Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu


Chemical Engineering, School of Engineering and Physical Sciences, Heriot Watt University, Edinburgh EH14 4AS, Scotland, UK
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(11), 2905; https://doi.org/10.3390/molecules23112905
Submission received: 7 October 2018
/
Revised: 4 November 2018
/
Accepted: 5 November 2018
/
Published: 7 November 2018
(This article belongs to the Special Issue Platform Chemical: Hydroxymethylfurfural (HMF))
Abstract
:We have investigated the synthesis and application of Au-Cu/CeO2 (Cu: Au = 2) in the continuous gas phase (P = 1 atm; T = 498 K) coupled hydrogenation of 5-hydroxymethyl-2-furaldehyde (HMF) with 2-butanol dehydrogenation. STEM-EDX analysis revealed a close surface proximity of both metals in Au-Cu/CeO2 post-TPR. XPS measurements suggest (support → metal) charge transfer to form Auδ− and strong metal-support interactions to generate Cu0 and Cu+. Au-Cu/CeO2 promoted the sole formation of 2,5-dihydroxymethylfuran (DHMF) and 2-butanone in the HMF/2-butanol coupling with full hydrogen utilisation. Under the same reaction conditions, Au/CeO2 was fully selective to DHMF in standard HMF hydrogenation (using an external hydrogen supply), but delivered a lower production rate and utilised less than 0.2% of the hydrogen supplied. Exclusive -C=O hydrogenation and -OH dehydrogenation is also demonstrated for the coupling of a series of m-substituted (-CH3, -CH2CH3, -CH2OH, -CF3, -N(CH3)2, -H) furaldehydes with alcohol (1-propanol, 1-butanol, 2-propanol, 2-butanol, cyclohexanol) dehydrogenation over Au-Cu/CeO2, consistent with a nucleophilic mechanism. In each case, we observed a greater hydrogenation rate and hydrogen utilisation efficiency with a 3–15 times lower E-factor in the coupling process relative to standard hydrogenation. Our results demonstrate the feasibility of using hydrogen generated in situ through alcohol dehydrogenation for the selective hydrogenation of m-furaldehydes with important industrial applications.
Keywords:
HMF; coupling hydrogenation/dehydrogenation; m-furaldehydes; alcohols; Au/CeO2; Au-Cu/CeO2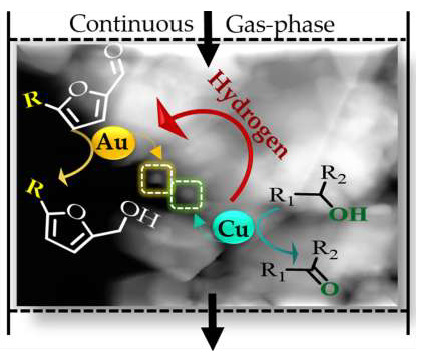
1. Introduction
5-hydroxymethyl-2-furaldehyde (HMF) has emerged as a promising biomass-derived platform chemical [1] that can be readily obtained from C6 carbohydrates (e.g., fructose, glucose) by dehydration in the presence of acid catalysts (e.g., metal halides, acid metal oxides) [2,3]. 2,5-Dihydroxymethylfuran (DHMF), a reduction product of HMF, is used as a precursor in the manufacture of polymer plastics (e.g., polyurethane foams, 16M tons per year [4]) resins and artificial fibres [5,6]. Conventional HMF→DHMF reduction involves standard catalytic hydrogenation in batch liquid phase [7] that suffers from safety and environmental issues due to the requirement for operating at high pressures (typically 5–100 bar) [7] under conditions with high excess of hydrogen in order to maximise H2 solubility and product yield. Hydrogen production requires non-renewable fossil fuel-based technologies such as steam and auto-thermal reforming [8]. Reaction selectivity is also challenging, as HMF hydrogen treatment can generate several products as shown in Figure 1(IA) [9,10,11] with Ru-catalysts delivering high combined activity/selectivity [7]. There is now a pressing demand for a more sustainable system that satisfies green chemistry principles to promote selective DHMF formation (i.e., #2, 7, 9 and 12 [12]). Several metrics have emerged to quantitatively assess green performance, most notably the environmental factor (E-factor, kgwaste kgproduct−1) [13]. E-factor values within the 5–100 range are characteristic of catalytic processes in the manufacture of pharmaceuticals and fine chemicals industries [13].
Hydrogenation can be effectively carried out in the absence of an external hydrogen supply through an alternative coupled process in which hydrogen generated in situ via (non-oxidative) dehydrogenation of alcohols (Figure 1(IB)) is utilised in a hydrogenation process (Figure 1(II)). This coupling strategy offers a series of advantages relative to stand-alone hydrogenation and dehydrogenation reactions including: (i) one-pot simultaneous production of two valuable chemicals (i.e., improved atom economy), (ii) good thermal efficiency (i.e. heat transfer from exothermic hydrogenation to endothermic dehydrogenation), (iii) enhanced yields (i.e. consumption in the hydrogenation step displaces the equilibrium of dehydrogenation increasing hydrogen production) and (iv) improved process safety (i.e. no need for pressurised H2) [14,15]. Despite the multiple advantages, it is a challenging system, with only a limited number of published studies on coupled heterogeneous catalytic systems. This is likely due to several factors, including: (i) a requirement for two active sites for dehydrogenation and hydrogenation (M1 and M2 in Figure 1(II)) that must be separated but in close proximity to facilitate hydrogen transport and utilisation, (ii) that sufficient hydrogen must be generated in the dehydrogenation step for the hydrogenation reaction and (iii) the possibility of cross reaction between reactants and/or products. It is nonetheless worth mentioning the promising results reported in the coupling of 1,4-butanediol [16] and n-butanol [17] dehydrogenation with maleic anhydride hydrogenation, the coupled of cyclohexanol/furfural [15,18,19,20] and 1,4-butanediol/furfural conversion [20,21,22,23]. Taking HMF → DHMF, the feasibility of coupling HMF with 1,4-butanediol over Cu-Al [24] and 2-butanol using zirconium hydroxides [25] has been explored only in batch liquid systems where the requirement of high pressure (16 bar) or low selectivity to target DHMF (SDHMF ≤ 90%) are decided drawbacks. We were unable to find any study in the open literature dealing with DHMF production through a coupling process with HMF in continuous gas-phase operation.
We have recently showed the feasibility of coupling 2-butanol with furfural over a physical mixture of Au/CeO2 + Cu/CeO2 [26]. Minimising the physical separation between Au and Cu in bimetallic catalysts should facilitate hydrogen transfer/utilisation. In this work we evaluate the continuous gas phase dehydrogenation of alcohols (to ketones/aldehydes) coupled with hydrogenation of HMF to DHMF over Au-Cu supported on (non-toxic, [27]) CeO2. The catalytic action of Au-Cu/CeO2 to promote -C=O group reduction for a series of m-substituted furaldehydes has been investigated to understand reaction mechanism and establish the potential of the bimetallic catalyst. The benefits of the coupled process are examined by comparison with (conventional) stand-alone hydrogenation using an external hydrogen supply over Au/CeO2 as a benchmark and employing the E-factor as a measure of environmental performance.
