; found 346.0649 [M + Na].
(E)-Dimethyl 2-(2-(4-chlorobenzoyl)hydrazono)succinate (7c): Following the general procedure, compound 7c was obtained as a white solid in a 99% yield. mp 138–140 °C. 1H-NMR (300 MHz, DMSO-d6) δ 3.64 (s, 3H), 3.76 (s, 3H), 3.89 (s, 2H), 7.60 (d, J = 8.6 Hz, 2H), 7.87 (d, J = 8.6 Hz, 2H), 11.47 (s, 1H). 13C-APT-NMR (75 MHz, DMSO-d6) δ 32.5 (1C), 52.2 (1C), 52.5 (1C), 128.4 (2C), 130.7 (2C), 131.7 (1C), 137.0 (1C), 164.5 (1C), 168.3 (3C). IR (neat) (cm–1) ν 3441, 3238, 2954, 1741, 1726, 1694, 1670, 1594, 1536, 1440, 1250, 1146, 1123, 1111, 1089, 1003, 889, 846, 756. HRMS (ESI+) calcd for C13H13ClN2NaO5 335.0405; found 335.0385 [M + Na].
(E)-Dimethyl 2-(2-(4-bromobenzoyl)hydrazono)succinate (7d): Following the general procedure, compound 7d was obtained as a white solid in a 99% yield. mp 114–116 °C. 1H-NMR (300 MHz, DMSO-d6) δ 3.64 (s, 3H), 3.76 (s, 3H), 3.89 (s, 2H), 7.77 (q, J = 8.4 Hz, 4H), 11.48 (s, 1H). 13C-APT-NMR (75 MHz, DMSO-d6) δ 30.9 (3C), 32.5 (1C), 52.2 (1C), 52.6 (1C), 125.9 (1C), 130.9 (2C), 131.3 (2C), 132.1 (2C), 164.5 (1C), 168.3 (2C). IR (neat) (cm–1) ν 3256, 2953, 1734, 1716, 1685, 1591, 1434, 1222, 1201, 1127, 1111, 1009, 889, 838, 752. HRMS (ESI+) calcd for C13H13BrN2NaO5 378.9900; found 378.9903 [M + Na].
(E)-Dimethyl 2-(2-(4-tert-butylbenzoyl)hydrazono)succinate (7e): Following the general procedure, compound 7e was obtained as a white solid in a 98% yield. mp 165–167 °C. 1H-NMR (300 MHz, DMSO-d6) δ 1.31(s, 9H), 3.64 (s, 3H), 3.77 (s, 3H), 3.89 (s, 2H), 7.54 (d, J = 8.6 Hz, 2H), 7.78 (d, J = 8.6 Hz, 2H), 11.33 (s, 1H). 13C-APT-NMR (75 MHz, DMSO-d6) δ 30.9 (3C), 32.4 (1C), 34.8 (1C), 52.1 (1C), 52.5 (1C), 125.1 (2C), 128.6 (2C), 130.3 (1C), 155.2(1C), 164.6 (1C), 168.4 (3C). IR (neat) (cm–1) ν 3232, 3201, 2964, 2949, 1741, 1721, 1671, 1609, 1536, 1432, 1246, 1205, 1150, 1126, 1118, 1021, 895, 857, 841, 707. HRMS (ESI+) calcd for C17H22N2NaO5 357.1421; found 357.1419 [M + Na].
(E)-Dimethyl 2-(2-(4-methoxybenzoyl)hydrazono)succinate (7f): Following the general procedure, compound 7f was obtained as a white solid in a 98% yield. mp 144–146 °C. 1H-NMR (300 MHz, DMSO-d6) δ 3.64 (s, 3H), 3.76 (s, 3H), 3.84 (s, 3H), 3.89 (s, 2H), 7.06 (d, J = 8.9 Hz, 2H), 7.87 (d, J = 8.9 Hz, 2H), 11.25 (s, 1H). 13C-APT-NMR (75 MHz, DMSO-d6) δ 32.4 (1C), 52.2 (1C), 52.5 (1C), 55.5 (1C), 113.6 (2C), 124.9 (1C), 130.9 (2C), 162.4 (1C), 164.6 (1C), 168.5 (3C). IR (neat) (cm–1) ν 3419, 3223, 3189, 1737, 1715, 1659, 1601, 1541, 1508, 1436, 1319, 1256, 1205, 1170, 1143, 1109, 1027, 996, 889, 849, 762. HRMS (ESI+) calcd for C14H16N2NaO6 331.0901; found 331.0888 [M + Na].
3.3. General Procedure for the Synthesis of 1,4-Dihydropyridines 10
To a mixture of β-isocupreidine catalyst X (20 mol%, 6.21 mg) and the corresponding benzylidenemalononitrile 9 (0.15 mmol) in tetrahydrofuran (0.5 mL), hydrazones 7 (0.1 mmol) were added. The reaction mixture was stirred for 5 days at room temperature. Then, the reaction crude was purified by column chromatography (SiO2, n-hexane:diethyl ether 20:80 to 0:100), giving rise to the corresponding final chiral adducts 10 (Scheme 6).
Dimethyl 6-amino-1-benzamido-5-cyano-4-phenyl-1,4-dihydropyridine-2,3-dicarboxylate (10aa): Following the general procedure, compound 10aa was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 61% yield (26.37 mg). mp 106–108 °C. The ee of the product was determined to be 50% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 237.7 nm): τmajor = 30.4 min; τminor = 11.1 min. [α]D24 = −25.4 (c = 0.07, MeOH, 50% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.51 (s, 3H), 3.57 (s, 0.75H), 3.64 (s, 2.25H), 4.36 (s, 0.75H), 4.48 (s, 0.25H), 6.38 (s, 1.5H), 6.46 (s, 0.5H), 7.24 (t, J = 7.3 Hz, 1.5H), 7.35 (t, J = 7.4 Hz, 2H), 7.45–7.59 (m, 3.5H), 7.63 (t, J = 7.1 Hz, 1H), 7.79–7.91 (m, 2H), 11.21 (s, 0.75H), 11.32 (s, 0.25H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.2 (1C), 51.8 (1C), 52.8 (1C), 58.6 (1C), 104.6 (1C), 120.9 (1C), 126.8 (2C), 127.7 (1C), 127.9 (2C), 128.3 (2C), 128.5 (2C), 131.3 (1C), 132.5 (1C), 142.6 (1C), 145.7 (1C), 151.1 (1C), 162.4 (1C), 164.7 (1C), 166.7 (1C). IR (neat) (cm–1) ν 3420, 3334, 3249, 3219, 2957, 2190, 1736, 1707, 1662, 1590, 1479, 1428, 1225, 1110, 717, 699, 689. HRMS (ESI+) calcd for C23H20N4NaO5 455.1326; found 455.1340 [M + Na].
