Heteroatom Substitution at Amide Nitrogen—Resonance Reduction and HERON Reactions of Anomeric Amides


Abstract
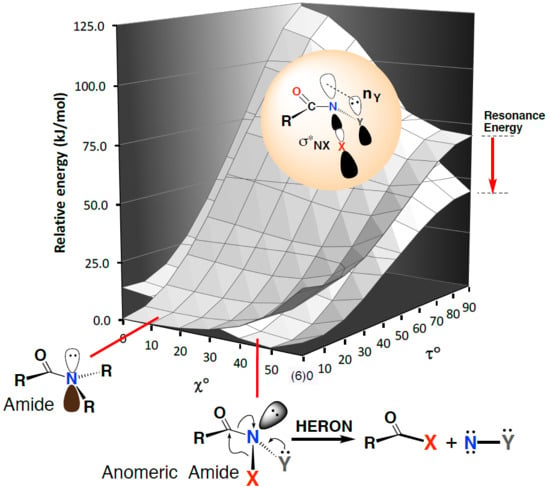
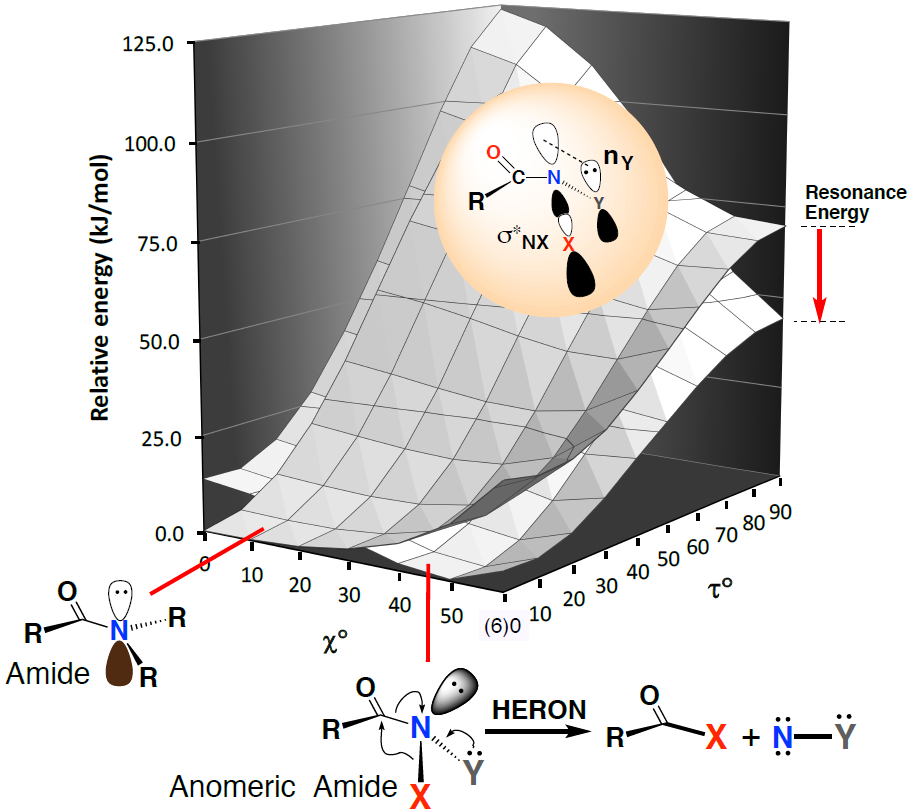
1. Introduction

2. Properties of Anomeric Amides
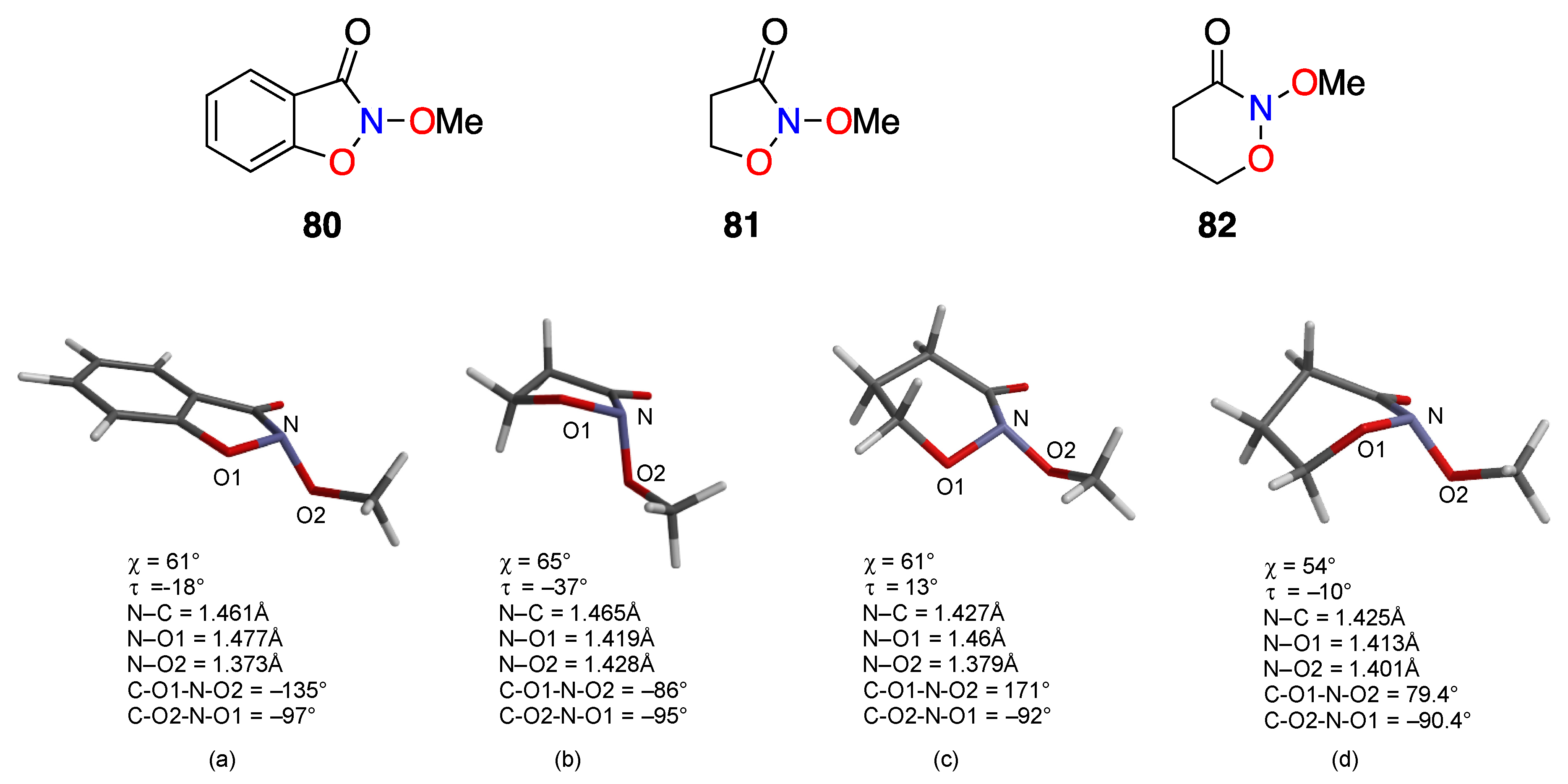
2.1. Structural Properties





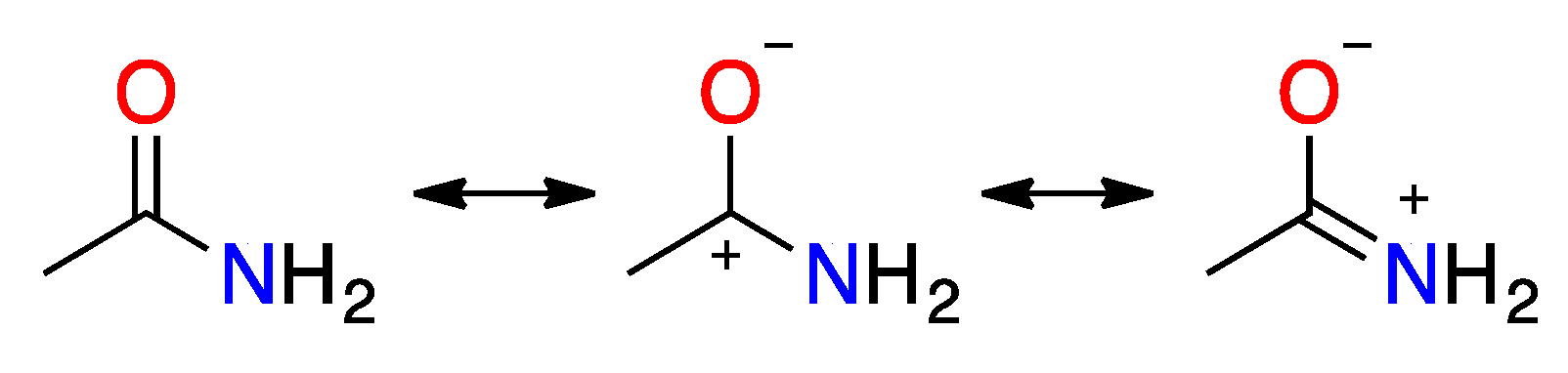
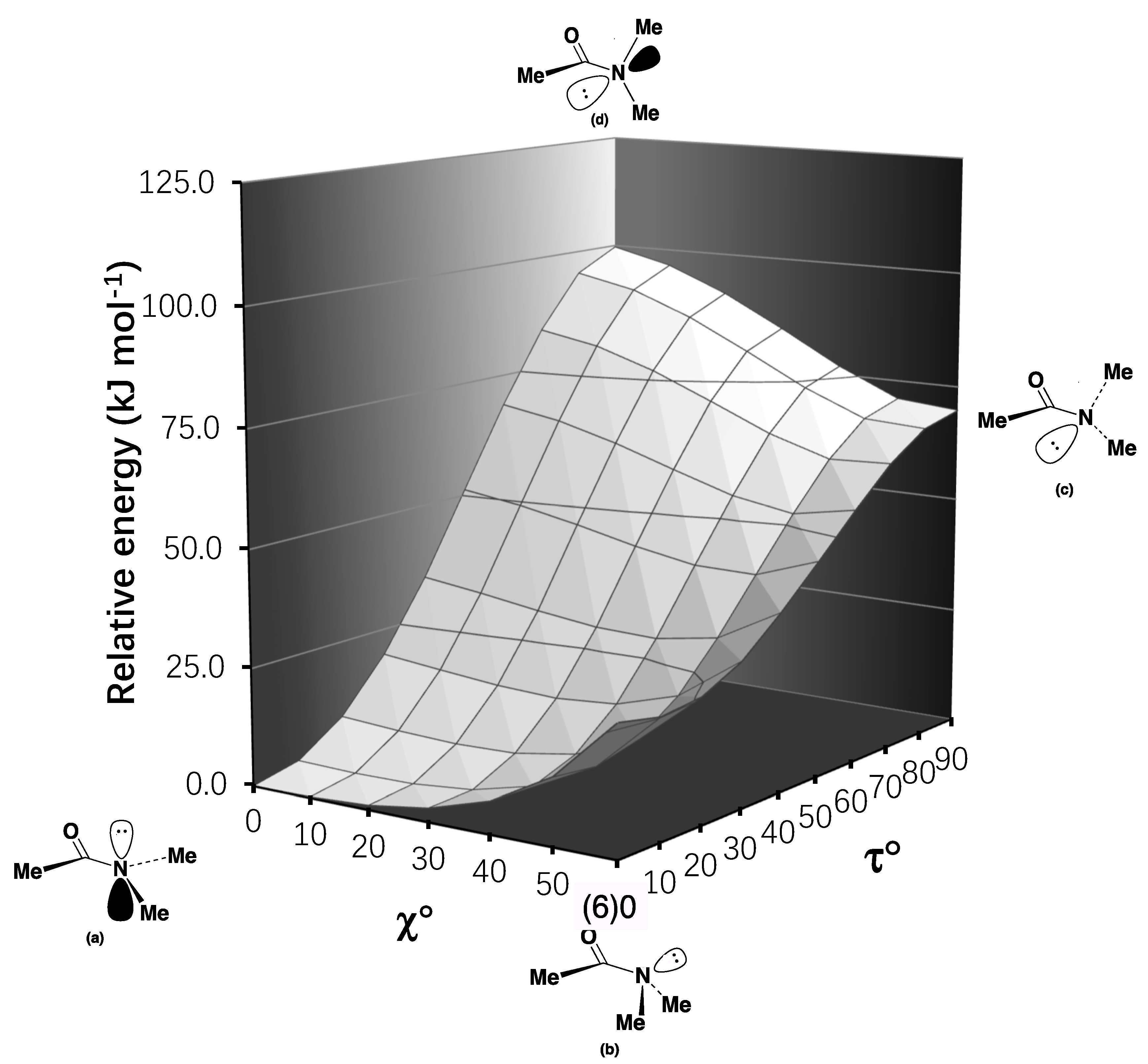
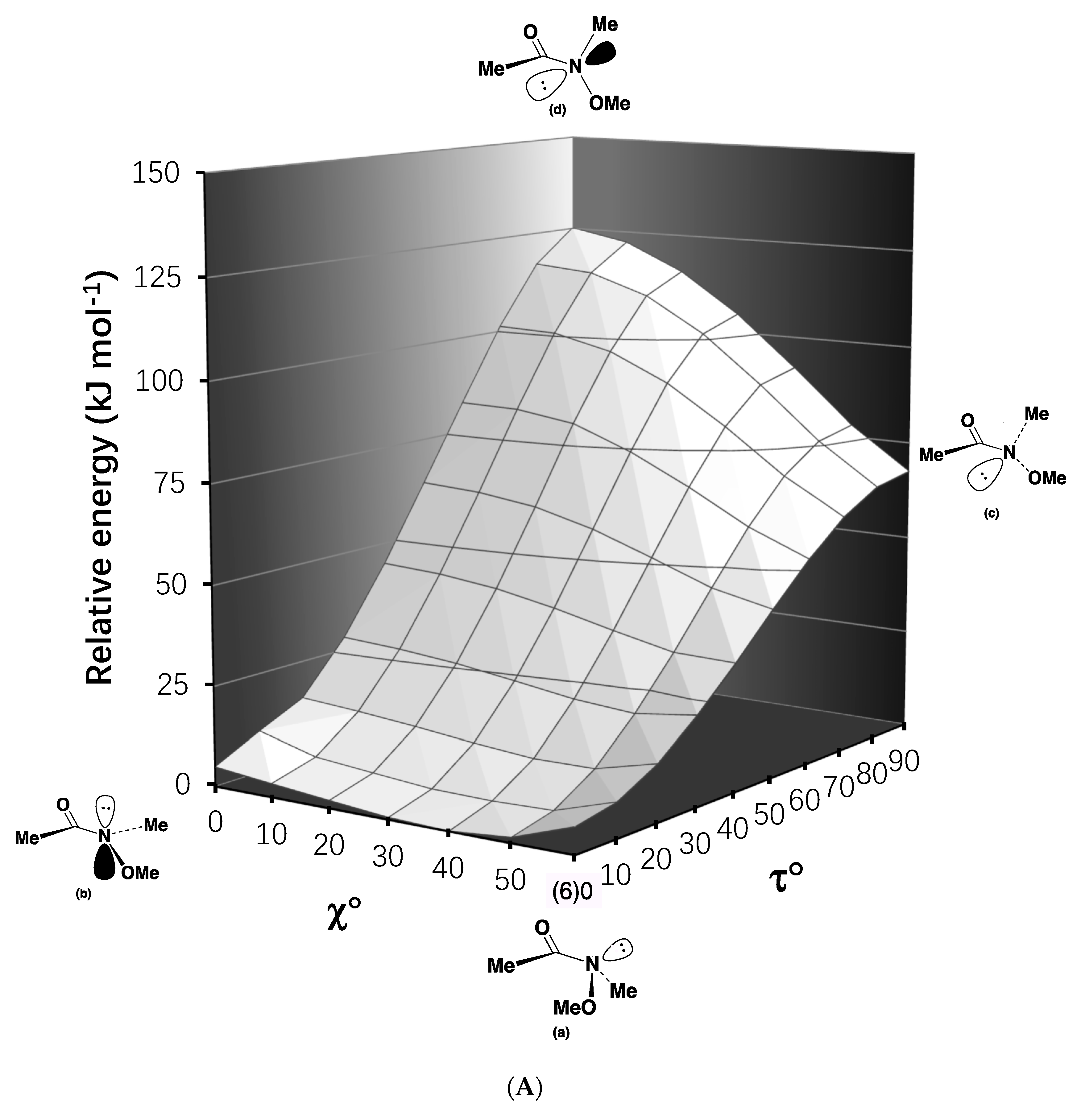
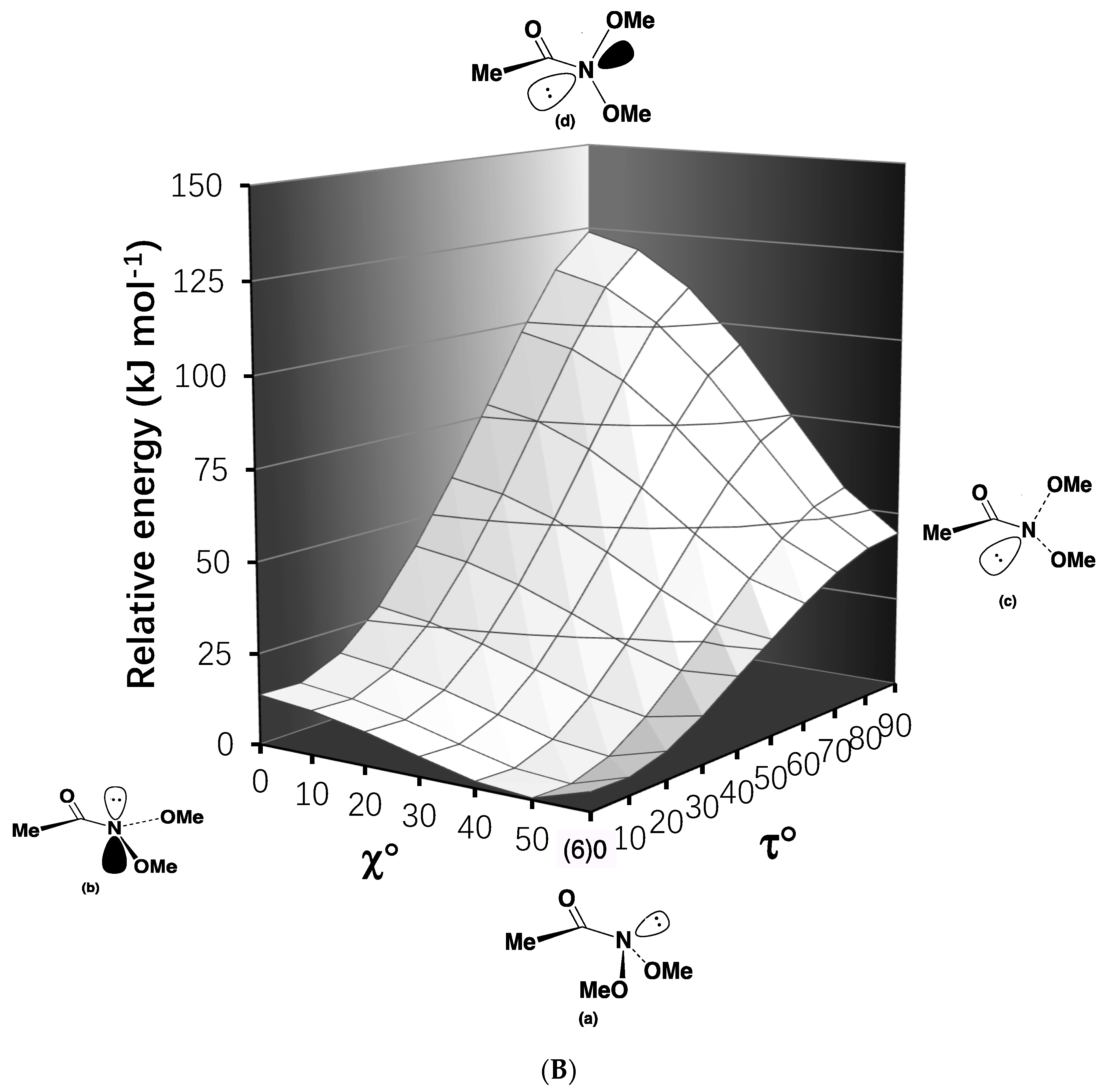
2.2. Resonance Energies and Amidicities
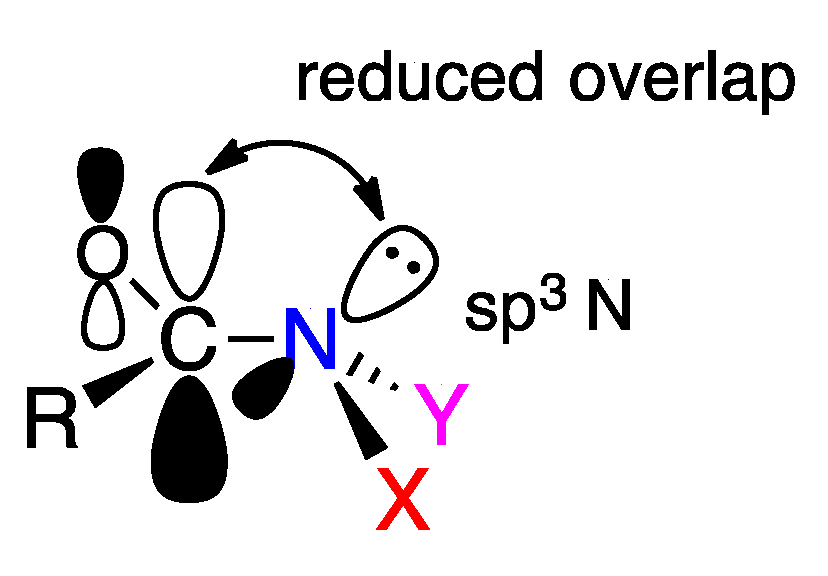
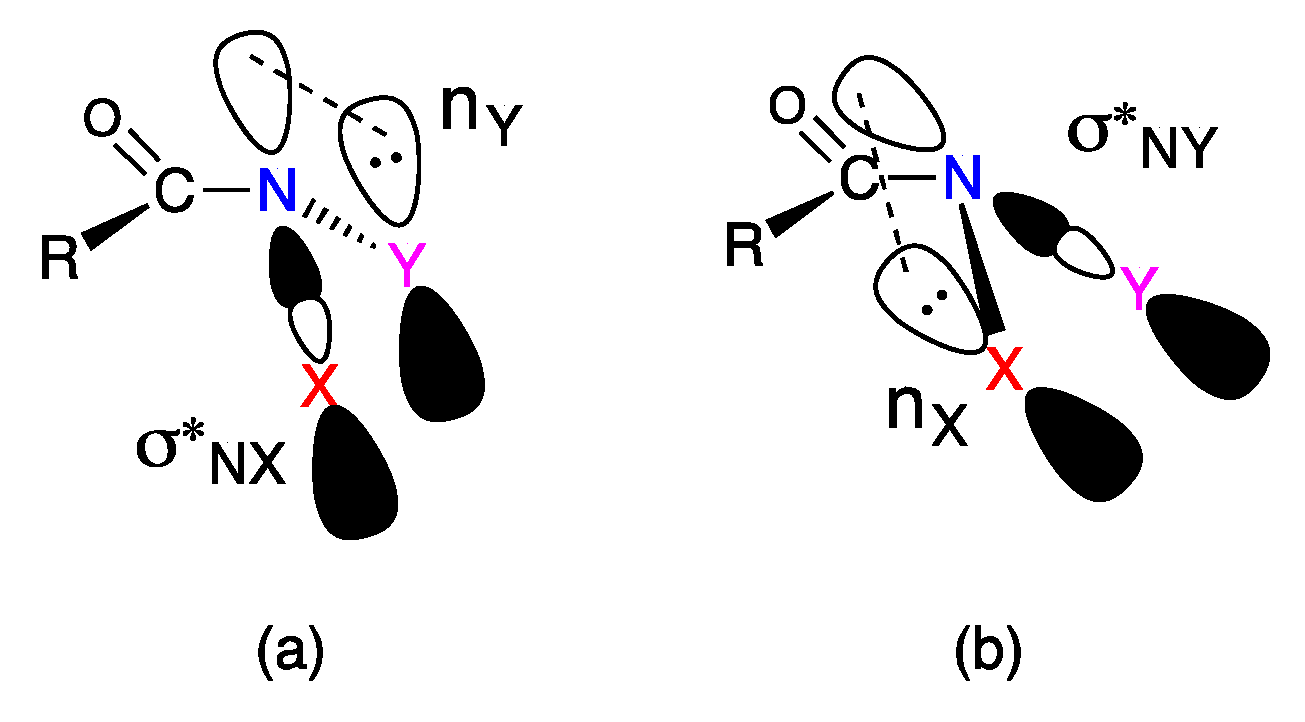
2.3. The Anomeric Effect
2.4. Spectroscopic Properties of Anomeric Amides

3. Reactivity of Anomeric Amides
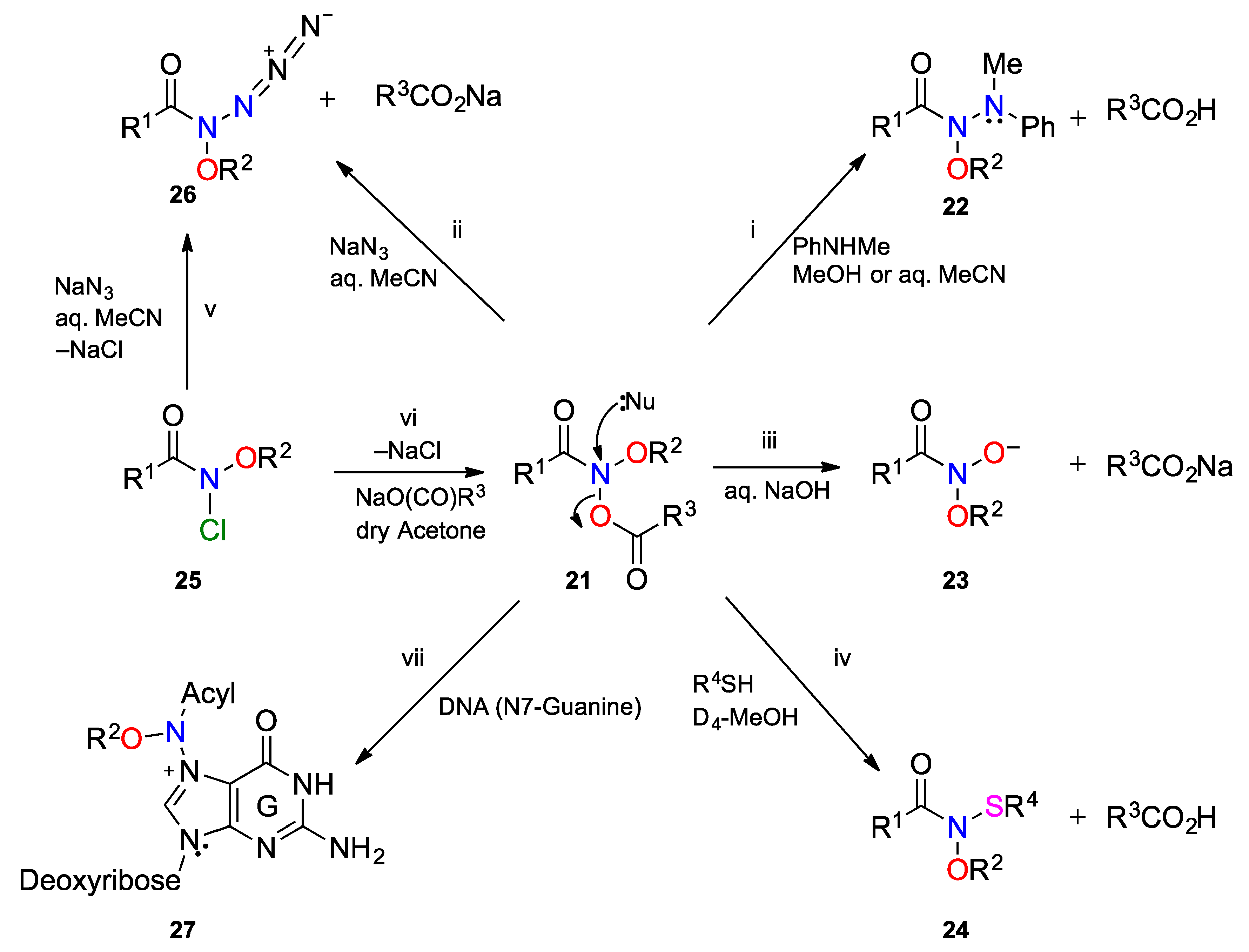
3.1. Reactivity at the Amide Nitrogen
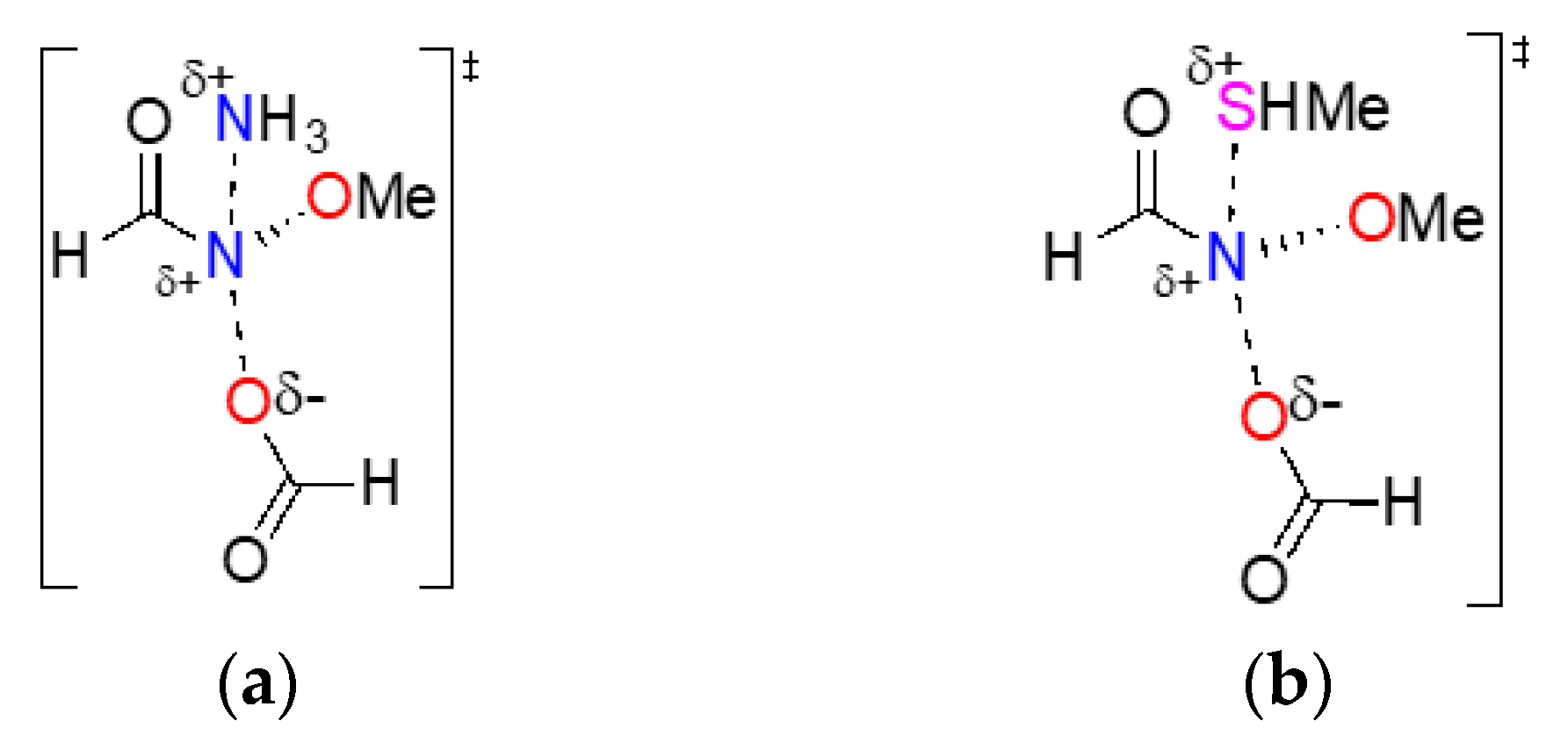
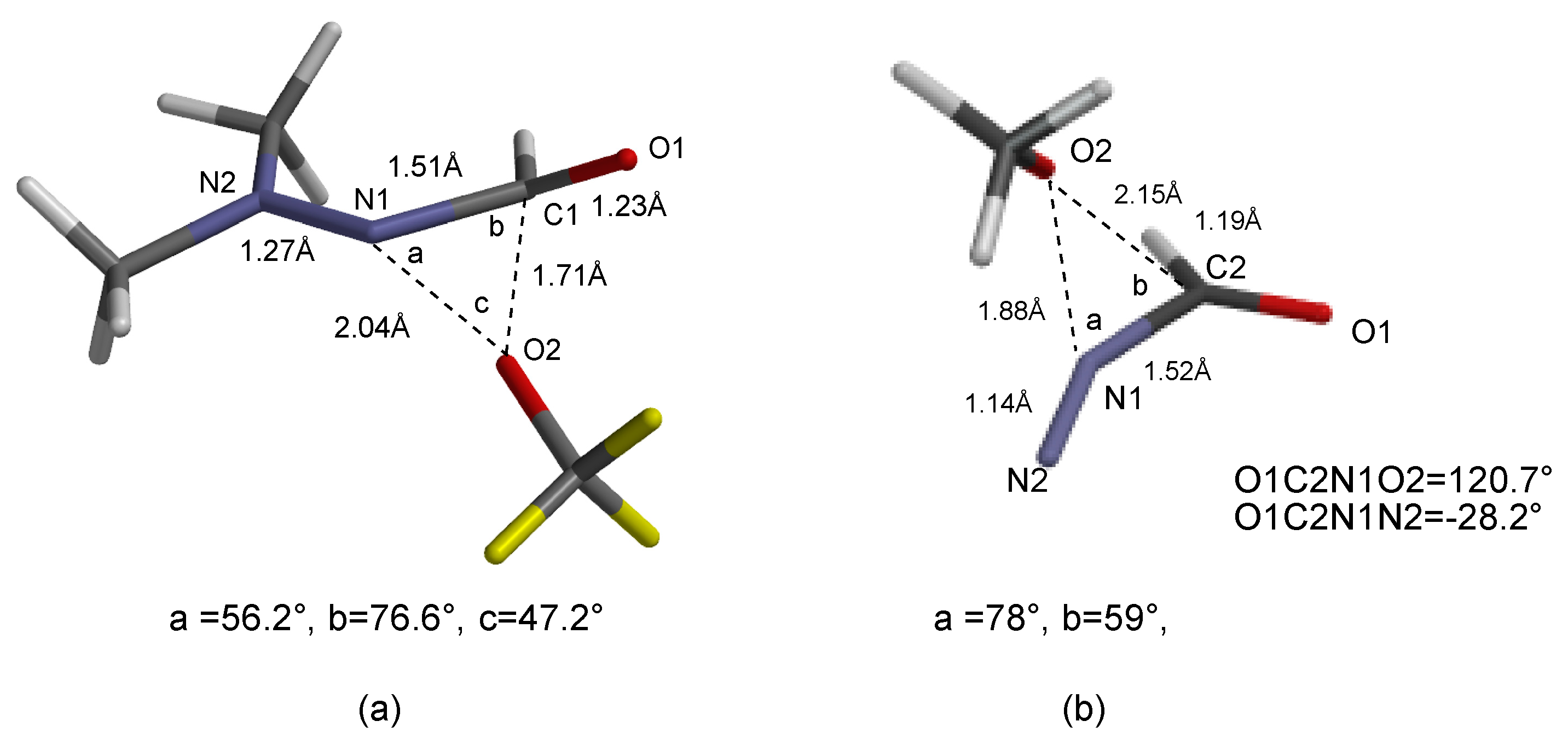
3.1.1. SN2 Reactions
3.1.2. Elimination Reactions
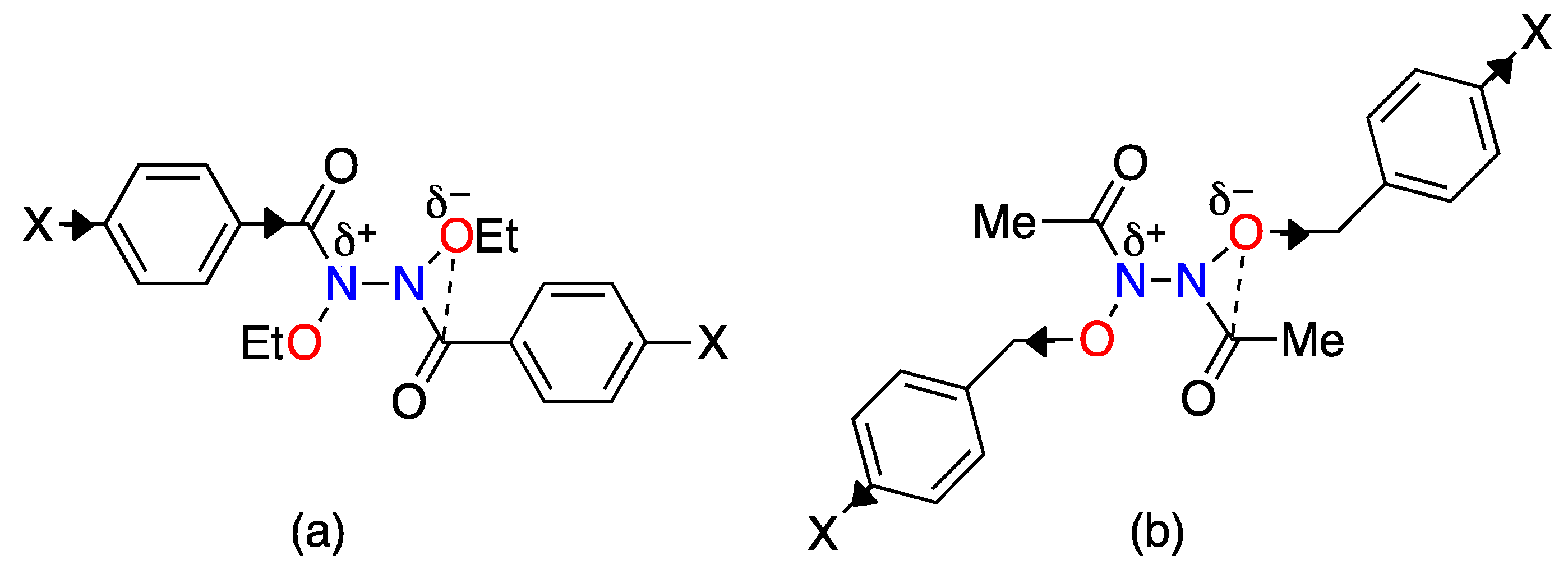
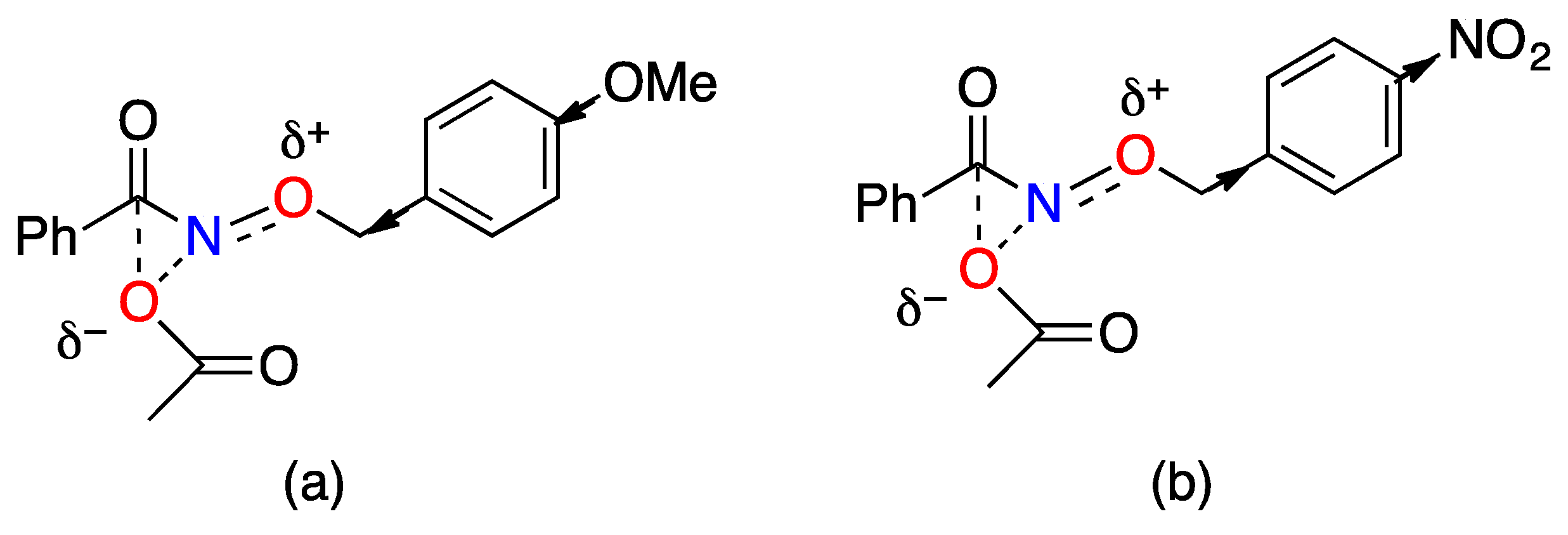
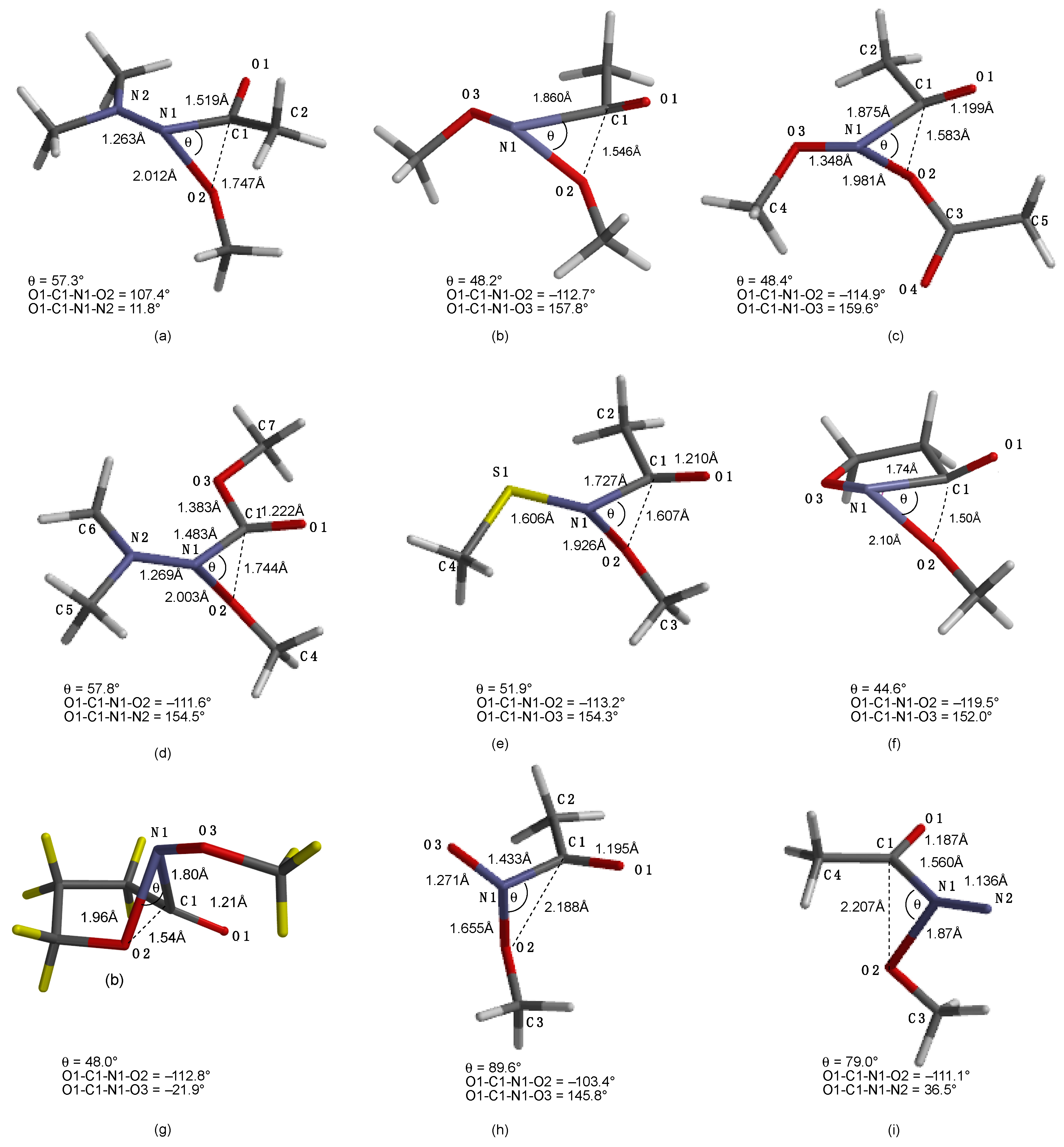
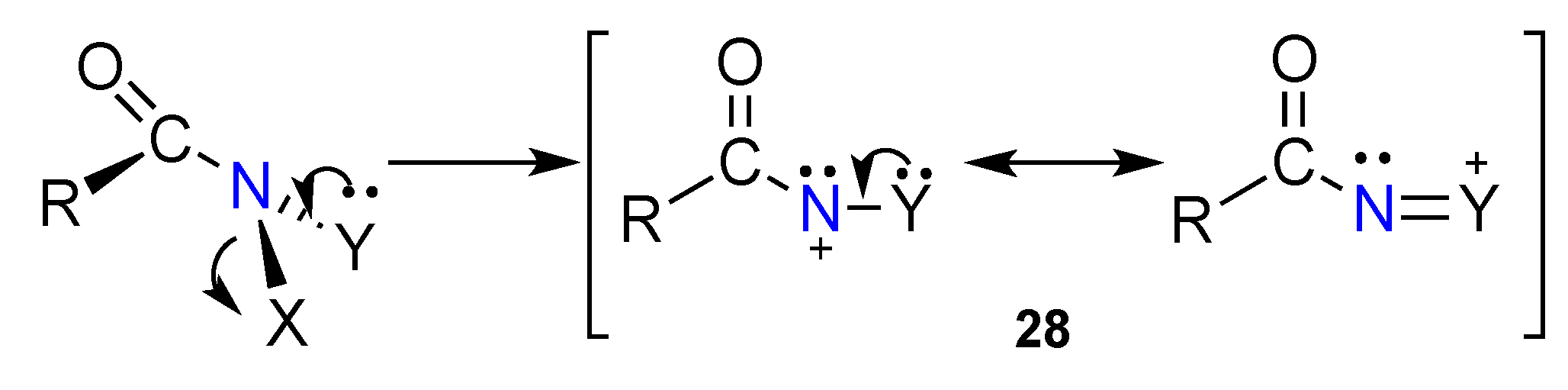
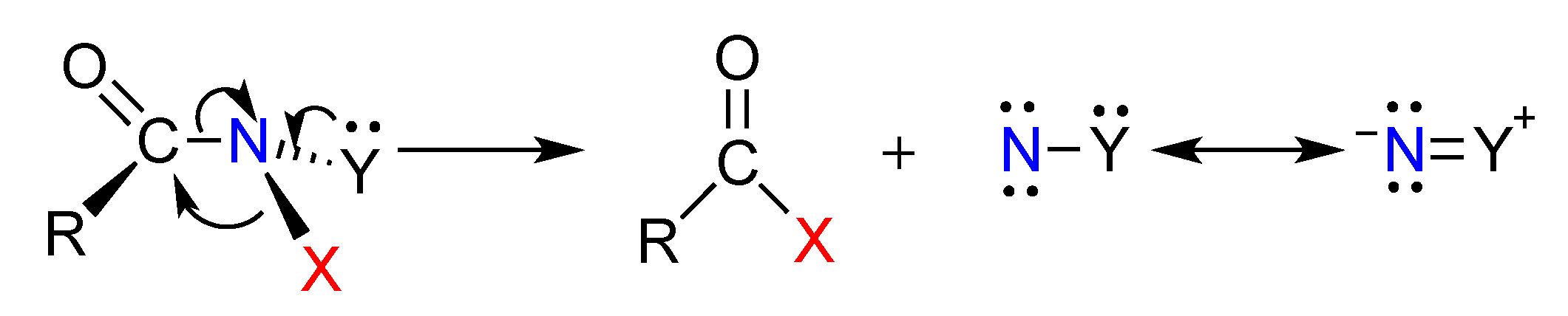
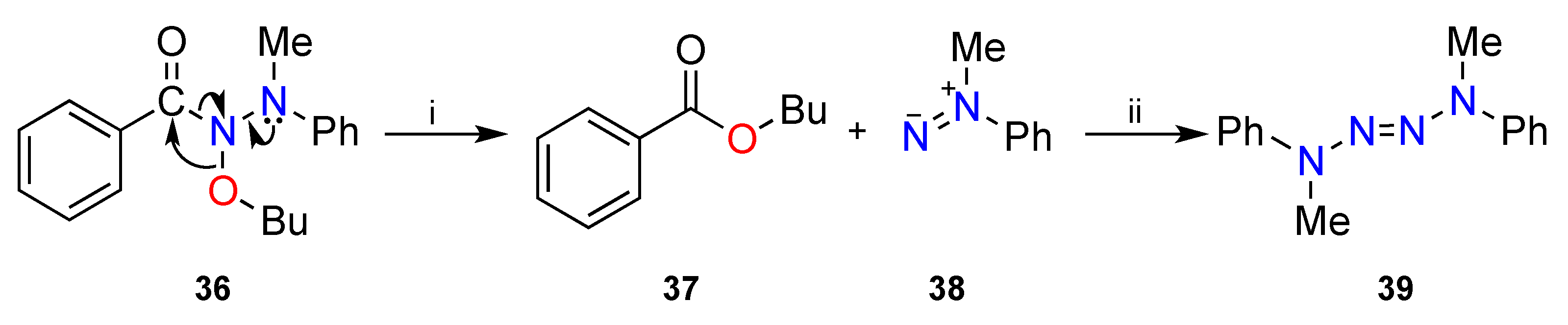
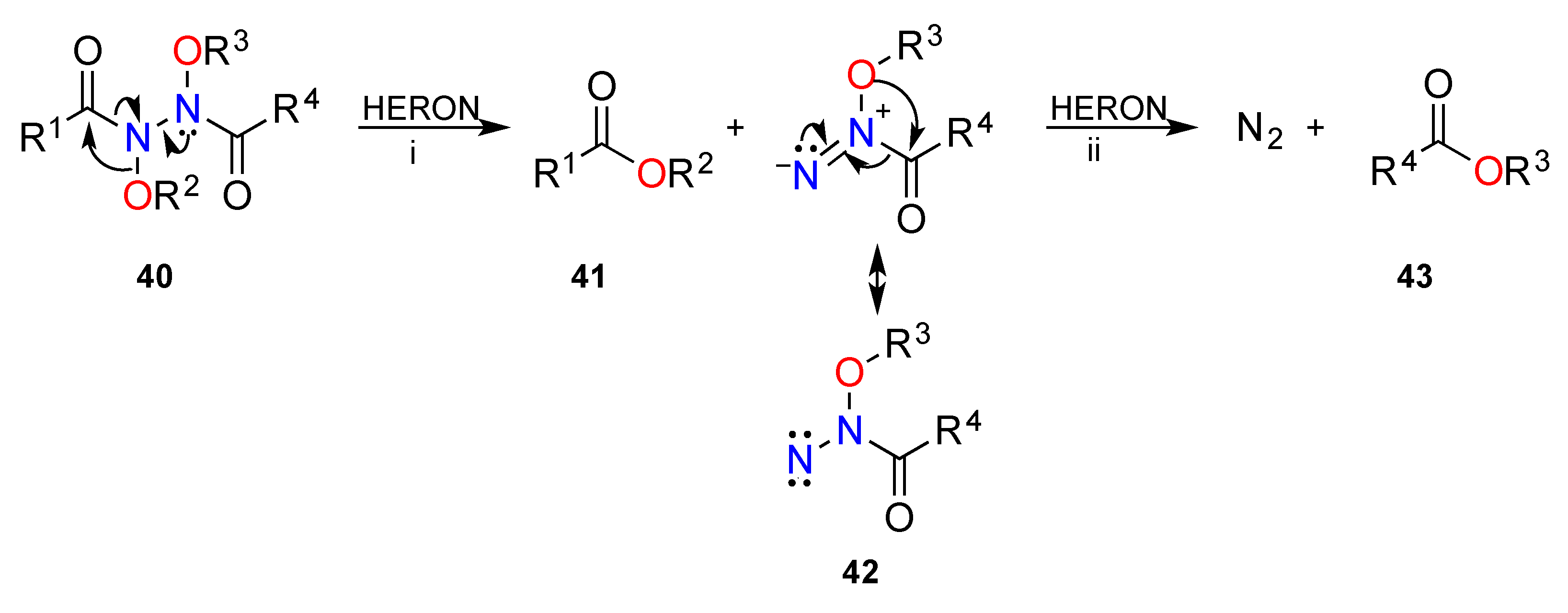
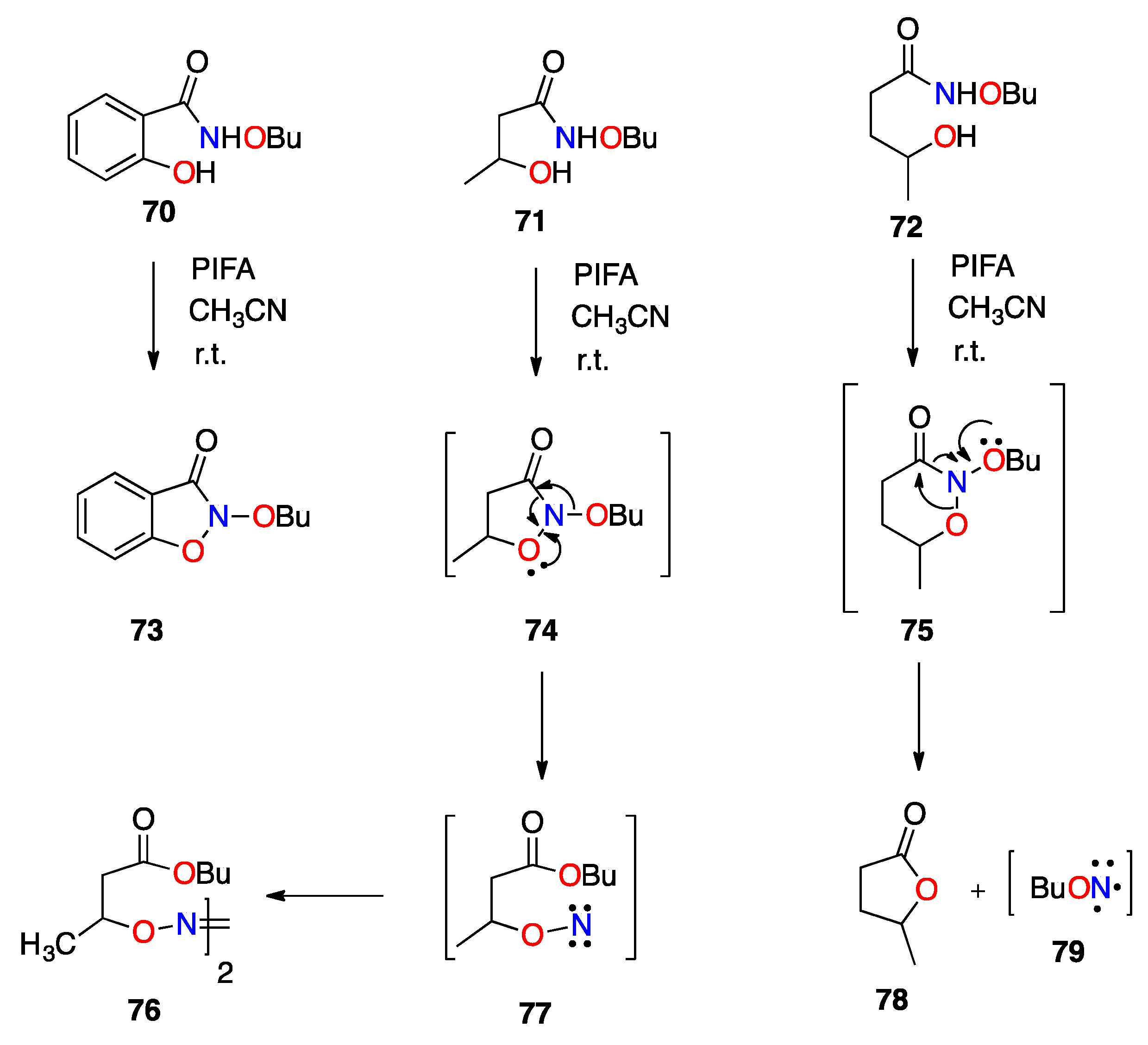
3.2. The HERON Reaction
3.2.1. HERON Reactions of N-Amino-N-Alkoxymides
3.2.2. Theoretical and Experimental Validation of the HERON Reaction
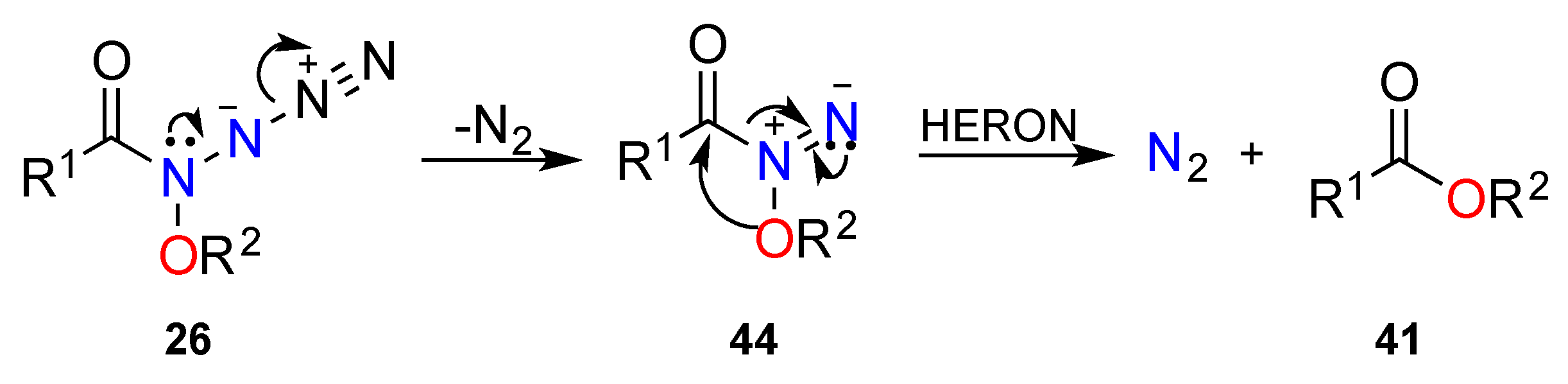
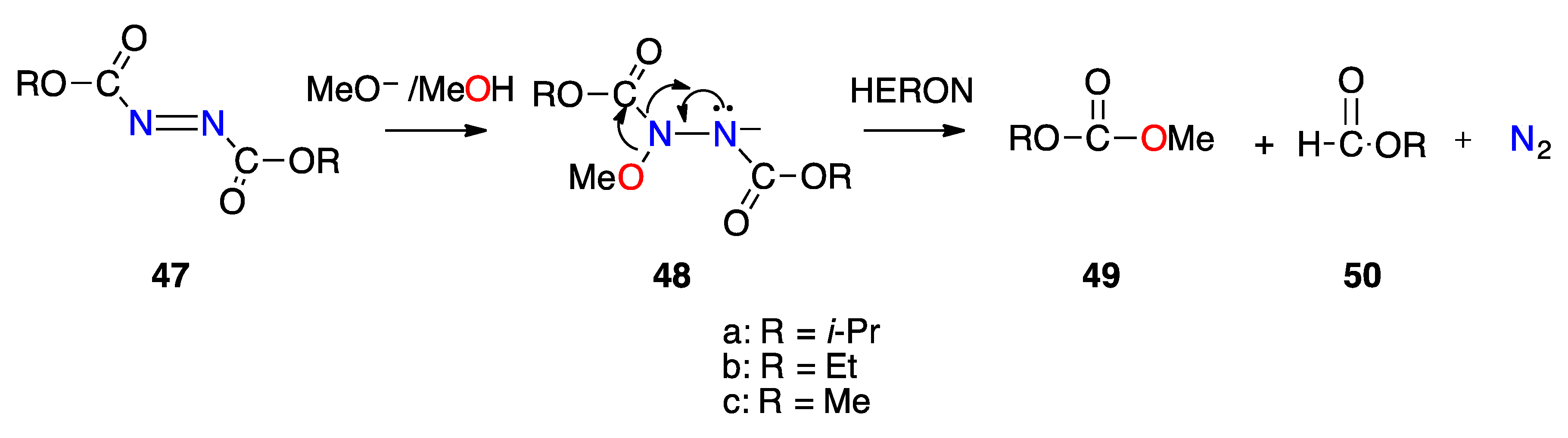
3.2.3. HERON Reaction of 1-Acyl-1-Alkoxydiazenes
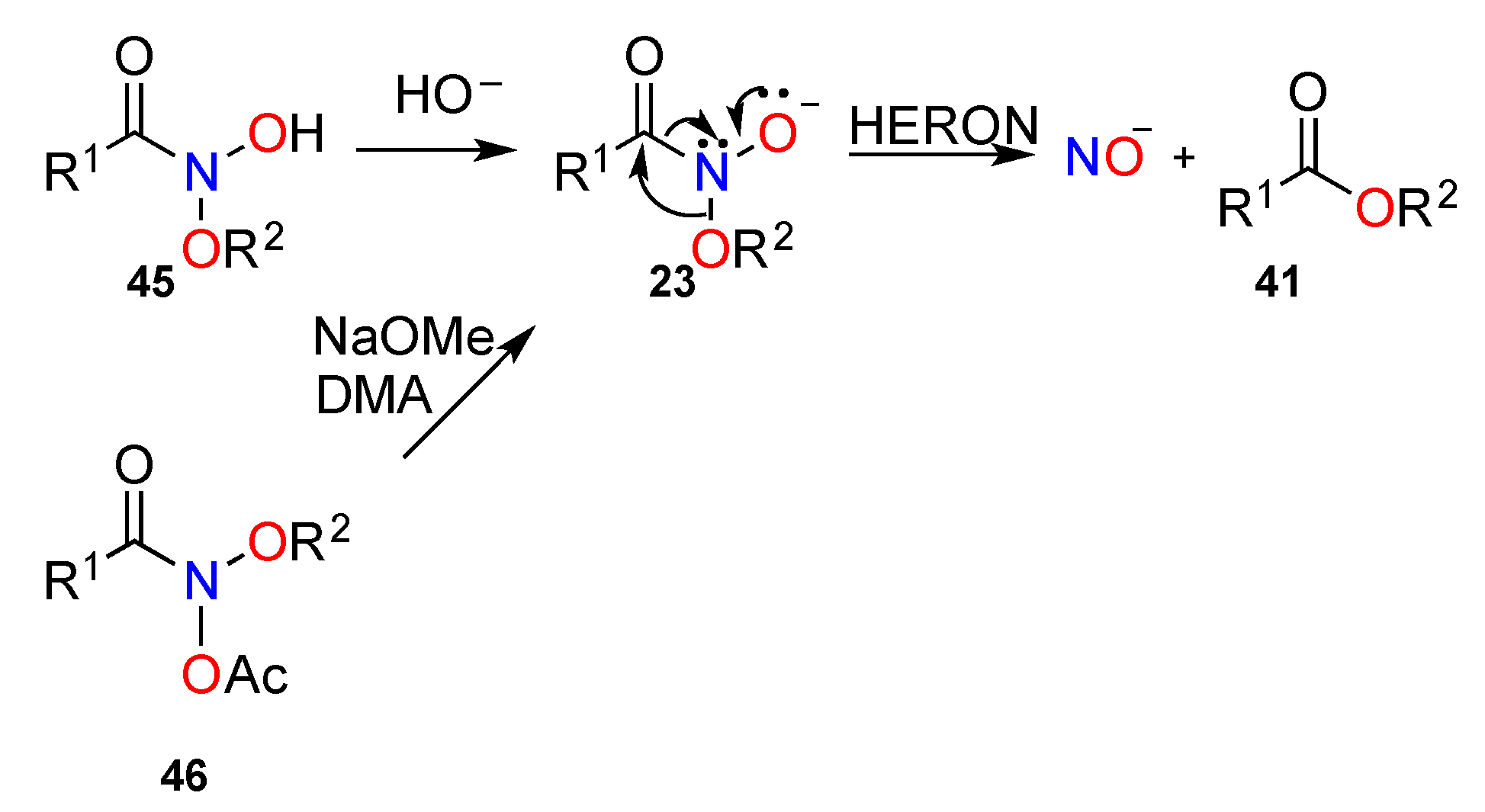
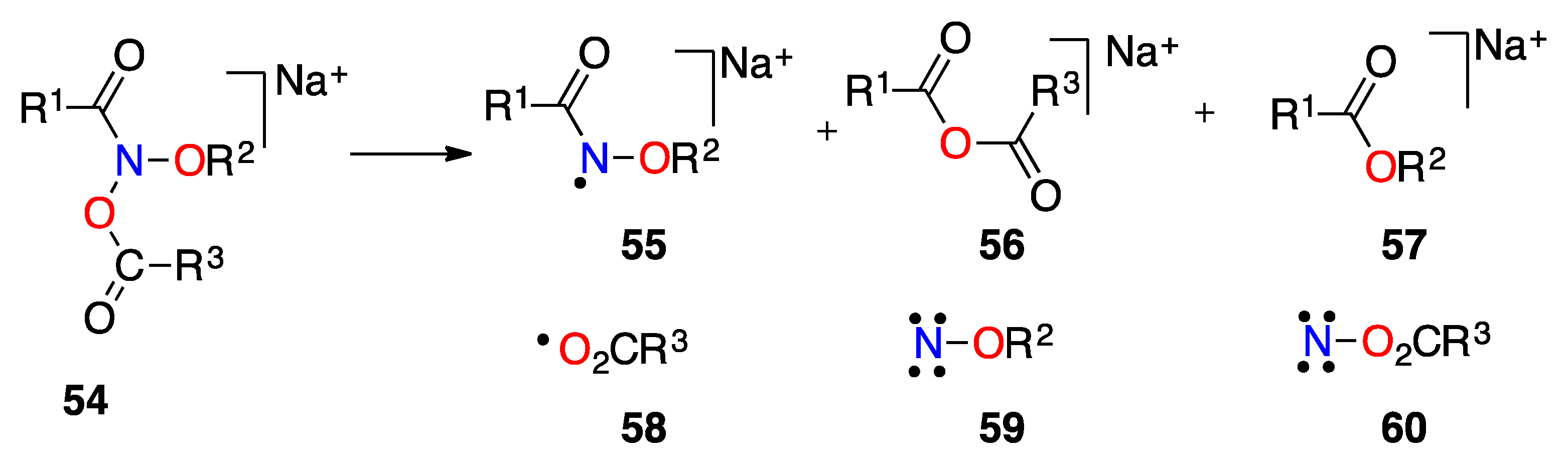
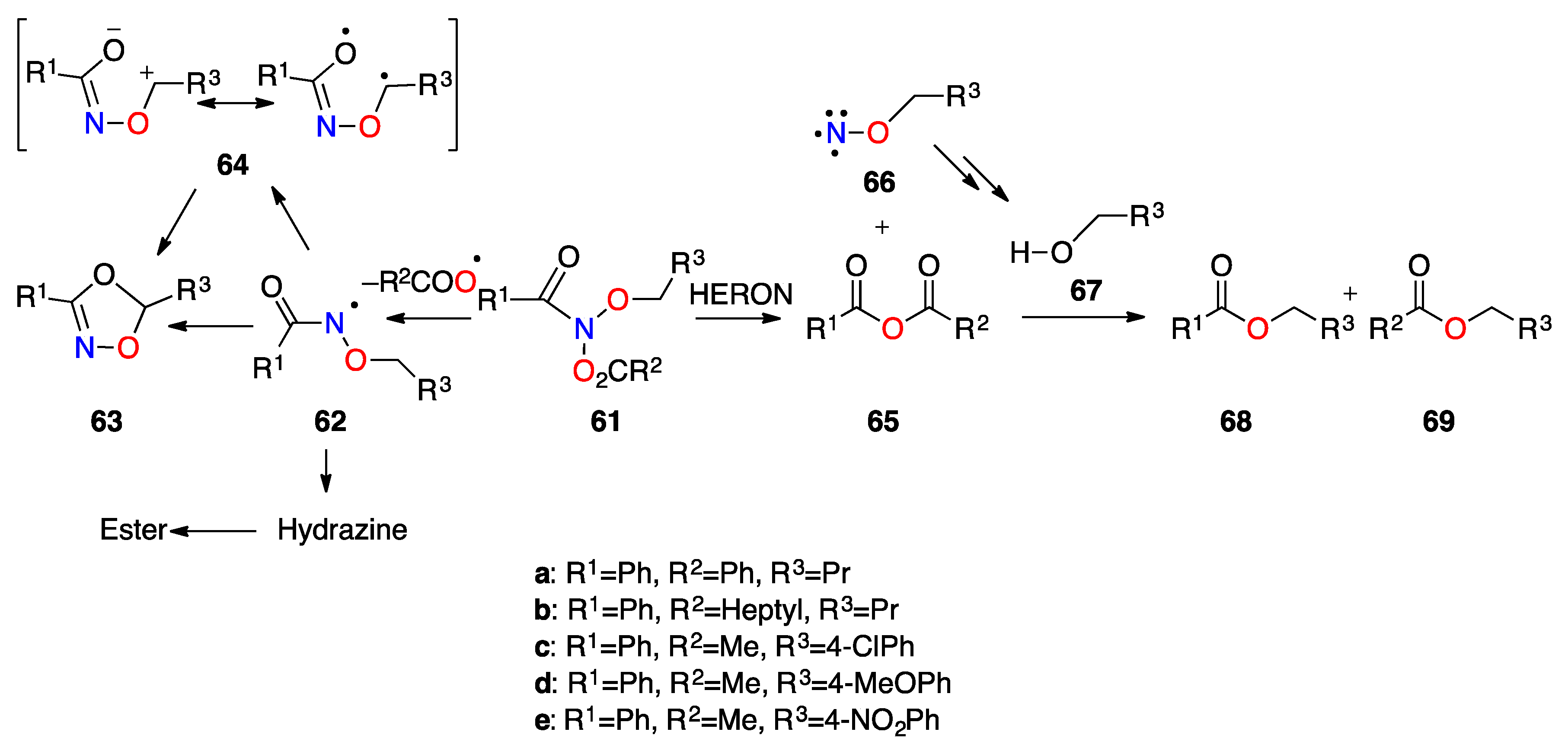
3.2.4. HERON Reactions of Anionic Systems
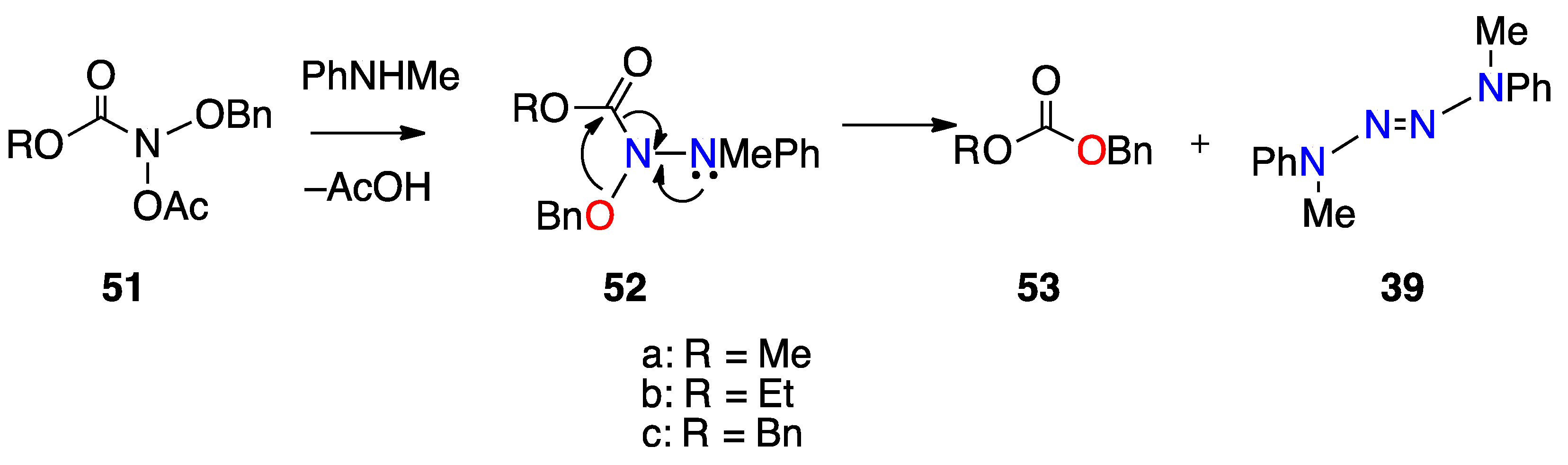
3.2.5. HERON Reactions of N-Alkoxy-N-Aminocarbamates
3.2.6. HERON Reactions of N-Acyloxy-N-Alkoxyamides
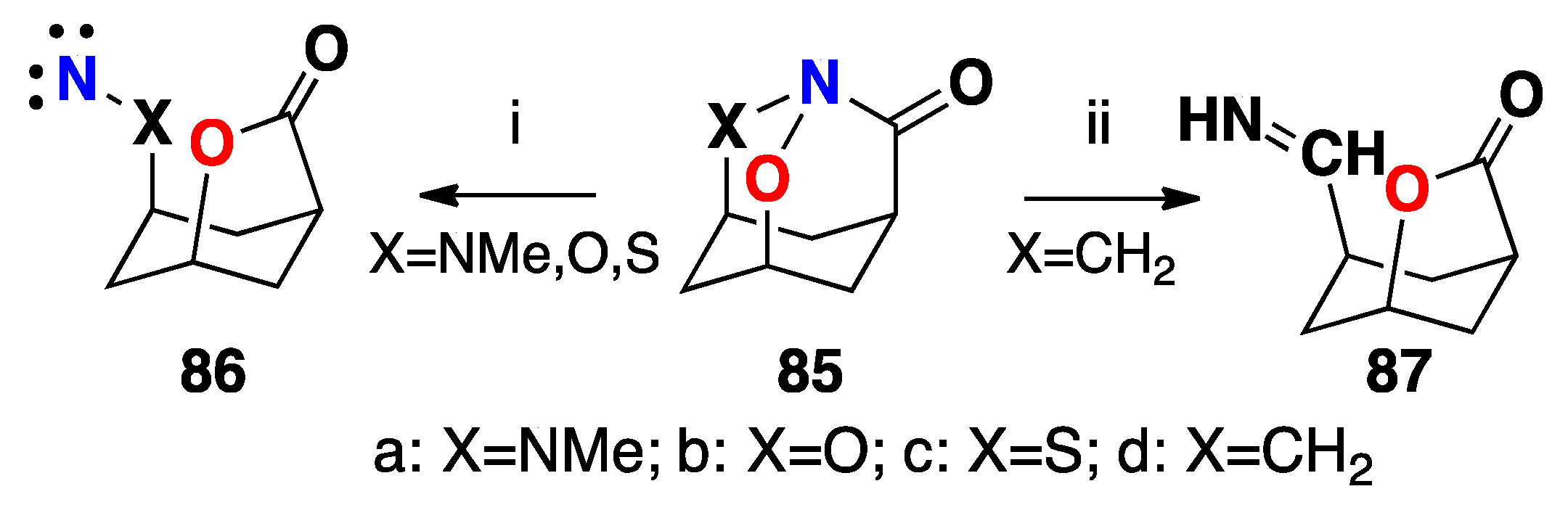
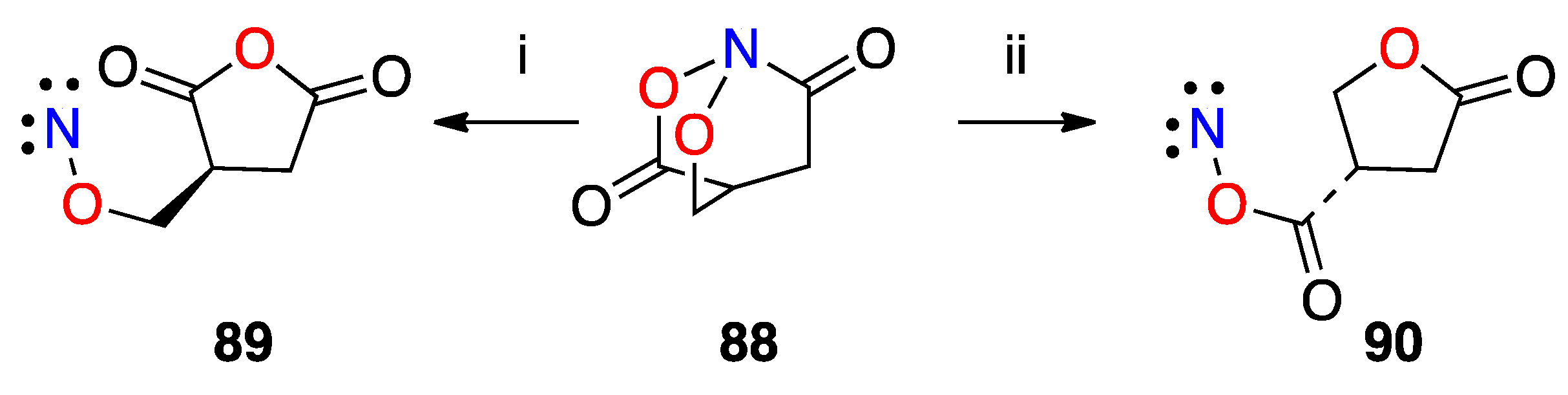
3.2.7. HERON Reactions of N,N-Dialkoxyamides
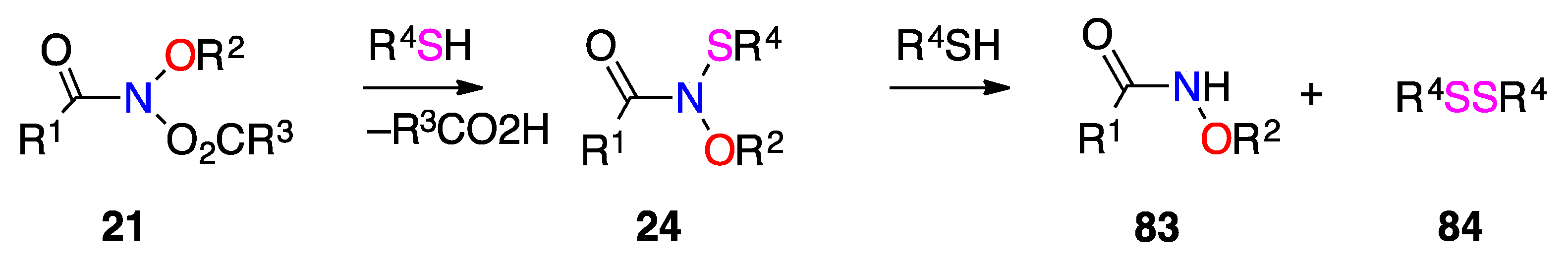
3.2.8. N-Alkoxy-N-Alkylthiylamides
3.3. Driving Force for the HERON Reaction
4. Conclusions
Conflicts of Interest
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Wiberg, K.B. Origin of the Amide Rotational Barrier. In The Amide Linkage. Structural Significance in Chemistry, Biochemistry and Materials Science; Greenberg, A., Breneman, C.M., Liebman, J.F., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2003; p. 33. [Google Scholar]
- Wiberg, K.B.; Breneman, C.M. Resonance interactions in acyclic systems. 3. Formamide internal rotation revisited. Charge and energy redistribution along the C–N bond rotational pathway. J. Am. Chem. Soc. 1992, 114, 831–840. [Google Scholar] [CrossRef]
- Glover, S.A.; Rosser, A.A. Reliable determination of amidicity in acyclic amides and lactams. J. Org. Chem. 2012, 77, 5492–5502. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D. X-ray Analysis and Structure of Organic Molecules; Cornell University Press: London, UK, 1979. [Google Scholar]
- Winkler, F.K.; Dunitz, J.D. The non-planar amide group. J. Mol. Biol. 1971, 59, 169–182. [Google Scholar] [CrossRef]
- Szostak, M.; Aube, J. Chemistry of bridged lactams and related heterocycles. Chem. Rev. 2013, 113, 5701–5765. [Google Scholar] [CrossRef] [PubMed]
- Szostak, M.; Aube, J. Medium-bridged lactams: A new class of non-planar amides. Org. Biomol. Chem. 2011, 9, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J.; Komarov, I.V.; Feeder, N. Spontaneous, millisecond formation of a twisted amide from the amino acid, and the crystal structure of a tetrahedral intermediate. J. Am. Chem. Soc. 1998, 120, 7101–7102. [Google Scholar] [CrossRef]
- Kirby, A.J.; Komarov, I.V.; Feeder, N. Synthesis, structure and reactions of the most twisted amide. J. Chem. Soc. Perk. Trans. 2001, 2, 522–529. [Google Scholar] [CrossRef]
- Kirby, A.J.; Komarov, I.V.; Kowski, K.; Rademacher, P. Distortion of the amide bond in amides and lactams. Photoelectron-spectrum and electronic structure of 3,5,7-trimethyl-1-azaadamantan-2-one, the most twisted amide. J. Chem. Soc. Perk. Trans. 1999, 2, 1313–1316. [Google Scholar] [CrossRef]
- Kirby, A.J.; Komarov, I.V.; Wothers, P.D.; Feeder, N. The most twisted amide: Structure and reactions. Angew. Chem. Int. Ed. 1998, 37, 785–786. [Google Scholar] [CrossRef]
