
Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides
by
 ,
,
Jing Tan
1, 
Jun-Jie Guo
1,*,
Ming-Yu Yin
1,
Huan Wang
1,
Wen-Pan Dong
2,
Jie Zeng
1 and
Shi-Liang Zhou
2 1
Research Institute of Tropical Forestry, Chinese Academy of Forestry, Guangzhou 510520, China
2
State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing 100093, China
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(11), 2963; https://doi.org/10.3390/molecules23112963
Submission received: 30 September 2018
/
Revised: 9 November 2018
/
Accepted: 11 November 2018
/
Published: 13 November 2018
(This article belongs to the Special Issue Discovery, Design, Synthesis, and Application of Nucleoside/Nucleotides)
Abstract
:Betula alnoides is a fast-growing valuable indigenous tree species with multiple uses in the tropical and warm subtropical regions in South-East Asia and southern China. It has been proved to be tetraploid in most parts of its distribution in China. In the present study, next generation sequencing (NGS) technology was applied to develop numerous SSR markers for B. alnoides, and 64,376 contig sequences of 106,452 clean reads containing 164,357 candidate SSR loci were obtained. Among the derived SSR repeats, mono-nucleotide was the main type (77.05%), followed by di- (10.18%), tetra- (6.12%), tri- (3.56%), penta- (2.14%) and hexa-nucleotide (0.95%). The short nucleotide sequence repeats accounted for 90.79%. Among the 291 repeat motifs, AG/CT (46.33%) and AT/AT (44.15%) were the most common di-nucleotide repeats, while AAT/ATT (48.98%) was the most common tri-nucleotide repeats. A total of 2549 primer sets were designed from the identified putative SSR regions of which 900 were randomly selected for evaluation of amplification successfulness and detection of polymorphism if amplified successfully. Three hundred and ten polymorphic markers were obtained through testing with 24 individuals from B. alnoides natural forest in Jingxi County, Guangxi, China. The number of alleles (NA) of each marker ranged from 2 to 19 with a mean of 5.14. The observed (HO) and expected (HE) heterozygosities varied from 0.04 to 1.00 and 0.04 to 0.92 with their means being 0.64 and 0.57, respectively. Shannon-Wiener diversity index (I) ranged from 0.10 to 2.68 with a mean of 1.12. Cross-species transferability was further examined for 96 pairs of SSR primers randomly selected, and it was found that 48.96–84.38% of the primer pairs could successfully amplify each of six related Betula species. The obtained SSR markers can be used to study population genetics and molecular marker assisted breeding, particularly genome-wide association study of these species in the future.
1. Introduction
Betula alnoides Buch. Ham. ex D. Don (Betulaceae) is a fast-growing valuable indigenous tree species with multiple uses in the tropical and warm subtropical regions in South-East Asia and southern China. In China, its main natural distribution is in Yunnan, Guangxi, and Guizhou provinces [1]. The wood has beautiful texture, moderate density, low crack and deformation rate, and easy processing, making it suitable for floor timber and high-grade furniture making, high-class interior decoration and overlaid veneer [2]. The bark has anti-inflammatory [3], weight-loss and lipid-lowering properties [4,5]. In addition, B. alnoides forests have high ecological values in water conservation, maintenance of biodiversity and soil fertility, and carbon sequestration [6]. In recent decades, more than 150,000 hectares of plantations have been established in Yunnan, Guangxi, Guangdong, and Fujian provinces of China [7]. However, the majority of these plantations are established with seedlings of poor genetic quality, resulting in large variation for some traits such as diameter at breast height, tree height, stem form, wood density, insect resistance, and medicinal ingredients. Conventional quantitative breeding methods may take a long time to obtain elite varieties whereas molecular marker assisted breeding can greatly accelerate the process.
Molecular marker technology is an important tool for genomic studies, and the polymorphism of markers can reflect the same and discrepant features among individuals or populations at a certain level [8,9,10]. Simple sequence repeat (SSR) markers are widely used in population genetics and molecular marker assisted breeding, such as genome-wide association study (GWAS) due to codominant character, high polymorphism, wide distribution in the genome, neutral selection, abundant genetic information, easy detection, good reproducibility, and less demand for DNA templates [11,12,13,14,15]. However, the development of SSR markers is usually limited in species with little information on their genomic sequence. The advent of next generation sequencing (NGS) technology has overcome this restriction, and greatly promoted the genome sequencing of a species [16,17]. NGS has three advantages: (1) it can measure hundreds of thousands to millions of DNA sequences at a time; (2) NGS makes it possible to carry out a detailed analysis of the transcriptome and genome of a species, wherefore it is also known as deep sequencing; and (3) it is cost effectively compared with the first generation sequencing technology [18,19,20,21]. Therefore, NGS technology is increasingly being applied for the development of SSR markers.
B. alnoides has been proved to be tetraploid (2n = 4x = 56) in most parts of its distribution in China [22]. In some previous studies, allozyme, RAPD and AFLP markers were applied to assess the genetic diversity and population genetic structure of B. alnoides [23,24,25]. Guo et al. [26] developed 19 microsatellite markers of this species, and Sui et al. [27] adopted four SSR primers of B. platyphylla to study genetic diversity of B. alnoides. However, a small number of markers cannot meet the demand for molecular breeding like GWAS. At present, the analysis of the genetic basis of important traits has become a hot topic. GWAS overcomes some limitations of traditional gene mapping methods, and improves accuracy of gene-trait association and possibility of discovering new genes [28]. However, GWAS needs a large number of genetic markers, such as SSR [29] and SNP (Single Nucleotide Polymorphisms) [30]. Considering the limited information on B. alnoides genome as mentioned above, developing a large amount of SSR markers would be an efficient way to solve this problem.
In the present study, DNA-seq of B. alnoides was carried out to produce contig sequences containing SSR loci through NGS in order to: (1) analyze the distribution and characteristics of SSR loci in the genome; (2) develop a large number of polymorphic SSR markers; and (3) evaluate cross-species transferability of these SSR markers. The obtained SSR markers could be useful to promote further studies on population genetics and molecular marker assisted breeding of this and related species.