Dimethyl 6-amino-5-cyano-1-(4-nitrobenzamido)-4-phenyl-1,4-dihydropyridine-2,3-dicarboxylate (10ba): Following the general procedure, compound 10ba was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a yellow solid in 56% yield (26.82 mg). mp 238–240 °C. The ee of the product was determined to be 45% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 249.6 nm): τmajor = 47.0 min; τminor = 21.4 min. [α]D24 = −6.6 (c = 0.13, MeOH, 45% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.51 (s, 3H), 3.58 (s, 0.78H), 3.65 (s, 2.22H), 4.37 (s, 0.74H), 4.44 (s, 0.26H), 6.55 (s, 1.48H), 6.60 (s, 0.52H), 7.24 (t, J = 7.3 Hz, 1.48H), 7.35 (t, J = 7.5 Hz, 2H), 7.52 (d, J = 7.2 Hz, 1.52H), 8.04 (d, J = 8.8 Hz, 0.52H), 8.10 (d, J = 8.8 Hz, 1.48H), 8.38 (d, J = 8.8 Hz, 2H), 11.62 (s, 0.74H), 11.68 (s, 0.26H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.2 (1C), 51.8 (1C), 52.9 (1C), 58.1 (1C), 104.9 (1C), 120.8 (1C), 123.5 (min), 123.6 (2C), 126.9 (1C), 127.6 (2C), 128.3 (2C), 128.6 (min), 129.4 (min), 129.5 (2C), 137.0 (1C), 142.2 (1C), 145.6 (1C), 149.7 (1C), 151.0 (1C), 162.4 (1C), 164.6 (1C), 165.4 (1C). IR (neat) (cm–1) ν 3420, 3331, 3208, 2955, 2923, 2852, 2193, 1722, 1710, 1679, 1660, 1589, 1527, 1430, 1346, 1231, 1117, 1081, 697. HRMS (ESI+) calcd for C23H19N5NaO7 500.1177; found 500.1175 [M + Na].
Dimethyl 6-amino-1-(4-chlorobenzamido)-5-cyano-4-phenyl-1,4-dihydropyridine-2,3-dicarboxylate (10ca): Following the general procedure, compound 10ca was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 75% yield (34.88 mg). mp 143–145 °C. The ee of the product was determined to be 43% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 254.0 nm): τmajor = 24.2 min; τminor = 11.6 min. [α]D24 = −26.7 (c = 0.10, MeOH, 43% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.49 (s, 0.9H), 3.51 (s, 2.1H), 3.56 (s, 0.9H), 3.63 (s, 2.1H), 4.35 (s, 0.7H), 4.47 (s, 0.3H), 6.46 (s, 1.4H), 6.50 (s, 0.6H), 7.24 (t, J = 7 Hz, 1.4H), 7.30-7.38 (m, 2.2H), 7.53 (d, J = 7,1 Hz, 1.4H), 7.63 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 8.6 Hz, 0.6H), 7.88 (d, J = 8.6 Hz, 1.4H), 11.33 (s, 0.7H), 11.42 (s, 0.3H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.1 (1C), 51.9 (1C), 52.8 (min), 52.9 (1C), 57.2 (min), 58.3 (1C), 104.7 (1C), 120.9 (1C), 121.0 (min), 126.9 (min), 127.0 (1C), 127.7 (2C), 128.4 (2C), 128.5 (min), 128.6 (2C), 129.9 (2C), 130.2 (1C), 130.3 (min), 137.2 (1C), 137.4 (1C), 142.5 (1C), 143.0 (1C), 145.7 (1C), 151.1 (1C), 151.6 (1C), 162.4 (1C), 164.7 (1C), 164.8 (1C), 165.3 (1C), 165.8 (1C). IR (neat) (cm–1) ν 3413, 3331, 3251, 3025, 2956, 2192, 1727, 1715, 1664, 1590, 1432, 1335, 1230, 1093, 1014, 698. HRMS (ESI+) calcd for C23H19ClN4NaO5 489.0912; found 489.0913 [M + Na].
Dimethyl 6-amino-1-(4-bromobenzamido)-5-cyano-4-phenyl-1,4-dihydropyridine-2,3-dicarboxylate (10da): Following the general procedure, compound 10da was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a yellow solid in 60% yield (30.81 mg). mp 134–136 °C. The ee of the product was determined to be 44% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 330.0 nm): τmajor = 24.6 min; τminor = 10.7 min. [α]D24 = −20.3 (c = 0.15, MeOH, 44% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.49 (s, 0.75H), 3.51 (s, 2.25H), 3.56 (s, 0.75H), 3.63 (s, 2.25H), 4.35 (s, 0.75H), 4.47 (s, 0.25H), 6.45 (s, 1.5 H), 6.50 (s, 0.50H), 7.24 (t, J = 7.3 Hz, 1.5H), 7.34 (t, J = 7.5 Hz, 2H), 7.53 (d, J = 7.1 Hz, 1.5H), 7.73–7.84 (m, 4H), 11.33 (s, 0.75H), 11.42 (s, 0.25H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.2 (1C), 51.9 (1C), 52.8 (1C), 58.3 (1C), 104.7 (1C), 120.9 (1C), 126.3 (1C), 126.8 (1C), 126.9 (min), 127.6 (2C), 128.3 (2C), 128.6 (min), 130.0 (2C), 130.5 (min), 131.4 (2C), 131.5 (1C), 142.4 (1C), 145.6 (1C), 151.1 (1C), 162.4 (1C), 164.6 (1C), 165.9 (1C). IR (neat) (cm–1) ν 3417, 3331, 3240, 2955, 2190, 1716, 1663, 1588, 1431, 1230, 1079, 1010, 698. HRMS (ESI+) calcd for C23H19BrN4NaO5 533.0418; found 533.0418 [M + Na].