- Morgan, J.; Greenberg, A. Novel bridgehead bicyclic lactams: (a) Molecules predicted to have O-protonated and N-protonated tautomers of comparable stability; (b) hyperstable lactams and their O-protonated tautomers. J. Chem. Thermodyn. 2014, 733, 206–212. [Google Scholar] [CrossRef]
- Tani, K.; Stoltz, B.M. Synthesis and structural analysis of 2-quinuclidonium tetrafluoroborate. Nature 2006, 441, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S. Chemistry of highly twisted amides. Rev. Heteroat. Chem. 1998, 19, 203–236. [Google Scholar]
- Yamada, S. Sterically Hindered Twisted Amides. In The Amide Linkage: Structural Aspects in Chemistry, Biochemistry, and Material Science; Greenberg, A., Breneman, C.M., Liebman, J.F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; pp. 215–246. [Google Scholar]
- Yamada, S. Structure and reactivity of an extremely twisted amide. Angew. Chem. 1993, 105, 1128–1130. [Google Scholar] [CrossRef]
- Cow, C.N.; Britten, J.F.; Harrison, P.H.M. X-ray crystal structure of 1,6-diacetyl-3,4,7,8-tetramethyl-2,5-dithioglycoluril a highly twisted acetamide. J. Chem. Soc. Chem. Commun. 1998, 1147–1148. [Google Scholar] [CrossRef]
- Anet, F.A.L.; Osyani, J.M. Nuclear magnetic resonance spectra and nitrogen inversion in 1-acylaziridinies. J. Am. Chem. Soc. 1967, 89, 352–356. [Google Scholar] [CrossRef]
- Boggs, G.R.; Gerig, J.T. Nitrogen inversion in N-Benzoylaziridines. J. Org. Chem. 1969, 34, 1484–1486. [Google Scholar] [CrossRef]
- Ohwada, T.; Achiwa, T.; Okamoto, I.; Shudo, K.; Yamaguchi, K. On the planarity of amide nitrogen. Intrinsic pyramidal nitrogen of N-acyl-7-azabicyclo 2.2.1 heptanes. Tetrahedron Lett. 1998, 39, 865–868. [Google Scholar] [CrossRef]
- Otani, Y.; Nagae, O.; Naruse, Y.; Inagaki, S.; Ohno, M.; Yamaguchi, K.; Yamamoto, G.; Uchiyama, M.; Ohwada, T. An Evaluation of amide group planarity in 7-azabicyclo[2.2.1]heptan amides. Low amide bond rotation barrier in solution. J. Am. Chem. Soc. 2003, 125, 15191–15199. [Google Scholar] [CrossRef] [PubMed]
- Otani, Y.; Futaki, S.; Kiwada, T.; Sugiura, Y.; Ohwada, T. Synthesis of non-planar peptides bearing the 7-azabicyclo[2.2.2]heptane skeleton, and possible self-organized structures. Pept. Sci. 2005, 41, 173–174. [Google Scholar]
- Ohwada, T.; Ishikawa, S.; Mine, Y.; Inami, K.; Yanagimoto, T.; Karaki, F.; Kabasawa, Y.; Otani, Y.; Mochizuki, M. 7-Azabicyclo[2.2.2]heptane as a structural motif to block mutagenicity of nitrosomines. Biorg. Med. Chem. 2011, 19, 2726–2741. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by x-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Glover, S.A. Anomeric amides—Structure, properties and reactivity. Tetrahedron 1998, 54, 7229–7272. [Google Scholar] [CrossRef]
- Rauk, A.; Glover, S.A. A computational investigation of the stereoisomerism in bisheteroatom-substituted amides. J. Org. Chem. 1996, 61, 2337–2345. [Google Scholar] [CrossRef]
- Glover, S.; Rauk, A. Conformational stereochemistry of the HERON amide, N-methoxy-N-dimethylaminoformamide: A theoretical study. J. Org. Chem. 1999, 64, 2340–2345. [Google Scholar] [CrossRef]
- Gillson, A.-M.E.; Glover, S.A.; Tucker, D.J.; Turner, P. Crystal structures and properties of mutagenic N-acyloxy-N-alkoxyamides—“Most pyramidal” acyclic amides. Org. Biomol. Chem. 2003, 1, 3430–3437. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.A.; Rosser, A.A.; Taherpour, A.; Greatrex, B.W. Formation and HERON reactivity of cyclic N,N-dialkoxyamides. Aust. J. Chem. 2014, 67, 507–520. [Google Scholar] [CrossRef]
- Glover, S.A. N-Acyloxy-N-alkoxyamide—Structure, properties, reactivity and biological activity. Adv. Phys. Org. Chem. 2007, 42, 35–123. [Google Scholar]
- Glover, S.A.; Mo, G.; Rauk, A.; Tucker, D.J.; Turner, P. Structure, conformation, anomeric effects and rotational barriers in the HERON amides, N,N′-diacyl-N,N′-dialkoxyhydrazines. J. Chem. Soc. Perkin Trans. 2 1999, 2053–2058. [Google Scholar] [CrossRef]
- Glover, S.A.; White, J.M.; Rosser, A.A.; Digianantonio, K.M. Structures of N,N-Dialkoxyamides—Pyramidal anomeric amides with low amidicity. J. Org. Chem. 2011, 76, 9757–9763. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.A.; Rosser, A.A.; Spence, R.M. Studies of the structure, amidicity and reactivity of N-chlorohydroxamic esters and N-chloro-β, β-dialkylhydrazides: Anomeric amides with low resonance energies. Aust. J. Chem. 2014, 67, 1344–1352. [Google Scholar] [CrossRef]