2. Results and Discussion
2.1. Catalytic Conversion of 5-Hydroxymethyl-2-Furaldehyde (HMF): Coupling Dehydrogenation-Hydrogenation vs. Conventional Stand-Alone Hydrogenation Using an External H2 Supply
In the gas-phase coupled 2-butanol dehydrogenation with HMF hydrogenation, Au-Cu/CeO2 was 100% selective towards the production of targets 2-butanone and DHMF (Figure 1(II)). These two products can be readily separated by distillation due to the (195 K) difference in boiling points [28]. The formation of DHMF demonstrates utilisation of hydrogen generated in the dehydrogenation step for the continuous conversion of HMF. We could not find any report on DHMF production through alcohol/HMF coupling dehydrogenation/hydrogenation in continuous gas phase operation. It is, nonetheless, worth mentioning the recent work of Hu et al. [25], who investigated the coupled HMF/2-butanol process in batch liquid phase over magnetic zirconium hydroxides although they reported the undesired formation of 5-methyl-2-furaldehyde (MF) and 5-methylfurfuryl alcohol (MFA) with selectivity towards the target DHMF ≤ 90%. Exclusive formation of DHMF has proved difficult to fully circumvent also in standard hydrogenation, with unwanted over-hydrogenation (to DHMTHF, Figure 1(IA)) and/or hydrogenolysis (to MFA and DMF) reported in the liquid phase hydrogen treatment of HMF over supported Ni [10], Pd [11] and Ru [9] catalysts. In addition to DHMF selectivity, hydrogen utilisation efficiency is a key parameter that must be optimised to guarantee process sustainability. Full hydrogen utilisation was achieved in the coupled system with all the amount generated via 2-butanol dehydrogenation being utilised in HMF hydrogenation, i.e., H2/HMF = 1. We recorded no conversion in the stand-alone dehydrogenation of 2-butanol (in N2) over Au/CeO2 while under similar reaction conditions (P = 1 atm; T = 498 K) Cu/SiO2 delivers negligible activity in the gas phase hydrogenation of furfural [29]. This suggests that Cu and Au are the active sites for 2-butanol dehydrogenation and HMF hydrogenation, respectively. Alcohol dehydrogenation over supported copper [30] proceeds through a two-step mechanism that involves sequential H abstraction from O-H bond and α-carbon [26,31]. HMF adsorption can proceed via -C=O bond π-back donation on Auδ− [32] or carbonyl O coordination with Auδ+ sites [33]. Spillover hydrogen species migrate from Cu → Au across the CeO2 surface [34] with auto-transfer in the catalytic conversion of HMF → DHMF.
We compared the catalytic response in the coupled vs. stand-alone process under the same reaction conditions of stoichiometric hydrogen supply (i.e. H2/HMF = 1). No conversion was detected over Au/CeO2 or Au-Cu/CeO2 in standard HMF hydrogenation using an external supply of H2. This lack of activity can be linked to the low capacity of Au for H2 chemisorption/activation [35,36], rate-limiting step in stand-alone hydrogenation [37]. Atomic hydrogen generated in situ in the coupled process during 2-butanol dehydrogenation is active for hydrogenation [38] and participates in HMF transformation, circumventing the issue of H2 activation by gold. An increase in hydrogen supply (H2/HMF = 80) in stand-alone hydrogenation enhanced the available surface H2 where both catalysts promote exclusive DHMF formation at similar production rates, but with less than 0.2% of the hydrogen supplied being utilised. This result demonstrates a requirement to work under conditions of hydrogen excess in the stand-alone process. Moreover, same activity/selectivity response over Au/CeO2 and Au-Cu/CeO2 further proves that the presence of Cu has no effect on the hydrogenation step which is governed by the gold component. We observed a 3.5-fold greater DHMF production rate in the coupling process relative to stand-alone hydrogenation over Au-Cu/CeO2. This demonstrates that the production of activated hydrogen over Cu by in situ alcohol dehydrogenation is more efficient than chemisorption of molecular H2 on Au via standard hydrogenation. Coupled reaction over Au-Cu/CeO2 was accompanied by lower E-factor (56) relative to standard hydrogenation in gas phase over Au/CeO2 (260) and batch mode using Ru catalysts (~100) [7]. Our results prove the benefits of the coupled process compared to standard hydrogenation for the transformation of HMF in terms of greater catalytic activity and full hydrogen utilisation efficiency with exclusive formation of target DHMF.
2.2. Catalyst Characterisation: Au-Cu/CeO2 and Au/CeO2
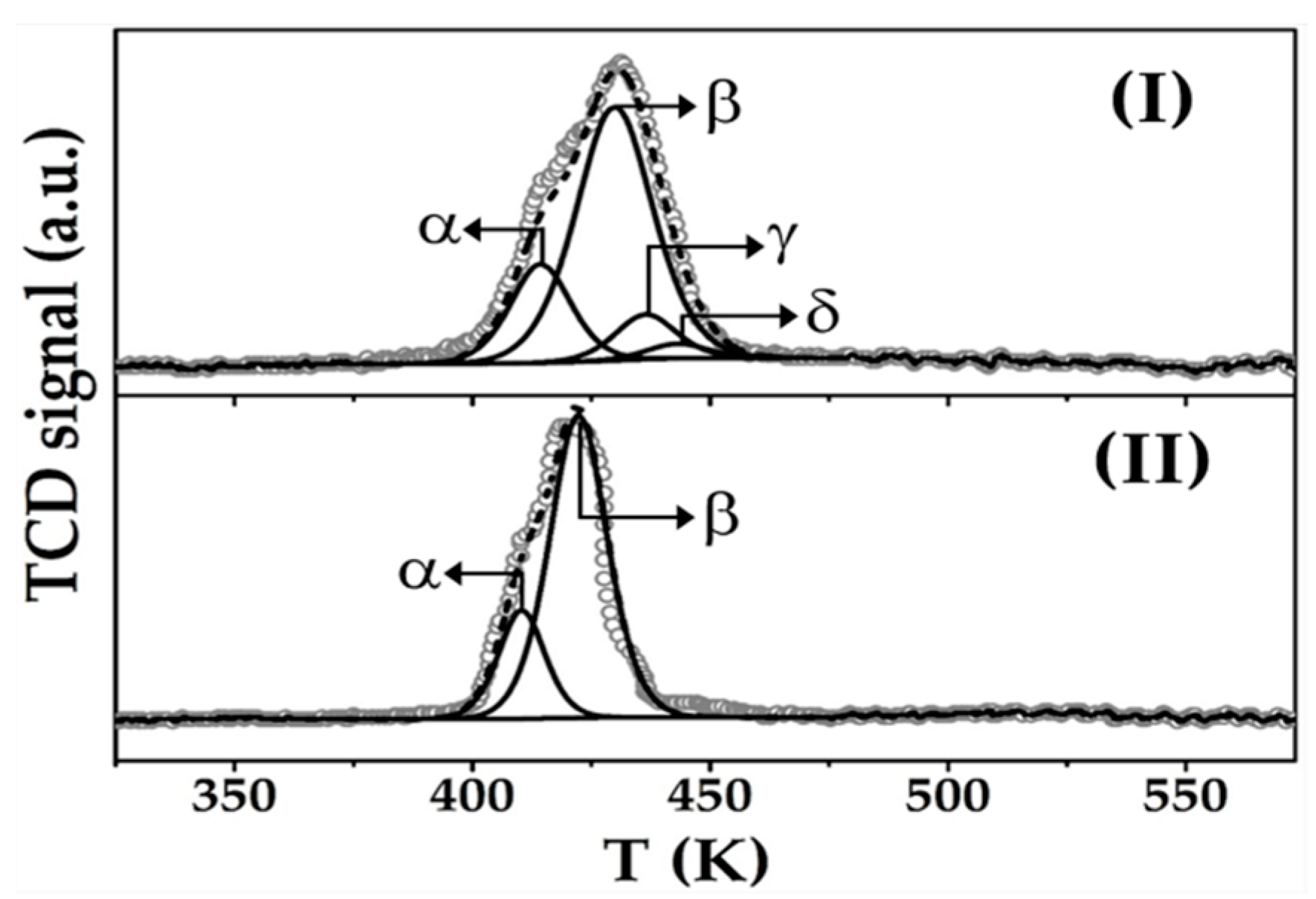
The critical properties of Au-Cu/CeO2 and Au/CeO2 are presented in Table 1. Both catalysts exhibit a similar TPR profile (Figure 2) characterised by a broad positive (hydrogen consumption) peak centred at 427 ± 5 K that is within the reported temperature range (408–550 K) for the reduction of Au3+ → Au0 and transformation of Cu2+ to zero valent Cu [39,40]. Total H2 consumption exceeded (by up to a factor of 4) the requirement for complete reduction of the metal precursor(s), indicative of partial support reduction at the metal-CeO2 interface by spillover hydrogen [41]. Both catalysts present peaks with Tmax at 412 ± 2 K (α) and 426 ± 3 K (β) characteristic of Au3+ → Au0 and Ce4+ → Ce3+, respectively [42,43]. The similar hydrogen consumption for α (67 ± 2 μmol g−1) and β (233 ± 5 μmol g−1) in both catalysts suggests same nature of Au and equivalent degree of CeO2 support reduction by spillover hydrogen promoted by gold. Limited capacity of Cu for H2 activation can account for the similar degree of CeO2 reduction and same catalytic response in stand-alone hydrogenation over Au-Cu/CeO2 and Au/CeO2. In addition, Au-Cu/CeO2 shows peaks characteristic of Cu2+ → Cu+ (γ centred at 437 K) and Cu+ → Cu0 (δ at 443 K) indicative of stepwise Cu reduction [44,45], where hydrogen consumption is consistent with a Cu+:Cu0 molar ratio = 3:1. The presence of cationic copper following TPR to 573 K can be linked to a stabilisation of Cu+ due to strong metal support interactions at Ce3+ sites [46,47]. Liu et al. [48], studying methanol steam reforming, showed (by XPS) the presence of Cu+ after reduction at T ≥ 573 K.