Dimethyl 6-amino-1-(4-(tert-butyl)benzamido)-5-cyano-4-phenyl-1,4-dihydropyridine-2,3-dicarboxylate (10ea): Following the general procedure, compound 10ea was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a yellow solid in 58% yield (28.33 mg). mp 141–143 °C. The ee of the product was determined to be 48% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 242.5 nm): τmajor = 13.2 min; τminor = 9.1 min. [α]D24 = −24.7 (c = 0.15, MeOH, 48% ee). 1H-NMR (400 MHz, DMSO-d6) δ 1.31 (s, 9H), 3.51 (s, 3H), 3.58 (s, 0.78H), 3.66 (s, 2.22H), 4.36 (s, 0.74H), 4.48 (s, 0.26H), 6.33 (s, 1.48H), 6.38 (s, 0.52H), 7.24 (t, J = 7.4 Hz, 1.48H), 7.35 (t, J = 7.4 Hz, 2H), 7.56 (d, J = 8.2 Hz, 3.52H), 7.74–7.84 (m, 2H), 11.13 (s, 0.74H), 11.13 (s, 0.26H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 30.9 (1C), 34.8 (1C), 39.2 (1C), 51.9 (1C), 52.8 (1C), 58.7 (1C), 104.5 (1C), 120.9 (1C), 125.2 (min), 125.3 (2C), 126.8 (1C), 127.0 (min), 127.7 (2C), 127.8 (2C), 128.3 (2C), 128.5 (1C), 128.6 (min), 142.7 (1C), 145.7 (1C), 151.1 (1C), 155.6 (1C), 162.4 (1C), 164.7 (1C), 166.4 (1C). IR (neat) (cm–1) ν 3261, 2958, 2192, 1731, 1638, 1608, 1438, 1270, 1238, 1118, 849, 698. HRMS (ESI+) calcd for C27H28N4NaO5 511.1948; found 511.1947 [M + Na].
Dimethyl 6-amino-5-cyano-1-(4-methoxybenzamido)-4-phenyl-1,4-dihydropyridine-2,3-dicarboxylate (10fa): Following the general procedure, compound 10fa was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a yellow solid in 55% yield (25.24 mg). mp 113–115 °C. The ee of the product was determined to be 42% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 253.2 nm): τmajor = 32.7 min; τminor = 14.8 min. [α]D24 = −25.6 (c = 0.08, MeOH, 42% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.51 (s, 3H), 3.55 (s, 0.75H), 3.62 (s, 2.25H), 3.84 (s, 3H), 4.35 (s, 0.75H), 4.47 (s, 0.25H), 6.33 (s, 1.50H), 6.38 (s, 0.5H), 7.07 (d, J = 8.7 Hz, 2H), 7.24 (t, J = 7.1 Hz, 1.5H), 7.34 (t, J = 7.4 Hz, 2H), 7.55 (d, J = 7.5 Hz, 1.5H), 7.77–7.89 (m, 2H), 11.04 (s, 0.75H), 11.16 (s, 0.25H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.3 (1C), 51.8 (1C), 52.7 (1C), 55.5 (1C), 58.6 (1C), 104.5 (1C), 113.6 (min), 113.8 (2C), 120.9 (1C), 123.4 (1C), 126.8 (1C), 127.0 (min), 127.7 (2C), 128.3 (2C), 128.6 (min), 129.9 (2C), 142.8 (1C), 145.7 (1C), 151.2 (1C), 162.4 (1C), 162.6 (1C), 164.7 (1C), 166.0 (1C).IR (neat) (cm–1) ν 3409, 3334, 3247, 3214, 2953, 2186, 1732, 1713, 1663, 1604, 1587, 1489, 1431, 1249, 1228, 1184, 1115, 1076, 1024, 841, 700. HRMS (ESI+) calcd for C24H22N4NaO6 485.1426; found 485.1425 [M+Na].
Dimethyl 6-amino-1-benzamido-5-cyano-4-(4-nitrophenyl)-1,4-dihydropyridine-2,3-dicarboxylate (10ab): Following the general procedure, compound 10ab was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 95% yield (45.3 mg). mp 158–160 °C. The ee of the product was determined to be 46% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 236.6 nm): τmajor = 26.1 min; τminor = 11.2 min. [α]D25 = +24.5 (c = 0.15, MeOH, 46% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.51 (s, 3H), 3.64 (s, 3H), 4.50 (s, 1H), 6.62 (s, 2H), 7.54 (t, J = 7.4 Hz, 2H), 7.59–7.83 (m, 3H), 7.87 (d, J = 7.3 Hz, 2H), 8.24 (d, J = 8.7 Hz, 2H), 11.31 (s, 1H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.1 (1C), 45.7 (1C), 52.1 (1C), 52.9 (1C), 103.8 (1C), 120.5 (1C), 123.8 (2C), 128.0 (2C), 128.5 (2C), 128.7 (2C), 131.2 (1C), 132.6 (1C), 146.5 (1C), 151.6 (1C), 152.9 (1C), 162.2 (1C), 164.4 (1C), 166.8 (1C). IR (neat) (cm–1) ν 3330, 3200, 2953, 2185, 1743, 1708, 1652, 1579, 1516, 1428, 1344, 1225, 1110, 823, 694. HRMS (ESI+) calcd for C23H19N5NaO7 500.1181; found 500.1181 [M + Na].
Dimethyl 6-amino-1-benzamido-5-cyano-4-(3-nitrophenyl)-1,4-dihydropyridine-2,3-dicarboxylate (10ac) [19]: Following the general procedure, compound 10ac was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 89% yield (42.4 mg). The ee of the product was determined to be 51% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 238.9 nm): τmajor = 18.6 min; τminor = 15.4 min. [α]D24 = −2.8 (c = 0.24, MeOH, 51% ee).
Dimethyl 6-amino-1-benzamido-4-(3-chlorophenyl)-5-cyano-1,4-dihydropyridine-2,3-dicarboxylate (10ad): Following the general procedure, compound 10ad was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 70% yield (32.6 mg). mp 134–136 °C. The ee of the product was determined to be 52% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 80:20, flow rate 1 mL min−1, λ = 236.6 nm): τmajor = 32.3 min; τminor = 18.6 min. [α]D24 = +23.4 (c = 0.13, MeOH, 52% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.53 (s, 3H), 3.65 (s, 3H), 4.41 (s, 1H), 6.49 (s, 2H), 7.31 (d, J = 7.4 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.54 (t, J = 7.6 Hz, 3H), 7.60–7.68 (m, 2H), 7.86 (d, J = 7.3 Hz, 2H), 11.27 (s, 1H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.0 (1C), 52.0 (1C), 52.9 (1C), 58.1 (1C), 104.3 (1C), 120.6 (1C), 126.4 (1C), 127.0 (1C), 127.6 (1C), 127.9 (2C), 128.5 (2C), 130.2 (1C), 131.3 (1C), 132.5 (1C), 133.2 (1C), 142.9 (1C), 148.1 (1C), 151.3 (1C), 162.3 (1C), 164.5 (1C), 166.8 (1C). IR (neat) (cm–1) ν 3330, 2953, 2922, 2850, 2184, 1736, 1707, 1654, 1578, 1430, 1227, 1115, 1079, 886, 781, 692. HRMS (ESI+) calcd for C23H19ClN4NaO5 489.0942; found 489.0941 [M + Na].
