
Reaction between Indazole and Pd-Bound Isocyanides—A Theoretical Mechanistic Study



1
Dipartimento di Scienze della Terra e del Mare, Università degli Studi di Palermo, Via Archirafi, 22, 90123 Palermo, Italy
2
Consorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici (C.I.R.C.M.S.B.), Piazza Umberto I, 70121 Bari, Italy
3
Dipartimento di Scienze Chimiche, Università di Padova, via F. Marzolo 1, 35131 Padova, Italy
4
International Group on Organometallic Chemistry, Institute of Chemistry, Saint Petersburg State University, Universitetskaya Nab., 7/9, Saint Petersburg 199034, Russia
5
Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
*
Author to whom correspondence should be addressed.
†
G.C. performed this work during his leave of absence period at the Dipartimento di Scienze Chimiche, Università di Padova, Padova, Italia at the Prof. Maurizio Casarin group.
Molecules 2018, 23(11), 2942; https://doi.org/10.3390/molecules23112942
Submission received: 8 October 2018
/
Accepted: 8 November 2018
/
Published: 10 November 2018
(This article belongs to the Special Issue Theoretical Investigations of Reaction Mechanisms)
Abstract
:The mechanism of the addition of indazole (Ind)—a bifunctional aromatic N,NH-nucleophile—to cyclohexyl isocyanide coordinated to the palladium(II) center in the model complex cis-[PdCl2(CNMe)(CNCy)] (1) to give the corresponding aminocarbene ligand was investigated in detail by theoretical (DFT) methods. The most plausible mechanism of this reaction is that of the associative type involving nucleophilic attack of Ind by its unprotonated N atom at the isocyanide carbon atom followed by the stepwise proton transfer from the nucleophile molecule to the isocyanide N atom via deprotonation/protonation steps. Two reaction channels based on two tautomeric forms of indazole were found. The channel leading to the experimentally isolated aminocarbene product is based on the less stable tautomeric form. Another channel based on the more stable tautomer of Ind is slightly kinetically more favorable but it is endergonic. Thus, the regioselectivity of this reaction is thermodynamically rather than kinetically driven. The bonding situation in key species was analyzed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The coupling of N-nucleophiles and isocyanides (C≡NR) is an important type of organic transformation yielding a great variety of chemical systems with newly formed C–N bonds [1,2,3,4]. In particular, these reactions represent an excellent route toward acyclic diaminocarbenes—an attractive alternative to N-heterocyclic carbene- and phosphine-based metal complexes used as catalysts in a number of important chemical processes [5,6,7,8,9,10,11], for example, Heck [12,13], Suzuki [12,14], Suzuki-Miyaura [13,15,16,17], and Sonogashira [13,18,19] cross-coupling reactions and some cyclizations/additions to substrates featuring C≡C and C=C bonds [20,21,22,23,24].
Due to the high chemical inertness of isocyanides toward nucleophilic addition (NA), they should usually be activated, e.g., by coordination to an appropriate metal center. Addition of NH-nucleophiles with the sp3-type nitrogen orbital hybridization (i.e., mono- or bifunctional amines [20,21,22,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51] and hydrazines [17,52,53,54,55,56,57,58,59]) to metal-bound isocyanides was extensively reported in the literature. Examples of NA of sp2-N-nucleophiles (e.g., imines [60]) and mixed sp2/sp3-N-nucleophiles (e.g., hydrazones [15] and amidines [18,61]) are much rarer.
Despite intensive experimental investigations of NA of N-nucleophiles to metal coordinated isocyanides, the mechanism of this addition started to become clear only recently. The experimental kinetic studies [43,62,63] indicated a negative activation entropy for the reaction between amines and isocyanides ligated to the Pt(II) or Pd(II) centers. However, as recently as 2017, it was established by theoretical calculations that the mechanism of NA of amines, imines, and hydrazones (HNMe2, HN=CPh2, and H2N–N=CPh2) to the platinum(II) complexes cis-[Pt(C≡NCy)(2-pyz)(dppe)]+ (2-pyz = 2-pyrazyl, dmpe = Me2PCH2CH2PMe2) and cis-[PtCl2(C≡NXyl)(C≡NMe)] is of the stepwise associative type including an attack of the nucleophile by the NH nitrogen atom at the isocyanide carbon atom followed by the amino or imino proton migration via stepwise deprotonation/protonation steps, as shown in Scheme 1 [64].
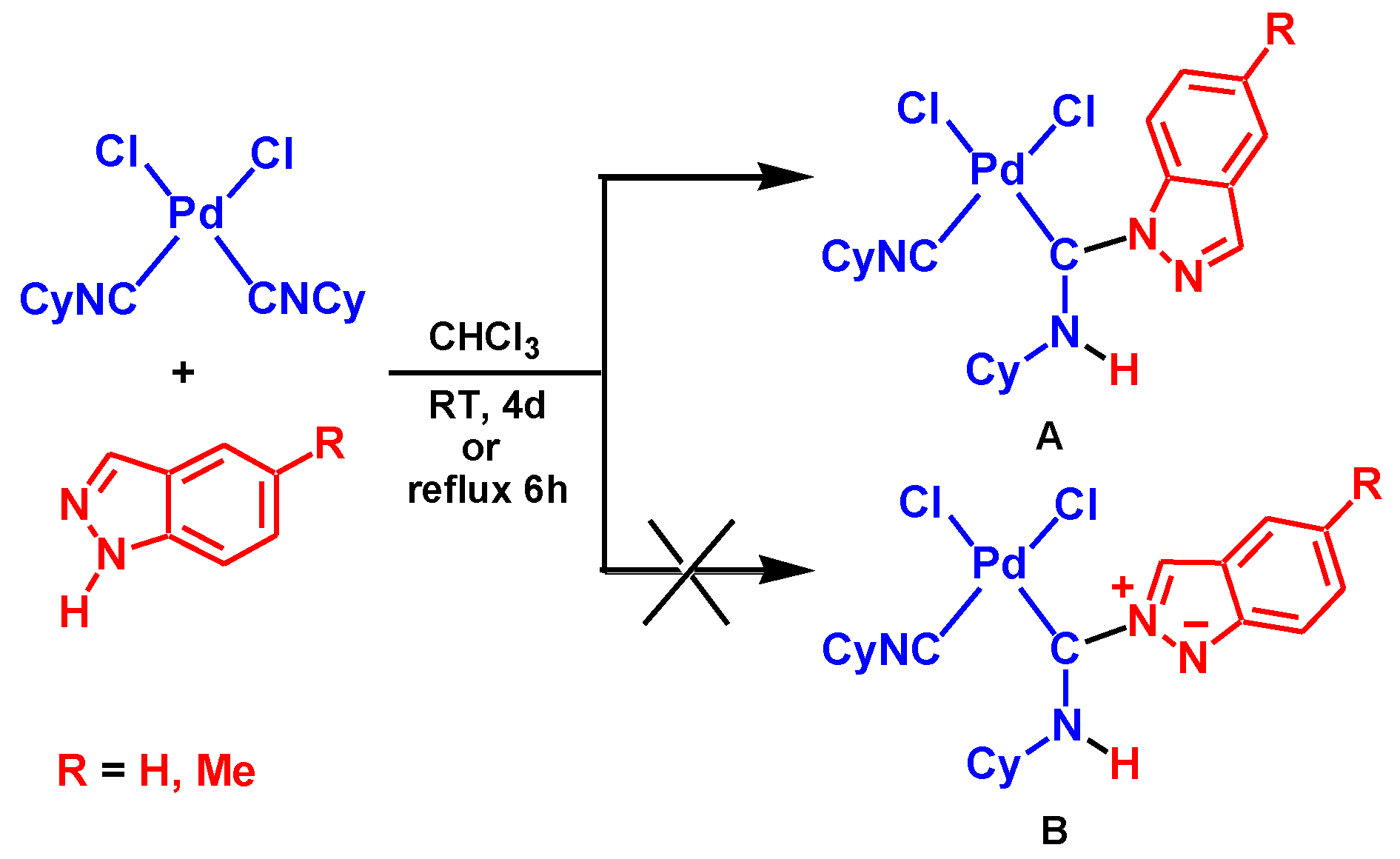
Coupling between aromatic heterocycles with the pyrazole unit and metal-activated isocyanides was reported only recently [65]. The reaction between equimolar amounts of indazole (Ind) or 5-methylindazole and the complex cis-[PdCl2(CNCy)2] (Cy = cyclohexyl) in chloroform yields aminocarbene species A, see Scheme 2. However, details of the mechanism for this reaction were not studied. In particular, the nature of the reaction regioselectivity is unclear. The Indazole molecule has two adjacent imine and amine nucleophilic centers. The former center exhibits stronger nucleophilic properties compared to the latter one. Indeed, in accord with DFT calculations (see Computational Details section), the cationic indazole molecule protonated at the imino N atom is more stable than Ind protonated at the amino nitrogen by 20.8 kcal/mol. Therefore, the preferable formation of a product of the B type, see Scheme 2, would be expected as a result of this reaction but only product A was isolated experimentally from the reaction mixture.
With the aim of interpreting these curious experimental findings and investigating in detail the mechanism of the reaction, theoretical calculations at the DFT (M06L) level of theory have been undertaken for the coupling of indazole and cyclohexyl isocyanide ligand in the model complex cis-[PdCl2(CNMe)(CNCy)] (1), and results of this study are reported here.
2. Computational Details
2.1. Calculations of the Reaction Mechanism
The full geometry optimization of all structures and transition states (TSs) was carried out at the DFT level of theory by using the M06L functional [66] with the help of the Gaussian 09 [67] program package. No symmetry operations were applied. The geometry optimization was carried out by using a quasi-relativistic Stuttgart pseudopotential that describes 28 core electrons and the appropriate contracted basis set (8s7p6d)/[6s5p3d] [68] for the palladium atom and the 6-31G* basis set for other atoms. Single-point calculations were then performed on the basis of the equilibrium geometries found by using the 6-311+G** basis set for non-metal atoms. The solvent effects were taken into account in both optimization and single-point calculations using the Solvation Model based on Density (SMD) [69] with chloroform taken as solvents. The energies discussed below are Gibbs free energies G(6-311+G**) calculated as G(6-311+G**) = E(6-311+G**) – E(6-31G*) + G(6-31G*) where the basis set used is indicated. This combination of method and basis sets was tested in our previous work for NA of amines, imines, and hydrazones to metal-bound isocyanides [64] and provided excellent agreement with available experimental data [62], the DFT calculated and experimental activation enthalpies being 6.9 and 7.0 ± 0.8 kcal/mol, respectively, and the corresponding Gibbs free energies of activation being 21.6 and 19.8 ± 1.7 kcal/mol for the reaction between cis-[Pt(C≡NCy)(2-pyz)(dppe)]+ and HNEt2.