2. Results and Discussion
2.1. Information of Sequencing Data
A total of 106,542 valid reads were generated using 1/6 of a run on the GS FLX plus platform. The raw sequencing dataset for B. alnoides was deposited into the NCBI SRA database (SRR7958615). The mean read length was 435 base pair (bp). Analysis on the length distribution of valid reads showed that 501–600 bp and 1101–1700 bp intervals had the maximum and minimum numbers of reads, respectively (Figure 1).
2.2. Distribution of SSR in the Genome
A total of 64,376 contig sequences, about 60.47%, of 106,452 clean reads, contained SSR loci. Among these, 25,431 sequences contained only one SSR locus, and the other 38,945 sequences contained at least two SSR loci. In total 164,357 candidate SSR loci were obtained (Table 1). The total size of examined sequences was 106,901,537 bp, and the size of the SSR repeat lengths ranged from 10 bp to 75 bp with a mean length of 16 bp. Mono-nucleotide repeats accounted for the largest percentage (77.05%) among all derived loci, followed by di-nucleotide (10.18%) and tetra-nucleotide (6.12%) repeats (Table 1). The frequency of tri-nucleotide, penta-nucleotide and hexa-nucleotide repeats was lower, being 3.56, 2.14 and 0.95%, respectively.
Our study found thousands of times more microsatellite sequences than those obtained by using the DNA library and the probe hybridization enrichment methods [31]. For example, only 58 microsatellite-containing fragments were obtained by screening genomic libraries with probes for B. alnoides [26] while 38 microsatellite sequences were obtained for B. pendula [32]. Among the abundant mono- to hexa-nucleotide repeats from the present study, mono-, di- and tetra-nucleotides were the major genomic microsatellite nucleotide types, while tri-, penta- and hexa-nucleotides were of relatively low proportion. Similar results have also been observed in other woody plants based on the same sequencing technology, such as Prunus persica, Carica papaya and Citrus sinensis [33].
2.3. Characteristics of SSR Sequences
It was found that 106,526 mono-nucleotide repeats (84.11% of the total) fell under 9–14 repeat number counts, and only four greater than 33 (Table 1). All the other nucleotide repeats were mainly within 3–8 repeat number category, 82.37% for di-, 96.91% for tri-, and nearly 100% for tetra-, penta- and hexa-nucleotide repeats, respectively.
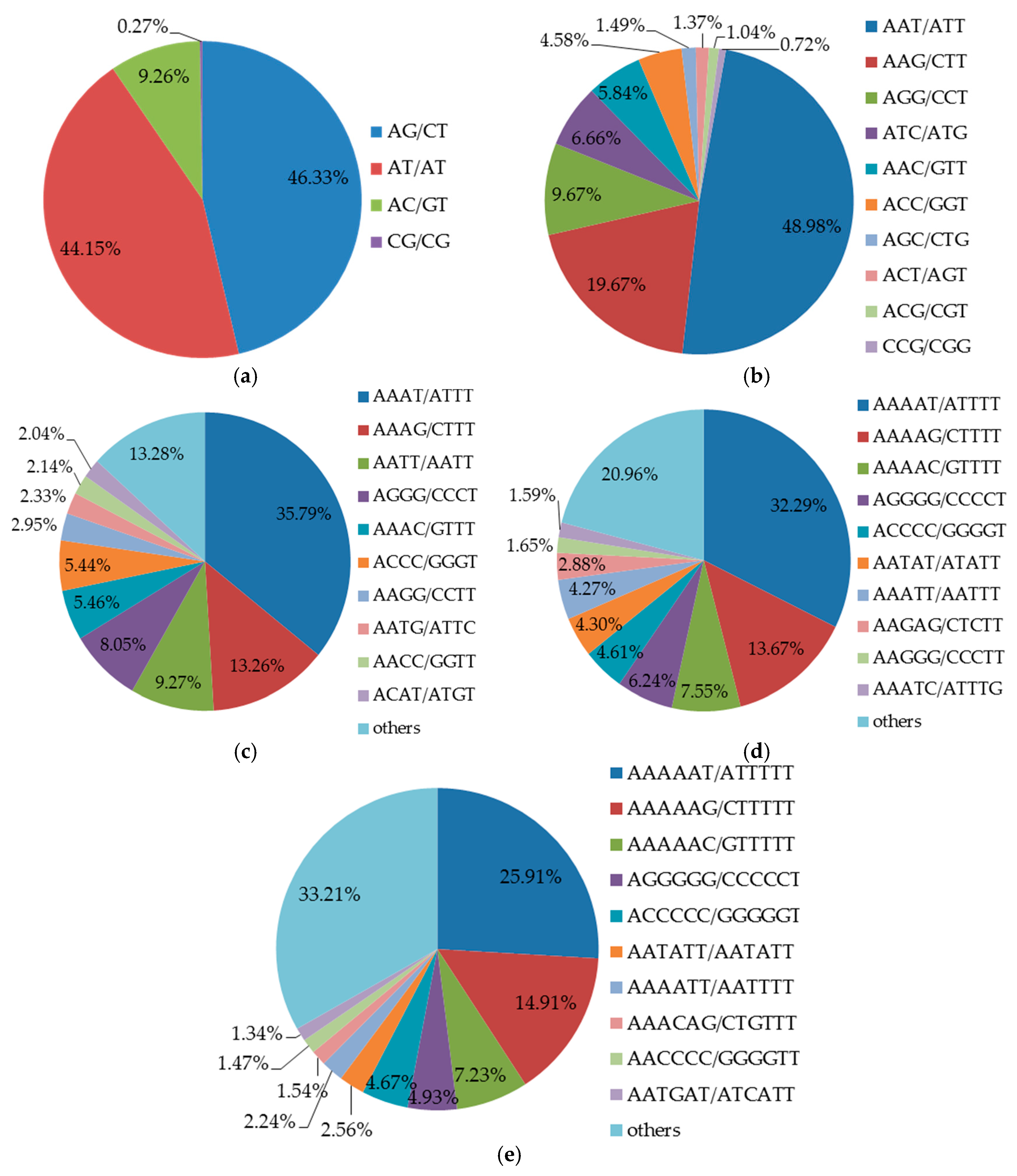
There were 291 types of repeat motifs in total. AG/CT (46.33%) and AT/AT (44.15%) were the most common repeat motifs in four types of di-nucleotide, whereas CG/CG accounted for only 0.27% (Figure 2a). These results are in accordance with previous studies reported by Gupta et al. [34] and Powell et al. [35]. In addition, the GC motif of lower frequency may be associated with cytosine methylation, which can be converted into a thymidine by deamination [36]. Among the ten types of repeat motifs in tri-nucleotide found in the present study (Figure 1b), the highest frequency repeat motif was AAT/ATT (48.98%), and the lowest was CCG/CGG (0.72%, Figure 2b). The main repeat motifs of tri-nucleotide were different from other plant species such as maize (CCG/GGC), rice (AGG/TCC) and barley (CCG/GGC) [37]. Repeat motifs AAAT/ATTT (60.85%), AAAAT/ATTTT (32.29%) and AAAAAT/ATTTTT (25.91%) observed in the present study were the most frequent in tetra-, penta- and hexa-nucleotide, respectively (Figure 2c–e).