Scanning transmission electron microscopy (STEM) measurements (Figure 3(I-II)) revealed a similar metal dispersion and mean size for both catalysts characterised by pseudo-spherical nano particles with an associated surface area weighted mean diameter of 3.5 ± 0.5 nm. The surface composition of the Au-Cu/CeO2 catalyst was probed by STEM/EDX elemental analysis. A detailed elemental analysis over small areas was carried out and the EDX spectra of three isolated metal particles (IIIAa–IIIAc) in Au-Cu/CeO2 are shown in Figure 3. The EDX spectra exhibit peaks due to nickel in the grid (7.5 and 8.3 keV) [49] and the CeO2 support (4.9–5.8 keV) [50]. The signals at 2.2, 9.7 and 11.5 keV in the EDX of particles a–b are characteristic of Au [50]. In contrast, particle c (IIIAc) shows only a peak at 8 keV consistent with the presence of Cu [50]. The detailed EDX analysis over single metal nanoparticles reveals a close proximity of Au and Cu on the surface. In the case of the Au-Cu system, the miscibility gap is such that bulk alloy formation is possible at T ≥ 490 K [51]. There is a dearth of literature dealing with Au-Cu systems; however, metal segregation for Au-Cu/CeO2 catalysts post-thermal treatment at T ≤ 573 has been proven theoretically [52] and experimentally [53] and attributed to strong interactions at the metal-CeO2 interphase. Zhang et al. [52] concluded that CeO2 support induces segregation for the Au-Cu system based on the CeO2-induced preferential segregation energy (Eseg,CeO2-Au), a parameter indicating the strength of the Cu-CeO2 vs. Cu-Au bond energy. Ta and co-workers [54] showed by atomic resolution environmental transmission electron microscopy that gold nanoparticles of 2–4 nm strongly anchored onto CeO2 and did not sinter after reduction at T ≤ 573 K.
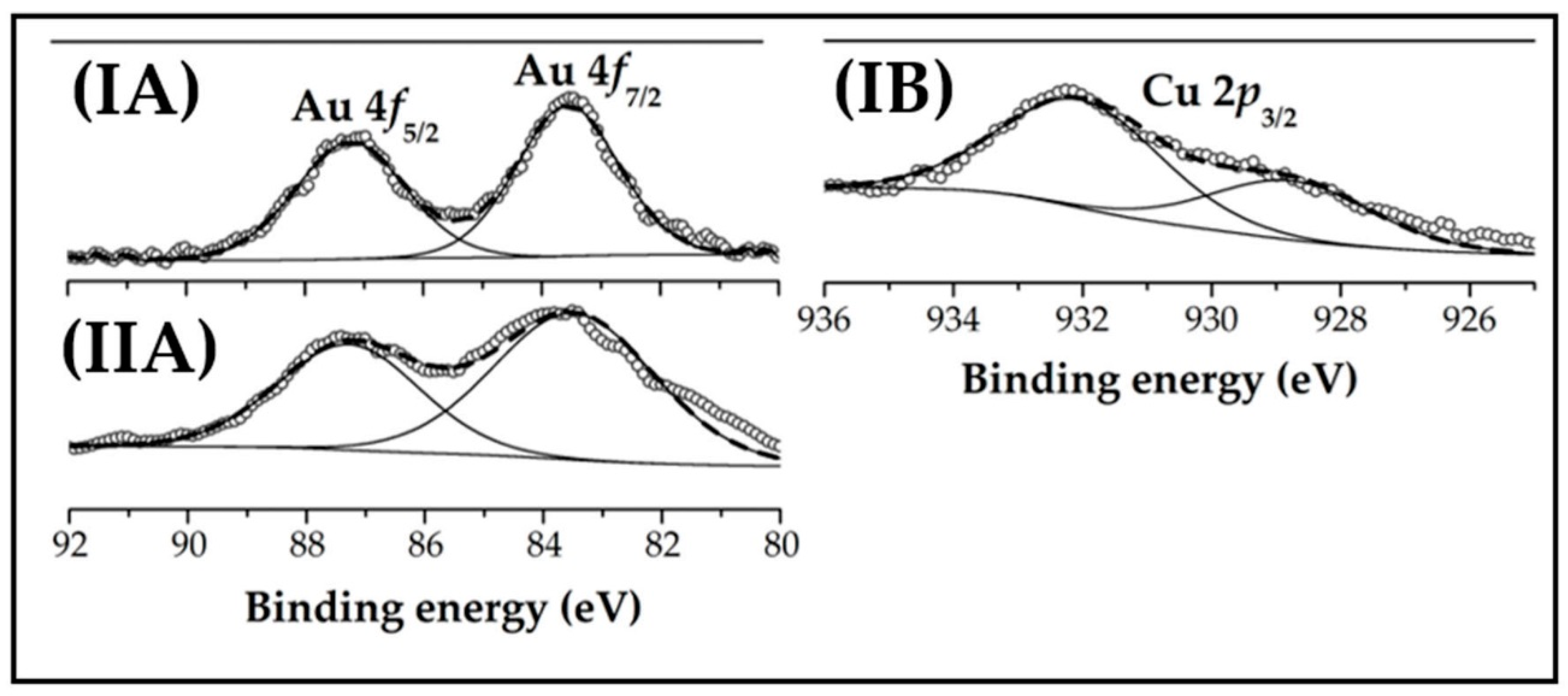
XPS spectra of in situ activated Au-Cu/CeO2 (I) and Au/CeO2 (II) over the Au 4f (A) and Cu 2p3/2 (B) binding energy (BE) regions are represented in Figure 4, and the results after deconvolution given in Table 1. The Au 4f profiles for Au-Cu/CeO2 and Au/CeO2 are equivalent suggesting same electronic properties for the gold phase with no measurable electron transfer in Au-Cu/CeO2 relative to Au/CeO2. The XPS spectrum presents two peaks with associated binding energies (BE) at 83.5 and 87.2 eV corresponding to 4f7/2 and 4f5/2 levels, respectively [55]. The BE values are lower than those reported for Au0 (Au 4f7/2 = 84.0 eV and Au 4f5/2 = 87.7 eV [55,56]), indicative of Au-support interactions that impact on Au electronic character [57]. A similar 0.4-1.0 eV downshift in BE for Au nanoparticles ≤6 nm on CeO2 was reported by Lai et al. [58] and attributed to electron transfer from the support. The presence of Auδ− nanoparticles is consistent with the adsorption/activation of the -C=O functionality in HMF via π-back donation [32]. The XPS spectrum for Au-Cu/CeO2 over the Cu 2p region (Figure 4(IB)) is characterised by a Cu 2p3/2 contribution at BE = 928.7 eV, corresponding to Cu0 (35%) [59]. A signal at higher BE (= 932.0 eV) with greater intensity can be linked to the presence of Cu+ (65%) [59]. The surface composition and oxidation state of copper from XPS measurements is consistent with TPR results. The XPS spectra over the Ce 3d region for CeO2, Au-Cu/CeO2 and Au/CeO2 are presented in Figure S1, Supplementary Materials. The as-received and H2-treated (to 573 K) CeO2 samples show an equivalent response characterised by a Ce3+ atomic ratio = 0.12 ± 0.01, indicative of a higher temperature requirement for Ce4+ → Ce3+ reduction (>625 K) [42]. A similar higher Ce3+ atomic ratio (0.26 ± 0.03) was recorded for both supported catalysts (Table 1). The presence of Au on CeO2 surface can induce a partial reduction of the support, due to hydrogen spillover [42,43], in agreement with H2 consumption during TPR analysis (Table 1).