The Hessian matrix was calculated analytically for the optimized structures to prove the location of correct minima (no imaginary frequencies) or saddle points (only one imaginary frequency) and to estimate the thermodynamic parameters, with the latter calculated at 25 °C. Transition states were calculated using a Berny geometry optimization and force constants calculated analytically for the first points. The starting geometries for the TS optimization were found with the help of the QST3 algorithm and/or potential energy scans. The nature of all transition states was investigated by analysis of the vectors associated with the imaginary frequency and by the calculations of the intrinsic reaction coordinates (IRC) by using the method developed by Gonzalez and Schlegel [70,71,72].
2.2. Bond Analysis
Single-point calculations on the previously optimized geometries of compounds 1, TS6, TS14, Z-P2, and Z-P1 were carried out by using the ADF package [73,74] in the DFT framework, at relativistic scalar ZORA level [75,76,77,78], by using the GGA BP86 functional [79,80,81] and Grimme3 BJ-DAMP dispersion corrections [82,83,84] in conjunction with the Slater-type triple ζ doubly polarized (TZ2P) basis set for all the atoms. Previously, it was shown that the GGA BP86 functional demonstrates very good performance in the analysis of the bonding nature in transition metal complexes [85], in the Voronoi Deformation Density (VDD) analysis [86], and in the Energy Decomposition Analysis (EDA) [87]. The effect of the solvent was taken into account according to the Conductor-like Screening Model (COSMO) formalism [88,89,90,91]. The bond analysis was carried out employing canonical Kohn-Sham molecular orbitals (KS-MO).
3. Results and Discussion
3.1. Reaction Mechanisms
In this work, we consider three global types of mechanisms for NA to isocyanides, i.e., dissociative, concerted, and associative, see Scheme 3. The dissociative mechanism starts with deprotonation of the nucleophile by a base that is present in the reaction mixture. The deprotonated nucleophile attacks the isocyanide C atom to give an anionic intermediate, which is protonated to give the final reaction product. The concerted mechanism occurs in one step via a cyclic transition state, which may be either 4- or 6-membered with the participation of a third molecule (solvent, one of the reactants, or an additive) playing the role of a proton shuttle. The associative mechanism includes the addition of a nucleophile in the molecular form to the isocyanide C atom followed by proton transfer (concerted or stepwise) to the N atom. In the following section, all these mechanisms are discussed for the reaction between indazole and complex 1.
3.2. Dissociative Mechanism
The strongest base in the reaction mixture is the imine N atom of indazole. Therefore, namely, this atom was considered as a base for the proton abstraction from indazole within the dissociative mechanism, see Reaction 1. The implicit solvation models (such as SMD used in this work) often fail to correctly describe solvent effects for the processes in which the number of species with the same charge is not preserved in the course of the reaction (as in the case of proton dissociation). Therefore, in order to estimate the energy of autodissociation of indazole (Reaction 1), the experimental pKa values of indazole and its protonated form, IndH+, in water solution were used (13.86 and 1.25, respectively [92]). The ΔG values of the Ind and IndH+ dissociation (ΔG2 and ΔG3, reactions (2) and (3)) may be determined using equation ΔG = 2.303RTpKa, and they are 18.9 and 1.7 kcal/mol for ΔG2 and ΔG3, respectively. Then, the ΔG of autodissociation of indazole in water is ΔGad = ΔG2 – ΔG3 = 17.2 kcal/mol. The theoretically calculated value of ΔGad for reaction 1 in water is 28.1 kcal/mol, and the difference between theoretical and experimental values is 10.9 kcal/mol. The DFT calculated ΔGad value for the chloroform solution is 49.8 kcal/mol. Assuming that the deviation from the experimental data is the same for both water and chloroform solutions, the corrected ΔGad value for the latter is 39.0 kcal/mol. This energy is too high for the efficient occurrence of NA; therefore, the dissociative mechanism may be ruled out.
 ΔGad
ΔGad
 ΔG2
ΔG2
 ΔG3
ΔG3
3.3. Concerted Mechanism
This one-step mechanism should include the formation of a 4-or 6-membered cyclic transition state, see Scheme 4. However, careful search of the potential energy surface found no minimum corresponding to TS, which directly connects the reactants Ind + 1 and the final reaction product P1. All attempts of its location led to transition states of the associative mechanism discussed below.
3.4. Associative Mechanisms
Since the indazole molecule has two potential nucleophilic centers—the amino and imino nitrogen atoms—several associative-type pathways are possible.
3.4.1. Nucleophilic Addition by the Amino Nitrogen Atom (Mechanism I)
This mechanism is similar to that established previously for the reactions of amines, imines, and hydrazones to metal-bound isocyanides [64]. It starts with the formation of a van der Waals pre-reaction complex OC1 which is then transformed into intermediate INT1 via transition state TS1 upon the attack of the amino N atom at the isocyanide C atom, see Scheme 5. The following proton transfer may occur either in a concerted- or a stepwise manner. Both 4- and 6-membered transition states were found for the concerted H-transfer (TS2 and TS3). Water inevitably existing in the reaction mixture as moisture plays the role of a proton shuttle in TS3.
The more favorable stepwise proton transfer includes deprotonation of the indazole moiety in complex INT1 by the second Ind molecule to give INT2 via TS4 and the subsequent protonation of the isocyanide N atom by IndH+ affording the final product P1 through TS5. The protonation step may occur via two channels leading to either the Z- or E-isomer of the final product. The calculations indicate that the Z-channel is both kinetically and thermodynamically more favorable than the E-channel. These results are in agreement with the experimental isolation of the Z-isomer of P1 from the reaction mixture [65].
As can be seen from Scheme 5, the proton abstraction step is the rate-limiting step for the whole process with the overall activation barrier of 36.4 kcal/mol (in terms of ΔGs≠). This value is too high to permit the realization of Mechanism I; therefore, the nucleophilic addition of Ind by the amino N atom may be ruled out. Thus, the mechanism of the reaction of indazole with the Pd-bound isocyanides is different from that when amines, imines, and hydrazones are employed as nucleophiles [64].
3.4.2. Nucleophilic Addition by the Imino Nitrogen Atom (Mechanism II)
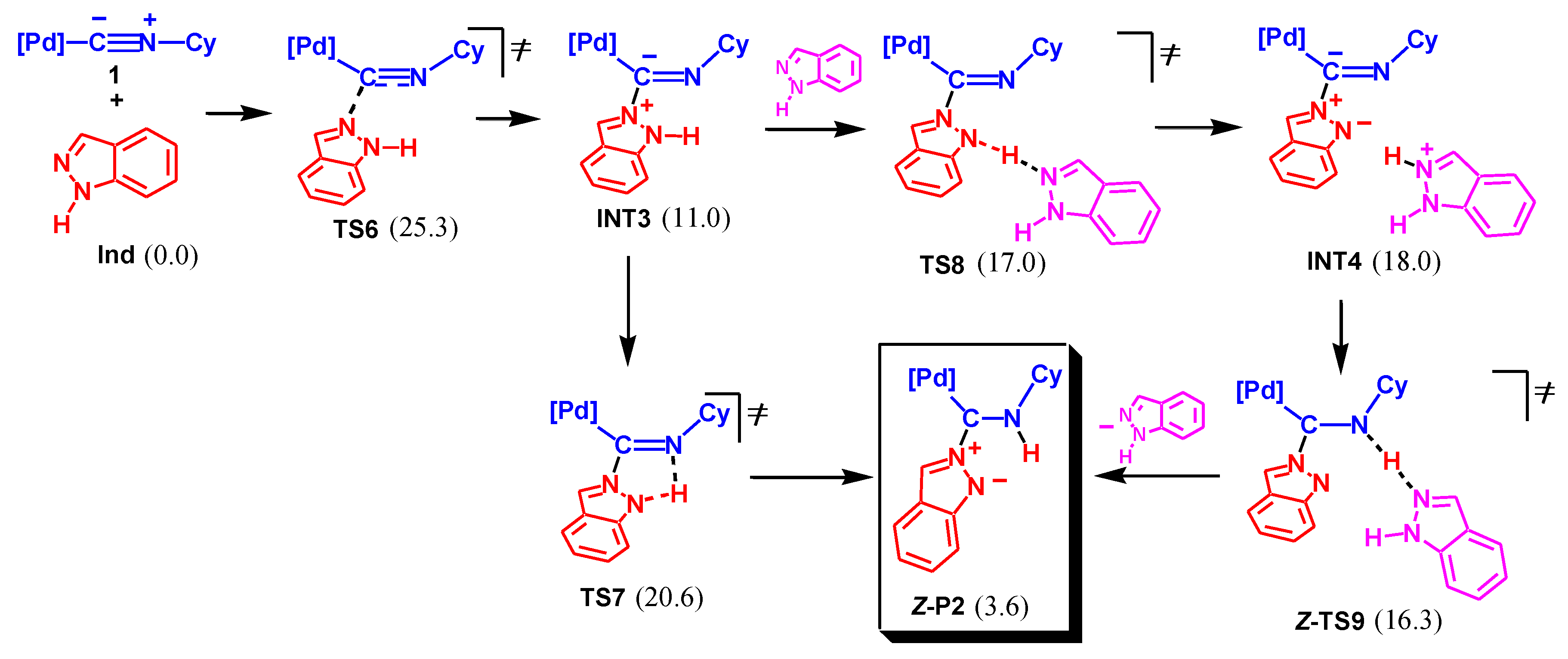
A similar associative mechanism was investigated for NA by the unprotonated imino N atom of Ind. The mechanism starts with the formation of intermediate INT3 via TS6, as shown in Scheme 6. As in the previous case, the following proton shift may occur either in a concerted mode via the 5-membered transition state TS7 or in a stepwise manner via TS8, intermediate INT4, and TS9. The latter pathway is slightly more energetically favorable than the former one (by 2.6 kcal/mol).
The rate-limiting step of this mechanism is the nucleophilic attack of Ind (via TS6), and the overall activation barrier is 25.3 kcal/mol. This value is quite reasonable and qualitatively corresponds to the experimental reaction conditions applied (20–25 °C for 4 days or under reflux in CHCl3 for 6 h [65]). However, the final product of this pathway is complex P2, which was not detected experimentally. In accord with the calculations, the formation of P2 from Ind and 1 is endergonic by 3.6 kcal/mol. Applying the experimental conditions (initial concentrations c0[1] = c0[Ind] ≈ 20 mmol/l), such a ΔGs value should correspond to the maximum concentration of P2 of ca. 8 × 10−4 mol/l. Thus, to detect the formation of P2 in a solution, a special instrumental technique, which was not applied in reference [65], should be used. Thus, despite favorable kinetics, the reaction Ind + 1 → P2 is not feasible thermodynamically.