- Glover, S.A. N-Heteroatom-Substituted Hydroxamic Esters. In The Chemistry of Hydroxylamines, Oximes and Hydroxamic; Rappoport, Z., Liebman, J.F., Eds.; Wiley: Chichester, UK, 2009; pp. 839–923. [Google Scholar]
- Alabugin, I.V.; Bresch, S.; Manoharan, M. Hybridization trends for main group elements and expanding the Bent’s rule beyond carbon: More than electronegativity. J. Phys. Chem. A 2014, 118, 3663–3677. [Google Scholar] [CrossRef] [PubMed]
- Bent, H.A. An appraisal of valence-bond structures and hybridization in compounds of the first-row elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Glover, S.A.; Schumacher, R.R. The effect of hydrophobicity upon the direct mutagenicity of N-acyloxy-N-alkoxyamides—Bilinear dependence upon LogP. Mutat. Res. 2016, 795, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Banks, T.M.; Clay, S.F.; Glover, S.A.; Schumacher, R.R. Mutagenicity of N-acyloxy-N-alkoxyamides as an indicator of DNA intercalation Part 1: Evidence for naphthalene as a DNA intercalator. Org. Biomol. Chem. 2016, 14, 3699–3714. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.A.; Schumacher, R.R.; Bonin, A.M.; Fransson, L.E. Steric effects upon the direct mutagenicity of N-acyloxy-N-alkoxyamides—Probes for drug-DNA interactions. Mutat. Res. 2011, 722, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.E.; Banks, T.M.; Bonin, A.M.; Clay, S.F.; Gillson, A.-M.E.; Glover, S.A. Mutagenic N-acyloxy-N-alkoxyamides—Probes for drug—DNA interactions. Aust. J. Chem. 2004, 57, 377–381. [Google Scholar] [CrossRef]
- Banks, T.M.; Bonin, A.M.; Glover, S.A.; Prakash, A.S. Mutagenicity and DNA damage studies of N-acyloxy-N-alkoxyamides—The role of electrophilic nitrogen. Org. Biomol. Chem. 2003, 1, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Bonin, A.M.; Banks, T.M.; Campbell, J.J.; Glover, S.A.; Hammond, G.P.; Prakash, A.S.; Rowbottom, C.A. Mutagenicity of electrophilic N-acyloxy-N-alkoxyamides. Mutat. Res. 2001, 494, 115–134. [Google Scholar] [CrossRef]
- Campbell, J.J.; Glover, S.A. (Synopsis) Bimolecular reactions of mutagenic N-(acyloxy)-N-alkoxybenzamides with aromatic amines. J. Chem. Res. 1999, 8, 474–475. [Google Scholar] [CrossRef]
- Glover, S.A.; Hammond, G.P.; Bonin, A.M. A comparison of the reactivity and mutagenicity of N-benzoyloxy-N-benzyloxybenzamides. J. Org. Chem. 1998, 63, 9684–9689. [Google Scholar] [CrossRef]
- Bonin, A.M.; Glover, S.A.; Hammond, G.P. Reactive intermediates from the solvolysis of mutagenic O-alkyl N-acetoxybenzohydroxamates. J. Chem. Soc. Perkin Trans. 2 1994, 6, 1173–1180. [Google Scholar] [CrossRef]
- Campbell, J.J.; Glover, S.A. Bimolecular reactions of mutagenic N-acetoxy-N-alkoxybenzamides and N-methylaniline. J. Chem. Soc. Perk. Trans. 2 1992, 10, 1661–1663. [Google Scholar] [CrossRef]
- Campbell, J.J.; Glover, S.A.; Hammond, G.P.; Rowbottom, C.A. Evidence for the formation of nitrenium ions in the acid-catalysed solvolysis of mutagenic N-acetoxy-N-alkoxybenzamides. J. Chem. Soc. Perkin Trans. 2 1991, 2067–2079. [Google Scholar] [CrossRef]
- Campbell, J.J.; Glover, S.A.; Rowbottom, C.A. Solvolysis and mutagenesis of N-acetoxy-N-alkoxybenzamides—Evidence for nitrenium ion formation. Tetrahedron Lett. 1990, 31, 5377–5380. [Google Scholar] [CrossRef]
- Andrews, L.E.; Bonin, A.M.; Fransson, L.E.; Gillson, A.-M.E.; Glover, S.A. The role of steric effects in the direct mutagenesis of N-acyloxy-N-alkoxyamides. Mutat. Res. 2006, 605, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, K.L.; Glover, S.A.; Price, H.L.; Schumacher, R.R. SN2 Substitution reactions at the amide nitrogen in the anomeric mutagens, N-acyloxy-N-alkoxyamides. Aust. J. Chem. 2009, 62, 700–710. [Google Scholar] [CrossRef]
- Glover, S.A.; Adams, M. Reaction of N-acyloxy-N-alkoxyamides with biological thiols. Aust. J. Chem. 2011, 64, 443–453. [Google Scholar] [CrossRef]
- Glover, S.A.; Rauk, A. A computational investigation of the structure of the novel anomeric amide N-azido-N-methoxyformamide and its concerted decomposition to methyl formate and nitrogen. J. Chem. Soc. Perk. Trans. 2 2002, 0, 1740–1746. [Google Scholar] [CrossRef]
- Glover, S.A.; Mo, G. Hindered ester formation by SN2 azidation of N-acetoxy-N-alkoxyamides and N-alkoxy-N-chloroamides—Novel application of HERON rearrangements. J. Chem. Soc. Perk. Trans. 2 2002, 10, 1728–1739. [Google Scholar] [CrossRef]