2.3. Dehydrogenation of Alcohols Coupled with Hydrogenation of meta-Substituted Furaldehydes
We examined the potential of the Au-Cu/CeO2 catalyst for coupling a range of alcohols with m-substituted furaldehydes and the results are presented in Figure 5. In each case, we achieved full selectivity to the target aldehyde/ketone (from dehydrogenation) and alcohol (from hydrogenation) with no evidence of undesired hydrogenolysis, ring reduction, dehydration or dimerisation. We first evaluated the effect of changing the nature of the alcohol in the dehydrogenation step and the results for the coupled process with HMF are shown in Figure 5(I). In each case, the rate of DHMF production was up to a 4-fold greater than that recorded over Au/CeO2 using an external hydrogen supply. The modified hydrogenation activity for the different alcohols (cyclohexanol < 1-propanol < 1-butanol < 2-propanol < 2-butanol) demonstrates that hydrogen generation (from dehydrogenation) is rate limiting in the coupling system. The greater hydrogenation rate for secondary vs. primary alcohols (2-butanol vs. 1-butanol and 2-propanol vs. 1-propanol) observed is in line with reports in the literature [60,61] and consistent with reaction thermodynamics where hydrogen extraction from 2-butanol (to methyl elthyl ketone; reduction potential = 69 kJ·mol−1 [62]) is more favourable than 1-butanol conversion (to butyl aldehyde, = 80 kJ·mol−1) [62]) as well as in case of 2-propanol (formation of acetone, = 70 kJ·mol−1 [62]) and 1-propanol (formation of propanal, = 86 kJ·mol−1 [62]). Greater (77 kJ·mol−1 [62]) and increased steric hindrance [63] can account for the lower dehydrogenation rate of (cyclic) cyclohexanol (to cyclohexanone) relative to secondary aliphatic alcohol conversions.
In the dehydrogenation of 2-butanol coupled with hydrogenation of substituted furaldehydes bearing -CH3, -CH2CH3, -CH2OH, -CF3, -N(CH3)2 and -H substituents in the meta-position, Au-Cu/CeO2 was again fully selective in generating 2-butanone and the corresponding alcohol product. The higher hydrogenation activity in the coupling process over Au-Cu/CeO2 relative to stand-alone hydrogenation using Au/CeO2 observed in the case of HMF extended to all the substituted furaldehydes (Figure 5(II)). The following activity sequence was established in the coupling process: 5-dimethylamino-2-furaldehyde < 5-ethyl-2-furaldehyde < 5-methyl-2-furaldehyde < furfural < HMF < 5-trifluoromethyl-2-furaldehyde. The hydrogenation of the carbonyl group has been proposed to occur through a nucleophilic mechanism [64] with formation of a negatively charged hydroxyalkyl intermediate [65] that results from the attack of the carbonyl oxygen by hydrogen that acts as a weak nucleophilic agent [66]. Electron withdrawing functionalities in meta-position of aromatic systems favour the delocalisation of the negative charge in the hydroxyalkyl intermediate lowering the activation energy barrier [65] of the first reaction step (i.e. addition of the first hydrogen), which, in turns increases hydrogenation rate. The σm factor (Hammet constant) is an empirical parameter that provides a measure of the electron donating/acceptor character of a functionality in meta-position on aromatic systems [67,68]. In a nucleophilic attack, reaction rate is increased by electron-withdrawing groups with σm > 0. The higher activity with increasing σm (from -N(CH3)2 σm = −0.16 to CF3 σm = 0.43) [63] (see Figure 5(II)) is consistent with a nucleophilic mechanism.
The hydrogen utilisation efficiency (see Material and Methods) in the conversion of the different m-furaldehydes by stand-alone hydrogenation (using conventional pressurised H2) and coupling is presented in Figure 5(III). The hydrogen utilisation for stand-alone hydrogenation of all the tested furaldehydes was 200–900 times greater than the stoichiometric (=1) requirements, a result that represents high inefficiency. This issue was circumvented in the coupled process where, in each case, hydrogen utilisation efficiency was close or equal to stoichiometry with an associated E-factor 3–15 times lower relative to stand alone hydrogenation.
3. Materials and Methods
3.1. Catalyst Preparation and Activation
The CeO2 support was purchased from Sigma-Aldrich and used as received. 1 mol% Au/CeO2 was prepared by deposition–precipitation using urea (Riedel-de Haën, 99%) as basification agent. An aqueous solution of urea (3 M) and HAuCl4·H2O (6 × 10-4 M, 300 cm3, Sigma-Aldrich, 99.995%) was added to the support (5 g) and the suspension was stirred (600 rpm) and heated to 353 K. The pH progressively increased to reach 7 as a result of thermal decomposition of urea with consequent formation of Au(III) surface complexes [69]. The (1 mol% Au and 2 mol% Cu) Au-Cu/CeO2 catalyst was prepared by stepwise deposition–precipitation of Cu followed by Au. NaOH (2 M, Fisher Scientific, ≥97%) as basification agent was added to an aqueous solution of the metal precursor (Cu(NO3)2·3H2O (1 × 10−2 M, 200 cm3 Sigma-Aldrich, 99%)) containing the support (5 g) until pH = 10, heated to 353 K and aged under vigorous stirring for 4 h to ensure homogeneous deposition of Cu(OH)2 [70]. Au incorporation to the Cu/CeO2 catalyst was performed by deposition–precipitation, method as above. The solid obtained was filtered, washed with distilled water until pH = 7 and dried at 393 K overnight. Prior to use in catalysis, the samples (sieved into a batch of 75 μm average diameter) were activated in 60 cm3 min−1 H2 at 2 K min−1 to 573 K, which was maintained for 1 h. The samples were cooled to ambient temperature and passivated in 1% v/v O2/N2 at 298 K for ex situ characterisation.
3.2. Catalyst Characterisation
The Au and Cu loading was measured by atomic absorption spectroscopy (AAS) using a Shimadzu AA-6650 spectrometer with an air-acetylene flame from the diluted extract in aqua regia (25% v/v HNO3/HCl). Temperature programmed reduction (TPR) was conducted on the CHEM-BET 3000 (Quantachrome Instrument) unit with data acquisition/manipulation using the TPR WinTM software. Samples were loaded into a U-shaped Pyrex glass cell (3.76 mm i.d.) and heated in 17 cm3 min−1 (Brooks mass flow controlled) 5% v/v H2/N2 to 573 K at 2 K min−1. The effluent gas passed through a liquid N2 trap and H2 consumption was monitored by a thermal conductivity detector (TCD). Curve fitting TPR profile (CasaXPS 2.3.17 software) was employed to analyse the reduction step(s) during thermal treatment [71]. Metal particle morphology (size and shape) was examined by STEM probe corrected on a JEOL ARM 200CF with an energy dispersive X-ray (EDX) detector operated at an accelerating voltage of 200 kV. The scanned images were collected using either Gatan 806 High Angle Annular Dark Field, Gatan 805 Annular Dark Field/Bright field or JEOL ADF1detectors under the control of a Gatan DigiScan II, employing Gatan DigitalMicrograph software (version 2.31) for data acquisition/manipulation. Samples were prepared for analysis by dry deposition on a holey carbon/Ni grid (300 Mesh). At least 250 individual metal nanoparticles were counted for each catalyst and the surface area-weighted mean metal particle size (dSTEM) was calculated from:
where ni is the number of particles of diameter di. X-ray photoelectron spectroscopy (XPS) measurements were performed using a monochromatised Al anode (Kα 1486.6 eV, 10 kV, 20 mA). Prior to analysis, the samples were activated in situ in a pre-chamber under H2 atmosphere (102 mbar) at 2 K min−1 to 573 K. The source power was maintained at 3.9 × 103 W and the emitted photoelectrons were sampled from an area of 13 mm2; the photoelectron take-off angle was normal emission (0°). The analyser pass energy was 150 eV for survey (0–1100 eV) and high-resolution spectra (over the Au 4f, Cu 2p and Ce 3d core levels). The C 1s peak was calibrated at 284.5 eV and used as internal standard to compensate for charging effects. Spectra curve fitting and quantification were performed with the CasaXPS software, using relative sensitivity factors provided by Kratos.