3.4.3. Isomerization of P2 into P1
Since the final product of the reaction is P1, it may be formed as a result of the isomerization of P2 into P1. Four possible pathways were considered for this transformation, as shown in Scheme 7.
(i) Monomolecular isomerization. Such isomerization occurs in one step via the formation of a three-membered cyclic transition state, TS10. However, the activation energy of this route is too high (45.1 kcal/mol).
(ii) Stepwise rebound. This pathway includes the cleavage of the C(1)N(2) bond in Z-P2 to give the cationic complex, INT5, and the deprotonated indazole, Ind-H–, followed by the formation of the C(1)N(3) bond. We were unable to locate any transition state for the C(1)N(2) bond cleavage. However, results of the energy scan toward this process, see Figure 1, indicate that the lowest limit of the activation energy is 70 kcal/mol (in terms of activation enthalpy at 0 K). Even considering the favorable entropic factor of this step, the activation barrier is too high, and this pathway may be excluded.
(iii) Proton-assisted stepwise rebound. The C(1)N(2) bond cleavage may be facilitated by the protonation of the indazole moiety in P2. However, the protonation step is energetically unfavorable, with the ΔGs value of formation of intermediate INT6 being 43.1 kcal/mol.
(iv) Stepwise rebound assisted by complex 1. The C(1)N(2) bond rupture may also be assisted by the second molecule of complex 1. This pathway includes the attack of P2 by the N(3) atom at the isocyanide carbon atom of the second molecule of 1 to give INT7 via TS11. The following proton transfer and C(1)N(2) bond cleavage leads to the final product P1 (via TS12, INT8, and TS13). However, the high activation barrier (30.7 kcal/mol) disproves this mechanism. Thus, the formation of the final product P1 through isomerization of P2 may be ruled out.
3.4.4. Mechanism Involving the Less Stable Tautomeric Form of Indazole (Mechanism III)
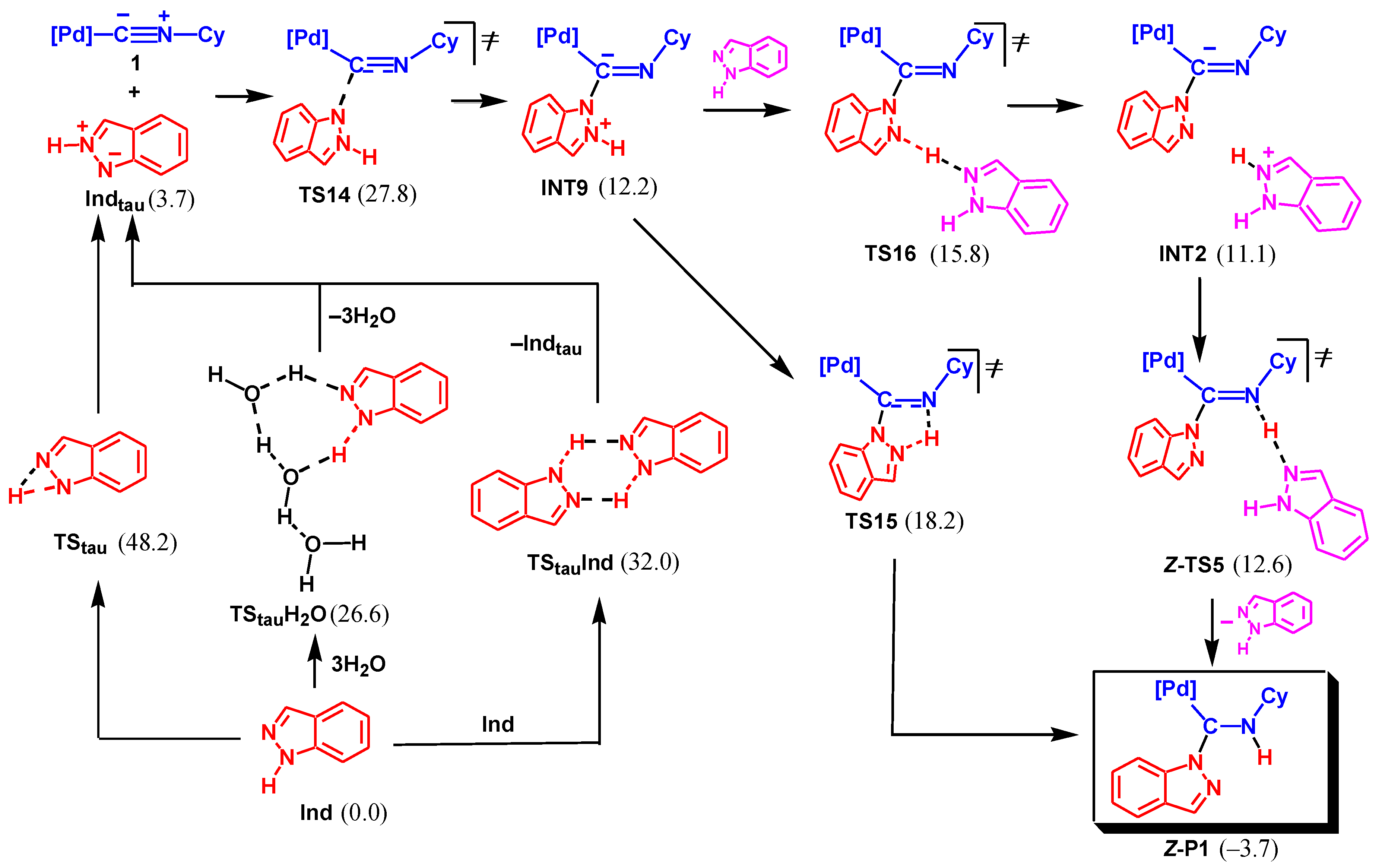
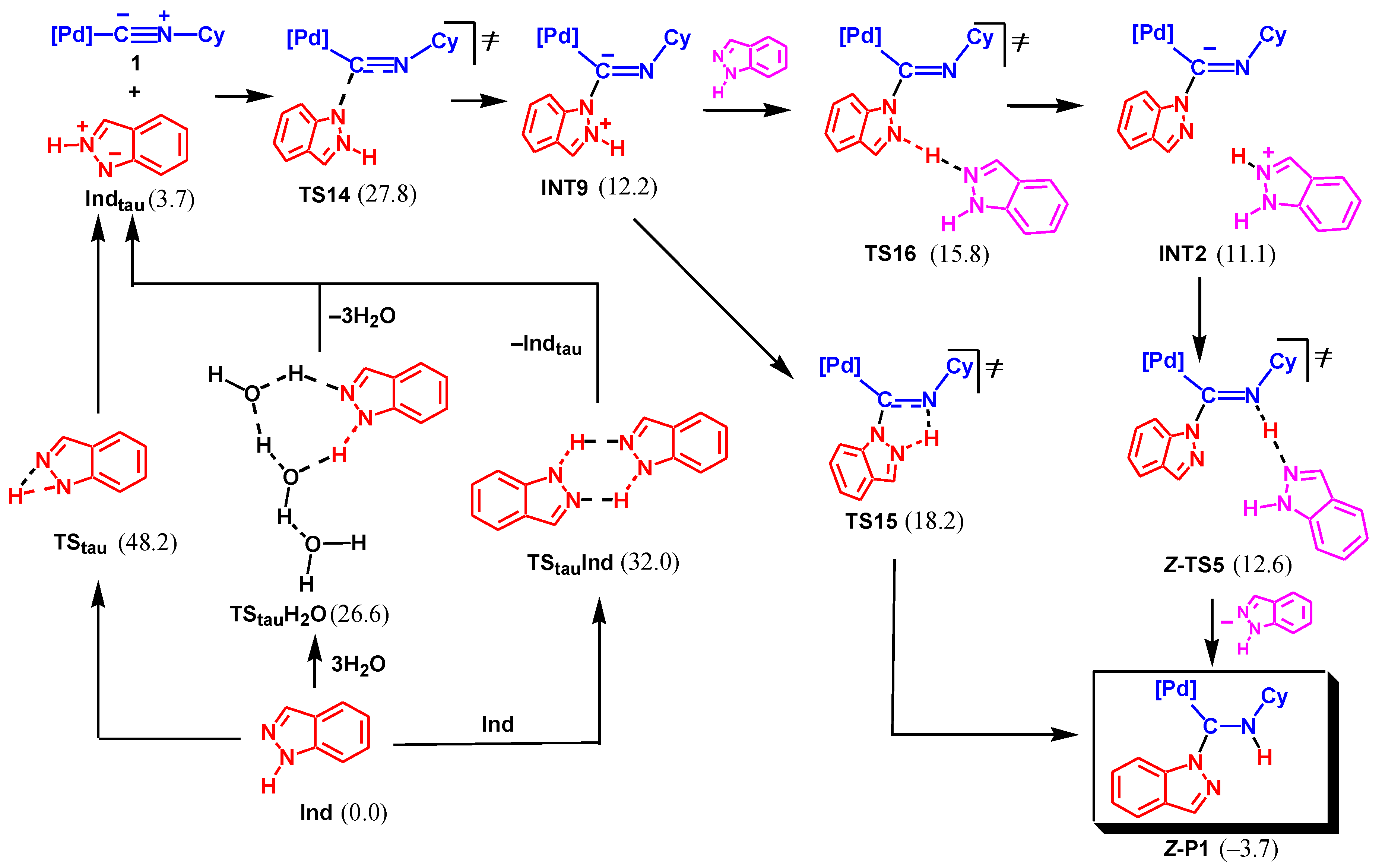
Another possible mechanism is associated with the initial tautomerization of indazole and nucleophilic attack of the less stable tautomeric form at the isocyanide C atom, see Scheme 8. The monomolecular tautomerization via TStau is not favorable with the activation barrier of 48.2 kcal/mol. However, the bimolecular tautomerization involving two Ind molecules via TStauInd requires significantly lower activation energy (32.0 kcal/mol). Finally, the proton shift assisted by water via TStauH2O is even more favorable (ΔGs≠ = 26.6 kcal/mol). The tautomeric form Indtau is only slightly endergonic relative to the most stable structure of Ind (by 3.7 kcal/mol).
The nucleophilic attack of Indtau at the isocyanide of 1 results in the formation of INT9 via TS14. The following proton transfer may occur either in a concerted fashion through the five-membered cyclic transition state, TS15, or in a stepwise manner via protonation/deprotonation involving Ind as a proton shuttle (INT9 → TS16 → INT2 → TS5 → P1).