2.4. Primer Screening and SSR Markers Polymorphism Detecting
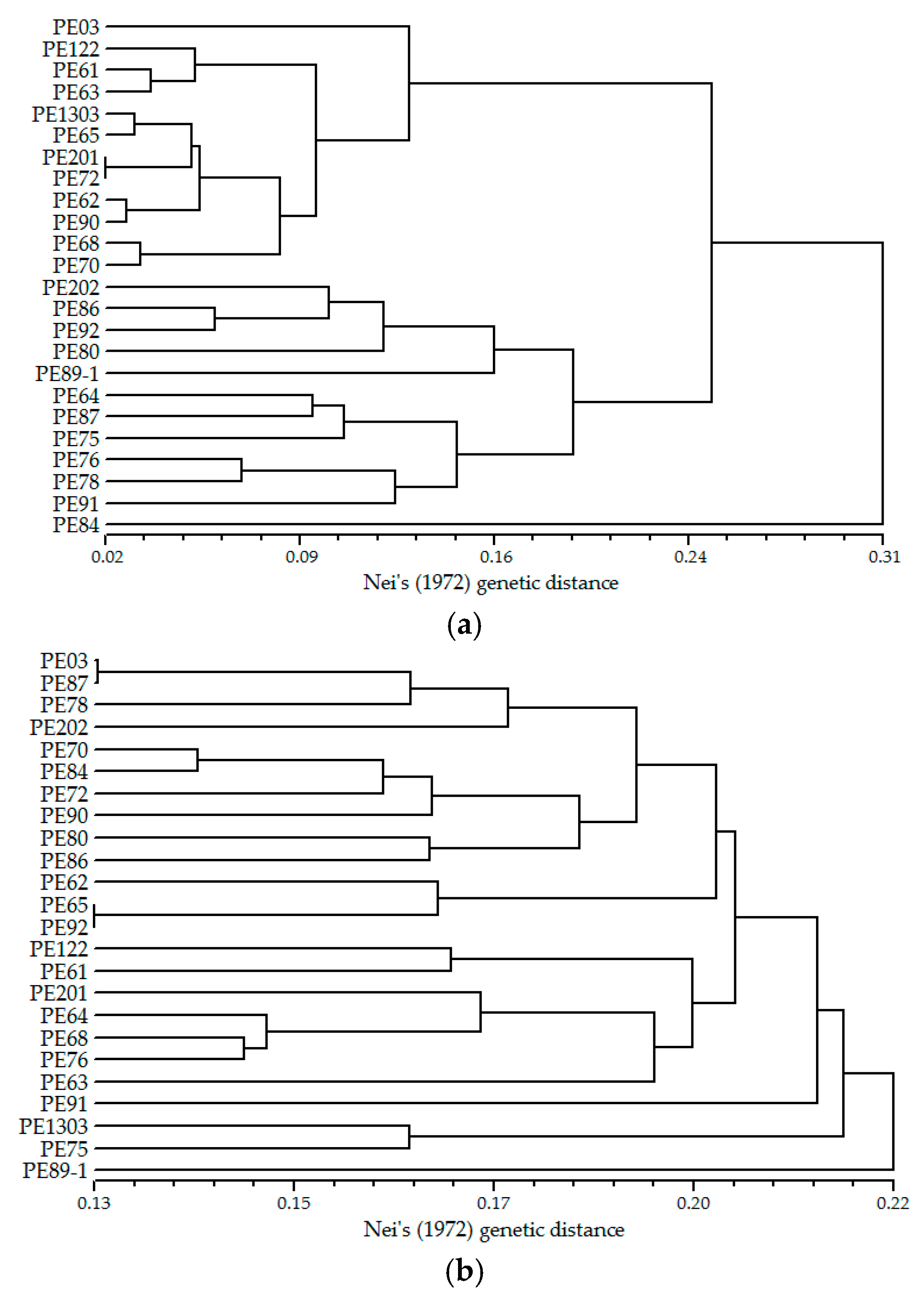
In total 2549 pairs of SSR primers were designed from all candidate SSR loci, of which 138 pairs were excluded due to the occurrence of compound formation in repeat regions of relevant SSR sequences. The majority of nucleotide repeats were di-nucleotide (75.53%), followed by tri-nucleotide (20.24%). A total of 900 pairs of primers were randomly selected for evaluating the successfulness of amplification, among them di-, tri-, tetra-, penta- and hexa-nucleotide repeats accounted for 74.11%, 21.00%, 3.33%, 0.67%, and 0.89%, respectively, and 580 pairs of primers (64.44%) were amplified successfully with clear and distinguishable microsatellite bands. The optimum annealing temperature of these SSR primers was 60 °C as determined by a temperature trial with three individuals. Eventually, 310 primers were shown to detect polymorphisms in 24 samples from a natural B. alnoides forest in Jingxi County, Guangxi, China and are therefore designated polymorphic SSR loci. Characterizations of 310 polymorphic SSR loci are presented in Table S1, and the remaining 270 pairs generated monomorphic markers or fixed heterozygosity in 24 samples or had weak fluorescence signals in amplified products of most samples (Table S2). The fragment length of the amplified products was from 105 bp to 306 bp, which was within the expectation range (100–400 bp). None of the 310 SSR markers developed in the present study was the same as the 19 pairs developed in a previous study [26]. When we tested the usefulness of the 19 pairs developed by Guo et al. [26] for our 24 B. alnoides samples, it was found that all of them were identified as polymorphic loci (Table S3). Based on the previously reported SSR markers (Figure 3a) and the SSR markers developed in the present study (Figure 3b), a phylogenetic relationship was constructed between the 24 individuals of B. alnoides. It could be seen that the larger number of markers resulted in very different phylogenetic relationships between the 24 samples.
