Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids †

 , ,
, ,  ,
,  , ,
, , 


Abstract
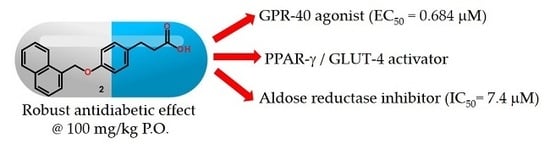
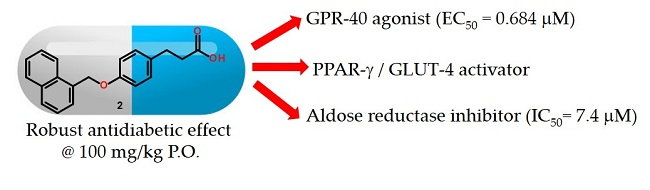
:
1. Introduction
2. Results and Discussion
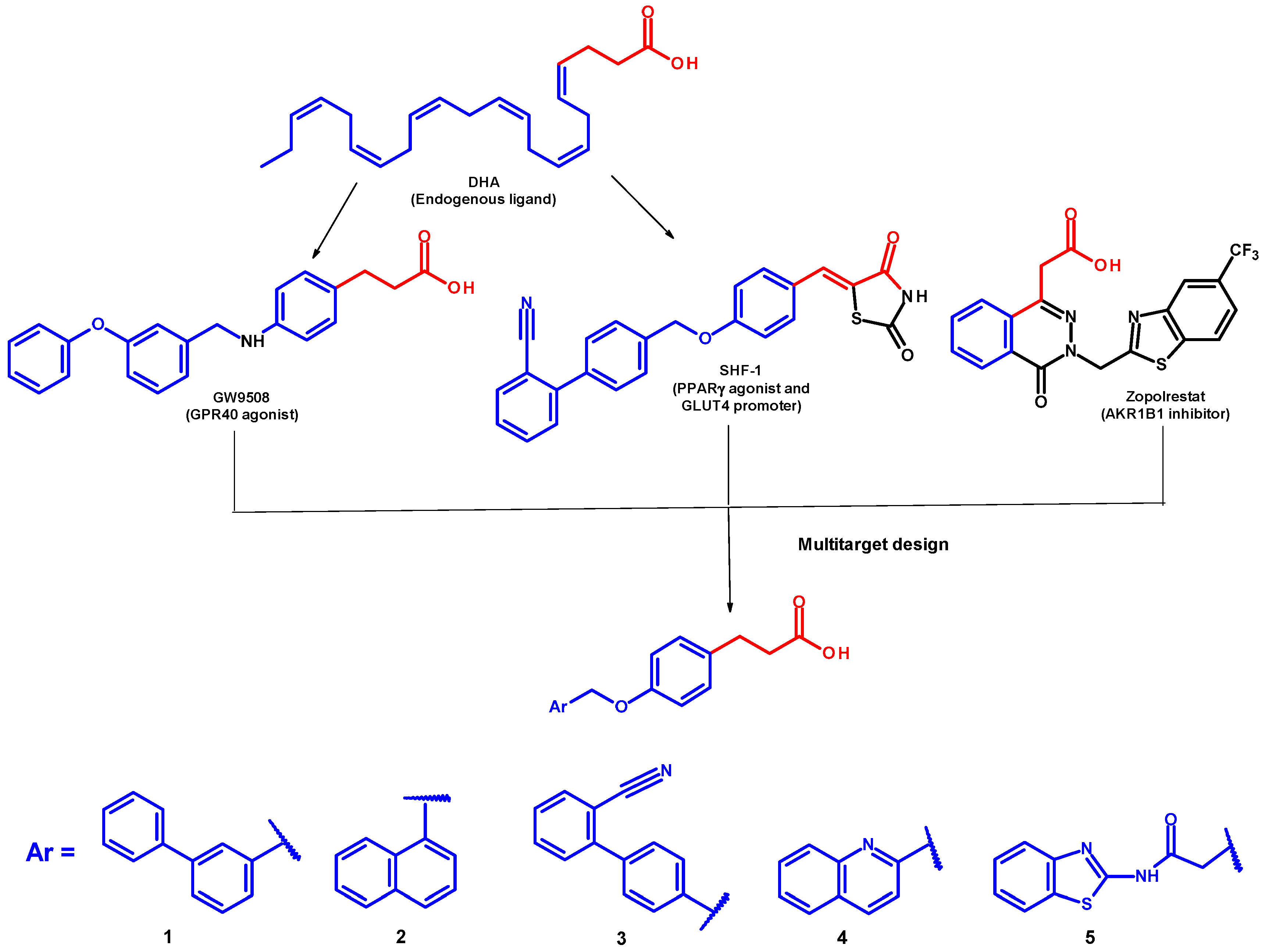