The rate-limiting step of the whole mechanism is NA of Indtau (via TS14) with an overall activation barrier of 27.8 kcal/mol. This value is only slightly higher than that found for Mechanism II (25.3 kcal/mol) leading to P2. However, the formation of P1 is exergonic (by –3.7 kcal/mol) while the formation of P2 is endergonic (by 3.6 kcal/mol). The difference of 7.3 kcal/mol for the ΔGs of the two reaction channels, 1 + Ind → P1 and 1 + Ind → P2, corresponds to the ratio of the equilibrium concentrations of P1 and P2 ca. 2.3 × 105. Such a huge ratio perfectly explains the lack of experimental detection of product P2 in the reaction mixture despite a slight kinetic preference for its formation over P1.
3.4.5. Mechanisms based on the coordinated indazole
This group of mechanisms includes the initial substitution of one of the isocyanide ligands for the indazole molecule to give complex INT10, as shown in Scheme 9. The substitution occurs via TS17 in a concerted fashion with a rather high activation barrier of 26.2 kcal/mol, and it is endergonic by 11.3 kcal/mol. The further reaction may follow several possible pathways most of them being directly mediated by Pd, see Scheme 9.
Pathway (i). The coordinated indazole may attack the C atom of free isocyanide liberated upon the ligand substitution to give intermediate INT11. However, all attempts to locate the equilibrium structure of this intermediate failed to lead to INT10a and separate C≡NCy. Thus, this pathway is not feasible.
Pathway (ii). In this pathway, the coordinated indazole interacts with the coordinated isocyanide ligand to give the cyclic intermediate INT12 followed by the Pd–N bond cleavage, coordination of the second isocyanide molecule, and proton transfer. However, the calculations indicated that there is no minimum corresponding to INT12, hence, this pathway may also be ruled out.
Pathway (iiia). In this and two following pathways, the deprotonation/proton transfer of/from the coordinated indazole precedes the C–N bond formation. The NH proton of indazole may be transferred to the N isocyanide atom via TS18. The calculations demonstrated that such a transfer is accompanied by the C–N bond formation to give INT14, while the acyclic intermediate INT13 does not exist. Complex INT14 is transformed to Z-P1 via TS19. Meanwhile, this pathway has a very high activation energy of 72.2 kcal/mol, thus, it may be excluded.
Pathway (iiib). The proton transfer may be directly assisted by the Pd atom to give the intermediate INT15. However, the energy of the latter is too high (69.4 kcal/mol) to permit the realization of this route.
Pathway (iiic). The coordination of Ind to the Pd center should increase the acidity of the NH group. This route includes the deprotonation of the ligated indazole by another free Ind molecule via TS20 to give INT16. The cyclization of INT16 to INT17 via TS21 and the protonation of the N isocyanide atom finally lead to Z-P1. The rate-limiting step of this route is the cyclization in INT16 which occurs with a too high activation barrier (49.2 kcal/mol relative to 1).
Pathway (iv). This pathway includes the addition of Ind to complex 1 to give the penta-coordinated intermediate INT18. However, the calculations indicated that such an intermediate does not exist. All attempts of its geometry optimization led to the extrusion of Ind to the second coordination sphere. These results are in agreement with those obtained previously for the substitution of the nitrile ligand in complexes trans-[MCl2(NCMe)2] (M = Pd, Pt) [93]. Therefore, all pathways considered in this part are not feasible either because the key intermediates do not exist or due to the very high activation barrier.
3.5. Bond Analysis
Previously [94,95], some of us demonstrated that the coordination of isocyanides to a metal center plays a tremendous role in the activation of C≡NR toward NA. Therefore, in this section we discuss results of the theoretical analysis of the coordination Pd–C bond nature in the starting Pd isocyanide complex 1, in two isomeric products Z-P1, and Z-P2 and in two rate-determining transition states leading to these products, TS6 and TS14. Additionally, the nature of the CN bond between 1 and Ind fragments in TS6 and TS14 was also analyzed.
The interaction between the {(MeN≡C)(Cl)2Pd} and carbene fragments was considered for the analysis of the Pd–C coordination bond, whereas the interaction between {Ind} and {1} with unrelaxed geometries corresponding to TS6 or TS14 were analyzed for the investigation of the PdC–NInd bond properties. The fragmentation of the model systems in Z-P1 and TS14 is shown in Figure 2.
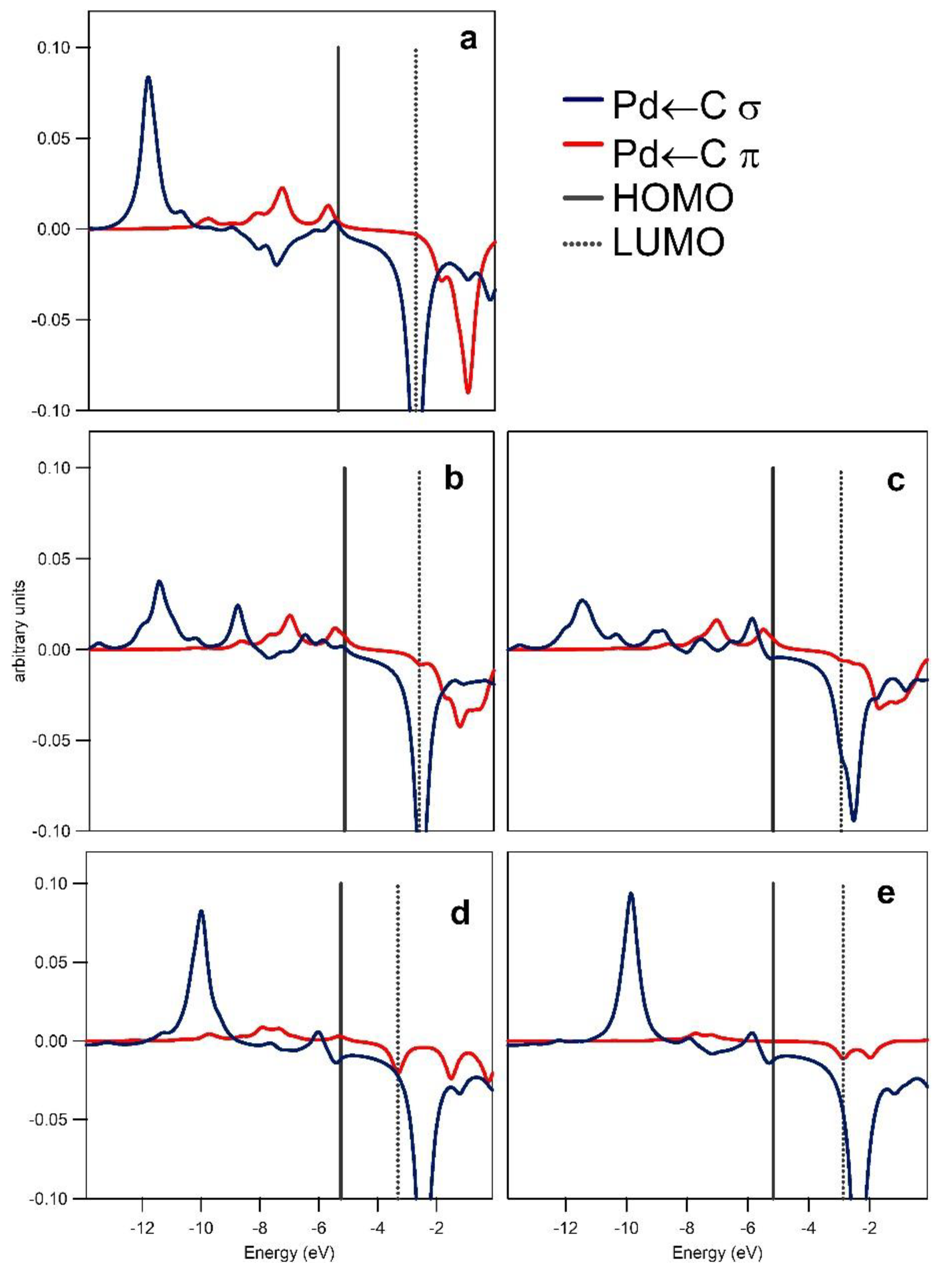
Crystal orbital overlap population (COOP) analysis and inspection of the relevant frontier molecular orbitals provided information on the Pd ← C (σ type; σ) and Pd → C (π type; π) interactions for the fragmentations (a) and (b) and on the PdC ← N (σ type; σ′) interactions for the fragmentation (c), see Figure 2. Overlap integrals for the interacting fragments and relevant populations are given in Table S1 in the Supplementary Material. Results of the COOP analysis depicted in a graphical form in Figure 3 and Figure 4 allow the interpretation of the bonding/anti-bonding nature of the discussed interactions. For the Pd–C bond, σ and π interactions show positive overlaps (bonding type) with the exceptions of the low anti-bonding σ interaction in the −8 to −5 eV energy range for 1. The π interactions give a non-negligible contribution to the Pd–C bond as inferred from the EDA analysis (see Supplementary Material). Finally, the corresponding COOPs for the σ′ interaction, see Figure 4, show very small overlap in agreement with the weak bonding between 1 and indazole in TS6 and TS14.
For all the models under study, the σ interactions mainly arise from the involvement of the CCN electron pair essentially localized at orbitals with the C2s and C2px character and a virtual orbital with the Pddz2, Pd5s, Pdd(x2-y2), and Clpx characters. Moreover, the π interactions reveal the back-donation from orbitals with the predominant Pddxz/dxy + Clpy/pz character, to the CCN virtual orbitals of the pz/y type. This effect is slightly more predominant in 1 compared to Z-P1 and Z-P2 (see VDD and EDA analyses in the Supplementary Material).