- Digianantonio, K.M.; Glover, S.A.; Johns, J.P.; Rosser, A.A. Synthesis and thermal decomposition of N,N-dialkoxyamides. Org. Biomol. Chem. 2011, 9, 4116–4126. [Google Scholar] [CrossRef] [PubMed]
- Buccigross, J.M.; Glover, S.A.; Hammond, G.P. Decomposition of N,N-diacyl-N,N-dialkoxyhydrazines revisited. Aust. J. Chem. 1995, 48, 353–361. [Google Scholar]
- Greenberg, A. The Amide Linkage as a Ligand: Its Properties and the Role of Distortion. In The Amide Linkage. Structural Significance in Chemistry, Biochemistry and Materials Science; Greenberg, A., Breneman, C.M., Liebman, J.F., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2003; pp. 47–83. [Google Scholar]
- Greenberg, A.; Moore, D.T.; DuBois, T.D. Small and medium-sized bridgehead lactams: A systematic ab initio molecular orbital study. J. Am. Chem. Soc. 1996, 118, 8658–8668. [Google Scholar] [CrossRef]
- Greenberg, A.; Venanzi, C.A. Structures and enrgetics of two bridgehead lactams and their N- and O-protonated froms: An ab initio molecular orbital study. J. Am. Chem. Soc. 1993, 115, 6951–6957. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.; Pople, J.A. Ab Initio Molecular Orbital Theory; John Wiley & Sons: New York, NY, USA, 1986. [Google Scholar]
- Glover, S.A.; Rosser, A.A. The role of substituents in the HERON reaction of anomeric amides. Can. J. Chem. 2016, 94, 1169–1180. [Google Scholar] [CrossRef]
- Glover, S.A.; Rosser, A.A. HERON reactions of anomeric amides: Understanding the driving force. J. Phys. Org. Chem. 2015, 28, 215–222. [Google Scholar] [CrossRef]
- Allen, F.H. The cambridge structural database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Rablen, P.R.; Rush, D.J.; Keith, T.A. Amides. 3. Experimental and theoretical studies of the effect of the medium on the rotational barriers for N,N-dimethylformamide and N,N-dimethylacetamide. J. Am. Chem. Soc. 1995, 117, 4261–4270. [Google Scholar] [CrossRef]
- Shtamburg, V.G.; Tsygankov, A.T.; Shishkin, O.V.; Zubatyuk, R.I.; Uspensky, B.V.; Shtamburg, V.V.; Mazepa, A.V.; Kostyanovsky, R.G. The properties and structure of N-chloro-N-methoxy-4-nitrobenzamide. Mendeleev Commun. 2012, 22, 164–166. [Google Scholar] [CrossRef]
- Shtamburg, V.G.; Shishkin, O.V.; Zubatyuk, R.I.; Kravchenko, S.V.; Tsygankov, A.V.; Mazepa, A.V.; Klots, E.A.; Kostyanovsky, R.G. N-Chloro-N-alkoxyureas: Synthesis, structure and properties. Mendeleev Commun. 2006, 16, 323–325. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Zubatyuk, R.I.; Shtamburg, V.G.; Tsygankov, A.V.; Klots, E.A.; Mazepa, A.V.; Kostyanovsky, R.G. Pyramidal amide nitrogen in N-acyloxy-N-alkoxyureas and N-acyloxy-N-alkoxycarbamates. Mendeleev Commun. 2006, 16, 222–223. [Google Scholar] [CrossRef]
- Shtamburg, V.G.; Shishkin, O.V.; Zubatyuk, R.I.; Kravchenko, S.V.; Tsygankov, A.V.; Shtamburg, V.V.; Distanov, V.B.; Kostyanovsky, R.G. Synthesis, structure and properties of N-alkoxy-N-(1-pyridinium)urea salts, N-alkoxy-N-acyloxyureas and N,N-dialkoxyureas. Mendeleev Commun. 2007, 17, 178–180. [Google Scholar] [CrossRef]
- Shtamburg, V.G.; Shishkin, O.V.; Zubatyuk, R.I.; Shtamburg, V.V.; Tsygankov, A.V.; Mazepa, A.V.; Kadorkina, G.K.; Kostanovsky, R.G. Synthesis and structure of N-alkoxyhydrazines and N-alkoxy-N′,N′, N′-trialkylhydrazinium salts. Mendeleev Commun. 2013, 23, 289–291. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Bresch, S.; dos Passos Gomez, G. Orbital hybridization: A key electronic factor in control of structure and reactivity. J. Phys. Org. Chem. 2015, 28, 147–162. [Google Scholar] [CrossRef]
- Alabugin, I.V. Stereoelectronic Effects: A Bridge between Structure and Reactivity, 1st ed.; John Wiley & Sons: Chichester, UK, 2016. [Google Scholar]
- Alabugin, I.V.; Zeidan, T.A. Stereoelectronic effects and general trends in hyperconjugative acceptor ability of σ bonds. J. Am. Chem. Soc. 2002, 124, 3175–3185. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. Orbital Interaction Theory of Organic Chemistry; John Wiley & Sons, Inc.: New York, NY, USA, 1994; p. 102. [Google Scholar]
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; John Wiley & Sons, Inc: New York, NY, USA, 1994; pp. 753–1191. [Google Scholar]
- Epiotis, N.D.; Cherry, W.R.; Shaik, S.; Yates, R.L.; Bernardi, F. Structural Theory of Organic Chemistry; Springer: Berlin, Germany, 1977. [Google Scholar]