Dimethyl 6-amino-1-benzamido-4-(4-chlorophenyl)-5-cyano-1,4-dihydropyridine-2,3-dicarboxylate (10ae) [19]: Following the general procedure, compound 10ae was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 70% yield (32.7 mg). The ee of the product was determined to be 50% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 80:20, flow rate 1 mL min−1, λ = 237.7 nm): τmajor = 22.5 min; τminor = 16.4 min. [α]D24 = −2.2 (c = 0.15, MeOH, 50% ee).
Dimethyl 6-amino-1-benzamido-4-(4-bromophenyl)-5-cyano-1,4-dihydropyridine-2,3-dicarboxylate (10af) [19]: Following the general procedure, compound 10af was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 61% yield (31.2 mg). The ee of the product was determined to be 52% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 80:20, flow rate 1 mL min−1, λ = 236.6 nm): τmajor = 23.0 min; τminor = 18.0 min. [α]D24 = −19.5 (c = 0.05, MeOH, 52% ee).
Dimethyl 6-amino-1-benzamido-5-cyano-4-(4-cyanophenyl)-1,4-dihydropyridine-2,3-dicarboxylate (10ag): Following the general procedure, compound 10ag was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 99% yield (45.09 mg). mp 154–156 °C. The ee of the product was determined to be 50% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 234.2 nm): τmajor = 22.2 min; τminor = 18.5 min. [α]D24 = +4.0 (c = 0.07, MeOH, 50% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.51 (s, 3H), 3.65 (s, 3H), 4.49 (s, 1H), 6.55 (s, 2H), 7.35–7.69 (m,4H), 7.70–7.91 (m, 5H), 11.30 (s, 1H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 39.4 (1C), 52.0 (1C), 52.9 (1C), 57.5 (1C), 103.9 (1C), 109.8 (1C), 118.9 (1C), 120.5 (1C), 127.9 (3C), 128.5 (2C), 128.6 (2C), 131.2 (1C), 132.5 (2C), 143.3 (1C), 151.0 (1C), 151.4 (1C), 162.2 (1C), 164.4 (1C), 166.8 (1C). IR (neat) (cm–1) ν 3414, 3313, 3210, 2947, 2231, 2192, 1750, 1704, 1682, 1665, 1590, 1433, 1360, 1272, 1253, 1222, 1115, 929, 846, 686. HRMS (ESI+) calcd for C24H19N5NaO5 480.1258; found 480.1256 [M + Na].
Dimethyl 6-amino-1-benzamido-5-cyano-4-(naphthalen-1-yl)-1,4-dihydropyridine-2,3-dicarboxylate (10ah): Following the general procedure, compound 10ah was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a yellow solid in 51% yield (24.61 mg). mp 122–124 °C. The ee of the product was determined to be 50% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 221.2 nm): τmajor = 27.9 min; τminor = 16.5 min. [α]D24 = −28.9 (c = 0.10, MeOH, 50% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.33 (s, 3H), 3.60 (s, 0.75H), 3.67 (s, 2.25H), 5.42 (s, 0.75H), 5.47 (s, 0.25H), 6.33 (s, 1.5H), 6.38 (s, 0.5H), 7.41–7.70 (m, 6.25H), 7.78–7.97 (m, 4H), 8.09 (d, J = 6.7 Hz, 0.75H), 8.46 (d, J = 8.7 Hz, 1H), 11.27 (s, 0.75H), 11.39 (s, 0.25H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 40.0 (1C), 51.7 (1C), 52.8 (1C), 59.2 (1C), 64.9 (1C), 105.4 (1C), 120.8 (1C), 123.5 (1C), 125.5 (1C), 125.7 (1C), 126.0 (1C), 127.0 (1C), 127.1 (1C), 127.9 (2C), 128.3 (1C), 128.5 (2C), 130.3 (1C), 131.4 (1C), 132.5 (1C), 133.1 (1C), 143.0 (1C), 151.1 (1C), 162.5 (1C), 164.8 (1C), 166.7 (1C). IR (neat) (cm–1) ν 3202, 2951, 2194, 1715, 1704, 1661, 1592, 1510, 1426, 1334, 1232, 1070, 782. HRMS (ESI+) calcd for C27H22N4NaO5 505.1482; found 505.1454 [M + Na].
Dimethyl 6-amino-1-benzamido-5-cyano-4-(p-tolyl)-1,4-dihydropyridine-2,3-dicarboxylate (10ai) [19]: Following the general procedure, compound 10ai was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 44% yield (19.6 mg). The ee of the product was determined to be 50% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 237.7 nm): τmajor = 15.9 min; τminor = 9.6 min. [α]D24 = −19.4 (c = 0.12, MeOH, 50% ee).
Dimethyl 6-amino-1-benzamido-5-cyano-4-(4-methoxyphenyl)-1,4-dihydropyridine-2,3-dicarboxylate (10aj): Following the general procedure, compound 10aj was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 40% yield (18.57 mg). mp 137–139 °C. The ee of the product was determined to be 40% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 237.7 nm): τmajor = 48.4 min; τminor = 19.2 min. [α]D24 = −24.0 (c = 0.05, MeOH, 40% ee). 1H-NMR (400 MHz, DMSO-d6) 3.52 (s, 3H), 3.55 (s, 0.75H), 3.63 (s, 2.25H), 3.75 (s, 3H), 4.30 (s, 0.75H), 4.42 (s, 0.25H), 6.32 (s, 1.5H), 6.38 (s, 0.50H), 6.89 (d, J = 8.5 Hz, 2.25H), 7.10–7.22 (m, 0.75H), 7.43–7.68 (m, 4H), 7.79–7.88 (m, 2H), 11.19 (s, 0.75H), 11.30 (s, 0.25H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 38.4 (1C), 51.8 (1C), 52.7 (1C), 55.0 (1C), 58.9 (1C), 104.9 (1C), 113.6 (2C), 113.9 (min), 120.9 (1C), 127.9 (2C), 128.5 (2C), 128.8 (2C), 131.3 (1C), 132.5 (1C), 137.9 (1C), 142.2 (1C), 151.0 (1C), 158.2 (1C), 162.5 (1C), 164.8 (1C), 166.7 (1C). IR (neat) (cm–1) ν 3313, 3201, 2952, 2838, 2186, 1742, 1707, 1651, 1606, 1509, 1428, 1227, 1175, 1110, 1028, 833, 691. HRMS (ESI+) calcd for C24H22N4NaO6 485.1438; found 485.1435 [M + Na].