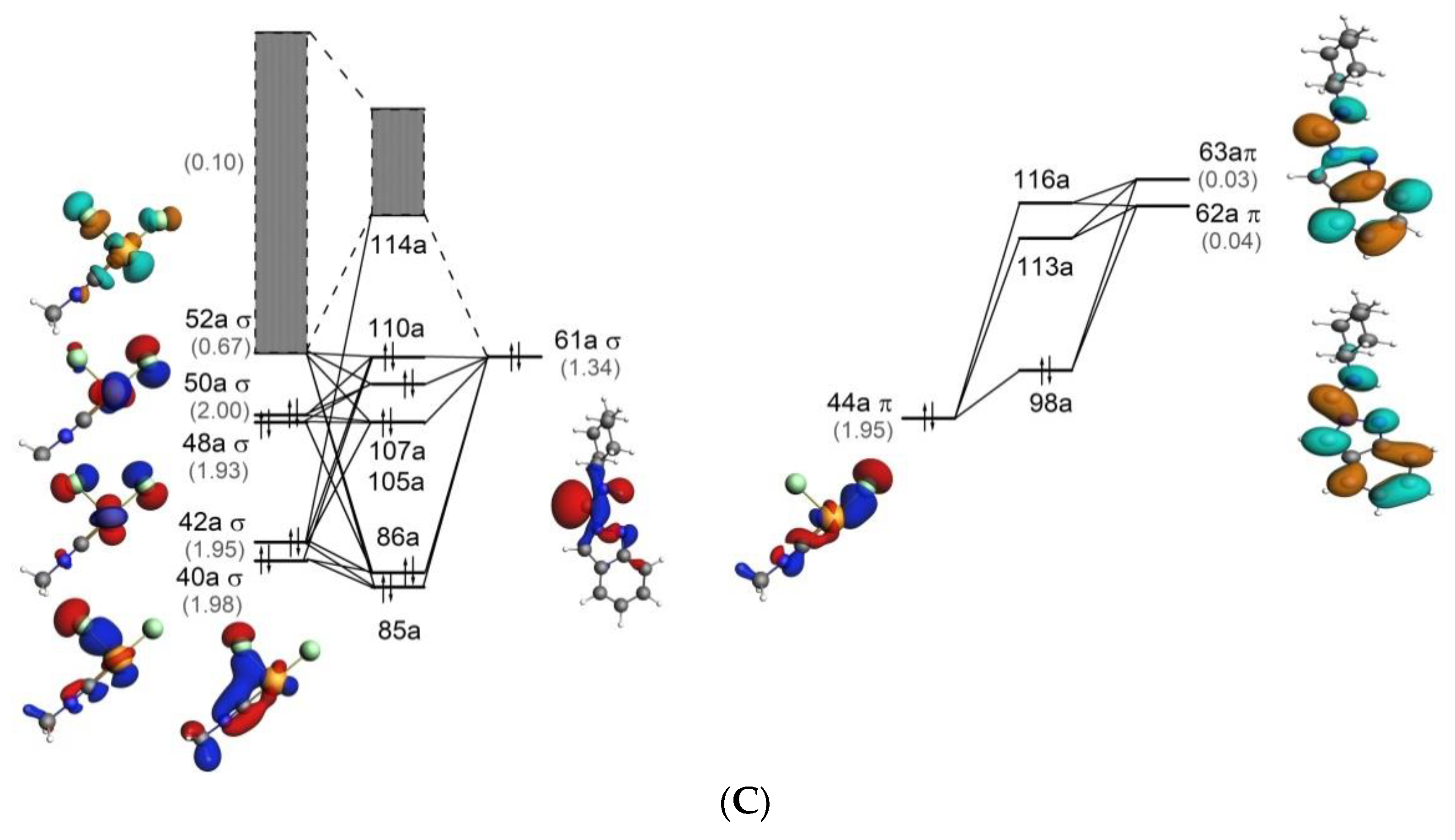
Molecular orbital (MO) diagrams for 1, Z-P1, and Z-P2 are shown in Figure 5. These diagrams demonstrate the relative energies of MOs of these structures and their composition in terms of the orbital interaction of the fragments shown in Figure 2. Comparison of 1 with Z-P1 and Z-P2, see Figure 5, shows appreciable differences in the MO composition. In particular, several σ highest occupied molecular orbitals (HOMOs) of Z-P1 and Z-P2 are composed by only one molecular orbital of the carbene ligand (61a), while three orbitals of C≡NCy (27a–29a) are involved in the σ bonding in 1.
The different bonding mechanisms characterizing the reactant and the product(s) implies a stronger Pd–C bond energy of both Z-P1 and Z-P2 of about 15 kcal/mol with respect to 1 (the detailed discussion of the EDA and VVD analyses is provided in Supplementary Material) determining a further stabilization of the Pd–C bond and, thus, of the newly formed carbene.
Finally, the bonding schemes illustrating the PdC ← N interactions in TS6 and TS14 are given in Figure 6. In contrast to TS6, the orbital 31a of indazole participates in the σ′ interaction in TS14. Despite the 31a orbital of indazole showing π character, the orientation of the interacting fragments provides the σ-type interaction between {Ind} and {1}.
4. Final Remarks
Mechanisms of the addition of N-nucleophiles to metal-bound isocyanides—a promising and efficient route toward practically important N-heteroatom stabilized carbenes—are still poorly explored. In this work, the mechanism of the reaction between indazole and the Pd-coordinated cyclohexyl isocyanide in the model complex cis-[PdCl2(CNMe)(CNCy)] (1) was investigated in detail by DFT (M06L) methods.
The amino N(H) atom of Ind was found to be inactive toward nucleophilic addition. Instead, indazole reacts with 1 by the unprotonated nitrogen atom. Therefore, the mechanism of this reaction is different from that previously established for NA of amines, imines, and hydrazones to Pt-bound isocyanides [64].
The mechanism of reaction between Ind and 1 includes (i) nucleophilic attack of Ind by the unprotonated N atom at the isocyanide C atom, (ii) deprotonation of the resulting intermediate by another Ind molecule, and (iii) protonation of the isocyanide N atom to give the carbene product.
Two reaction channels based on different tautomeric forms of Ind were found. The first channel is based on the less stable tautomer of Ind and it leads to the experimentally isolated aminocarbene product P1 (Mechanism III, Scheme 8). The second channel results in the formation of the isomeric carbene product P2 which was not detected experimentally and is based on the most stable tautomer of Ind (Mechanism II, Scheme 6).
The second channel is slightly more favorable kinetically compared to the first one (by 2.5 kcal/mol). However, P1 is thermodynamically more stable than P2 (by 7.3 kcal/mol), the latter being endergonic relative to the reactants by 3.6 kcal/mol. Thus, the regioselectivity of this reaction is thermodynamically rather than kinetically driven. The nucleophilic addition of Ind to 1 by the amino N atom as well as the isomerization of P2 into P1 are not feasible.
The bonding nature in 1, Z-P1, Z-P2, and the rate-limiting TS6 and TS14 were analyzed in detail.
Supplementary Materials
The following are available online, additional computational details and bond analysis discussion, Figures S1, S2, S4, S5, S8, S9: bonding schemes, Figure S3: VDD charges, Figure S6: optimized structures of R1 and RP, Figure S7: COOPs for R1 and RP, Figures S10–S14: energy profiles, Tables S1, S3, S5: VDD charges, Table S2: results of the EDA analysis, Tables S4 and S6: overlaps and relevant orbital populations, Table S7: calculated total energies, enthalpies, Gibbs free energies, and entropies, Table S8: Cartesian atomic coordinates of the equilibrium structures.
Author Contributions
G.C. performed calculations and wrote the bond analysis section; M.C. analyzed the data and discussed results; V.Y.K. analyzed the data and discussed results. M.L.K. initiated the work, performed calculations, analyzed the data, and wrote the paper.
Funding
This research was funded by [Russian Science Foundation] grant number [14-43-00017P].
Acknowledgments
The authors are grateful to the Russian Science Foundation (grant 14-43-00017P) for support of this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Y. Metal-mediated and metal-catalyzed reactions of isocyanides. Chem. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef] [PubMed]
- Hulme, C.; Lee, Y.-S. Emerging approaches for the syntheses of bicyclic imidazo[1,2-x]-heterocycles. Mol. Divers. 2008, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sadjadi, S.; Heravi, M.M. Recent Application of Isocyanides in Synthesis of Heterocycles. Tetrahedron 2011, 67, 2707–2752. [Google Scholar] [CrossRef]
- Michelin, R.A.; Pombeiro, A.J.L.; Guedes da Silva, M.F.C. Aminocarbene complexes derived from nucleophilic addition to isocyanide ligands. Coord. Chem. Rev. 2001, 218, 75–112. [Google Scholar] [CrossRef]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic Carbenes: Synthesis and Coordination Chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Boyarskiy, V.P.; Luzyanin, K.V.; Kukushkin, V.Y. Acyclic diaminocarbenes (ADCs) as a promising alternative to N-heterocyclic carbenes (NHCs) in transition metal catalyzed organic transformations. Coord. Chem. Rev. 2012, 256, 2029–2056. [Google Scholar] [CrossRef]
- Nolan, S.P.; Navarro, O. C–C Bond Formation by Cross-coupling, in Comprehensive Organometallic Chemistry III; Canty, A., Ed.; Elsevier: Oxford, England, UK, 2007; Volume 11, pp. 1–38. [Google Scholar]
- Marion, N.; Nolan, S.P. Well-Defined N-Heterocyclic Carbenes−Palladium(II) Precatalysts for Cross-Coupling Reactions. Acc. Chem. Res. 2008, 41, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-Heterocyclic carbenes in late transition metal catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, L.M. Acyclic aminocarbenes in catalysis. ACS Catal. 2012, 2, 1802–1816. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Kremzow, D.; Seidel, G.; Lehmann, C.W.; Furstner, A. Diaminocarbene- and Fischer-carbene complexes of palladium and nickel by oxidative insertion: Preparation, structure, and catalytic activity. Chem. Eur. J. 2005, 11, 1833–1853. [Google Scholar] [CrossRef] [PubMed]
- Dhudshia, B.; Thadani, A.N. Acyclic diaminocarbenes: simple, versatile ligands for cross-coupling reactions. Chem. Commun. 2006, 6, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Miltsov, S.; Karavan, V.; Boyarsky, V.; Gómez-de Pedro, S.; Alonso-Chamarro, J.; Puyol, M. New acyclic Pd–Diaminocarbene catalyst for suzuki arylation of meso-chlorosubstituted tricarboindocyanine dyes. Tetrahedron Lett. 2013, 54, 1202–1204. [Google Scholar] [CrossRef]
- Luzyanin, K.V.; Tskhovrebov, A.G.; Carias, M.C.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L.; Kukushkin, V.Y. Novel metal–mediated (M = Pd, Pt) coupling between isonitriles and benzophenone hydrazone as a route to aminocarbene complexes exhibiting high catalytic activity (M = Pd) in the suzuki–miyaura reaction. Organometallics 2009, 28, 6559–6566. [Google Scholar] [CrossRef]
- Slaughter, L.M. Covalent self-assembly of acyclic diaminocarbene ligands at metal centers. Comments Inorg. Chem. 2008, 29, 46–72. [Google Scholar] [CrossRef]
- Moncada, A.I.; Manne, S.; Tanski, J.M.; Slaughter, L.M. Modular chelated palladium diaminocarbene complexes: synthesis, characterization, and optimization of catalytic suzuki−miyaura cross–coupling activity by ligand modification. Organometallics 2006, 25, 491–505. [Google Scholar] [CrossRef]
- Tskhovrebov, A.G.; Luzyanin, K.V.; Kuznetsov, M.L.; Sorokoumov, V.N.; Balova, I.A.; Haukka, M.; Kukushkin, V.Y. Substituent R-dependent regioselectivity switch in nucleophilic addition of N-phenylbenzamidine to PdII– and PtII–complexed isonitrile RN≡C giving aminocarbene-like species. Organometallics 2011, 30, 863–874. [Google Scholar] [CrossRef]
- Ryabukhin, D.S.; Sorokoumov, V.N.; Savicheva, E.A.; Boyarskiy, V.P.; Balova, I.A.; Vasilyev, A.V. Catalytic activity of palladium acyclic diaminocarbene complexes in the synthesis of 1,3–diarylpropynones via sonogashira reaction: cross– versus homo–coupling. Tetrahedron Lett. 2013, 54, 2369–2372. [Google Scholar] [CrossRef]
- Bartolomé, C.; Ramiro, Z.; García-Cuadrado, D.; Pérez-Galán, P.; Raducan, M.; Bour, C.; Echavarren, A.M.; Espinet, P. Nitrogen acyclic gold(I) carbenes: excellent and easily accessible catalysts in reactions of 1,6-enynes. Organometallics 2010, 29, 951–956. [Google Scholar] [CrossRef]
- Bartolomé, C.; García-Cuadrado, D.; Ramiro, Z.; Espinet, P. Synthesis and catalytic activity of gold chiral nitrogen acyclic carbenes and gold hydrogen bonded heterocyclic carbenes in cyclopropanation of vinyl arenes and in intramolecular hydroalkoxylation of allenes. Inorg. Chem. 2010, 49, 9758–9764. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Hengst, T.; Lothschütz, C.; Rominger, F. New and easily accessible nitrogen acyclic gold(I) carbenes: structure and application in the gold–catalyzed phenol synthesis as well as the hydration of alkynes. Adv. Synth. Catal. 2010, 352, 1315–1337. [Google Scholar] [CrossRef]
- Seo, H.; Snead, D.R.; Abboud, K.A.; Hong, S. Bulky acyclic aminooxycarbene ligands. Organometallics 2011, 30, 5725–5730. [Google Scholar] [CrossRef]