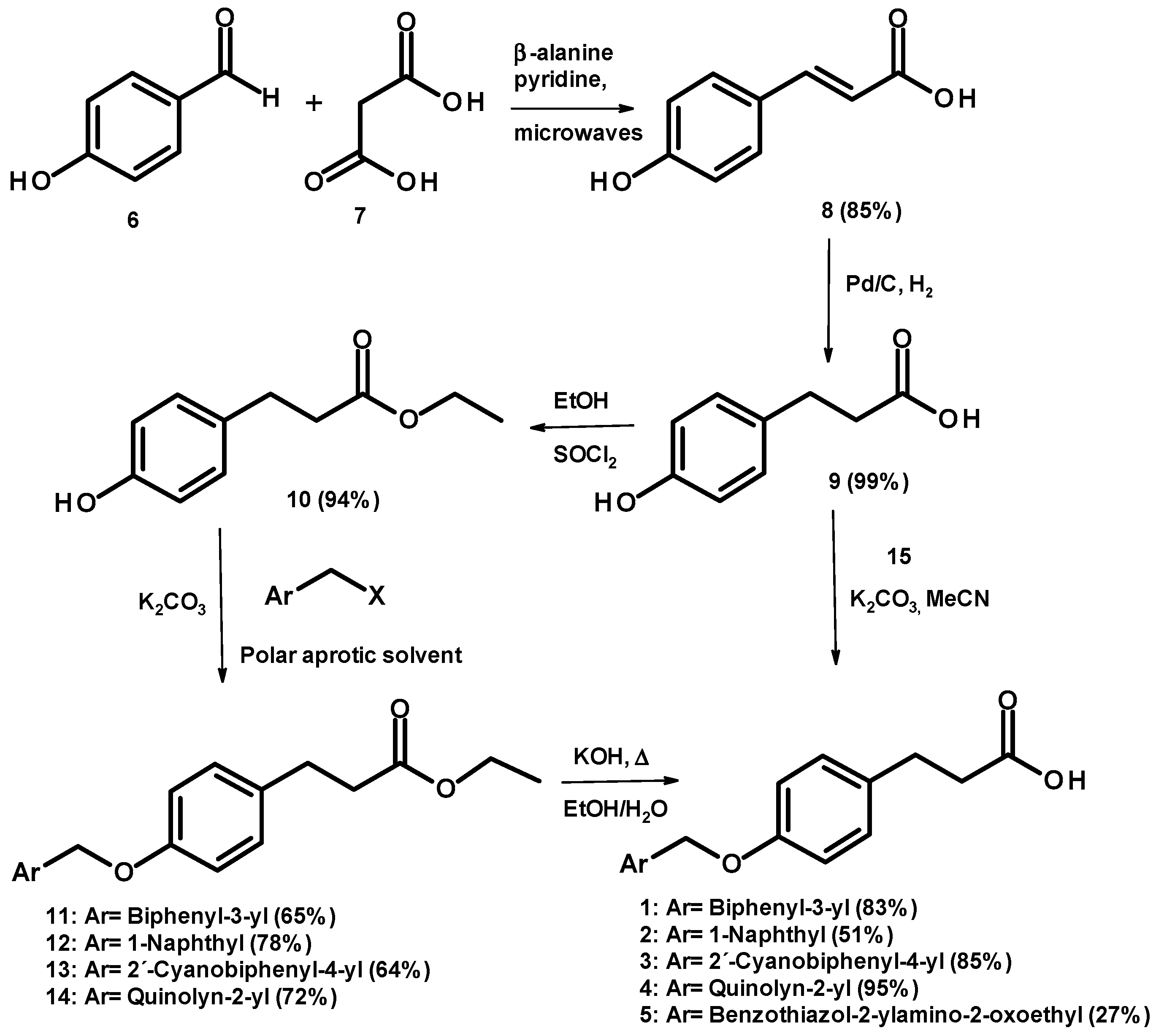
2.1. Chemical Synthesis
2.2. In Vitro GPR40 Activity