Dimethyl 6-amino-1-benzamido-5-cyano-4-(furan-2-yl)-1,4-dihydropyridine-2,3-dicarboxylate (10ak): Following the general procedure, compound 10ak was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 70% yield (29.77 mg). mp 151–153 °C. The ee of the product was determined to be 54% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 80:20, flow rate 1 mL min−1, λ = 236.6 nm): τmajor = 16.4 min; τminor = 12.4 min. [α]D24 = −15.3 (c = 0.10, MeOH, 54% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.55 (s, 1.35H), 3.58 (s, 1.35H), 3.59 (s, 1.65H), 3.65 (s, 1.65H), 4.51 (s, 0.55H), 4.64 (s, 0.45H), 6.11 (d, J = 3.1 Hz, 0.45H), 6.33 (d, J = 3.1 Hz, 0.55H), 6.37–6.45 (m, 1H), 6.50 (s, 1.1H), 6.57 (s, 0.9H), 7.46–7.67 (m, 4H), 7.77–7.86 (m, 2H), 11.20 (s, 0.55H), 11.38 (s, 0.45H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 32.4 (1C), 33.0 (min), 52.0 (min), 52.1 (1C), 52.7 (min), 52.9 (1C), 53.8 (min), 55.5 (1C), 101.8 (min), 102.3 (1C), 105.3 (min), 106.0 (1C), 110.6 (min), 110.8 (1C), 120.8 (1C), 120.9 (min), 127.8 (1C), 128.0 (min), 128.3 (min), 128.5 (1C), 131.3 (1C), 131.4 (min), 132.3 (min), 132.5 (1C), 141.6 (1C), 142.3 (min), 143.1 (1C), 143.7 (min), 152.0 (1C), 152.6 (min), 156.4 (min), 157.6 (1C), 162.1 (min), 162.2 (1C), 164.5 (1C), 164.7 (min), 166.1 (min), 166.2 (1C). IR (neat) (cm–1) ν 3527, 3406, 3328, 2175, 1735, 1707, 1652, 1578, 1431, 1336, 1225, 1013, 939, 754, 705. HRMS (ESI+) calcd for C21H18N4NaO6 445.1125; found 445.1125 [M + Na].
Dimethyl 6-amino-1-benzamido-5-cyano-4-(thiophen-2-yl)-1,4-dihydropyridine-2,3-dicarboxylate (10al): Following the general procedure, compound 10al was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100), as a white solid in 76% yield (33.18 mg). mp 124–126 °C. The ee of the product was determined to be 43% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 236.6 nm): τmajor = 27.1 min; τminor = 16.1 min. [α]D24 = −12.6 (c = 0.10, MeOH, 43% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.55 (s, 1.11H), 3.60 (s, 3H), 3.64 (s, 1.89H), 4.66 (s, 0.63H), 4.82 (s, 0.37H), 6.50 (s, 1.26H), 6.57 (s, 0.74H), 6.88 (s, 0.37H), 7.00 (s, 1H), 7.19 (s, 0.63H), 7.39 (d, J = 4.7 Hz, 1H), 7.52 (t, J = 6.4 Hz, 2H), 7.62 (t, J = 6.7 Hz, 1H), 7.83 (d, J = 7.4 Hz, 2H), 11.21 (s, 0.63H), 11.38 (d, 0.37H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 33.7 (1C), 52.0 (1C), 52.8 (1C), 57.9 (1C), 104.3 (1C), 120.8 (1C), 124.5 (1C), 124.9 (1C), 127.1 (1C), 127.9 (2C), 128.3 (min), 128.5 (2C), 131.4 (1C), 132.4 (1C), 142.3 (1C), 148.7 (1C), 151.6 (1C), 162.2 (1C), 164.5 (1C), 166.2 (1C). IR (neat) (cm–1) ν 3415, 3332, 3248, 3213, 2954, 2191, 1719, 1685, 1660, 1587, 1434, 1337, 1230, 941, 707, 687. HRMS (ESI+) calcd for C21H18N4NaO5S 461.0898; found 461.0897 [M + Na].
Dimethyl 6′-amino-1′-benzamido-5′-cyano-1′,4′-dihydro-[3,4′-bipyridine]-2′,3′-dicarboxylate (10am): Following the general procedure, compound 10am was obtained after 120 h of reaction at room temperature and was purified by column chromatography (n-hexane:diethyl ether 20:80 to 0:100 to ethyl acetate 100), as a white solid in 92% yield (39.66 mg). mp 157–159 °C. The ee of the product was determined to be 40% by HPLC using a Daicel Chiralpak IC column (n-hexane/i-PrOH = 70:30, flow rate 1 mL min−1, λ = 336.0 nm): τmajor = 32.9 min; τminor = 50.6 min. [α]D24 = −12.6 (c = 0.17, MeOH, 40% ee). 1H-NMR (400 MHz, DMSO-d6) δ 3.52 (s, 3H), 3.64 (s, 3H), 4.44 (s, 1H), 6.53 (s, 2H), 7.42 (dd, J = 8 Hz, 5 Hz, 1H), 7.52 (t, J = 7.5 Hz, 2H), 7.63 (t, J = 7.3 Hz, 1H), 7.87 (d, J = 7.3 Hz, 2H), 8.05 (s, 1H), 8.47 (dd, J = 4.7 Hz, 1.6 Hz, 1H), 8.67 (s, 1H), 11.31 (s, 1H). 13C-APT-NMR (100 MHz, DMSO-d6) δ 36.9 (1C), 45.8 (1C), 52.0 (1C), 52.9 (1C), 104.1 (1C), 120.6 (1C), 123.8 (1C), 127.9 (2C), 128.5 (2C), 131.2 (1C), 132.6 (1C), 135.3 (1C), 141.0 (1C), 143.1 (1C), 148.2 (1C), 148.7 (1C), 151.5 (1C), 162.2 (1C), 164.4 (1C), 166.8 (1C). IR (neat) (cm–1) ν 3317, 3226, 2952, 2185, 1749, 1707, 1685, 1654, 1578, 1425, 1330, 1275, 1221, 1115, 1029, 703. HRMS (ESI+) calcd for C22H20N5O5 434.1468; found 434.1467 [M + H].