- Glover, S.A.; Mo, G.; Rauk, A. HERON rearrangement of N,N′-diacyl-N,N′-dialkoxyhydrazines—A theoretical and experimental study. Tetrahedron 1999, 55, 3413–3426. [Google Scholar] [CrossRef]
- De Almeida, M.V.; Barton, D.H.R.; Bytheway, I.; Ferriera, J.A.; Hall, M.B.; Liu, W.; Taylor, D.K.; Thomson, L. Preparation and thermal decomposition of N,N′-diacyl-N,N′-dialkoxyhydrazines: Synthetic applications and mechanitic insights. J. Am. Chem. Soc. 1995, 117, 4870–4874. [Google Scholar] [CrossRef]
- Cooley, J.H.; Mosher, M.W.; Khan, M.A. Preparation and reactions of N,N′-diacyl-N,N′-dialkoxyhydrazines. J. Am. Chem. Soc. 1968, 90, 1867–1871. [Google Scholar] [CrossRef]
- Banks, T.M. Reactivity, Mutagenicity and DNA Damage of N-Acyloxy-N-Alkoxyamides. Ph.D. Thesis, University of New England, Armidale, Australia, 2003. [Google Scholar]
- Gerdes, R.G.; Glover, S.A.; Ten Have, J.F.; Rowbottom, C.A. N-Acetoxy-N-alkoxyamides—A new class of nitrenium ion precursors which are mutagenic. Tetrahedron Lett. 1989, 30, 2649–2652. [Google Scholar] [CrossRef]
- Taherpour, A. University of New England, Armidale, New South Wales, Australia. Personal communication, 2012. [Google Scholar]
- Williams, D.H.; Fleming, I. Spectroscopic Methods in Organic Chemistry, 3rd ed.; McGraw Hill Ltd.: Maidenhead, UK, 1980. [Google Scholar]
- Jackman, L.M. Rotation about Partial Double Bonds in Organic Molecules. In Dynamic Nuclear Magnetic Resonance Spectroscopy; Jackman, L.M., Cotton, F.A., Eds.; Academic Press: Cambridge, MA, USA, 1975. [Google Scholar]
- Günther, H. NMR Spectroscopy—Basic Principles, Concepts, and Applications in Chemistry, 2nd ed.; John Wiley & Sons Ltd.: Chichester, UK, 1995. [Google Scholar]
- Johns, J.P.; van Losenoord, A.; Mary, C.; Garcia, P.; Pankhurst, D.S.; Rosser, A.A.; Glover, S.A. Thermal decomposition of N-acyloxy-N-alkoxyamides—A new HERON reaction. Aust. J. Chem. 2010, 63, 1717–1729. [Google Scholar] [CrossRef]
- Glover, S.A. SN2 reactions at amide nitrogen—Theoretical models for reactions of mutagenic N-acyloxy-N-alkoxyamides with bionucleophiles. Arkivoc 2002, 2001, 143–160. [Google Scholar] [CrossRef]
- Campbell, J.J.; Glover, S.A. (Microfiche) Bimolecular reactions of mutagenic N-(acyloxy)-N-alkoxybenzamides with aromatic amines. J. Chem. Res. 1999, 8, 2075–2096. [Google Scholar]
- Isaacs, N.S. Physical Organic Chemistry, 2nd ed.; Longman Scientific and Technical: New York, NY, USA, 1995; p. 422. [Google Scholar]
- Forster, W.; Laird, R.M. The mechanism of alkylation reactions. Part 1. The effect of ubstituents on the reaction of phenacyl bromide with pyridine in methanol. J. Chem. Soc. Perk. Trans. 2 1982, 2, 135–138. [Google Scholar] [CrossRef]
- De Kimpe, N.; Verhé, R. The Chemistry of α-Haloketones, α-Haloaldehydes and α-Haloimines; John Wiley & Sons: Chichester, UK, 1988; p. 38. [Google Scholar]
- Thorpe, J.W.; Warkentin, J. Stereochemical and steric effects in nucleophilic substitution of α-halo ketones. Can. J. Chem. 1973, 51, 927–935. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised methods for salmonella mutagenicity tests. Mutat. Res. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Mortelmans, K.; Zeiger, E. The ames salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Pullman, A.; Pullman, B. Electrostatic effect of macromolecular structure on the biochemical reactivity of the nucleic acids. Significance for chemical carcinogenesis. Int. J. Quantum Chem. Symposia 1980, 18, 245–259. [Google Scholar] [CrossRef]
- Kohn, K.W.; Hartley, J.A.; Mattes, W.B. Mechanism of sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Nucleic Acids Res. 1987, 15, 10531–10544. [Google Scholar] [CrossRef] [PubMed]
- Warpehoski, M.A.; Hurley, L.H. Sequence selectivity of DNA covalent modification. Chem. Res. Toxicol. 1988, 1, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.S.; Denny, W.A.; Gourdie, T.A.; Valu, K.K.; Woodgate, P.D.; Wakelin, L.P.G. DNA-directed alkylating ligands as potential antitumor agents: Sequence specificity of alkylation by aniline mustards. Biochemistry 1990, 29, 9799–9807. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.A.; Scott, A.P. MNDO properties of heteroatom and phenyl substituted nitrenium ions. Tetrahedron 1989, 45, 1763–1776. [Google Scholar] [CrossRef]