2.3. In Vitro Aldose Reductase (AKR1B1) Inhibition
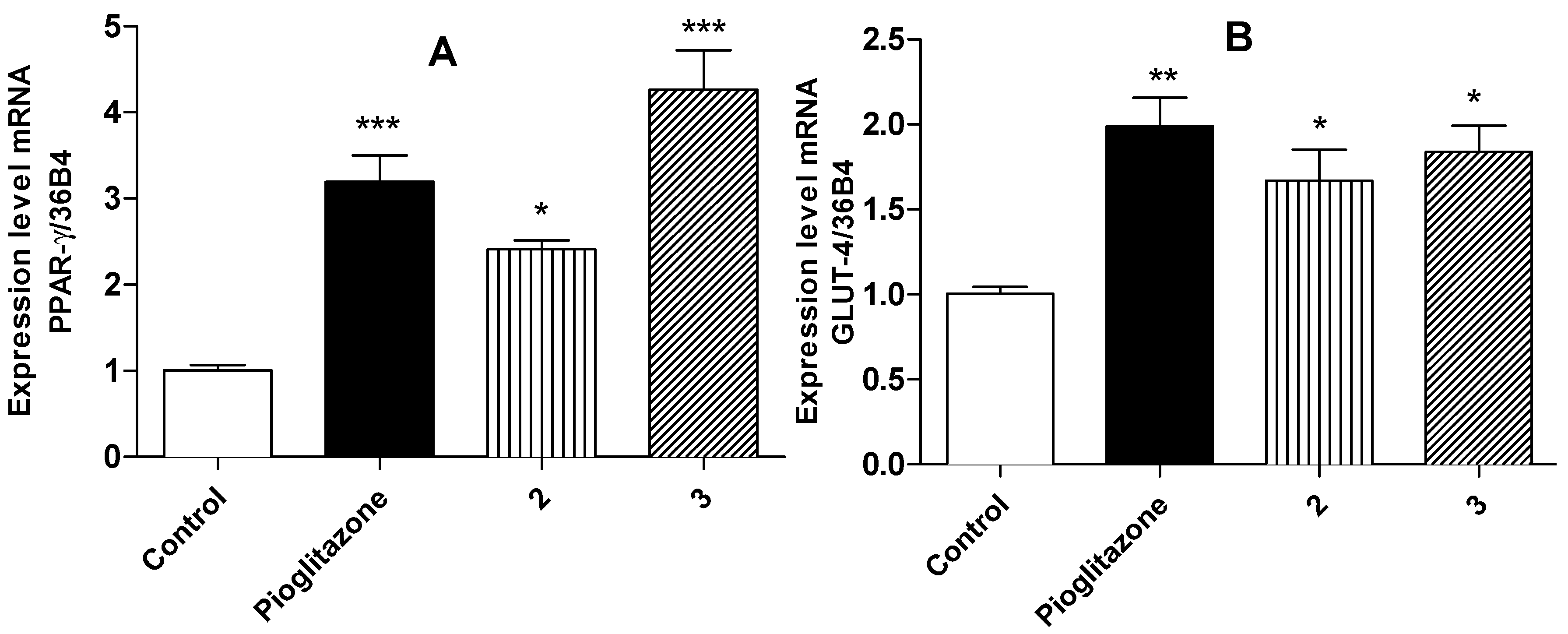
2.4. Relative Expression of PPARγ and GLUT-4