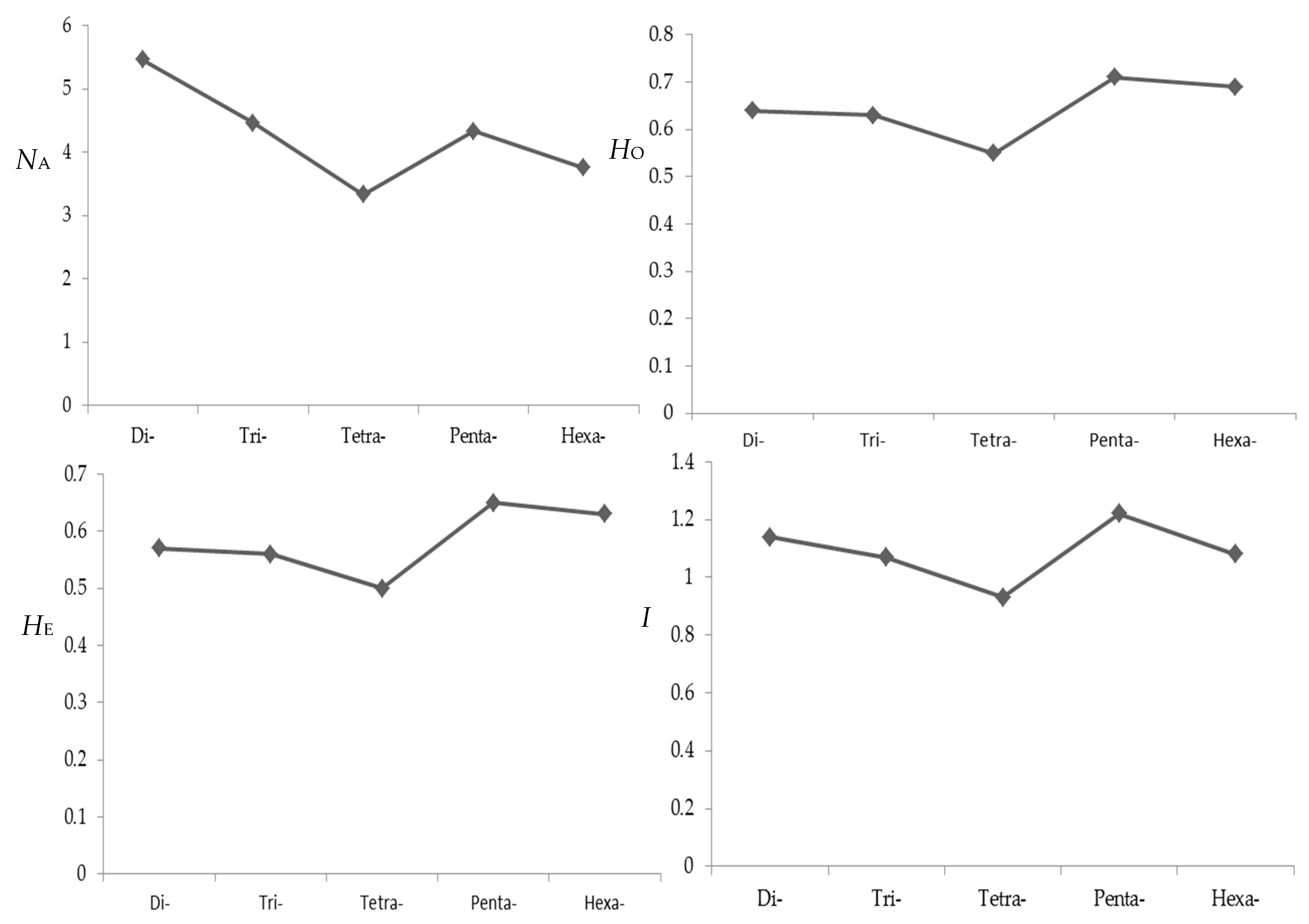
The genetic variation revealed by all polymorphic SSR loci for 24 individuals is also demonstrated in Table S1. The number of alleles (NA) of each locus ranged from 2 to 19 with a mean of 5.14, and 86.45% of loci had 2 to 8 alleles. The observed (HO) and expected (HE) heterozygosities of each locus varied from 0.04 to 1.00 (mean 0.64) and 0.04 to 0.92 (mean 0.57) respectively. The Shannon–Wiener diversity index (I) ranged from 0.10 to 2.68 with a mean of 1.12. The parameters of polymorphism decreased as nucleotide repeat varying from di- to tetra-nucleotides (Figure 4). However, this trend could not be seen for penta- and hexa-nucleotides due perhaps to their less occupancy. As a whole, the percentage of polymorphic loci with di-nucleotide repeats was larger than that with other nucleotide repeats. It was indicated that a selection of di-nucleotide repeats might be more effective to screen out polymorphic microsatellite markers, which is similar to the results of a previous study on B. platyphylla [38].