- Cavarzan, A.; Scarso, A.; Sgarbossa, P.; Strukul, G.; Reek, J.N.H. Supramolecular control on chemo- and regioselectivity via encapsulation of (NHC)-Au catalyst within a hexameric self-assembled host. J. Am. Chem. Soc. 2011, 133, 2848–2851. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, C.; Carrasco-Rando, M.; Coco, S.; Cordovilla, C.; Martín-Alvarez, J.M.; Espinet, P. Luminescent gold(I) carbenes from 2-pyridylisocyanide complexes: structural consequences of intramolecular versus intermolecular hydrogen-bonding interactions. Inorg. Chem. 2008, 47, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-W.; Chan, M.C.-W.; Cheung, K.-K.; Che, C.-M. Carbene and isocyanide ligation at luminescent cyclometalated 6-phenyl-2,2‘-bipyridyl platinum(II) complexes: Structural and spectroscopic studies. Organometallics 1999, 18, 3327–3336. [Google Scholar] [CrossRef]
- Crociani, B.; Boschi, T.; Belluco, U. Synthesis and reactivity of novel palladium(II)–isocyanide complexes. Inorg. Chem. 1970, 9, 2021–2025. [Google Scholar] [CrossRef]
- Michelin, R.A.; Zanotto, L.; Braga, D.; Sabatino, P.; Angelici, R.J. Transition–metal–promoted cyclization reactions of isocyanide ligands. synthesis of cyclic diaminocarbenes from isocyanide complexes of palladium(II) and platinum(II) and X-ray structure of cis-Br2Pt[CN(C6H4-p-Me)CH2CH2N(H)](PPh3). Inorg. Chem. 1988, 27, 93–99. [Google Scholar] [CrossRef]
- Yu, I.; Wallis, C.J.; Patrick, B.O.; Diaconescu, P.L.; Mehrkhodavandi, P. Phosphine–tethered carbene ligands: template synthesis and reactivity of cyclic and acyclic functionalized carbenes. Organometallics 2010, 29, 6065–6076. [Google Scholar] [CrossRef]
- Arias, J.; Bardají, M.; Espinet, P. Luminescence and mesogenic properties in crown-ether-isocyanide or carbene gold(I) complexes: Luminescence in solution, in the solid, in the mesophase, and in the isotropic liquid state. Inorg. Chem. 2008, 47, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Crociani, B.; Uguagliati, P.; Belluco, U. Steric role of aromatic ring ortho-substituents in the mechanism of carbene formation from palladium(II) arylisocyanide complexes and anilines. J. Organomet. Chem. 1976, 117, 189–199. [Google Scholar] [CrossRef]
- Lazar, M.; Zhu, B.; Angelici, R.J. Non-nanogold catalysis of reactions of isocyanides, secondary amines, and oxygen to give ureas. J. Phys. Chem. C 2007, 111, 4074–4076. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Levi, C.; Bertolasi, V. Synthesis and mechanism of formation of novel NHC−NAC bis-carbene complexes of gold(I). Organometallics 2011, 30, 875–883. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, J.; García, L.; Mejuto, C.; Vivanco, M.; Díaz, M.R.; García-Granda, S. Strong electron-donating metalla-N-heterocyclic carbenes. Chem. Commun. 2014, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Anding, B.J.; Ellern, A.; Woo, L.K. Comparative study of rhodium and iridium porphyrin diaminocarbene and N-heterocyclic carbene complexes. Organometallics 2014, 33, 2219–2229. [Google Scholar] [CrossRef]
- Vicente, J.; Chicote, M.T.; Huertas, S.; Jones, P.G. 1,1-Ethylenedithiolato complexes of palladium(II) and platinum(II) with isocyanide and carbene ligands. Inorg. Chem. 2003, 42, 4268–4274. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huynh, H.V. Mixed carbene–isocyanide Pd(II) complexes: Synthesis, structures and reactivity towards nucleophiles. Dalton Trans. 2009, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martínez, A.-J.; Chicote, M.-T.; Bautista, D.; Vicente, J. Synthesis of palladium(II), -(III), and -(IV) complexes with acyclic diaminocarbene ligands. Organometallics 2012, 31, 3711–3719. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Böhling, C.; Lothschütz, C.; Rominger, F. From isonitriles to carbenes: Synthesis of new NAC− and NHC−palladium(II) compounds and their catalytic activity. Organometallics 2011, 30, 2411–2417. [Google Scholar] [CrossRef]
- Heathcote, R.; Howell, J.A.S.; Jennings, N.; Cartlidge, D.; Cobden, L.; Coles, S.; Hursthouse, M. Gold(I)–isocyanide and gold(I)–carbene complexes as substrates for the laser decoration of gold onto ceramic surfaces. Dalton Trans. 2007, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, C.; Carrasco-Rando, M.; Coco, S.; Cordovilla, C.; Espinet, P.; Martín-Alvarez, J.M. Gold(I)–carbenes derived from 4-pyridylisocyanidecomplexes: supramolecular macrocycles supported by hydrogen bonds, and luminescent behavior. Dalton Trans. 2007, 5339–5345. [Google Scholar] [CrossRef]
- Handa, S.; Slaughter, L.M. Enantioselective alkynylbenzaldehyde cyclizations catalyzed by chiral gold(I) acyclic diaminocarbene complexes containing weak Au–arene interactions. Angew. Chem. Int. Ed. 2012, 51, 2912–2915. [Google Scholar] [CrossRef] [PubMed]
- Uguagliati, P.; Crociani, B.; Belluco, U.; Calligaro, L. Solvent, ligand and temperature effects on the rates of reactions of cis-[PdCl2(CNR)(L)] with secondary amines. J. Organomet. Chem. 1976, 112, 111–121. [Google Scholar] [CrossRef]
- Spallek, M.J.; Riedel, D.; Rominger, F.A.; Hashmi, A.S.K.; Trapp, O. Six–membered, chiral NHCs derived from camphor: Structure–reactivity relationship in asymmetric oxindole synthesis. Organometallics 2012, 31, 1127–1132. [Google Scholar] [CrossRef]
- Ruiz, J.; Perandones, B.F.; García, G.; Mosquera, M.E.G. Synthesis of N-heterocyclic carbene complexes of manganese(I) by coupling isocyanide ligands with propargylamines and propargylic alcohols. Organometallics 2007, 26, 5687–5695. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Lothschütz, C.; Graf, K.; Häffner, T.; Schuster, A.; Rominger, F. A short way to switchable carbenes. Adv. Synth. Catal. 2011, 353, 1407–1412. [Google Scholar] [CrossRef]
- Wanniarachchi, Y.A.; Slaughter, L.M. One-step assembly of a chiral palladium bis(acyclic diaminocarbene) complex and its unexpected oxidation to a bis(amidine) complex. Chem. Commun. 2007, 3294–3296. [Google Scholar] [CrossRef] [PubMed]
- Zanella, R.; Boschi, T.; Crociani, B.; Belluco, U. Reactions of palladium(II) isocyanide complexes with bifunctional amines. J. Organomet. Chem. 1974, 71, 135–143. [Google Scholar] [CrossRef]
- Zanella, R.; Boschi, T.; Nicolini, M.; Belluco, U. The reactions of bis-isocyanide derivatives of PdII with bidentate ligands. J. Organomet. Chem. 1973, 49, C91–C94. [Google Scholar] [CrossRef]
- Crociani, B.; Di Bianca, F.; Fontana, A.; Forsellini, E.; Bombieri, G. Nucleophilic attack at co-ordinated isocyanides promoted by the 2-pyridyl ligand. J. Chem. Soc. Dalton Trans. 1994, 407–414. [Google Scholar] [CrossRef]
- Belluco, U.; Michelin, R.A.; Ros, R.; Bertani, R.; Facchin, G.; Mozzon, M.; Zanotto, L. Synthesis of heterocyclic systems by activation of isocyanide, carbonyl, trifluoromethyl and nitrile ligands in platinum(II) and palladium(II) complexes. Inorg. Chim. Acta 1992, 198–200, 883–897. [Google Scholar] [CrossRef]
- Tschugajeff, L.; Skanawy-Grigorjewa, M. Reaction of K2PtCl4 with isonitriles and hydrazine. J. Russ. Chem. Soc. 1915, 47, 776. [Google Scholar]
- Tschugajeff, L.; Grigorjewa, M.; Posnjak, A. Über die hydrazine–carbilamin–komplexe des platins. Z. Anorg. Allg. Chem. 1925, 148, 37–42. [Google Scholar] [CrossRef]
- Rouschias, G.; Shaw, B.L. The chemistry and structure of Chugaev’s salt and related compounds containing a cyclic carbene ligand. J. Chem. Soc. A 1971, 2097–2104. [Google Scholar] [CrossRef]
- Burke, A.; Balch, A.L.; Enemark, J.H. Palladium and platinum complex resulting from the addition of hydrazine to coordinated isocyanide. J. Am. Chem. Soc. 1970, 92, 2555–2557. [Google Scholar] [CrossRef]
- Butler, W.M.; Enemark, J.H.; Parks, J.; Balch, A.L. Chelative addition of hydrazines to coordinated isocyanides. structure of Chugaev’s red salt. Inorg. Chem. 1973, 12, 451–457. [Google Scholar] [CrossRef]
- Badley, M.; Chatt, J.; Richards, R.L. Isonitrile complexes of platinum(II) and their reactions with alcohols and amines to give carbene complexes. J. Chem. Soc. A 1971, 21–25. [Google Scholar] [CrossRef]
- Moncada, A.I.; Tanski, J.M.; Slaughter, L.M. Sterically controlled formation of monodentate versus chelating carbene ligands from phenylhydrazine. J. Organomet. Chem. 2005, 690, 6247–6251. [Google Scholar] [CrossRef]
- Wanniarachchi, Y.A.; Slaughter, L.M. Reversible chelate ring opening of a sterically crowded palladium bis(acyclic diaminocarbene) complex. Organometallics 2008, 27, 1055–1062. [Google Scholar] [CrossRef]
- Luzyanin, K.V.; Guedes da Silva, M.F.C.; Kukushkin, V.Y.; Pombeiro, A.J.L. First example of an imine addition to coordinated isonitrile. Inorg. Chim. Acta 2009, 362, 833–838. [Google Scholar] [CrossRef]
- Balch, A.L.; Parks, J.E. Platinum and palladium complexes formed by chelative addition of amines to isocyanides. J. Am. Chem. Soc. 1974, 96, 4114–4121. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Uguagliati, P.; Crociani, B.; Di Bianca, F. Mechanism of aminocarbene formation by nucleophilic attack on isocyanide ligands in platinum(II) 2-pyrazyl and 4-pyridyl complexes. J. Organomet. Chem. 1997, 535, 69–75. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Uguagliati, P.; Crociani, B. Mechanism of formation of (methoxy)(amino)–and bis(amino) carbene complexes by nucleophilic attack of methoxide ion and amines on platinum(II)-coordinated isocyanide in anhydrous methanol. J. Organomet. Chem. 1997, 543, 145–151. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Kukushkin, V.Y. Metal-mediated addition of N-nucleophiles to isocyanides: mechanistic aspects. Molecules 2017, 22, 1141. [Google Scholar] [CrossRef] [PubMed]
- Kinzhalov, M.A.; Boyarskiy, V.P.; Luzyanin, K.V.; Dolgushin, F.M.; Kukushkin, V.Y. Metal-mediated coupling of a coordinated isocyanide and indazoles. Dalton Trans. 2013, 42, 10394–10397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main–group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef] [PubMed]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2013, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, Netherlands. Available online: http://www.scm.com (accessed on 30 October 2018).