4. Conclusions
In summary, we have developed the first organocatalytic enantioselective approach for the obtainment of chiral 1-benzamido-1,4-dihydropyridines. Final 1,4-dihydropyridines were reached with high yields and showed promising results of enantioselectivity for the first time, using mild conditions and following a simple approach. Further mechanistic studies are required in order to understand and to prove the role of the β-isocupreidine catalyst in this process. Moreover, additional studies with the aim of improving the enantioselectivity of the method are actively ongoing in our laboratory.
Supplementary Materials
The following are available online. Figures S1–S5: 1H and 13C-APT NMR spectra of hydrazones 7. Figures S6–S19: 1H and 13C-APT NMR spectra of 1,4-dihydropyridines 10 Figures S20–S57: HPLC chromatograms of 1,4-dihydropyridines 10.
Author Contributions
All the authors designed the experiments and analyzed the data; F.A.-L. performed the experiments; E.M.-L. and R.P.H. wrote the paper; all authors read and approved the final manuscript.
Funding
This research was funded by Ministerio de Economía, Industria y Competitividad [CTQ2017-88091-P], Universidad de Zaragoza [JIUZ-2017-CIE-05] and Gobierno de Aragón-Fondo Social Europeo [Research Group E07_17R]
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eisner, U.; Kuthan, J. The chemistry of dihydropyridines. Chem. Rev. 1972, 72, 1–42. [Google Scholar] [CrossRef]
- Stout, D.M.; Meyers, A.I. Recent advances in the chemistry of dihydropyridines. Chem. Rev. 1982, 82, 223–243. [Google Scholar] [CrossRef]
- Sausins, A.; Duburs, G. Synthesis of 1,4-dihydropyridines by cyclocondensation reactions. Heterocycles 1988, 27, 269–289. [Google Scholar] [CrossRef]
- Lavilla, R. Recent developments in the chemistry of dihydropyridines. J. Chem. Soc. Perkin Trans. 2002, 1, 1141–1156. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, S.K. Synthesis, utility and medicinal importance of 1,2- & 1,4-dihydropyridines. RSC Adv. 2017, 7, 2682–2732. [Google Scholar] [Green Version]
- Reddy, G.M.; Shiradkar, M.; Chakravarthy, A.K. Chemical and pharmacological significance of 1,4-dihydropyridines. Curr. Org. Chem. 2007, 11, 847–852. [Google Scholar] [CrossRef]
- Mai, A.; Valente, S.; Meade, S.; Carafa, V.; Tardugno, M.; Nebbioso, A.; Galmozzi, A.; Mitro, N.; Fabiani, E.D.; Altucci, L.; et al. Study of 1,4-dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 2009, 52, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Ioan, P.; Carosati, E.; Micucci, M.; Cruciani, G.; Broccatelli, F.; Zhorov, B.S.; Chiarini, A.; Budriesi, R. 1,4-Dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 1): Action in ion channels and GPCRs. Curr. Med. Chem. 2011, 18, 4901–4922. [Google Scholar] [CrossRef] [PubMed]
- Carosati, E.; Ioan, P.; Micucci, M.; Broccatelli, F.; Cruciani, G.; Zhorov, B.S.; Chiarini, A.; Budriesi, R. 1,4-dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 2): Action in other targets and antitargets. Curr. Med. Chem. 2012, 19, 4306–4323. [Google Scholar] [CrossRef] [PubMed]
- Loev, B.; Goodman, M.; Snader, K.; Tedeschi, R.; Macko, E. Hantzsch-type dihydropyridine hypotensive agents. J. Med. Chem. 1974, 17, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Bruncko, M. Bioactive Heterocyclic Compound Classes: Pharmaceuticals; Lamberth, C., Dinges, J., Eds.; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2012; pp. 135–151. [Google Scholar]
- Edraki, N.; Mehdipour, A.R.; Khoshneviszadeh, M.; Miri, R. Dihydropyridines: Evaluation of their current and future pharmacological applications. Drug Discov. Today 2009, 14, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.-P.; Liu, Y. Recent advances in new multicomponent synthesis of structurally diversified 1,4-dihydropyridines. RSC Adv. 2012, 2, 9763–9777. [Google Scholar] [CrossRef]
- Pham, H.T.; Chataigner, I.; Renaud, J.-L. New approaches to nitrogen containing heterocycles: enantioselective organocatalyzed synthesis of dihydropyridines (DHP’s), quinolizidine derivatives and dihydropyrimidines (DHPM’s). Curr. Org. Chem. 2012, 16, 1754–1775. [Google Scholar] [CrossRef]
- Auria-Luna, F.; Marqués-López, E.; Herrera, R.P. Organocatalytic enantioselective synthesis of 1,4-dihydropyridines. Adv. Synth. Catal. 2017, 359, 2161–2175. [Google Scholar] [CrossRef]
- Auria-Luna, F.; Marqués-López, E.; Gimeno, M.C.; Heiran, R.; Mohammadi, S.; Herrera, R.P. Asymmetric organocatalytic synthesis of substituted chiral 1,4-dihydropyridine derivatives. J. Org. Chem. 2017, 82, 5516–5523. [Google Scholar] [CrossRef] [PubMed]
- Auria-Luna, F.; Marqués-López, E.; Mohammadi, S.; Heiran, R.; Herrera, R.P. New organocatalytic asymmetric synthesis of highly substituted chiral 2-oxospiro-[indole-3,4′-(1′,4′-dihydropyridine)] derivatives. Molecules 2015, 20, 15807–15826. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, Y.-H.; Yan, C.-G. One-pot four-component reaction for convenient synthesis of functionalized 1-benzamidospiro[indoline-3,4′-pyridines]. Beilstein J. Org. Chem. 2014, 10, 2671–2676. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shen, G.; Xie, Y.; Yan, C.-G. Synthesis of functionalized 1-benzamido-1,4-dihydropyridines via a one-pot two-step four-component reaction. Chin. J. Chem. 2014, 32, 1143–1150. [Google Scholar] [CrossRef]
- Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R.P.; Sgarzani, V. Organocatalytic enantioselective decarboxylative addition of malonic half thioesters to imines. Adv. Synth. Catal. 2007, 349, 1037–1040. [Google Scholar] [CrossRef]
- Pettersen, D.; Marcolini, M.; Bernardi, L.; Fini, F.; Herrera, R.P.; Sgarzani, V.; Ricci, A. Direct access to enantiomerically enriched α-amino phosphonic acid derivatives by organocatalytic asymmetric hydrophosphonylation of imines. J. Org. Chem. 2006, 71, 6269–6272. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Fini, F.; Herrera, R.P.; Ricci, A.; Sgarzani, V. Enantioselective aza-Henry reaction using cinchona organocatalysts. Tetrahedron 2006, 62, 375–380. [Google Scholar] [CrossRef]
- Miyabe, H.; Takemoto, Y. Discovery and application of asymmetric reaction by multi-functional thioureas. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar] [CrossRef]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Sonsona, I.G.; Marqués-López, E.; Herrera, R.P. The aminoindanol core as a key scaffold in bifunctional organocatalysts. Beilstein J. Org. Chem. 2016, 12, 505–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanabadi, A.; Hosseini-Tabatabaei, M.R.; Shahraki, M.; Anary-Abbasinejad, M.; Ghoroghchian, S. An efficient one-pot synthesis of 1,3,5-substituted-1H-pyrazoles derivatives. J. Chem. Res. 2011, 35, 11–14. [Google Scholar] [CrossRef]
- Postole, G.; Chowdhury, B.; Karmakar, B.; Pinki, K.; Banerji, J.; Auroux, A. Knoevenagel condensation reaction over acid–base bifunctional nanocrystalline CexZr1−xO2 solid solutions. J. Catal. 2010, 269, 110–121. [Google Scholar] [CrossRef]
- Hosseini-Sarvari, M.; Sharghi, H.; Etemad, S. Solvent-free knoevenagel condensations over TiO2. Chin. J. Chem. 2007, 25, 1563–1567. [Google Scholar] [CrossRef]
- Yamashita, K.; Tanaka, T.; Hayashi, M. Use of isopropyl alcohol as a solvent in Ti(O-i-Pr)4-catalyzed Knoëvenagel reactions. Tetrahedron 2005, 61, 7981–7985. [Google Scholar] [CrossRef]
- Kharas, G.B.; Hanawa, E.; Lachenberg, J.; Brozek, B.; Miramon, P.; Mojica, A.C.; Colbert, A.C.; Crowell, B.T.; Madison, A.; Martinez, A.P. Novel copolymers of vinyl acetate and some ring-substituted 2-phenyl-1,1-dicyanoethylenes. J. Macromol. Sci. Part A Pure Appl. Chem. 2008, 45, 420–424. [Google Scholar] [CrossRef]
- Ying, A.-G.; Wang, L.-M.; Wang, L.-L.; Chen, X.-Z.; Ye, W.-D. Green and efficient knoevenagel condensation catalysed by a DBU based ionic liquid in water. J. Chem. Res. 2010, 34, 30–33. [Google Scholar] [CrossRef]
- Kacprzak, K.; Gawroński, J. Cinchona Alkaloids and their derivatives: Versatile catalysts and ligands in asymmetric synthesis. Synthesis 2001, 961–998. [Google Scholar]
- Amberg, W.; Bennani, Y.L.; Chadha, R.K.; Crispino, G.A.; Davis, W.D.; Hartung, J.; Jeong, K.-S.; Ogino, Y.; Shibata, T.; Sharpless, K.B. Syntheses and crystal structures of the cinchona alkaloid derivatives used as ligands in the osmium-catalyzed asymmetric dihydroxylation of olefins. J. Org. Chem. 1993, 58, 844–849. [Google Scholar] [CrossRef]
- Papageorgiou, C.D.; Cubillo de Dios, M.A.; Ley, S.V.; Gaunt, M.J. Enantioselective organocatalytic cyclopropanation via ammonium ylides. Angew. Chem. Int. Ed. 2004, 43, 4641–4644. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wang, C.-J. Asymmetric N-allylic alkylation of hydrazones with morita–baylis–hillman carbonates. Adv. Synth. Catal. 2015, 357, 384–388. [Google Scholar] [CrossRef]
- Iwabuchi, Y.; Nakatani, M.; Yokoyama, N.; Hatakeyama, S. Chiral amine-catalyzed asymmetric baylis-hillman reaction: A reliable route to highly enantiomerically enriched (α-methylene-β-hydroxy)esters. J. Am. Chem. Soc. 1999, 121, 10219–10220. [Google Scholar] [CrossRef]
- Yang, W.; Du, D.-M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, Y.; Deng, L. The Mannich reaction of malonates with simple imines catalyzed by bifunctional cinchona alkaloids: Enantioselective synthesis of β-amino acids. J. Am. Chem. Soc. 2006, 128, 6048–6049. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 7a–7f, 9a–9m and 10 are available from the authors. |
Figure 1.
1,4-Dihydropyridine based drugs as calcium channel blockers.


Scheme 2.
Synthesis of highly substituted benzamidospiro[indoline-3,4′-pyridines] 4 [18] and benzamido-1,4-dihydropyridines 5 [19].

Scheme 3.
Preparation of hydrazones 7a–f.

Scheme 4.
Synthesis of alkylidenemalononitriles 9a–m.

Scheme 5.
Chiral organocatalysts I-X tested to synthesize chiral 1,4-DHPs 10aa. Rac. = racemic mixture. N.d. = not determined.
Scheme 5.
Chiral organocatalysts I-X tested to synthesize chiral 1,4-DHPs 10aa. Rac. = racemic mixture. N.d. = not determined.

Scheme 6.
Scope of the reaction to obtain 1-benzamido-1,4-dihydropyridine derivatives 10.