2.5. Cross-Species Transferability
Ninety six pairs of SSR primers were randomly selected to examine cross-species transferability among six related species in the genus Betula. The results showed that 48.96% of these SSR primers could be amplified in B. platyphylla, 73.96% in B. austro-sinensis, 82.29% in B. cylindrostachya, 78.13% in B. fujianensis, 79.17% in B. hainanensis, and 84.38% in B. luminifera (Table S4). These results thus indicated that these SSR primers had a high rate of transferability among the six species. High rate of transferability between species in the same genus has also been verified by Wang et al. [39] who discovered that 86.10% of SSR primer pairs developed from other Prunus species was transferable to P. virginiana. This is because the transferability of SSR primers across species is determined by the conservativeness of flanking sequences of microsatellite and their evolutionary stability [40]. The genomic differences between the related species are relatively small, making SSR primers highly transferable across specie. Thus, a large number of available primer pairs can be used in the genetic studies and molecular assistant breeding for the species in the genus Betula.
3. Materials and Methods
3.1. Plant Material and DNA Isolation
In total 24 individuals of B. alnoides were sampled from natural forest in Jingxi County, Guangxi, China. Among them, one sample was randomly selected for de novo genome sequencing, three samples were applied to screen out the successfully amplified SSR primers and optimum annealing temperatures, and all 24 individuals were used to evaluate polymorphism of the developed SSR loci. Six species in the genus Betula (Table 2) were used to assess cross-species transferability of these SSR loci. Fresh leaves were collected from each individual and dried separately with silica gel. Total genomic DNA was then extracted by modified CTAB method [41] and stored at −20 °C. The concentration and purity of DNA were determined by NanoDrop 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA).
3.2. Genome Sequencing, SSR Finding and Survey
De novo genome sequencing for B. alnoides was conducted using 1/6 of a run on the Roche 454 GS FLX + platform (454 Life Sciences, Roche Company, Branford, CT, USA) according to the process of Guo et al. [42]. For each sequence of raw data, the quality-control processes were conducted with the software Qiime (version 1.17, http://qiime.org/), which included: (1) the sequencing adapters were trimmed; (2) low-quality bases were trimmed with the index of the Q20 bases percent more than 90%; and (3) the ambiguous bases were deleted.
The MISA software was used to search SSR with following conditions: (1) the repeat number of the mono-nucleotide was at least ten; (2) the repeat number of the di-nucleotide was at least five; (3) the repeat number of the tri-nucleotide was at least four; and (4) the repeat numbers of the te-, penta-, and hexa-nucleotide were at least three. The characteristics of SSR were analyzed at the whole genome level.
3.3. SSR Primer Design and Screening
Primers were designed by Primer 3 with the following parameters: primer length = 18–30 bp; GC = 40–70%; temperature = 55–60 °C; product size range = 100–400 bp; and without secondary structure and dimer. SSR primers were synthesized by Sangon Biotech Co., Ltd., (Shanghai, China).
The optimum annealing temperature of SSR primers was screened out from 56 °C, 58 °C and 60 °C. The PCR reaction mixture (10 μL) contained 50 ng of DNA template, 150 μM dNTPs, 2.0 μM MgCl2, 0.5 μM forward and reverse primers, 1× PCR buffer (Tiangen Biotech Ltd., Beijing, China), and 0.04 U/μL of Taq DNA polymerase (Tiangen Biotech Ltd., Beijing, China). PCR was performed on the PCR system (Applied Biosystems Veriti) according to the following program: 4 min for initial denaturation at 94 °C; 31 cycles of 30 s for denaturation at 94 °C, 30 s at annealing temperature and 30 s at 72 °C; and 10 min at 72 °C. The amplified products were stored at 4 °C, and detected by 1% agarose gel electrophoresis. Successful amplification was judged by clear and distinguishable microsatellite bands.
3.4. SSR Markers Polymorphism Detection
Polymorphism of SSR loci was detected with the M13-tailed primers method [43]. SSR forward primers were synthesized and labeled with M13 sequence (5′-CACGACGTTGTAAAACGAC-3′) at 5′ end, and the reverse primers were not labeled. The M13 primer was labeled with a fluorescent dye FAM, NED, VIC, or ROX. The PCR was performed with a reaction mixture (10 μL) containing 50 ng of DNA template, 150 μM dNTPs, 2.0 μM MgCl2, 0.5 μM fluorescent forward M13-labeled primers and reverse primers, 1× PCR buffer (Tiangen Biotech Ltd., Beijing, China), and 0.04 U/μL of Taq DNA polymerase (Tiangen Biotech Ltd., Beijing, China). The PCR program was used as mentioned above.
The PCR products were detected using 3730 XL automatic sequencer (ABI Co., Foster, CA, USA). The genotyping of products was performed using GeneMarker V2.2.0 [44]. The number of alleles (NA) and observed heterozygosity (HO) were calculated with the results of genotyping in Excel 2010 (Microsoft Corporation, Redmond, WA, USA), the expected heterozygosity (HE) and Shannon–Wiener diversity index (I) were generated using ATETRA [45], which is specially applied for the analysis of tetraploid. The Nei’s genetic distance between the 24 samples was computed by NTSYS 2.1 [46] software. The software was also used to obtain clustering graph of 24 samples based on Nei’s genetic distance by the unweighted pair group method with arithmetic (UPGMA) method.
Supplementary Materials
The supplementary materials are available online. Table S1: 310 polymorphic SSR markers validated in 24 samples of Betula alnoides. Table S2: The PCR results for 270 SSR primer pairs in 24 samples of Betula alnoides. Table S3: Validation of 19 SSR markers developed previously in 24 samples of Betula alnoides. Table S4: Amplification rate of 96 SSR markers for Betula alnoides in six related species of genus Betula.
Author Contributions
J.-J.G., J.Z. and S.-L.Z. designed the experiment. J.T., J.-J.G., M.-Y.Y. and H.W. performed the experiments and collected the data. J.T., J.-J.G., M.-Y.Y. and W.-P.D. analyzed the data. J.T., J.-J.G., J.Z. and S.-L.Z. contributed to writing the manuscript.