3.3. Catalytic Procedure
Reactions (independent hydrogenation of m-furaldehyde in H2, alcohol dehydrogenation in N2 and coupled alcohol dehydrogenation/m-furaldehyde hydrogenation in N2) were carried out under atmospheric pressure, in situ immediately after activation, in a fixed bed vertical continuous flow glass reactor (i.d. = 15 mm) at 498 K. Operating conditions ensured negligible internal/external mass and heat transfer limitations [72]. A layer of borosilicate glass beads served as preheating zone where the organic reactant(s) was(were) vaporised and reached reaction temperature before contacting the catalyst bed. Isothermal conditions (± 1 K) were maintained by thoroughly mixing the catalyst with ground glass (75 µm). Reaction temperature was continuously monitored by a thermocouple inserted in a thermowell within the catalyst bed. The reactant(s) was(were) delivered to the reactor via a glass/Teflon air-tight syringe and Teflon line using a microprocessor-controlled infusion pump (Model 100 kd Scientific). Stand-alone hydrogenation and dehydrogenation were carried out in a co-current flow of H2 with m-furaldehyde (GHSV = 3.4 × 103 h− 1; molar Au to reactant feed rate (nAu/Fm-furaldehyde) = 1 × 10− 3 h) or N2 with alcohol (GHSV = 3.4 × 103 h−1, nAu/Falcohol = 3 × 10−2 h). The coupled reaction was carried out in N2 (GHSV = 3.4 × 103 h− 1, nAu/Fm-furaldehyde = 2 × 10− 4 – 1 × 10− 3 h). The catalytic response over Au-Cu/CeO2 vs. Au/CeO2 in the stand-alone hydrogenation of HMF (using an external hydrogen supply) was examined by using an aprotic solvent (anisole) to avoid any contribution to hydrogenation from in situ hydrogen production via alcohol dehydrogenation over Cu. In a series of blank tests, passage of each reactant in a stream of H2 or N2 through the empty reactor or over the (CeO2) support alone did not result in any detectable conversion. The reactor effluent was condensed in a liquid nitrogen trap for subsequent analysis using a Perkin-Elmer Auto System XL gas chromatograph equipped with a programmed split/splitless injector and a flame ionisation detector, employing a DB-1 (50 m × 0.33 mm i.d., 0.20 μm film thickness) capillary column (J&W Scientific). Data acquisition and manipulation were performed using the TurboChrom Workstation Version 6.3.2 (for Windows) chromatography data system. Furfural (Sigma-Aldrich, 99%), 5-methyl-2-furaldehyde (Sigma-Aldrich, >98%), 5-ethyl-2-furaldehyde (Sigma-Aldrich, 98%), 5-hydroxymethyl-2-furaldehyde HMF (Sigma-Aldrich, 99%), 5-trifluoromethyl-2-furaldehyde (Sigma-Aldrich), 5-dimethylamino-2-furaldehyde (Sigma-Aldrich), 1-butanol (Sigma-Aldrich, 99.8%), 2-butanol (Sigma-Aldrich, 99.5%) 1-propanol (Fisher Scientific, >99%), 2-propanol (Fisher Scientific, >99.5%), cyclohexanol (Fisher Scientific, >98%) and anisole (Sigma-Aldrich, 99.7%) were used as supplied without further purification. All gases (O2, H2, N2 and He) were of ultra-high purity (>99.99%, BOC). Reactant (i) fractional conversion (Xi) is defined by:
while selectivity to product j (Sj) is defined as:
where subscripts in and out refer to the inlet and outlet streams. The catalysts exhibited a decline in conversion to reach a pseudo-steady state after 2 h on-stream. Catalytic activity is also quantified in terms initial rate (rC = hydrogenation in coupling process and rSA = hydrogenation in stand-alone reaction; mol molAu−1 h−1), determined from time on-stream measurements as described elsewhere [72] according to:
Hydrogen utilisation efficiency in the stand-alone hydrogenation vs. coupled process was assessed by:
where H2 supply is the molar hydrogen provided ((i) from an external gas cylinder supply or (ii) via alcohol dehydrogenation) while H2 consumed is the amount utilised in the conversion of m-furaldehyde. Repeated reactions with different samples from the same batch of catalyst delivered raw data reproducibility and carbon mass balance within ± 5%.
4. Conclusions
Activation in hydrogen of Au-Cu/CeO2 (Cu/Au mol ratio = 2) prepared by stepwise deposition–precipitation generated metal nanoparticles in the range 1–7 nm (mean = 3.5 nm). STEM-EDX analysis of Au-Cu/CeO2 has revealed that Au and Cu nanoparticles are in close proximity on the surface while XPS results are consistent with formation of Auδ−, Cu0 and Cu+. Au/CeO2 (bearing Auδ−nanoparticles with mean size = 3 nm) promoted the gas phase continuous hydrogenation of HMF (P = 1 atm, T = 498 K) exclusively to DHMF with only a small fraction of the hydrogen supplied being utilised. Under the same reaction conditions, Au-Cu/CeO2 delivered a higher DHMF production rate and full hydrogen utilisation in the coupled hydrogenation/dehydrogenation of HMF/2-butanol with a lower E-factor. Exclusive carbonyl-group hydrogenation and hydroxyl-group dehydrogenation with (up to a 6-fold) increase hydrogenation rate and hydrogen utilisation efficiency with a lower E-factor (relative to conventional stand-alone hydrogenation) extends to the coupling of a series of m-substituted (-CH3, -CH2CH3, -CH2OH, -CF3, -N(CH3)2 and -H) furaldehydes with alcohol (1-propanol, 1-butanol, 2-propanol, 2-butanol, cyclohexanol) dehydrogenation over Au-Cu/CeO2. Hydrogen generation is rate limiting and furaldehyde hydrogenation proceeds via a nucleophilic mechanism where the presence of electron withdrawing substituents (in the meta-position) is shown to increase hydrogenation rate. Our results open new possibilities for the application of bimetallic Au-Cu catalysts for sustainable hydrogenation/dehydrogenation coupling directed at production of high value chemicals from furaldehydes.
Supplementary Materials
The following are available online, Figure S1: XPS spectra over the Ce 3d region for (I) fresh CeO2, (II) CeO2 thermally treated in H2 to 573 K, (III) Au-Cu/CeO2 and (IV) Au/CeO2. Note: Raw data are shown as open symbols (⚬) while curve fitted and envelope is represented by solid and dashed lines.
Author Contributions
M.A.K. and F.C.L. conceived the idea and were in charge of overall direction and planning of the project; L.C. and C.P. synthesised and characterised the samples; C.P. carried out the experiments and wrote the manuscript with input from all the authors.
Funding
This research was funded by the Engineering and Physical Sciences Research Council EPRSC grant number EP/L016419/1 [Ph.D. studentship to Chiara Pischetola, CRITICAT program].