{kind=link}
{kind=link}
{kind=link}
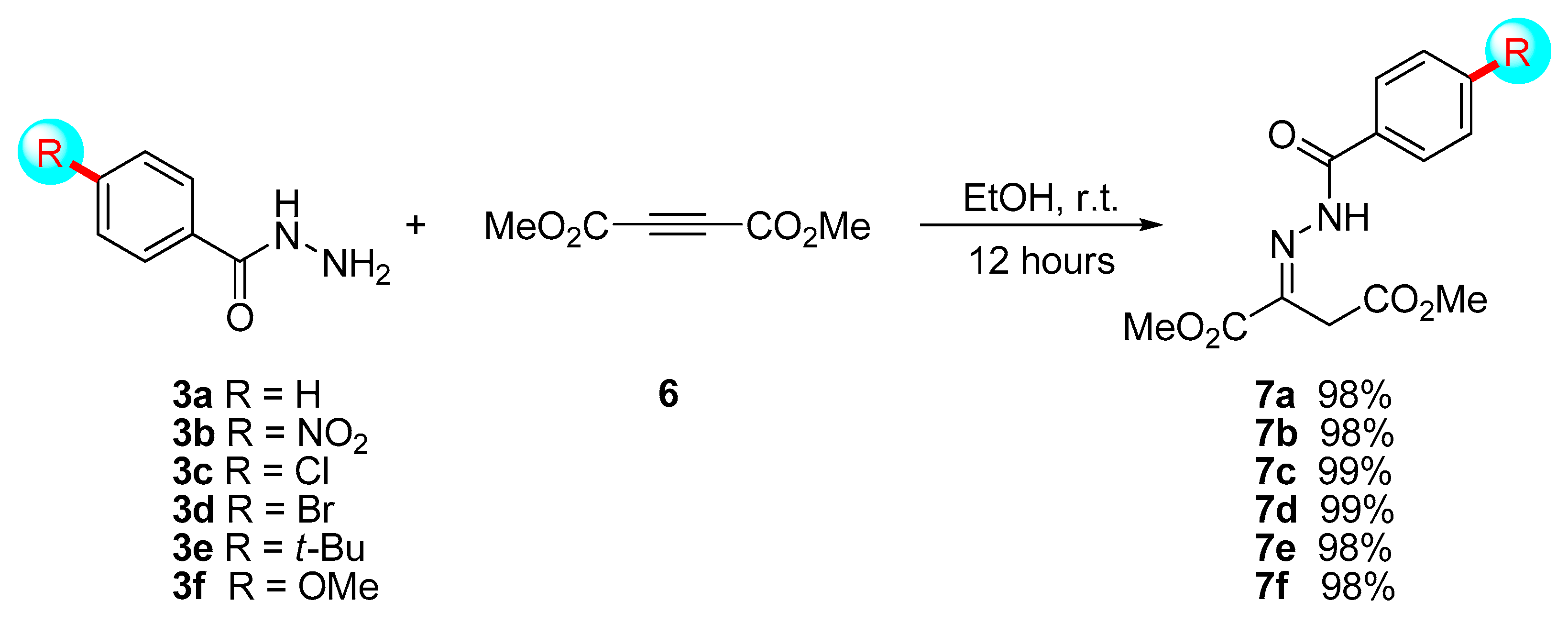{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Screening of the reaction conditions for the synthesis of chiral 1-benzamido-1,4-dihydropyridine 10aa a.
Table 1.
Screening of the reaction conditions for the synthesis of chiral 1-benzamido-1,4-dihydropyridine 10aa a.

| Entry | Solvent (mL) | 7a (mmol) | 9a (mmol) | Cat. (mol%) | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|---|
| 1 | MeCN (0.5) | 0.1 | 0.1 | VI (20%) | >95 | 7 |
| 2 | MeCN (0.5) | 0.1 | 0.1 | X (20%) | 56 | 10 |
| 3 | AcOEt (0.5) | 0.1 | 0.1 | VI (20%) | 72 | 20 |
| 4 | AcOEt (0.5) | 0.1 | 0.1 | X (20%) | 54 | 40 |
| 5 | CH2Cl2 (0.5) | 0.1 | 0.1 | VI (20%) | 67 | 15 |
| 6 | CH2Cl2 (0.5) | 0.1 | 0.1 | X (20%) | 35 | Rac. d |
| 7 | CHCl3 (0.5) | 0.1 | 0.1 | VI (20%) | 44 | 24 |
| 8 | CHCl3 (0.5) | 0.1 | 0.1 | X (20%) | 49 | 33 |
| 9 | Et2O (0.5) | 0.1 | 0.1 | VI (20%) | 26 | 25 |
| 10 | Et2O (0.5) | 0.1 | 0.1 | X (20%) | 39 | 40 |
| 11 | THF (0.5) | 0.1 | 0.1 | VI (20%) | 54 | 17 |
| 12 | THF (0.5) | 0.1 | 0.1 | X (20%) | 54 | 50 |
| 13 | MeOH (0.5) | 0.1 | 0.1 | VI (20%) | >95 | Rac. d |
| 14 | MeOH (0.5) | 0.1 | 0.1 | X (20%) | 77 | Rac. d |
| 15 | THF (0.5) | 0.1 | 0.15 | X (20%) | 61 | 50 |
| 16 | THF (0.5) | 0.15 | 0.1 | X (20%) | 51 | 49 |
| 17 | THF (0.25) | 0.1 | 0.15 | X (20%) | 58 | 46 |
| 18 | THF (1) | 0.1 | 0.15 | X (20%) | 48 | 52 |
| 19 | THF (0.5) | 0.1 | 0.15 | X (10%) | 57 | 48 |
| 20 | THF (0.5) | 0.1 | 0.15 | X (30%) | 61 | 46 |
a Otherwise indicated: To a mixture of catalyst VI or X (20 mol%) and hydrazone 7a (0.1 mmol), in the corresponding solvent (0.5 mL), alkylidenemalononitrile 9a (0.1 mmol) was added. b Isolated yield after column chromatography (SiO2, Hex:Et2O 20:80 to Hex:Et2O 0:100). c Determined by chiral HPLC analysis (Daicel Chiralpak IC, Hex:iPrOH 70:30, 1 mL/min). d Racemic mixture.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Auria-Luna, F.; Marqués-López, E.; P. Herrera, R. First Organocatalytic Asymmetric Synthesis of 1-Benzamido-1,4-Dihydropyridine Derivatives. Molecules 2018, 23, 2692. https://doi.org/10.3390/molecules23102692
AMA Style
Auria-Luna F, Marqués-López E, P. Herrera R. First Organocatalytic Asymmetric Synthesis of 1-Benzamido-1,4-Dihydropyridine Derivatives. Molecules. 2018; 23(10):2692. https://doi.org/10.3390/molecules23102692
Chicago/Turabian StyleAuria-Luna, Fernando, Eugenia Marqués-López, and Raquel P. Herrera. 2018. "First Organocatalytic Asymmetric Synthesis of 1-Benzamido-1,4-Dihydropyridine Derivatives" Molecules 23, no. 10: 2692. https://doi.org/10.3390/molecules23102692