Funding
This research was supported by the National Nonprofit Institute Research Grant of Chinese Academy of Forestry, China (CAFYBB2017SY019) and Forestry Science and Technology Innovation Project of Guangdong, China (2017KJCX032).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zeng, J.; Zhen, H.S.; Weng, Q.J. Geographic distributions and ecological conditions of Betula alnoides in China. For. Res. 1999, 12, 479–484, (In Chinese with English Abstract). [Google Scholar]
- Wang, C.S.; Zhao, Z.G.; Hein, S.; Zeng, J.; Schuler, J.; Guo, J.J. Effect of planting density on knot attributes and branch occlusion of Betula alnoides under natural pruning in southern China. Forests 2015, 6, 1343–1361. [Google Scholar] [CrossRef]
- Sur, T.K.; Pandit, S.; Battacharyya, D.; Kumar, C.K.A.; Lakshmi, S.M.; Chatttopadhyay, D.; Mandal, S.C. Studies on the anti-inflammatory activity of Betula alnoides bark. Phytother. Res. 2002, 16, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.D.A.; Malarvili, T.; Velavan, S. Restorative effect of Betula alnoides bark on hepatic metabolism in high fat diet fed Wistar rats. Int. J. Pharma BioSci. 2015, 6, 1281–1288. [Google Scholar]
- Velavan, S. Antioxidant activity of Betula alnoides bark extract in high fat diet fed Wistar rats. Int. J. Chemtech. Res. 2015, 7, 2391–2398. [Google Scholar]
- Zeng, J.; Guo, W.F.; Zhao, Z.G.; Weng, Q.J.; Yin, G.T.; Zhen, H.S. Domestication of Betula alnoides in China: Current Status and Perspectives. For. Res. 2006, 19, 379–384, (In Chinese with English Abstract). [Google Scholar]
- Wang, C.S.; Hein, S.; Zhao, Z.G.; Guo, J.J.; Zeng, J. Branch occlusion and discoloration of Betula alnoides, under artificial and natural pruning. For. Ecol. Manag. 2016, 375, 200–210. [Google Scholar] [CrossRef]
- Sakiyama, N.S.; Ramos, H.C.C.; Caixeta, E.T.; Pereira, M.G. Plant breeding with marker-assisted selection in Brazil. Crop Breed. Appl. Biotechnol. 2014, 14, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Zargar, S.M.; Raatz, B.; Sonah, H.; Bhat, J.A.; Dar, Z.A.; Agrawal, G.K.; Rakwal, R. Recent advances in molecular marker techniques: Insight into QTL mapping, GWAS and genomic selection in plants. J. Crop Sci. Biotechnol. 2015, 18, 293–308. [Google Scholar] [CrossRef]
- Zhao, D.W.; Yang, J.B.; Yang, S.X.; Kato, K.; Luo, J.P. Genetic diversity and domestication origin of tea plant Camellia taliensis (Theaceae) as revealed by microsatellite markers. BMC Plant Biol. 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, P.; Kang, D.; Mo, X.; Tian, E.; Hu, Y.; Huang, R. Development and characterization of High-throughput Est-based SSR markers for Pogostemon cablin using transcriptome sequencing. Molecules 2018, 23, 2014. [Google Scholar] [CrossRef] [PubMed]
- Payn, K.G.; Dvorak, W.S.; Janse, B.J.H.; Myburg, A.A. Microsatellite diversity and genetic structure of the commercially important tropical tree species Eucalyptus urophylla, endemic to seven islands in eastern Indonesia. Tree Genet. Genomes 2008, 4, 519–530. [Google Scholar] [CrossRef]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic diversity and population structure of Chinese White poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.W.; Zhao, T.T.; Ma, Q.H.; Liang, L.S.; Wang, G.X. Assessment of genetic diversity and population genetic structure of Corylus mandshurica in China using SSR markers. PLoS ONE 2015, 10, e0137528. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Zhang, M.Y.; Liu, Q.Z.; Li, L.T.; Song, Y.; Wang, L.F.; Zhang, S.L.; Wu, J. Transferability of newly developed pear SSR markers to other Rosaceae species. Plant Mol. Biol. Rep. 2013, 31, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, N.; Zhao, K.G.; Chen, Y.X.; Tang, R.J.; Chen, L.Q. Development and primer selection of EST-SSR molecular markers based on transcriptome sequencing of Chimonanthus praecox. J. Beijing For. Univ. 2013, 35, 25–32, (In Chinese with English Abstract). [Google Scholar]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X.; Li, Y. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genom. 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Singh, A.K.; Singh, S.; Singh, B.D. Next-generation sequencing (NGS) tools and impact in plant breeding. In Advances in Plant Breeding Strategies: Breeding, Biotechnology and Molecular Tools; Springer: Cham, Switzerland, 2015; pp. 563–612. [Google Scholar]