- Schroeder, D.; Grandinetti, F.; Hrusak, J.; Schwarz, H. Experimental and ab initio MO studies on [H2,N,O]+ ions in the gas phase: Characterization of the isomers H2NO+, HNOH+ and NOH2+ and the mechanism of unimolecular dehydrogenation of [H2,N,O]+. J. Phys. Chem. 1992, 96, 4841–4845. [Google Scholar] [CrossRef]
- Glover, S.A.; Goosen, A.; McCleland, C.W.; Schoonraad, J.L. N-alkoxy-N-acylnitrenium ions as possible intermediates in intramolecular aromatic substitution: Novel formation of N-acyl-3,4-dihydro-1H-2,1-benzoxazines and N-acyl-4,5-dihydro-1H-2,1-benzoxazepine. J. Chem. Soc. Perkin Trans. 1 1984, 2255–2260. [Google Scholar] [CrossRef]
- Glover, S.A.; Rowbottom, C.A.; Scott, A.P.; Schoonraad, J.L. Alkoxynitrenium ion cyclisations: Evidence for difference mechanisms in the formation of benzoxazines and benzoxazepines. Tetrahedron 1990, 46, 7247–7262. [Google Scholar] [CrossRef]
- Kikugawa, Y.; Kawase, M. Electrophilic aromatic substitution with a nitrenium ion generated from N-chloro-N-methoxyamides. Application to the synthesis of 1-methoxy-2-oxindoles. J. Am. Chem. Soc. 1984, 106, 5728–5729. [Google Scholar] [CrossRef]
- Kawase, M.; Kitamura, T.; Kikugawa, Y. Electrophilic aromatic substitution with N-methoxy-N-acylnitrenium ions generated from N-chloro-N-methoxy amides: Syntheses of nitrogen heterocyclic compounds bearing a N-methoxy amide group. J. Org. Chem. 1989, 54, 3394–3403. [Google Scholar] [CrossRef]
- Kikugawa, Y.; Shimada, M.; Kato, M.; Sakamoto, T. A new synthesis of N-alkoxy-2-ethoxyarylacetamides from N-alkoxy-N-chloroarylacetamides with triethylamine in ethanol. Chem. Pharm. Bull. 1993, 41, 2192–2194. [Google Scholar] [CrossRef]
- Miyazawa, E.; Sakamoto, T.; Kikugawa, Y. Syntheis of spirodienones by intramolecular ipso-cyclization of N-methoxy-(4-halegenophenyl)amides using [hydroxy(tosyloxy)iodo]benzene in trifluoroethanol. J. Org. Chem. 2003, 68, 5429–5432. [Google Scholar] [CrossRef] [PubMed]
- Kikugawa, Y.; Kawase, M.; Miyake, Y.; Sakamoto, T.; Shimada, M. A convenient synthesis of eupolauramine. Tetrahedron Lett. 1988, 29, 4297–4298. [Google Scholar] [CrossRef]
- Kikugawa, Y.; Shimado, M.; Matsumoto, K. Cyclization with nitrenium ions generated from N-methoxy- or N-allyloxy-N-chloroamides with anhydrous zinc acetate. Synthesis of N-hydroxy- and N-methoxynitrogen heterocyclic compounds. Heterocycles 1994, 37, 293–301. [Google Scholar] [CrossRef]
- Kikugawa, Y. Uses of hydroxamic acids and N-alkoxyimidoyl halides in organic synthesis. Rev. Heteroat. Chem. 1996, 15, 263–299. [Google Scholar]
- Greenstein, G. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 14nd ed.; O’Neil, M.J., Heckelman, P.E., Koch, C.B., Roman, K.J., Eds.; Emerald Group Publishing Limited: Whitehouse Station, NJ, USA, 2006; p. 40. [Google Scholar]
- The Heron Reaction—Merck Index Named Reaction Index. Available online: https://www.rsc.org/Merck-Index/reaction/r197/ (accessed on 30 October 2018).
- Buccigross, J.M.; Glover, S.A. Molecular orbital studies of N to C migrations in N,N-bisheteroatom-substituted amides—HERON rearrangements. J. Chem. Soc. Perk. Trans. 2 1995, 595–603. [Google Scholar] [CrossRef]
- Glover, S.A.; Rauk, A.; Buccigross, J.M.; Campbell, J.J.; Hammond, G.P.; Mo, G.; Andrews, L.E.; Gillson, A.-M.E. Review: The HERON reaction: Origin, theoretical background and prevalence. Can. J. Chem. 2005, 83, 1492–1509. [Google Scholar] [CrossRef]
- Glover, S.A. Development of the HERON reaction: A historical account. Aust. J. Chem. 2017, 70, 344–361. [Google Scholar] [CrossRef]
- Hinsberg, W.D., III; Dervan, P.B. Synthesis and direct spectroscopic observation of a 1,1-dialkyldiazene. Infrared and electronic spectrum of N-(2,2,6,6-tetramethylpiperidyl)nitrene. J. Am. Chem. Soc. 1978, 100, 1608–1610. [Google Scholar] [CrossRef]
- Hinsberg, W.D., III; Dervan, P.B. Kinetics of the thermal decomposition of a 1,1-dialkyldiazene, N-(2,2,6,6-tetramethylpiperidyl)nitrene. J. Am. Chem. Soc. 1979, 101, 6142–6144. [Google Scholar] [CrossRef]
- Hinsberg, W.D., III; Schultz, P.G.; Dervan, P.B. Direct studies of 1,1-diazenes. Syntheses, infrared and electronic spectra, and kinetics of the thermal decomposition of N-(2,2,6,6-tetramethylpiperidyl)nitrene and N-(2,2,5,5-tetramethylpyrrolidyl)nitrene. J. Am. Chem. Soc. 1982, 104, 766–773. [Google Scholar] [CrossRef]