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two–component hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- van Lenthe, E.; van Leeuwen, R.; Baerends, E.J.; Snijders, J.G. Relativistic regular two–component hamiltonians. Int. J. Quantum Chem. 1996, 57, 281–293. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density–functional exchange–energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density–functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P. Erratum: Density–functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 34, 7406. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.A. Consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Bertani, R.; Mozzon, M.; Sgarbossa, P.; Tamburini, S.; Casarin, M.; Mangione, G.; Casella, G.; Venzo, A.; Rizzato, S.; Albinati, A. Pt(II) nitrile complexes: New insights on old complexes from a combined experimental and theoretical study. Inorg. Chim. Acta 2017, 455, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.F.; Handgraaf, J.-W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Orenha, R.P.; Rocha, M.V.J.; Poater, J.; Galembeck, S.E.; Bickelhaupt, F.M. Nature of the Ru–NO coordination bond: Kohn–Sham molecular orbital and energy decomposition analysis. ChemistryOpen 2017, 6, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996, 105, 9972–9981. [Google Scholar] [CrossRef]
- Pye, C.C.; Ziegler, T. An implementation of the conductor–like screening model of solvation within the amsterdam density functional package. Theor. Chem. Acc. 1999, 101, 396–408. [Google Scholar] [CrossRef]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structure, Reactions, Synthesis and Applications; Wiley–VCH: Weinheim, Germany, 2012. [Google Scholar]
- Kuznetsov, M.L.; Kukushkin, V. Yu.; Pombeiro, A.J.L. Reactivity of Pt– and Pd–bound nitriles towards nitrile oxides and nitrones: Substitution vs. cycloaddition. Dalton Trans. 2008, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Menezes, F.M.C.; Kuznetsov, M.L.; Pombeiro, A.J.L. Isocyanide complexes with platinum and palladium and their reactivity toward cycloadditions with nitrones to form aminooxycarbenes: A theoretical study. Organometallics 2009, 28, 6593–6602. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L. Theoretical study of Re(IV) and Ru(II) bis–isocyanide complexes and their reactivity in cycloaddition reactions with nitrones. Inorg. Chim. Acta 2012, 380, 78–89. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Scheme 1.
The mechanism of nucleophilic addition (NA) of amines, imines, and hydrazones to isocyanides coordinated to the Pt(II) center.
Scheme 1.
The mechanism of nucleophilic addition (NA) of amines, imines, and hydrazones to isocyanides coordinated to the Pt(II) center.

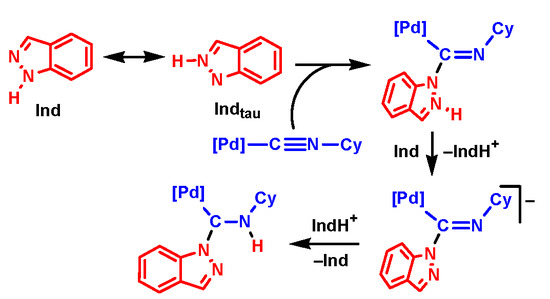
Scheme 2.
Reaction between cis-[PdCl2(C≡NCy)2] and the indazoles.
Scheme 3.
Mechanisms of nucleophilic addition to metal-bound isocyanides.

Scheme 4.
Concerted mechanism of the reaction between Ind and 1.

Scheme 5.
Associative mechanism of NA of indazole to 1 by amino nitrogen atom (Mechanism I, Gibbs free energies are indicated in kcal/mol relative to 1 + Ind, final product is boxed).
Scheme 5.
Associative mechanism of NA of indazole to 1 by amino nitrogen atom (Mechanism I, Gibbs free energies are indicated in kcal/mol relative to 1 + Ind, final product is boxed).

Scheme 6.
Associative mechanism of NA of indazole to 1 by imino nitrogen atom (Mechanism II, Gibbs free energies are indicated in kcal/mol relative to 1 + Ind, final product is boxed, only the Z-isomeric pathway is shown).
Scheme 6.
Associative mechanism of NA of indazole to 1 by imino nitrogen atom (Mechanism II, Gibbs free energies are indicated in kcal/mol relative to 1 + Ind, final product is boxed, only the Z-isomeric pathway is shown).

Scheme 7.
Possible mechanisms of isomerization of P2 into P1 (Gibbs free energies relative to 1 + Ind are shown in kcal/mol).
Scheme 7.
Possible mechanisms of isomerization of P2 into P1 (Gibbs free energies relative to 1 + Ind are shown in kcal/mol).
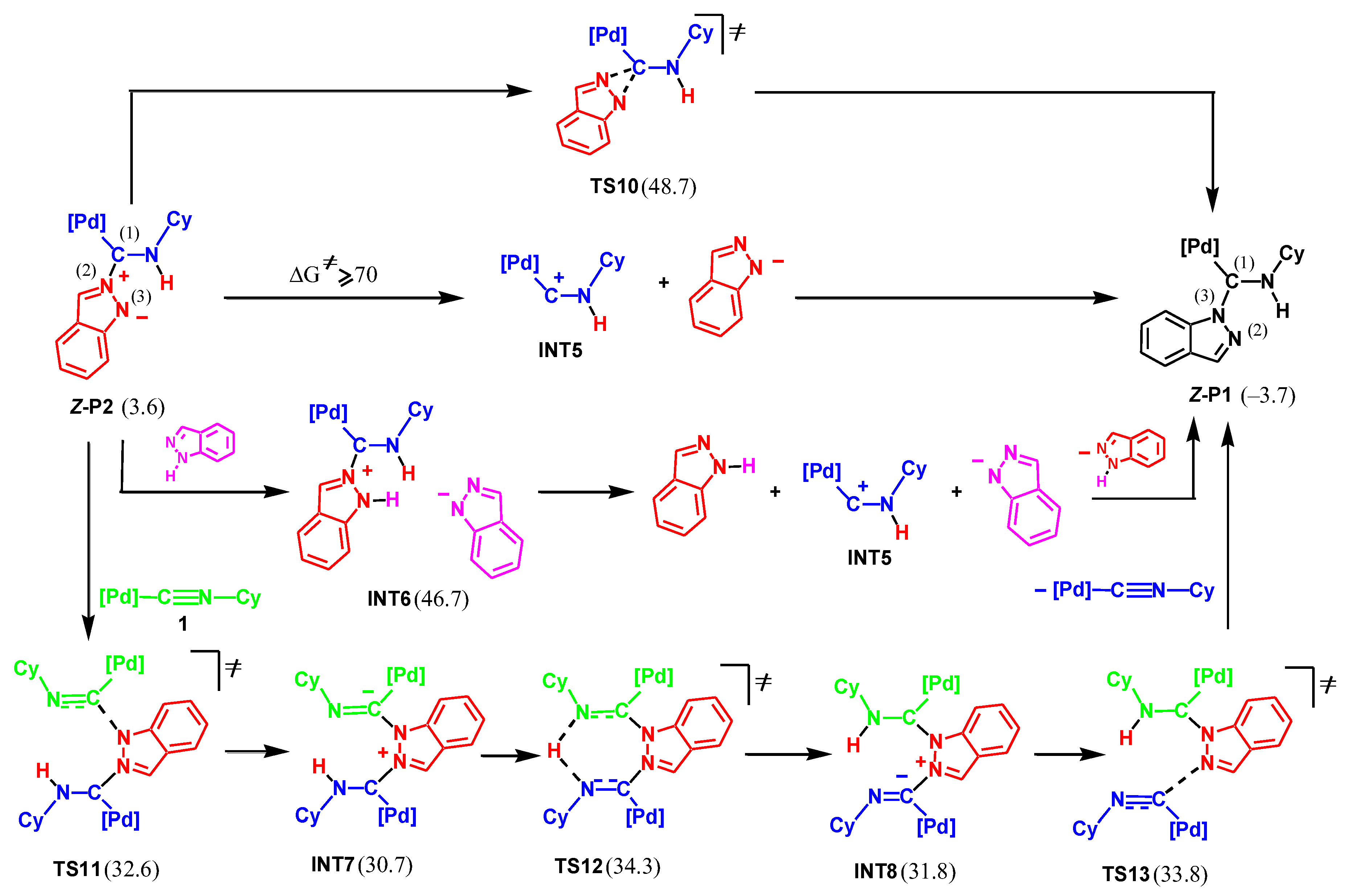
Figure 1.