- Taheri, S.; Abdullah, T.L.; Jain, S.M.; Sahebi, M.; Azizi, P. TILLING, high-resolution melting (HRM), and next-generation sequencing (NGS) techniques in plant mutation breeding. Mol. Breed. 2017, 37, 40. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Mcallister, H.A.; Bartlett, P.R.; Buggs, R.J.A. Molecular phylogeny and genome size evolution of the genus Betula (Betulaceae). Ann. Bot. 2016, 117, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zou, Y.; Bai, J.; Zheng, H. RAPD analysis of genetic variation in natural populations of Betula alnoides from Guangxi, China. Euphytica 2003, 134, 33–41. [Google Scholar] [CrossRef]
- Zeng, J.Z.R.; Wang, S.L.; Zhou, J.Y.; Bai, J.; Zheng, H.S. Allozyme variation and population genetic structure of Betula alnoides from Guangxi, China. Biochem. Genet. 2003, 41, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Huang, S.X.; Chen, R.; Gui, R.Y. Study on genetic diversity of Betula alnoides. J. Zhejiang For. Sci. Technol. 2010, 30, 50–52, (In Chinese with English Abstract). [Google Scholar]
- Guo, J.J.; Zeng, J.; Zhou, S.L.; Zhan, Z.G. Isolation and characterization of 19 microsatellite markers in a tropical and warm-subtropical birch, Betula alnoides Buch.-Ham. Ex D. Don. Mol. Ecol. Resour. 2008, 8, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.Z.; Zhen, Y.; Qiu, Y.P.; Nie, Y.L. Genetic diversity analysis of four strains of Betula alnoides. J. Southwest For. Univ. 2017, 37, 21–25, (In Chinese with English Abstract). [Google Scholar]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.H.; Huang, C.; You, C.Y.; Li, W.; Zhao, W.X.; Shen, C.; Zhang, B.B.; Wang, H.T.; Yan, Z.H.; Dai, B.S.; et al. Genome-wide SSR-based association mapping for fiber quality in nation-wide upland cotton inbreed cultivars in China. BMC Genom. 2016, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yan, J.; Zhao, J.; Song, W.; Zhang, X.; Xiao, Y.; Zheng, Y. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 2012, 196, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Csencsics, D.; Brodbeck, S.; Holderegger, R. Cost-effective, species-specific microsatellite development for the endangered dwarf bulrush (Typha minima) using next-generation sequencing technology. J. Hered. 2010, 101, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Kkm, K.; Pekkinen, M.; Varvio, S. Twenty-three microsatellite primer pairs for Betula pendula (Betulaceae). Mol. Ecol. Resour. 2004, 4, 471–473. [Google Scholar]
- Shi, J.; Dai, X.; Chen, Y.; Chen, J.; Shi, J.; Yin, T. Discovery and experimental analysis of microsatellites in an oil woody plant Camellia chekiangoleosa. Plant Syst. Evol. 2013, 299, 1387–1393. [Google Scholar] [CrossRef]
- Gupta, P.K.; Balyan, H.S.; Sharma, P.C.; Ramesh, B. Microsatellites in plants: A new class of molecular markers. Curr. Sci. India 1996, 70, 45–54. [Google Scholar]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Schorderet, D.F.; Gartler, S.M. Analysis of CpG suppression in methylated and nonmethylated species. Proc. Natl. Acad. Sci. USA 1992, 89, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Kantety, R.V.; La, R.M.; Matthews, D.E.; Sorrells, M.E. Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol. Biol. 2002, 48, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, S.J.; Liu, H.J.; Zhou, B.; Wang, X.; Jiang, T. Development of SSR markers and genetic diversity in white birch (Betula platyphylla). PLoS ONE 2015, 10, e0129758. [Google Scholar]
- Wang, H.X.; Walla, J.A.; Zhong, S.B.; Huang, D.Q.; Dai, W.H. Development and cross-species genera transferability of microsatellite markers discovered using 454 genome sequencing in chokecherry (Prunus virginiana L.). Plant Cell Rep. 2012, 31, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Baird, R.E.; Wadl, P.A.; Allen, T.; McNeill, D.; Wang, X.; Moulton, J.K.; Rinehart, T.A.; Abbas, H.K.; Shier, T.; Trigiano, R.N. Variability of united states isolates of Macrophomina phaseolina based on simple sequence repeats and cross genus transferability to related genera within Botryosphaeriaceae. Mycopathologia 2010, 170, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zou, Y.P.; Bai, J.Y.; Zheng, H.S. Preparation of Total DNA from “Recalcitrant Plant Taxa”. Acta Bot. Sin. 2002, 44, 694–697. [Google Scholar]
- Guo, J.J.; Shang, S.B.; Wang, C.S.; Zhao, Z.G.; Zeng, J. Twenty microsatellite markers for the endangered Vatica mangachapoi (Dipterocarpaceae). Appl. Plant Sci. 2017, 5, 1600134. [Google Scholar] [CrossRef] [PubMed]