- Schultz, P.G.; Dervan, P.B. Synthesis and direct spectroscopic observation of N-(2,2,5,5-tetramethylpyrrolidyl)nitrene. Comparison of five- and six-membered cyclic 1,1-dialkyldiazenes. J. Am. Chem. Soc. 1980, 102, 878–880. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, R.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Direct conversion of N-alkoxyamides to carboxylic esters through tandem NBS-mediated oxidative homocoupling and thermal denitrogenation. J. Org. Chem. 2014, 78, 8705–8711. [Google Scholar] [CrossRef] [PubMed]
- Thomson, L.M.; Hall, M.B. Theoretical study of the thermal decomposition of N,N′-diacyl-N,N′-dialkoxyhydrazines: A comparison of HF, MP2, and DFT. J. Phys. Chem. A 2000, 104, 6247–6252. [Google Scholar] [CrossRef]
- Novak, M.; Glover, S.A. Generation and trapping of the 4-biphenylyloxenium ion by water and azide: Comparisons with the 4-biphenylylnitrenium ion. J. Am. Chem. Soc. 2004, 126, 7748–7749. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Rajagopal, S. N-arylnitrenium ions. Adv. Phys. Org. Chem. 2001, 36, 167–254. [Google Scholar] [CrossRef]
- Shtamburg, V.G.; Klots, E.A.; Pleshkova, A.P.; Avramenko, V.I.; Ivonin, S.P.; Tsygankov, A.V.; Kostyanovsky, R.G. Geminal systems. 50. Synthesis and alcoholysis of N-acyloxy-N-alkoxy derivatives of ureas, carbamates, and benzamides. Russ. Chem. Bull. 2003, 52, 2251–2260. [Google Scholar] [CrossRef]
- Crawford, R.J.; Raap, R. The Synthesis and reaction of N,N-dicarboalkoxy-N,N-dialkoxyhydrazines and some observations on carbalkoxylium ions. J. Org. Chem. 1963, 28, 2419–2424. [Google Scholar] [CrossRef]
- Fahr, E.; Lind, H. Chemisry of a-carbonyl azo compounds. Angew. Chem. 1966, 5, 372–384. [Google Scholar] [CrossRef]
- Mo, G. Properties and Reactions of Anomeric Amides. Ph.D. Thesis, University of New England, Armidale, Australia, 1999. [Google Scholar]
- Cavanagh, K.L.; Glover, S.A. Heron reactivity of N-acetoxycarbamates with anilines. Unpublished work.





































{kind=link}
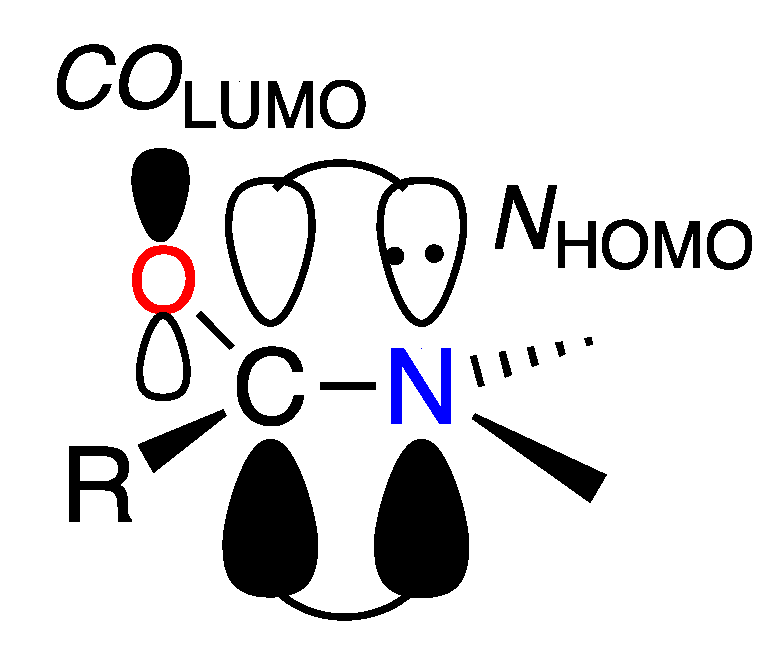{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
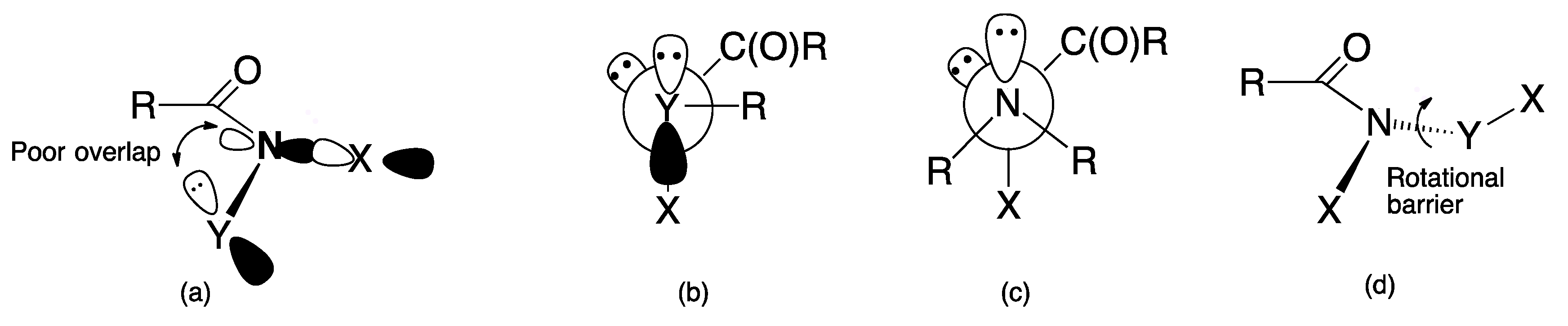{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
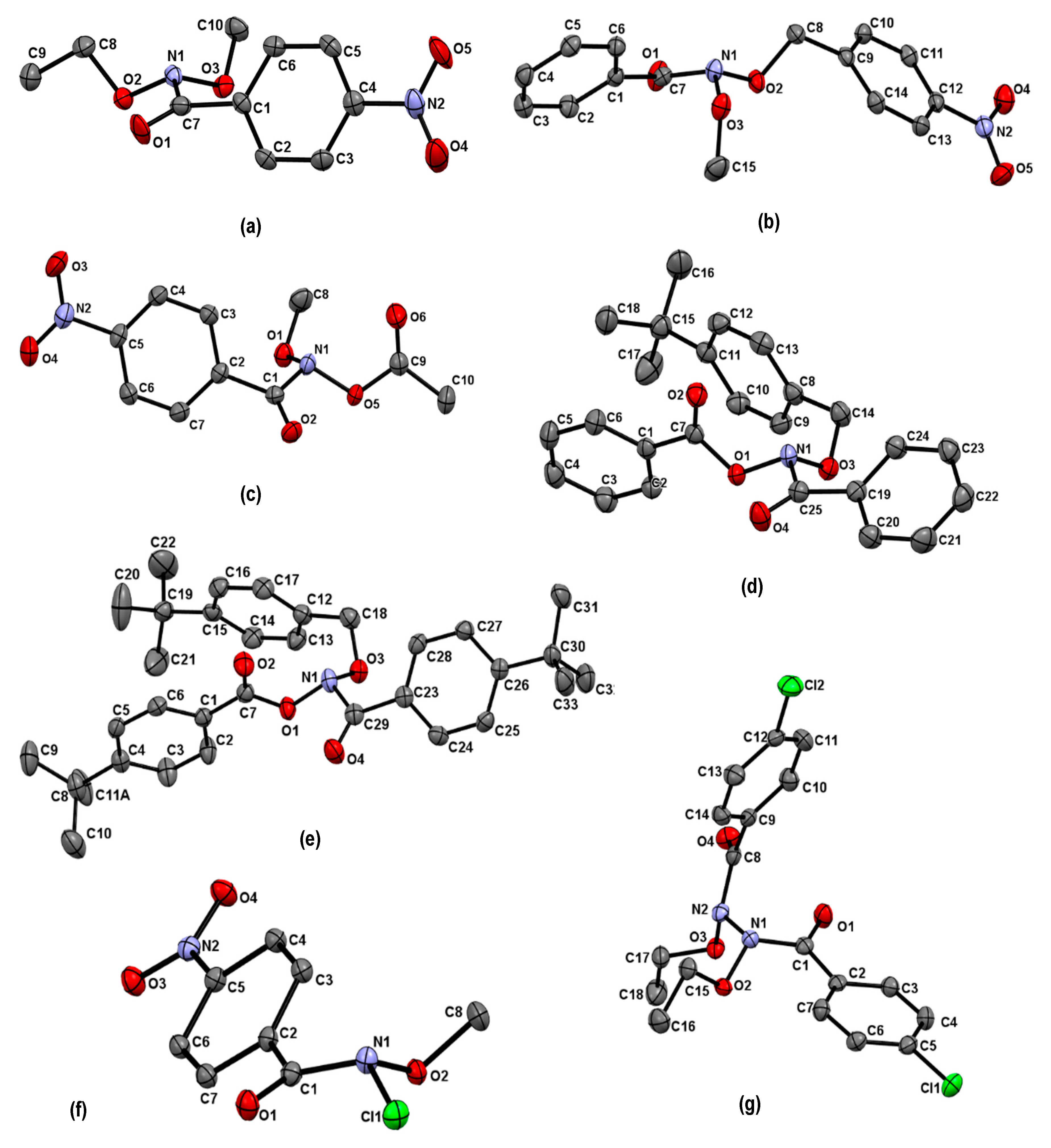
| Structure | N–C(O)/Å | (N)C=O/Å | θ/° | χN/° | τ/° | Anomeric Twist 1/° |
|---|---|---|---|---|---|---|
| 5a [33] | 1.409 | 1.206 | 331.2 | 58.3 | 6.7 | C10-O3-N1-O2: 95.9 C8-O2-N1-O3: −114.1 |
| 5b [33] | 1.421 | 1.211 | 334.5 | 55.6 | 13.9 | C8-O2-N1-O3: −101.6 C15-O3-N1-O2: −63.8 |
| 6a [29] | 1.439 | 1.205 | 323.5 | 65.3 | 15.5 | C18-O3-N1-O1:96.2 C7-O1-N1-O3: −141.6 |
| 6b [29] | 1.441 | 1.207 | 324.1 | 65.6 | 13.9 | C14-O3-N1-O1:96.7 C7-O1-N1-O3: −137.6 |
| 7 [65] | 1.411 | 1.203 | 330.3 | 59.7 | −14.0 | C8-O1-N1-O5:−91.8 C9-O5-N1-O1: 116.2 |
| 8(N1) [32] | 1.412 | 1.213 | 343.2 | 47.3 | −8.5 | LP (N2)-N2 -N1-O2: 47.3 |
| 8(N2) | 1.410 | 1.207 | 341.1 | 48.9 | −11.4 | LP (N1)-N1-N2-O3: 178.6 |
| 9 [65] | 1.408 | 1.204 | 337.5 | 52.5 | −13.3 | C8-O2-N1-Cl1: −84.2 |
| Structure | N–C(O)/Å | (N)C=O/Å | θ/° | χN/° | τ/° | Anomeric Twist 2/° |
|---|---|---|---|---|---|---|
| 10a [66] | 1.443 1 | 1.226 | 329.1 | 59.9 | 8.2 | C-O-N-Cl: –90.9 |
| 10b [66] | 1.472 1 | 1.210 | 325.8 | 61.9 | −13.4 | C-O-N-Cl: –100.1 |
| 11a [67] | 1.426 1 | 1.222 | 333.6 | −57.1 | −6.8 | C-O-N-Oacyl: –104.0 |
| 11b [68] | 1.441 1 | 1.233 | 323.7 | 64.6 | 11.8 | C-O-N-Oacyl: –98.2 |
| 12 [68] | 1.438 1 | 1.220 | 331.8 | 57.4 | 0.8 | C-O2-N1-O1:–89.3 C-O1-N1-O2: 55.2 |
| 13 [67] | 1.424 | 1.198 | 334.2 | −56.3 | 2.9 | C-O-N-Oacyl: –95.5 |
| 14 [69] (N1)/(N2) 3 | 1.408 | 1.194 | 340.0 | 59.8 | −9.4 | LP (N1)-N1-N2-O: 189.2 |
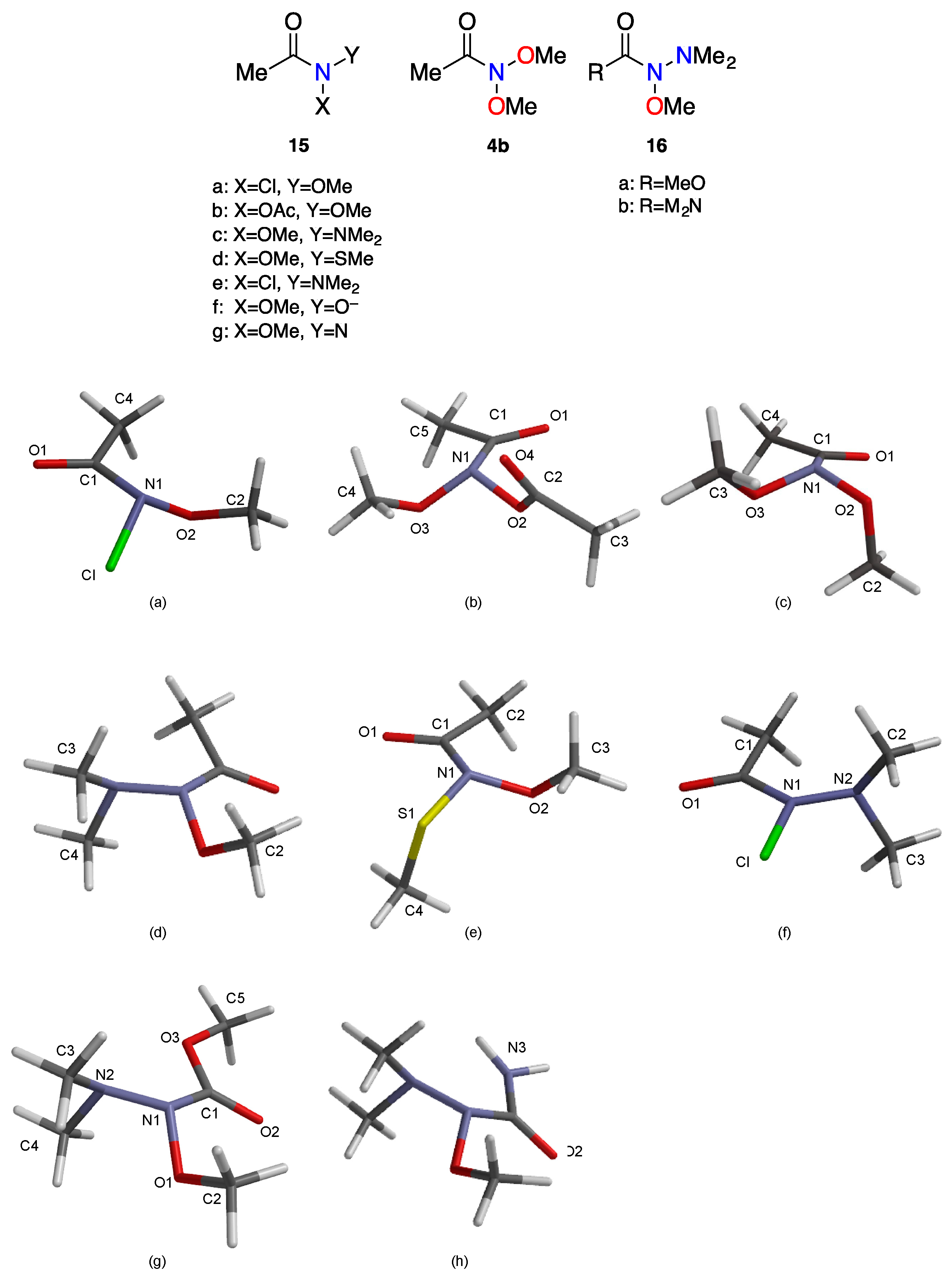
| Structure | N–C(O)/Å | (N)C=O/Å | (N–X,N–Y)/Å | θ/° | χN/° | τ/° | Anomeric Twist Angles 1/° |
|---|---|---|---|---|---|---|---|
| 15a ONCl [34] | 1.432 | 1.207 | Cl:1.787 O2:1.389 | 337.6 | 52.3 | −5.3 | C2-O2-N1-Cl: 88.8 |
| 15b ONOAc [61] | 1.429 | 1.209 | O2:1.423 O3:1.395 | 332.1 | 58.0 | 2.3 | C4-O3-N1-O2: 101.1 |
| 4b ONO [33] | 1.417 | 1.212 | O2:1.387 O3:1.412 | 342.9 | 48.1 | 8.5 | C2-O2-N1-O3: 66.6 C3-O3-N1-O2: 83.8 |
| 15c NNO [62] | 1.404 | 1.217 | O1:1.430 N2:1.387 | 346.5 | 41.8 | 5.4 | LP (N2)-N2-N1-O1: 190 |
| 15d ONS [61] | 1.408 | 1.215 | S1:1.717 O2:1.420 | 352.4 | 31.7 | −4.6 | C4-S1-N1-O2: −79 C3-O2-N1-S1: −86.6 |
| 15e NNCl [34] | 1.414 | 1.209 | Cl:1.820 N2:1.351 | 360.0 | 0 | 0 | LP(N2)N2-N1-Cl: 180 |