Energy scan for the C(1)N(2) bond cleavage in Z-P2.

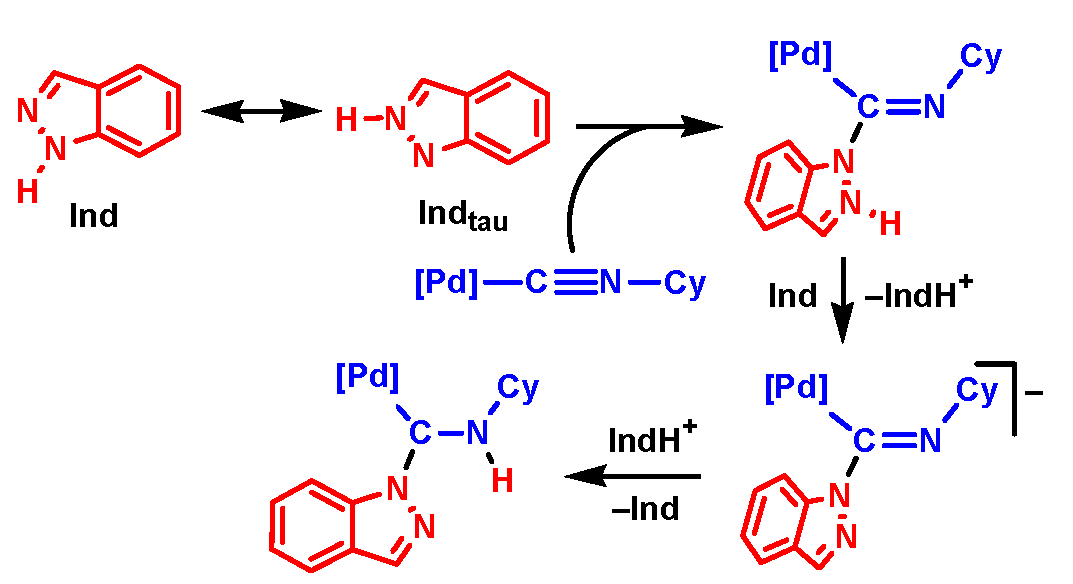
Scheme 8.
Associative mechanism of NA of indazole to 1 based on the less stable tautomeric form of Ind (Mechanism III, Gibbs free energies are indicated in kcal/mol relative to 1 + Ind, final product is boxed, only the Z-isomeric pathway is shown).
Scheme 8.
Associative mechanism of NA of indazole to 1 based on the less stable tautomeric form of Ind (Mechanism III, Gibbs free energies are indicated in kcal/mol relative to 1 + Ind, final product is boxed, only the Z-isomeric pathway is shown).
Scheme 9.
Mechanisms of NA of indazole to 1 based on the coordinated indazole (Gibbs free energies are indicated in kcal/mol relative to 1 + Ind).
Scheme 9.
Mechanisms of NA of indazole to 1 based on the coordinated indazole (Gibbs free energies are indicated in kcal/mol relative to 1 + Ind).
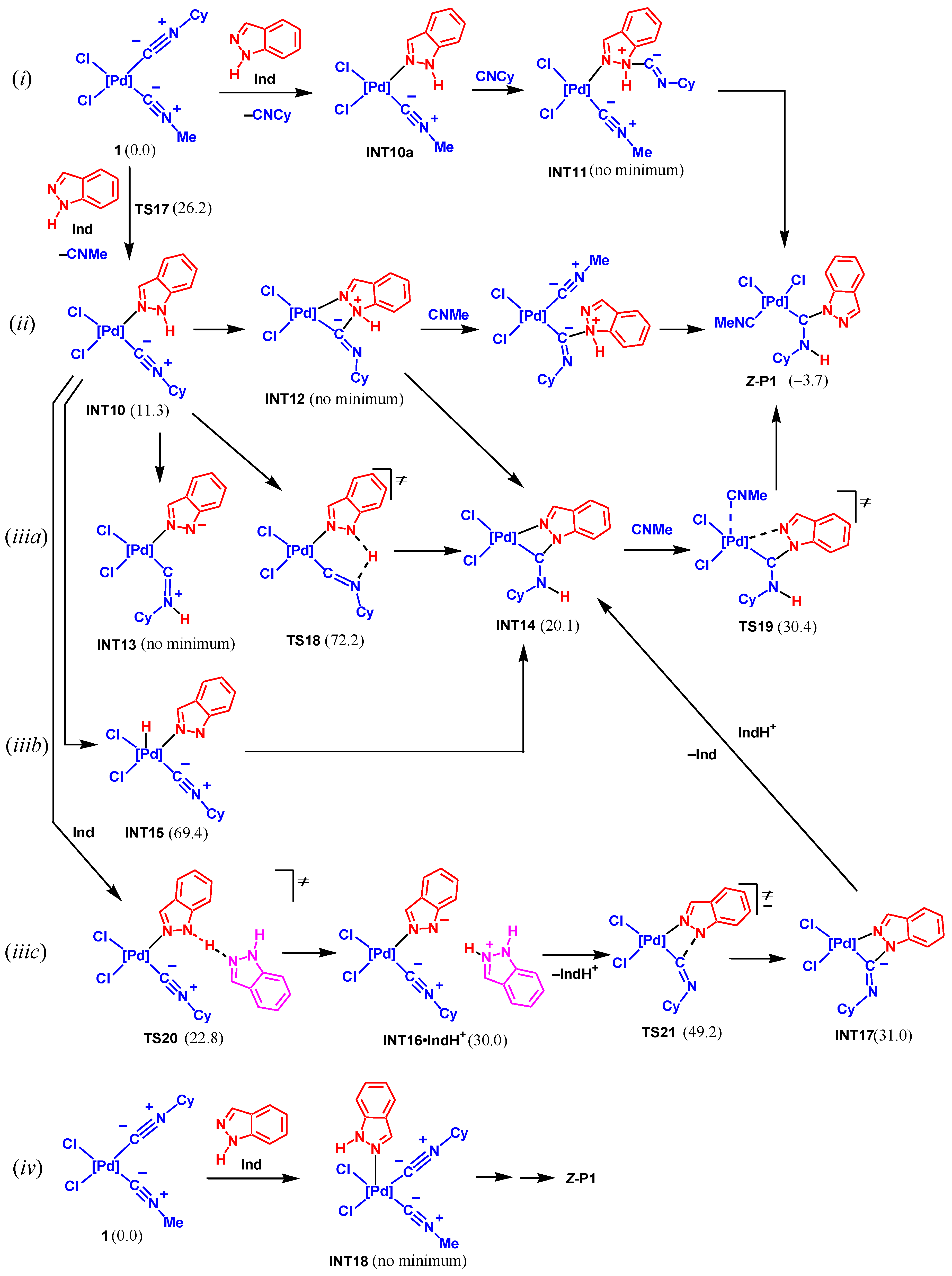
Figure 2.
Schematic depictions of the fragments considered for the Pd–C interaction in Z-P1 (a) and TS14 (b) and for the PdC–NInd interaction in TS14 (c). Analogous fragmentation schemes were used for 1 (a), Z-P2 (a), and TS6 (b,c).
Figure 2.
Schematic depictions of the fragments considered for the Pd–C interaction in Z-P1 (a) and TS14 (b) and for the PdC–NInd interaction in TS14 (c). Analogous fragmentation schemes were used for 1 (a), Z-P2 (a), and TS6 (b,c).

Figure 3.
Crystal orbital overlap populations (COOPs) for the Pd–C interaction in 1 (a), TS6 (b), TS14 (c), Z-P2 (d), and Z-P1 (e). Bonding/antibonding interactions correspond to the positive/negative peaks, respectively. Solid and dotted vertical lines correspond to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively.
Figure 3.
Crystal orbital overlap populations (COOPs) for the Pd–C interaction in 1 (a), TS6 (b), TS14 (c), Z-P2 (d), and Z-P1 (e). Bonding/antibonding interactions correspond to the positive/negative peaks, respectively. Solid and dotted vertical lines correspond to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively.

Figure 4.
COOPs for the PdC–N interaction in TS14 (a) and TS6 (b). Solid and dotted vertical lines correspond to the HOMO and LUMO, respectively.
Figure 4.
COOPs for the PdC–N interaction in TS14 (a) and TS6 (b). Solid and dotted vertical lines correspond to the HOMO and LUMO, respectively.

Figure 5.
Molecular orbital diagrams for 1 (A), Z-P1 (B), and Z-P2 (C) for the Pd ← C (σ type) (left) and Pd → C (π type) (right) interactions. Corresponding orbitals populations (in e) are given in parentheses. Isosurface values are 0.05 e1/2/Å3/2. Gray bands indicate the overall participation of the virtual orbitals with populations other than 0.00.
Figure 5.
Molecular orbital diagrams for 1 (A), Z-P1 (B), and Z-P2 (C) for the Pd ← C (σ type) (left) and Pd → C (π type) (right) interactions. Corresponding orbitals populations (in e) are given in parentheses. Isosurface values are 0.05 e1/2/Å3/2. Gray bands indicate the overall participation of the virtual orbitals with populations other than 0.00.


Figure 6.
Molecular orbital diagrams for the PdC ← N (σ-type) interaction in TS14 (a) and TS6 (b). Corresponding orbitals populations (in e) are given in parentheses. Isosurface values are 0.05 e1/2/Å3/2.
Figure 6.
Molecular orbital diagrams for the PdC ← N (σ-type) interaction in TS14 (a) and TS6 (b). Corresponding orbitals populations (in e) are given in parentheses. Isosurface values are 0.05 e1/2/Å3/2.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Casella, G.; Casarin, M.; Kukushkin, V.Y.; Kuznetsov, M.L. Reaction between Indazole and Pd-Bound Isocyanides—A Theoretical Mechanistic Study. Molecules 2018, 23, 2942. https://doi.org/10.3390/molecules23112942
AMA Style
Casella G, Casarin M, Kukushkin VY, Kuznetsov ML. Reaction between Indazole and Pd-Bound Isocyanides—A Theoretical Mechanistic Study. Molecules. 2018; 23(11):2942. https://doi.org/10.3390/molecules23112942
Chicago/Turabian StyleCasella, Girolamo, Maurizio Casarin, Vadim Yu. Kukushkin, and Maxim L. Kuznetsov. 2018. "Reaction between Indazole and Pd-Bound Isocyanides—A Theoretical Mechanistic Study" Molecules 23, no. 11: 2942. https://doi.org/10.3390/molecules23112942