Development of an LC–Tandem Mass Spectrometry Method for the Quantitative Analysis of Hercynine in Human Whole Blood

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Optimization of Chromatographic Conditions
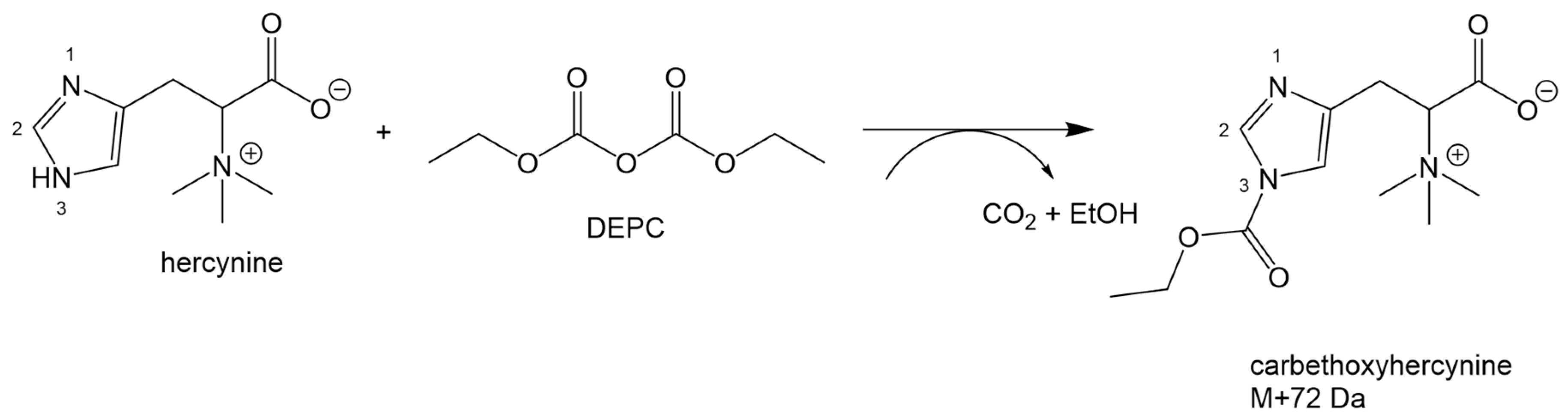
2.2. Optimization of the Derivatization Procedure
2.3. Recovery and Matrix Effect
2.4. Linearity, Calibration Range, Precision, LOD and LOQ
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Solutions
4.3. Sample Treatment and Derivatization
4.4. Participants to Study and Samples Collection
4.5. LC Equipment, Chromatographic and Mass Spectrometer Conditions
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paul, B.D.; Snyder, S.H. The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death Differ. 2010, 17, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Grundemann, D.; Harlfinger, S.; Golz, S.; Geerts, A.; Lazar, A.; Berkels, R.; Jung, N.; Rubbert, A.; Schömig, E. Discovery of the ergothioneine transporter. Proc. Natl. Acad. Sci. USA 2005, 102, 5256–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundemann, D. The ergothioneine transporter controls and indicates ergothioneine activity—A review. Prev. Med. 2012, 54, S71–S74. [Google Scholar] [CrossRef] [PubMed]
- Ey, J.; Schömig, E.; Taubert, D. Dietary sources and antioxidant effects of ergothioneine. J. Agric. Food Chem. 2007, 55, 6466–6474. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.; Bauer, T.; Surek, B.; Schömig, E.; Gründemann, D. Cyanobacteria produce high levels of ergothioneine. Food Chem. 2001, 129, 1766–1769. [Google Scholar] [CrossRef]
- Schauss, A.G.; Vértesi, A.; Endres, J.R.; Hirka, G.; Clewell, A.; Qureshi, I.; Pasics, I. Evaluation of the safety of the dietary antioxidant ergothioneine using the bacterial reverse mutation assay. Toxicology 2010, 278, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Cheah, I.K.; Drum, C.L. Ergothioneine, an adaptive antioxidant for the protection of injured tissues? A hypothesis. Biochem. Biophys. Res. Commun. 2016, 470, 245–250. [Google Scholar] [CrossRef]
- Servillo, L.; Castaldo, D.; Casale, R.; D’Onofrio, N.; Giovane, A.; Cautela, D.; Balestrieri, M.L. An uncommon redox behavior sheds light on the cellular antioxidant properties of ergothioneine. Free Radic. Biol. Med. 2015, 79, 228–236. [Google Scholar] [CrossRef]
- Servillo, L.; D’Onofrio, N.; Balestrieri, M.L. Ergothioneine Antioxidant Function: From Chemistry to Cardiovascular Therapeutic Potential. J. Cardiovasc. Pharmacol. 2017, 69, 183–191. [Google Scholar] [CrossRef]
- Reglinski, J.; Smith, W.E.; Wilson, R.; Buchanan, L.M.; McKillop, J.H.; Thomson, J.A.; Brzeski, M.; Marabani, M.; Sturrock, R.D. Clinical analysis in intact erythrocytes using 1H spin echo NMR. Clin. Chim. Acta 1991, 201, 45–57. [Google Scholar] [CrossRef]
- Taubert, D.; Lazar, A.; Grimberg, G.; Jung, N.; Rubbert, A.; Delank, K.S.; Perniok, A.; Erdmann, E.; Schömig, E. Association of rheumatoid arthritis with ergothioneine levels in red blood cells: A case control study. J. Rheumatol. 2006, 33, 2139–2145. [Google Scholar] [PubMed]
- Cheah, I.K.; Halliwell, B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta 2012, 822, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, S.; Pisanu, E.; Pintus, G.; Erre, G.L.; Pinna, G.A.; Deiana, L.; Carru, C.; Zinellu, A. Plasma L-ergothioneine measurement by high-performance liquid chromatography and capillary electrophoresis after a pre-column derivatization with 5-iodoacetamidofluorescein (5-IAF) and fluorescence detection. PLoS ONE 2013, 8, e70374. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, S.; Zinellu, A.; Pintus, G.; Pinna, G.A.; Deiana, L.; Carru, C. Quantification of L-ergothioneine in whole blood by hydrophilic interaction ultra-performance liquid chromatography and UV-detection. J. Sep. Sci. 2013, 36, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Grigat, S.; Harlfinger, S.; Pal, S.; Striebinger, R.; Golz, S.; Geerts, A.; Lazar, A.; Schömig, E.; Gründemann, D. Probing the substrate specificity of the ergothioneine transporter with methimazole, hercynine, and organic cations. Biochem. Pharmacol. 2007, 74, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, S.; Mangoni, A.A.; Forteschi, M.; Murphy, R.B.; Elliot, D.; Sotgiu, E.; Pintus, G.; Carru, C.; Zinellu, A. Identification of the main intermediate precursor of l-ergothioneine biosynthesis in human biological specimens. Molecules 2016, 21, 1298. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Vitiello, M.; Casale, R.; Servillo, L.; Giovane, A.; Balestrieri, M.L. Sirtuins in vascular diseases: Emerging roles and therapeutic potential. Biochim. Biophys. Acta 2015, 1852, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Cheah, I.K.; Tang, R.M.; Yew, T.S.; Lim, K.H.; Halliwell, B. Administration of pure ergothioneine to healthy human subjects: Uptake, metabolism, and effects on biomarkers of oxidative damage and inflammation. Antioxid. Redox Signal. 2017, 26, 193–206. [Google Scholar] [CrossRef]
- Zinellu, A.; Sotgia, S.; Usai, M.F.; Zinellu, E.; Deiana, L.; Carru, C. GSH depletion after erythrocytes acidic treatment is related to intracellular hemoglobin levels. Clin. Chim. Acta 2006, 366, 363. [Google Scholar] [CrossRef]
- Carru, C.; Zinellu, A.; Pes, G.M.; Marongiu, G.; Tadolini, B.; Deiana, L. Ultrarapid capillary electrophoresis method for the determination of reduced and oxidized glutathione in red blood cells. Electrophoresis 2002, 23, 1716–1721. [Google Scholar] [CrossRef]
- Hnízda, A.; Santrůcek, J.; Sanda, M.; Strohalm, M.; Kodícek, M. Reactivity of histidine and lysine side-chains with diethylpyrocarbonate—A method to identify surface exposed residues in proteins. J. Biochem. Biophys. Methods 2008, 70, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, S.; Zinellu, A.; Mangoni, A.A.; Pintus, G.; Attia, J.; Carru, C.; McEvoy, M. Clinical and biochemical correlates of serum l-ergothioneine concentrations in community-dwelling middle-aged and older adults. PLoS ONE 2014, 9, e84918. [Google Scholar] [CrossRef] [PubMed]
- Kumosani, T.A. L-ergothioneine level in red blood cells of healthy human males in the Western province of Saudi Arabia. Exp. Mol. Med. 2001, 33, 20–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C0 (nmol/L ± SD) | C1 (nmol/L) | C2 (nmol/L ± SD) | Recovery% |
|---|---|---|---|---|
| Spike 1 | 119.5 ± 2.4 | 440 | 558.6 ± 15.6 | 99.8 |
| Spike 2 | 119.5 ± 2.4 | 220 | 345.9 ± 9.0 | 102.9 |
| Spike 3 | 119.5 ± 2.4 | 110 | 231.4 ± 11.7 | 101.7 |
| Spike 4 | 119.5 ± 2.4 | 55 | 177.6 ± 9.8 | 105.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sotgia, S.; Murphy, R.B.; Zinellu, A.; Elliot, D.; Paliogiannis, P.; Pinna, G.A.; Carru, C.; Mangoni, A.A. Development of an LC–Tandem Mass Spectrometry Method for the Quantitative Analysis of Hercynine in Human Whole Blood. Molecules 2018, 23, 3326. https://doi.org/10.3390/molecules23123326
Sotgia S, Murphy RB, Zinellu A, Elliot D, Paliogiannis P, Pinna GA, Carru C, Mangoni AA. Development of an LC–Tandem Mass Spectrometry Method for the Quantitative Analysis of Hercynine in Human Whole Blood. Molecules. 2018; 23(12):3326. https://doi.org/10.3390/molecules23123326
Chicago/Turabian StyleSotgia, Salvatore, Rhys B. Murphy, Angelo Zinellu, David Elliot, Panagiotis Paliogiannis, Gerard Aimè Pinna, Ciriaco Carru, and Arduino A. Mangoni. 2018. "Development of an LC–Tandem Mass Spectrometry Method for the Quantitative Analysis of Hercynine in Human Whole Blood" Molecules 23, no. 12: 3326. https://doi.org/10.3390/molecules23123326