Acknowledgments
We acknowledge C. Baddeley, F. Grillo and S. Francis for assistance with XPS measurements and are grateful to N. Tomás Arias for her contribution to the work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Van Putten, R.-J.; Van der Waal, J.C.; De Jong, E.D.; Rasrendra, C.B.; Heeres, H.J.; De Vries, J.G. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nolte, M.W.; Shanks, B.H. Catalytic dehydration of C6 carbohydrates for the production of hydroxymethylfurfural (HMF) as a versatile platform chemical. Green Chem. 2014, 16, 548–572. [Google Scholar] [CrossRef]
- Zieleniewska, M.; Szczepkowski, L.; Krzyżowska, M.; Leszczyński, M.; Ryszkowska, J. Rigid polyurethane foam composites with vegetable filler for application in the cosmetics industry. Polimery 2016, 61, 807–814. [Google Scholar] [CrossRef]
- Kong, X.; Zhu, Y.; Zheng, H.; Dong, F.; Zhu, Y.; Li, Y.-W. Switchable synthesis of 2,5-dimethylfuran and 2,5-dihydroxymethyltetrahydrofuran from 5-hydroxymethylfurfural over Raney Ni catalyst. RSC Adv. 2014, 4, 60467–60472. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ishizaka, T.; Kawanami, H. Selective hydrogenation of 5-hydroxymethylfurfural to 2,5-bis-(hydroxymethyl) furan using Pt/MCM-41 in an aqueous medium: A simple approach. Green Chem. 2014, 16, 4734–4739. [Google Scholar] [CrossRef]
- Hu, L.; Xu, J.; Zhou, S.; He, A.; Tang, X.; Lin, L.; Xu, J.; Zhao, Y. Catalytic advances in the production and application of biomass-derived 2,5-dihydroxymethylfuran. ACS Catal. 2018, 8, 2959–2980. [Google Scholar] [CrossRef]
- Schüth, F. Hydrogen: Economics and its role in biorefining. In Catalytic Hydrogenation for Biomass Valorization; The Royal Society of Chemistry: London, UK, 2014; pp. 1–21. ISBN 1-84973-801-7. [Google Scholar]
- Hu, L.; Tang, X.; Xu, J.; Wu, Z.; Lin, L.; Liu, S. Selective transformation of 5-hydroxymethylfurfural into the liquid fuel 2,5-dimethylfuran over carbon-supported ruthenium. Ind. Eng. Chem. Res. 2014, 53, 3056–3064. [Google Scholar] [CrossRef]
- Cai, H.; Li, C.; Wang, A.; Zhang, T. Biomass into chemicals: One-pot production of furan-based diols from carbohydrates via tandem reactions. Catal. Today 2014, 234, 59–65. [Google Scholar] [CrossRef]
- Audemar, M.; Vigier, K.D.O.; Clacens, J.-M.; De Campo, F.; Jérôme, F. Combination of Pd/C and Amberlyst-15 in a single reactor for the acid/hydrogenating catalytic conversion of carbohydrates to 5-hydroxy-2,5-hexanedione. Green Chem. 2014, 16, 4110–4114. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Metrics of green chemistry and sustainability: Past, present, and future. ACS Sustain. Chem. Eng. 2017, 6, 32–48. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Xu, B. Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Padmasri, A.H.; Seetharamulu, P.; Reddy, K.H.P.; Raju, B.D.; Rao, K.R. A highly active Cu-MgO-Cr2O3 catalyst for simultaneous synthesis of furfuryl alcohol and cyclohexanone by a novel coupling route—Combination of furfural hydrogenation and cyclohexanol dehydrogenation. J. Mol. Catal. Chem. 2007, 278, 29–37. [Google Scholar] [CrossRef]
- Zhu, Y.-L.; Xiang, H.-W.; Wu, G.-S.; Bai, L.; Li, Y.-W. A novel route for synthesis of γ-butyrolactone through the coupling of hydrogenation and dehydrogenation. Chem. Commun. 2002, 254–255. [Google Scholar] [CrossRef]
- Zhang, D.; Yin, H.; Zhang, R.; Xue, J.; Jiang, T. Gas phase hydrogenation of maleic anhydride to γ-butyrolactone by Cu–Zn–Ce catalyst in the presence of n-butanol. Catal. Lett. 2008, 122, 176–182. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Padmasri, A.H.; Raju, B.D.; Rao, K.S.R. Production of hydrogen through the coupling of dehydrogenation and hydrogenation for the synthesis of cyclohexanone and furfuryl alcohol over different promoters supported on Cu–MgO catalysts. Int. J. Hydrogen Energy 2011, 36, 3417–3425. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Zhu, Y.-L.; Bai, Z.-Q.; Huang, L.; Xiang, H.-W.; Li, Y.-W. An environmentally benign process for the efficient synthesis of cyclohexanone and 2-methylfuran. Green Chem. 2006, 8, 107–109. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Zhu, Y.-L.; Huang, L.; Zeng, Z.-Y.; Wan, H.-J.; Li, Y.-W. Study on Cu–Mn–Si catalysts for synthesis of cyclohexanone and 2-methylfuran through the coupling process. Catal. Commun. 2008, 9, 342–348. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Yang, J.; Zhu, Y.-L.; Zhao, G.-W. Synthesis of γ-butyrolactone and 2-methylfuran through the coupling of dehydrogenation and hydrogenation over copper-chromite catalyst. React. Kinet. Catal. Lett. 2004, 82, 263–269. [Google Scholar] [CrossRef]
- Zhu, Y.-L.; Xiang, H.-W.; Li, Y.-W.; Jiao, H.; Wu, G.-S.; Zhong, B.; Guo, G.-Q. A new strategy for the efficient synthesis of 2-methylfuran and γ-butyrolactone. New J. Chem. 2003, 27, 208–210. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, H.-Y.; Zhu, Y.-L.; Zhao, G.-W.; Zhang, C.-H.; Teng, B.-T.; Xiang, H.-W.; Li, Y. Effects of calcination temperature on performance of Cu–Zn–Al catalyst for synthesizing γ-butyrolactone and 2-methylfuran through the coupling of dehydrogenation and hydrogenation. Catal. Commun. 2004, 5, 505–510. [Google Scholar] [CrossRef]
- Aellig, C.; Jenny, F.; Scholz, D.; Wolf, P.; Giovinazzo, I.; Kollhoff, F.; Hermans, I. Combined 1, 4-butanediol lactonization and transfer hydrogenation/hydrogenolysis of furfural-derivatives under continuous flow conditions. Catal. Sci. Technol. 2014, 4, 2326–2331. [Google Scholar] [CrossRef]
- Hu, L.; Yang, M.; Xu, N.; Xu, J.; Zhou, S.; Chu, X.; Zhao, Y. Selective transformation of biomass-derived 5-hydroxymethylfurfural into 2,5-dihydroxymethylfuran via catalytic transfer hydrogenation over magnetic zirconium hydroxides. Korean J. Chem. Eng. 2018, 35, 99–109. [Google Scholar] [CrossRef]
- Pischetola, C.; Collado, L.; Aguado-Molina, R.; Treceno-Martìn, S.; Cárdenas-Lizana, F.; Keane, M.A. Coupled dehydrogenation-hydrogenation over supported Cu and Au catalysts for continuous furfuryl alcohol production: Support effects. 2018; submitted. [Google Scholar]
- Li, Z.X.; Zhang, C.; Sun, L.D.; Zhang, Y.W.; Yan, C.H. Catalysis by ceria and related materials. In Catalytic Science Series; Imperial College Press: London, UK, 2013; Volume 12, pp. 295–359. ISBN 978-1-84816-963-0. [Google Scholar]
- Green, D.W. Perry’s Chemical Engineers’; McGraw Hill: New York, NY, USA, 2008; ISBN 9780071422949. [Google Scholar]
- Keane, M.A.; Li, M.; Collado, L.; Cárdenas-Lizana, F. Overcoming the limitations of gold catalysts in hydrogenation: Enhanced activity with full hydrogen utilization. React. Kinet. Mech. Catal. 2018, 1–12. [Google Scholar] [CrossRef]
- Crivello, M.E.; Pérez, C.F.; Mendieta, S.N.; Casuscelli, S.G.; Eimer, G.A.; Elías, V.R.; Herrero, E.R. n-Octyl alcohol dehydrogenation over copper catalysts. Catal. Today 2008, 133–135, 787–792. [Google Scholar] [CrossRef]
- Wang, F.; Shi, R.; Liu, Z.-Q.; Shang, P.-J.; Pang, X.; Shen, S.; Feng, Z.; Li, C.; Shen, W. Highly efficient dehydrogenation of primary aliphatic alcohols catalyzed by Cu nanoparticles dispersed on rod-shaped La2O2CO3. ACS Catal. 2013, 3, 890–894. [Google Scholar] [CrossRef]
- Ohyama, J.; Esaki, A.; Yamamoto, Y.; Arai, S.; Satsuma, A. Selective hydrogenation of 2-hydroxymethyl-5-furfural to 2,5-bis(hydroxymethyl)furan over gold sub-nano clusters. RSC Adv. 2013, 3, 1033–1036. [Google Scholar] [CrossRef]
- Gallezot, P.; Richard, D. Selective hydrogenation of α, β-unsaturated aldehydes. Catal. Rev. 1998, 40, 81–126. [Google Scholar] [CrossRef]
- Conner, W.C., Jr.; Falconer, J.L. Spillover in heterogeneous catalysis. Chem. Rev. 1995, 95, 759–788. [Google Scholar] [CrossRef]
- Li, M.; Collado, L.; Cárdenas-Lizana, F.; Keane, M.A. Role of support oxygen vacancies in the gas phase hydrogenation of furfural over gold. Catal. Lett. 2018, 148, 90–96. [Google Scholar] [CrossRef]