| 16a NNO [61] | 1.406 | 1.212 | O1:1.404 N2:1.383 | 342 | 46.3 | 0 | LP (N2)-N2-N1-O1: 169.4 |
| 16b NNO | 1.428 | 1.217 | O1:1.397 N2:1.428 | 340.0 | 49.6 | −1.7 | LP (N2)-N2-N1-O1: 167.5 |
| Amide (R = Me) | ΔECOSNAR1 /kJ mol−1 | ΔETA /kJ mol−1 | ΔEind /kJ mol−1 | RETA1, 2 /kJ mol−1 |
|---|---|---|---|---|
| 15a (ONCl) [34] | −29.6(38) | 69.9 | 23.4 | −29.5(39) |
| 15a3 | −27.2 (36) | |||
| 15a4 | −34.4(45) | |||
| 15b (ONOAc) [61] | −39.7(52) | 65.3 | 29.7 | −40.5(53) |
| 15b4 | −39.5 (52) | |||
| 4b (ONO) [35] | −36.0(47) | 58.2 | 18.0 | −36.0(47) |
| 4b4 | −39.5(53) | - | ||
| 15c (ONN) [62] | −52.3(69) | 35.1 | 10.0 | −51.0(67) |
| 15c4 | −55.7(73) | |||
| 15d (ONS) [61] | −48.6(64) | 26.8 | 5.0 | −48.6(64) |
| 15d4 | −47.3(62) | |||
| 15e (NNCl) [34] | −28.7(38) | 64.5 | 17.6 | −29.0(37) |
| 15e3 | −28.6(38) | |||
| 15e4 | −48.7(64) | |||
| 16a (ONN) [61] | −47.7(63) | 31.8 | 14.6 | −50.6(67) |
| 1 | −75.9(100) | |||
| 14 [34] | −75.9(100) |
| System | X | Y | R | Amide ν/cm−1 (δ13C) | Hydroxamic Ester ν/cm−1 (δ13C) |
|---|---|---|---|---|---|
| 2a [79] | Cl | OBu | Me | 1740 1(175.3) | 1678 (167.9) |
| 2a [35] | Cl | OBu | Ph | 1719 (174.2) | 1654 (165.7) |
| 2b [79] | OAc | OBu | Me | 1746 (176.2) | 1678 (167.9) |
| 2b [80] | OAc | OBu | Ph | 1732 (173.9) | 1654 (165.7) |
| 2c [81] | OBu | OBu | Me | 1707 (174.1) | 16781 (167.9) |
| 2c [55] | OMe | OMe | Ph | 1711 (174.3) | 1683 (166.4) |
| 2d [76] | 4-MeBnONAc | 4-MeBnO | Me | 1734/1700 (171.3) | 1693 (168.0) |
| 2d [76] | BzNOEt | OEt | Ph | 1708 (170.0) | 1685 (166.5) |
| Series | System | EA/kJ mol−1 | ΔS‡/J K−1 mol−1 | 106.k298/s−1 |
|---|---|---|---|---|
| Series 19: σ+ reaction constant ρ = −0.35 1 | 19a | 99.2 (4.3) | −21.2 (13) | 5.56 |
| 19b | 100.7 (8.2) | −19.6 (25) | 3.72 | |
| 19c | 100.4 (5.1) | −23.8 (15) | 2.52 | |
| 19d | 107.7 (1.7) | 0.9 (5) | 2.50 | |
| 19e | 104.0 (1.7) | −15.4 (5) | 1.57 | |
| Series 20: σ reaction constant ρ = +1.02 1 | 108.k298/s−1 | |||
| 20a | 111.4 (0.9) | −21.1 (2) | 4.02 | |
| 20b | 125.9 (1.0) | 24.5 (3) | 2.76 | |
| 20c | 114.1 (3.9) | −8.7 (11) | 6.21 | |
| 20d | 125.1 (8.5) | 27.4 (24) | 5.44 | |
| 20e | 98.8 (0.3) | −46.4 (1) | 31.9 |
| R | R′ | Isolated Crude Yield/% |
|---|---|---|
| Ph | (CH3)3C | 87 |
| (CH3)3C | (CH3)3C | 30 |
| 1-adamantyl | (CH3)3C | 82 |
| (CH3)3C | cyclohexyl | 97 |
| Ph | (CH3)2CH | 92 |
| Ph | PhCH2 | 93 |
| CH3 | PhCH2 | 92 |
| p-NO2C6H4 | Et | 94 |
| Ph | Et | 94 |
| Migratory Mode | EA | XNY 1 | RETA 2 | Erearr |
|---|---|---|---|---|
| MeO from anti 15c | 95.0 | ONN | −51.5 | 43.5 |
| MeO from 4b | 156.5 | ONO | −35.9 | 120.5 |
| MeO from syn 16a | 92.4 | ONN | −50.7 | 41.8 |
| AcO from syn 15b | 181.0 | AcONO | −40.5 | 140.5 |
| MeO from syn 15b | 207.9 | ONOAc | −40.5 | 167.4 |
| AcO from anti 15b | 181.5 | AcONO | −40.5 | 141.0 |
| MeO from anti 15b | 182.8 | ONOAc | −40.5 | 142.3 |
| MeO from anti 15d | 174.1 | ONS | −48.6 | 125.4 |
| MeO from 15f 3 | 48.0 | ONO– | −32.4 | 15.6 |
| MeO from 15g | 8.8 | ONNitrene | −14.8 | −6.0 |
| Ring opening 81 | 113.0 | ONO | −19.2 | 93.8 |
| Ring contraction 82 | 145.2 | ONO | −36.7 | 108.4 |
| Ring opening of 82 | 136.4 | ONO | −36.7 | 99.7 |
| Scheme 17 i X=NMe | 59.0 | ONN | 0.0 | 59.0 |
| Scheme 17 i X=O | 146.9 | ONO | 0.0 | 146.9 |
| Scheme 17 i X=S | 152.7 | ONS | 0.0 | 152.7 |
| Scheme 17 ii X=CH2 | 242.7 | ONCH2 | 0.0 | 242.7 |
| Scheme 18 i | 133.5 | AcONO | 0.0 | 133.5 |
| Scheme 18 ii | 161.5 | ONOAc | 0.0 | 161.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glover, S.A.; Rosser, A.A. Heteroatom Substitution at Amide Nitrogen—Resonance Reduction and HERON Reactions of Anomeric Amides. Molecules 2018, 23, 2834. https://doi.org/10.3390/molecules23112834
Glover SA, Rosser AA. Heteroatom Substitution at Amide Nitrogen—Resonance Reduction and HERON Reactions of Anomeric Amides. Molecules. 2018; 23(11):2834. https://doi.org/10.3390/molecules23112834
Chicago/Turabian StyleGlover, Stephen A., and Adam A. Rosser. 2018. "Heteroatom Substitution at Amide Nitrogen—Resonance Reduction and HERON Reactions of Anomeric Amides" Molecules 23, no. 11: 2834. https://doi.org/10.3390/molecules23112834
APA StyleGlover, S. A., & Rosser, A. A. (2018). Heteroatom Substitution at Amide Nitrogen—Resonance Reduction and HERON Reactions of Anomeric Amides. Molecules, 23(11), 2834. https://doi.org/10.3390/molecules23112834