- Boutinganache, I.; Raposo, M.; Raymond, M.; Deschepper, C.F. M13-tailed primers improve the readability and usability of microsatellite analyses performed with two different allele-sizing methods. Biotechniques 2001, 31, 24–28. [Google Scholar]
- Hulce, D.; Li, X.; Snyderleiby, T.; Johathan-Liu, C.S. Genemarker® genotyping software: Tools to increase the statistical power of DNA fragment analysis. J. Biomol. Tech. 2011, 22, S35. [Google Scholar]
- Puyvelde, K.V.; Geert, AV.; Triest, L. Atetra, a new software program to analyse tetraploid microsatellite data: Comparison with TETRA and TETRASAT. Mol. Ecol. Resour. 2010, 10, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Rohlf, F.J. NTSYS-pc numerical taxonomy and multivariate analysis system. Am. Stat. 2000, 41, 330. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Length distribution of valid reads sequenced on GS FLX plus platform for Betula alnoides.
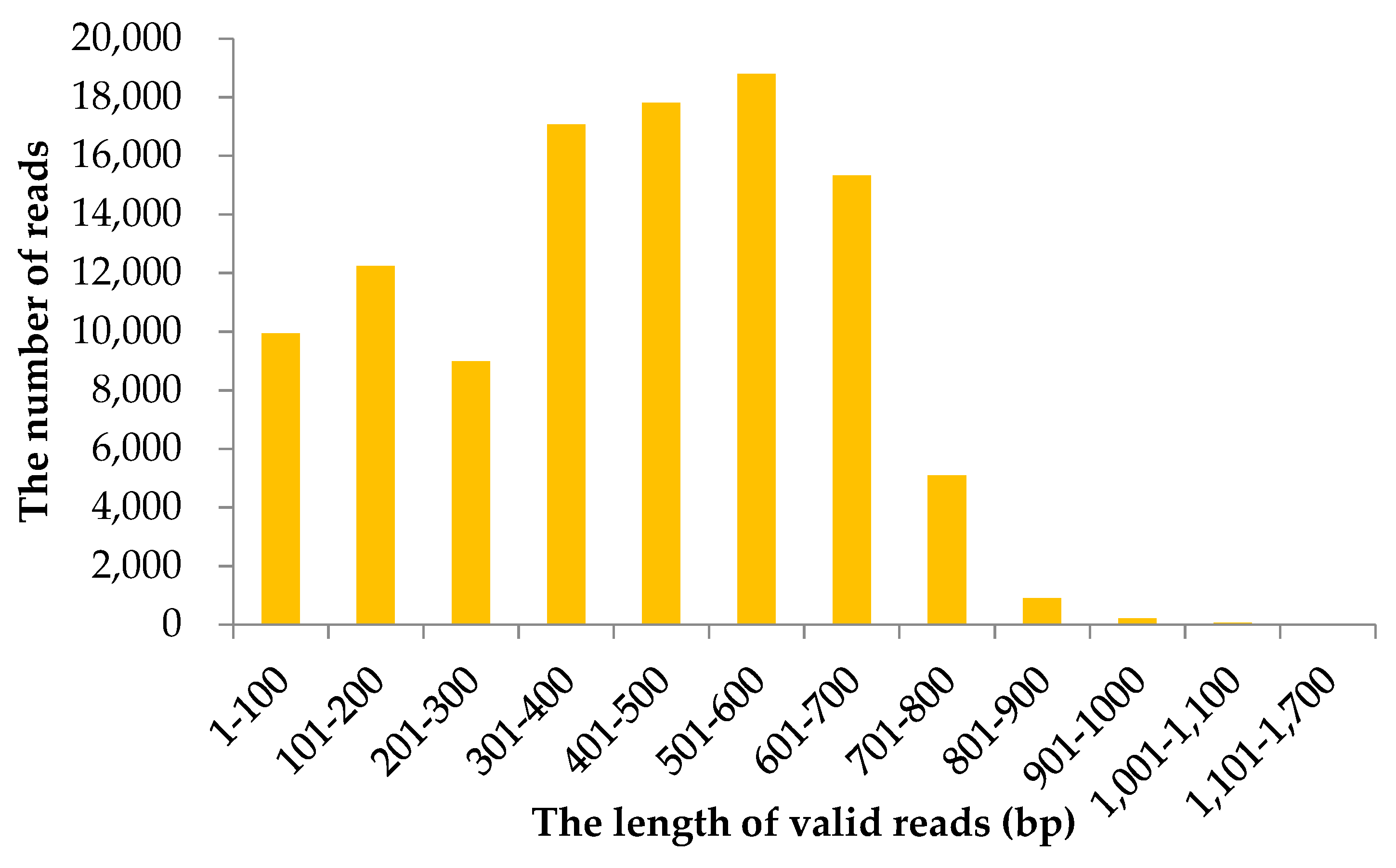
Figure 2.
Percentages of different types of repeat motifs in the genome of Betula alnoides. (a–e) refer to di-, tri-, tetra-, penta- and hexa-nucleotide repeat motifs, respectively.
Figure 2.
Percentages of different types of repeat motifs in the genome of Betula alnoides. (a–e) refer to di-, tri-, tetra-, penta- and hexa-nucleotide repeat motifs, respectively.

Figure 3.
Phylogenetic relationship between 24 Betula alnoides individuals generated from UPGMA cluster analysis based on 19 (a) and 310 SSR markers (b).
Figure 3.
Phylogenetic relationship between 24 Betula alnoides individuals generated from UPGMA cluster analysis based on 19 (a) and 310 SSR markers (b).

Figure 4.
Influence of repeat type on polymorphism level of microsatellite markers. NA, number of alleles; HO, observed heterozygosity; HE, expected heterozygosity; I, Shannon-Wiener diversity index.
Figure 4.
Influence of repeat type on polymorphism level of microsatellite markers. NA, number of alleles; HO, observed heterozygosity; HE, expected heterozygosity; I, Shannon-Wiener diversity index.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Distribution of the SSR loci in the genome of Betula alnoides.
| Repeat Type | Repeat Number | Total | Frequency (%) | |||||
|---|---|---|---|---|---|---|---|---|
| 3–8 | 9–14 | 15–20 | 21–26 | 27–32 | ≥33 | |||
| Mono-nucleotide | - | 106,526 | 16,966 | 2328 | 821 | 4 | 126,645 | 77.05 |
| Di-nucleotide | 13,777 | 2601 | 296 | 37 | 15 | - | 16,726 | 10.18 |
| Tri-nucleotide | 5675 | 163 | 15 | 3 | - | - | 5856 | 3.56 |
| Tetra-nucleotide | 10,053 | 2 | - | - | - | - | 10,055 | 6.12 |
| Penta-nucleotide | 3509 | 1 | 2 | - | - | - | 3512 | 2.14 |
| Hexa-nucleotide | 1562 | 1 | - | - | - | - | 1563 | 0.95 |
| Total | 34,576 | 109,294 | 17,279 | 2368 | 836 | 4 | 164,357 | 100 |
| Frequency (%) | 21.04 | 66.50 | 10.51 | 1.44 | 0.51 | 0.24 × 10−4 | ||
Table 2.
List of six Betula species in China used for cross-species transferability of developed SSR markers.
Table 2.
List of six Betula species in China used for cross-species transferability of developed SSR markers.
| Related Species | Collection Locality | Longitude (°E) | Latitude (°N) |
|---|---|---|---|
| B. platyphylla | Baihuashan Mountain, Beijing | 115.57 | 39.85 |
| B. austro-sinensis | Yangdongshan Shierdushui Provincial Nature Reserve, Lechang, Guangdong | 113.39 | 25.28 |
| B. cylindrostachya | Hill behind Cizhong Church, Weixi County, Yunnan | 98.92 | 28.48 |
| B. fujianensis | Luoboyan Natural Reserve, Fujian | 117.57 | 27.43 |
| B. hainanensis | Jianfengling Natural Reserve, Hainan | 109.17 | 18.73 |
| B. luminifera | Qinwang Mountain, Tianlin County, Guangxi | 106.62 | 23.91 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tan, J.; Guo, J.-J.; Yin, M.-Y.; Wang, H.; Dong, W.-P.; Zeng, J.; Zhou, S.-L. Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides. Molecules 2018, 23, 2963. https://doi.org/10.3390/molecules23112963
AMA Style
Tan J, Guo J-J, Yin M-Y, Wang H, Dong W-P, Zeng J, Zhou S-L. Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides. Molecules. 2018; 23(11):2963. https://doi.org/10.3390/molecules23112963
Chicago/Turabian StyleTan, Jing, Jun-Jie Guo, Ming-Yu Yin, Huan Wang, Wen-Pan Dong, Jie Zeng, and Shi-Liang Zhou. 2018. "Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides" Molecules 23, no. 11: 2963. https://doi.org/10.3390/molecules23112963