- Zanella, R. Crotonaldehyde hydrogenation by gold supported on TiO2: Structure sensitivity and mechanism. J. Catal. 2004, 223, 328–339. [Google Scholar] [CrossRef]
- Wang, X.; Perret, N.; Delannoy, L.; Louis, C.; Keane, M.A. Selective gas phase hydrogenation of nitroarenes over Mo2C-supported Au–Pd. Catal. Sci. Technol. 2016, 6, 6932–6941. [Google Scholar] [CrossRef]
- Shi, R.; Wang, F.; Tana; Li, Y.; Huang, X.; Shen, W. A highly efficient Cu/La2O3 catalyst for transfer dehydrogenation of primary aliphatic alcohols. Green Chem. 2010, 12, 108–113. [Google Scholar] [CrossRef]
- Ying, F.; Wang, S.; Au, C.-T.; Lai, S.-Y. Highly active and stable mesoporous Au/CeO2 catalysts prepared from MCM-48 hard-template. Microporous Mesoporous Mater. 2011, 142, 308–315. [Google Scholar] [CrossRef]
- Huang, Z.; Cui, F.; Xue, J.; Zuo, J.; Chen, J.; Xia, C. Cu/SiO2 catalysts prepared by hom- and heterogeneous deposition–precipitation methods: Texture, structure, and catalytic performance in the hydrogenolysis of glycerol to 1, 2-propanediol. Catal. Today 2012, 183, 42–51. [Google Scholar] [CrossRef]
- Liao, X.; Chu, W.; Dai, X.; Pitchon, V. Bimetallic Au–Cu supported on ceria for PROX reaction: Effects of Cu/Au atomic ratios and thermal pretreatments. Appl. Catal. B Environ. 2013, 142, 25–37. [Google Scholar] [CrossRef]
- Nevanperä, T.K.; Ojala, S.; Bion, N.; Epron, F.; Keiski, R.L. Catalytic oxidation of dimethyl disulfide (CH3SSCH3) over monometallic Au, Pt and Cu catalysts supported on γ-Al2O3, CeO2 and CeO2-Al2O3. Appl. Catal. B Environ. 2016, 182, 611–625. [Google Scholar] [CrossRef]
- Scire, S.; Minico, S.; Crisafulli, C.; Satriano, C.; Pistone, A. Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts. Appl. Catal. B Environ. 2003, 40, 43–49. [Google Scholar] [CrossRef]
- Dow, W.-P.; Huang, T.-J. Effect of chlorine on TPR and TPO behavior of an YSZ/γ-Al2O3 supported copper oxide catalyst. Appl. Catal. A Gen. 1996, 141, 17–29. [Google Scholar] [CrossRef]
- Jacono, M.L.; Cimino, A.; Inversi, M. Oxidation states of copper on alumina studied by redox cycles. J. Catal. 1982, 76, 320–332. [Google Scholar] [CrossRef]
- Chen, J.; Zhan, Y.; Zhu, J.; Chen, C.; Lin, X.; Zheng, Q. The synergetic mechanism between copper species and ceria in NO abatement over Cu/CeO2 catalysts. Appl. Catal. A Gen. 2010, 377, 121–127. [Google Scholar] [CrossRef]
- Dhas, N.A.; Raj, C.P.; Gedanken, A. Synthesis, characterization, and properties of metallic copper nanoparticles. Chem. Mater. 1998, 10, 1446–1452. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S. Production of hydrogen by steam reforming of methanol over Cu/CeO2 catalysts derived from Ce1− xCuxO2− x precursors. Catal. Commun. 2001, 2, 195–200. [Google Scholar] [CrossRef]
- Chen, H.; Hu, L.; Chen, M.; Yan, Y.; Wu, L. Nickel–cobalt layered double hydroxide nanosheets for high–performance supercapacitor electrode materials. Adv. Funct. Mater. 2014, 24, 934–942. [Google Scholar] [CrossRef]
- Huang, X.-S.; Sun, H.; Wang, L.-C.; Liu, Y.-M.; Fan, K.-N.; Cao, Y. Morphology effects of nanoscale ceria on the activity of Au/CeO2 catalysts for low-temperature CO oxidation. Appl. Catal. B: Environ. 2009, 90, 224–232. [Google Scholar] [CrossRef]
- Wei, S.-H.; Mbaye, A.; Ferreira, L.; Zunger, A. First-principles calculations of the phase diagrams of noble metals: Cu-Au, Cu-Ag, and Ag-Au. Phys. Rev. B 1987, 36, 4163–4185. [Google Scholar] [CrossRef]
- Zhang, L.; Kim, H.Y.; Henkelman, G. CO oxidation at the Au–Cu interface of bimetallic nanoclusters supported on CeO2 (111). J. Phys. Chem. Lett. 2013, 4, 2943–2947. [Google Scholar] [CrossRef]
- Scirè, S.; Fiorenza, R.; Crisafulli, C. AuAg/CeO2 and AuCu/CeO2 catalysts for CO preferential oxidation. In Proceedings of the Hydrogen Power Theoretical and Engineering Solutions International Symposium 2015, Toledo, Spain, 6–9 September 2015. [Google Scholar]
- Ta, N.; Liu, J.; Chenna, S.; Crozier, P.A.; Li, Y.; Chen, A.; Shen, W. Stabilized gold nanoparticles on ceria nanorods by strong interfacial anchoring. J. Am. Chem. Soc. 2012, 134, 20585–20588. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.D.; Riggs, W.M.; Davis, L.E.; Moulder, J.F.; Muilenberg, G.E. Handbook of X-ray Photoelectron Spectroscopy; Perkin-Elmer Corporation: Waltham, MA, USA, 2015; pp. 183–184. ISBN 55344. [Google Scholar]
- Delannoy, L.; Thrimurthulu, G.; Reddy, P.S.; Méthivier, C.; Nelayah, J.; Reddy, B.M.; Ricolleau, C.; Louis, C. Selective hydrogenation of butadiene over TiO2 supported copper, gold and gold–copper catalysts prepared by deposition–precipitation. Phys. Chem. Chem. Phys. 2014, 16, 26514–26527. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Lemire, C.; Shaikhutdinov, S.K.; Freund, H.-J. Surface chemistry of catalysis by gold. Gold Bull. 2004, 37, 72–124. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.-Y.; Qiu, Y.; Wang, S. Effects of the structure of ceria on the activity of gold/ceria catalysts for the oxidation of carbon monoxide and benzene. J. Catal. 2006, 237, 303–313. [Google Scholar] [CrossRef]
- Li, M.; Hao, Y.; Cárdenas-Lizana, F.; Yiu, H.H.P.; Keane, M.A. “Hydrogen-free” hydrogenation of nitrobenzene over Cu/SiO2 via coupling with 2-butanol dehydrogenation. Top. Catal. 2015, 58, 149–158. [Google Scholar] [CrossRef]
- Bertero, N.M.; Trasarti, A.F.; Apesteguía, C.R.; Marchi, A.J. Solvent effect in the liquid-phase hydrogenation of acetophenone over Ni/SiO2: A comprehensive study of the phenomenon. Appl. Catal. A Gen. 2011, 394, 228–238. [Google Scholar] [CrossRef]
- Dunbar, R.E.; Arnold, M.R. Catalytic dehydrogenation of primary and secondary alcohols with copper-chromium oxide. J. Org. Chem. 1945, 10, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Van der Waal, J.C.; Kunkeler, P.J.; Tan, K.; Van Bekkum, H. Zeolite titanium beta: A selective catalyst for the gas-phase Meerwein–Ponndorf–Verley, and Oppenauer reactions. J. Catal. 1998, 173, 74–83. [Google Scholar] [CrossRef]
- Di Cosimo, J.; Acosta, A.; Apesteguía, C. Allylic alcohol synthesis by gas-phase hydrogen transfer reduction of unsaturated ketones. J. Mol. Catal. Chem. 2005, 234, 111–120. [Google Scholar] [CrossRef]
- Cárdenas-Lizana, F.; Lamey, D.; Gómez-Quero, S.; Perret, N.; Kiwi-Minsker, L.; Keane, M.A. Selective three-phase hydrogenation of aromatic nitro-compounds over β-molybdenum nitride. Catal. Today 2011, 173, 53–61. [Google Scholar] [CrossRef]
- Sitthisa, S.; Sooknoi, T.; Ma, Y.; Balbuena, P.B.; Resasco, D.E. Kinetics and mechanism of hydrogenation of furfural on Cu/SiO2 catalysts. J. Catal. 2011, 277, 1–13. [Google Scholar] [CrossRef]
- Jacobs, G.; Keogh, R.A.; Davis, B.H. Steam reforming of ethanol over Pt/ceria with co-fed hydrogen. J. Catal. 2007, 245, 326–337. [Google Scholar] [CrossRef]
- Remya, G.S.; Suresh, C.H. Quantification and classification of substituent effects in organic chemistry: A theoretical molecular electrostatic potential study. Phys. Chem. Chem. Phys. 2016, 18, 20615–20626. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Baatz, C.; Thielecke, N.; Prüße, U. Influence of the preparation conditions on the properties of gold catalysts for the oxidation of glucose. Appl. Catal. B Environ. 2007, 70, 653–660. [Google Scholar] [CrossRef]
- Nino, E.; Lapena, A.; Martinez, J.; Gutierrez, J.M.; Mendioroz, S.; Fierro, J.L.G.; Pajares, J.A. Cu/Kieselguhr catalysts for hydration of acrylonitrile. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1983; Volume 16, pp. 747–755. ISBN 0167-2991. [Google Scholar]
- Kirumakki, S.R.; Shpeizer, B.G.; Sagar, G.V.; Chary, K.V.; Clearfield, A. Hydrogenation of naphthalene over NiO/SiO2–Al2O3 catalysts: Structure–activity correlation. J. Catal. 2006, 242, 319–331. [Google Scholar] [CrossRef]
- Li, M.; Hao, Y.; Cárdenas-Lizana, F.; Keane, M.A. Selective production of furfuryl alcohol via gas phase hydrogenation of furfural over Au/Al2O3. Catal. Commun. 2015, 69, 119–122. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds ...... are available from the authors. |
Figure 1.
(I) Reaction scheme for (A) HMF hydrogenation to the target DHMF (bold arrow, dashed frame) and undesired by-products from hydrogenolysis (MF, MFA, DMF), ring reduction (DHMTHF, MTHFA, DMTHF) and ring opening (2-HXOL) and (B) 2-butanol dehydrogenation to the target 2-butanone (bold arrow, dashed frame) and undesired by-products from dehydration (butene) and dimerisation (octanone and/or octanol isomers). (II) Schematic representation of the coupling system. Note: HMF = 5-hydroxymethyl-2-furaldehyde, MF = 5-methyl-2-furaldehyde, 2-HXOL = 2-hexanol, DHMF = 2,5-di-(hydroxymethyl)-furan, MFA = 5-methyl furfuryl alcohol, DMF = 2,5-dimethylfuran, DHMTHF = 2,5-di-(hydroxymethyl)-tetrahrydrofuran, MTHFA = 5-methyltetrahydro furfuryl alcohol, DMTHF = 2,5-dimethyltetrahydrofuran.
Figure 1.
(I) Reaction scheme for (A) HMF hydrogenation to the target DHMF (bold arrow, dashed frame) and undesired by-products from hydrogenolysis (MF, MFA, DMF), ring reduction (DHMTHF, MTHFA, DMTHF) and ring opening (2-HXOL) and (B) 2-butanol dehydrogenation to the target 2-butanone (bold arrow, dashed frame) and undesired by-products from dehydration (butene) and dimerisation (octanone and/or octanol isomers). (II) Schematic representation of the coupling system. Note: HMF = 5-hydroxymethyl-2-furaldehyde, MF = 5-methyl-2-furaldehyde, 2-HXOL = 2-hexanol, DHMF = 2,5-di-(hydroxymethyl)-furan, MFA = 5-methyl furfuryl alcohol, DMF = 2,5-dimethylfuran, DHMTHF = 2,5-di-(hydroxymethyl)-tetrahrydrofuran, MTHFA = 5-methyltetrahydro furfuryl alcohol, DMTHF = 2,5-dimethyltetrahydrofuran.

Figure 2.
H2-Temperature programmed reduction (TPR) profiles for (I) Au-Cu/CeO2 and (II) Au/CeO2. Note: Raw data is shown as open symbols (⚬) while curve fitted and envelope is represented by solid and dashed lines, respectively.
Figure 2.
H2-Temperature programmed reduction (TPR) profiles for (I) Au-Cu/CeO2 and (II) Au/CeO2. Note: Raw data is shown as open symbols (⚬) while curve fitted and envelope is represented by solid and dashed lines, respectively.

Figure 3.
(I) Representative STEM images with (II) associated metal size distribution histogram for (A) Au-Cu/CeO2 (solid bars) and (B) Au/CeO2 (hatched bars). Note: STEM-EDX analyses of (a–c) isolated metal particles in (IA) are shown in (IIIAa–IIIAc).
Figure 3.
(I) Representative STEM images with (II) associated metal size distribution histogram for (A) Au-Cu/CeO2 (solid bars) and (B) Au/CeO2 (hatched bars). Note: STEM-EDX analyses of (a–c) isolated metal particles in (IA) are shown in (IIIAa–IIIAc).

Figure 4.
XPS spectra over the (A) Au 4f and (B) Cu 2p3/2 regions for (I) Au-Cu/CeO2 and (II) Au/CeO2. Note: Raw data is shown as open symbols (⚬) while curve fitted and envelope is represented by solid and dashed lines, respectively.
Figure 4.
XPS spectra over the (A) Au 4f and (B) Cu 2p3/2 regions for (I) Au-Cu/CeO2 and (II) Au/CeO2. Note: Raw data is shown as open symbols (⚬) while curve fitted and envelope is represented by solid and dashed lines, respectively.

Figure 5.
Comparison of hydrogenation performance over Au-Cu/CeO2 in the coupled process vs. stand-alone reaction using Au/CeO2: Hydrogenation rate in the coupling reaction (rC, bars) and rate enhancement (in the coupling vs. stand-alone hydrogenation given as rC/rSA, ◇) for the conversion of (I) a series of alcohols + HMF and (II) 2-butanol + m-substituted furaldehydes; (III) H2 utilisation efficiency in coupling system (●) and in stand-alone hydrogenation using an external H2 supply (⚬). Note: horizontal dashed line in (III) represents full H2 utilisation under stoichiometric conditions. Reaction conditions: P = 1 atm, T = 498 K.
Figure 5.
Comparison of hydrogenation performance over Au-Cu/CeO2 in the coupled process vs. stand-alone reaction using Au/CeO2: Hydrogenation rate in the coupling reaction (rC, bars) and rate enhancement (in the coupling vs. stand-alone hydrogenation given as rC/rSA, ◇) for the conversion of (I) a series of alcohols + HMF and (II) 2-butanol + m-substituted furaldehydes; (III) H2 utilisation efficiency in coupling system (●) and in stand-alone hydrogenation using an external H2 supply (⚬). Note: horizontal dashed line in (III) represents full H2 utilisation under stoichiometric conditions. Reaction conditions: P = 1 atm, T = 498 K.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Physico-chemical characteristics of CeO2 supported Au-Cu and Au catalysts.
| Au-Cu/CeO2 | Au/CeO2 | |||
|---|---|---|---|---|
| Metal loading (mol%) | 1 (Au)/2 (Cu) | 1 | ||
| TPR | Tmax (K) | α | 414 | 410 |
| β | 429 | 423 | ||
| γ | 437 | - | ||
| δ | 443 | - | ||
| H2 consumption (µmol g−1) | 340 a/63 b/61 c | 307 a/69 b | ||
| dSTEM(nm) d | 4 | 3 | ||
| XPS | Binding energies (eV) | Au0 (%) | 83.6 (100) | 83.5 (100) |
| Cu0 (%) | 928.7 (35) | |||
| Cu+ (%) | 932.0 (65) | |||
| Ce3+ atomic ratio e | 0.24 | 0.29 | ||
a experimental value; b theoretical hydrogen requirement for Au3+ → Au0; c theoretical amount of hydrogen for Cu2+ → Cu+ → Cu0; d mean metal particle size from scanning transmission electron microscopy (STEM) analysis (Equation (1)); e Ce3+ atomic ratio = Ce3+/(Ce4+ + Ce3+) from XPS measurements (see Figure S1 in Supplementary Materials). Note: α, β, γ and δ represent hydrogen consumption peaks during TPR associated with transition of Au3+ → Au0, Ce4+ → Ce3+, Cu2+ → Cu+ and Cu+ → Cu0, respectively.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pischetola, C.; Collado, L.; Keane, M.A.; Cárdenas-Lizana, F. Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu. Molecules 2018, 23, 2905. https://doi.org/10.3390/molecules23112905
AMA Style
Pischetola C, Collado L, Keane MA, Cárdenas-Lizana F. Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu. Molecules. 2018; 23(11):2905. https://doi.org/10.3390/molecules23112905
Chicago/Turabian StylePischetola, Chiara, Laura Collado, Mark A. Keane, and Fernando Cárdenas-Lizana. 2018. "Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu" Molecules 23, no. 11: 2905. https://doi.org/10.3390/molecules23112905