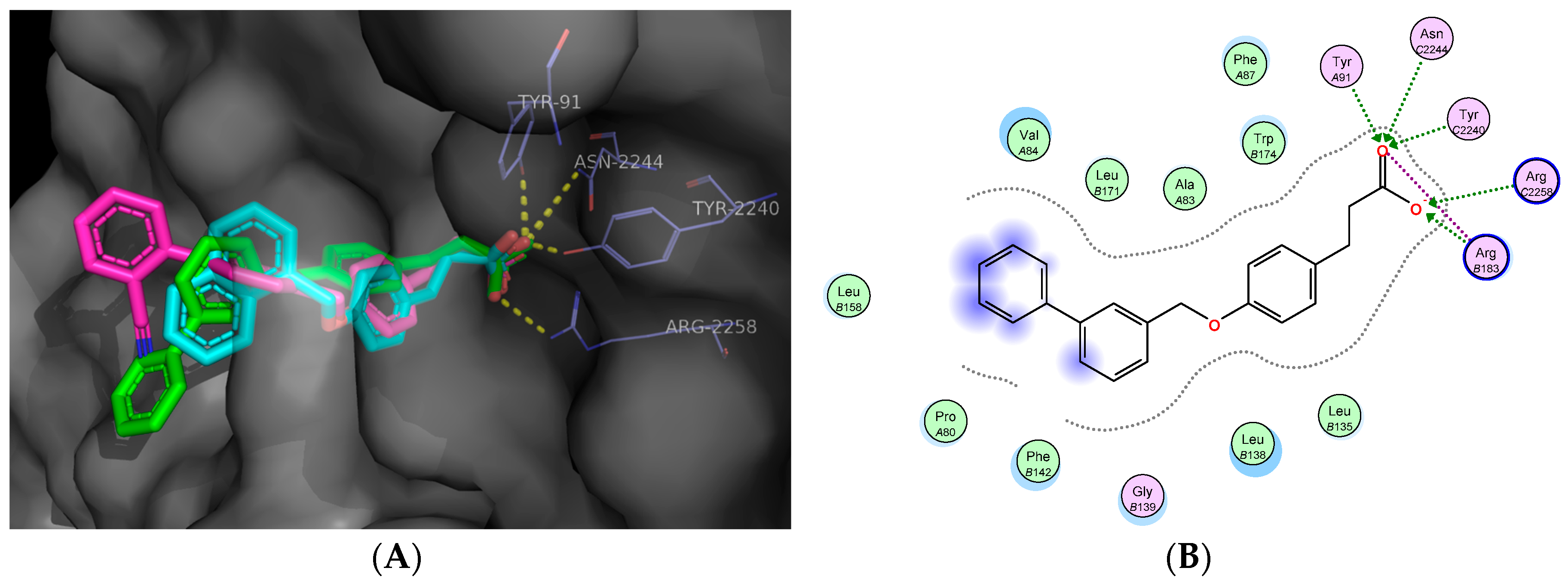
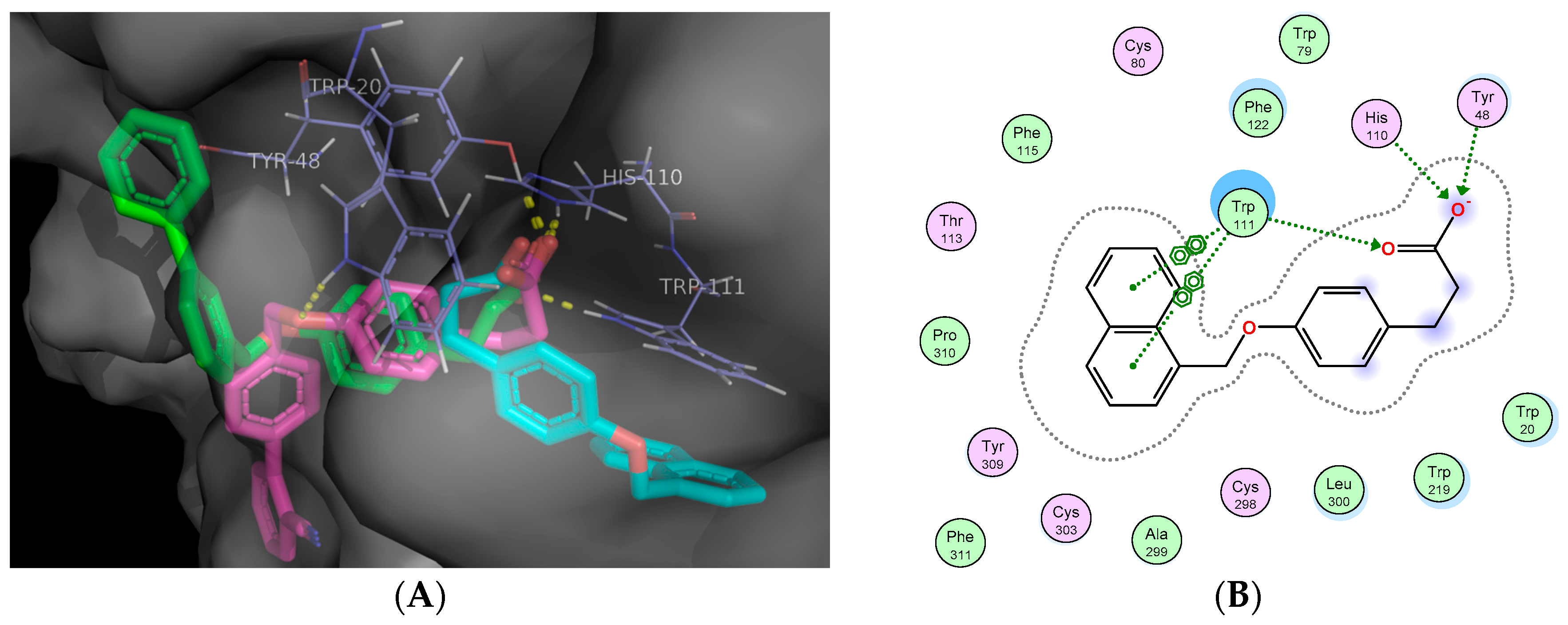
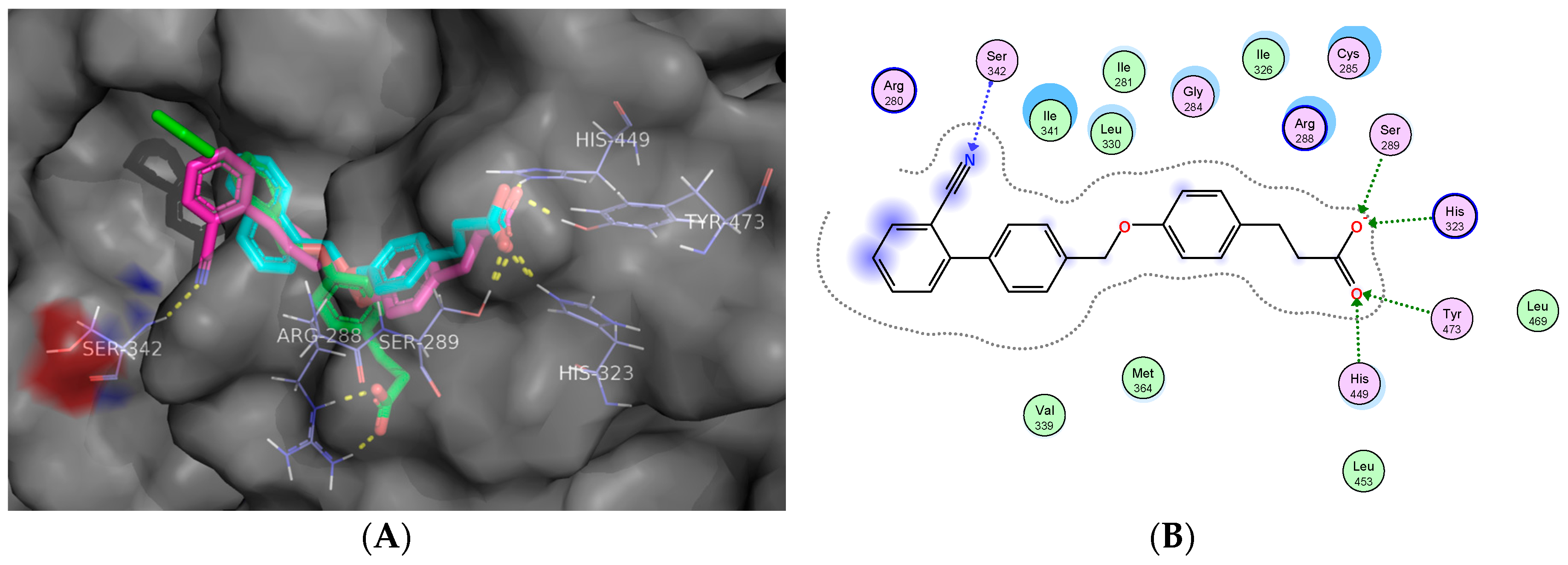
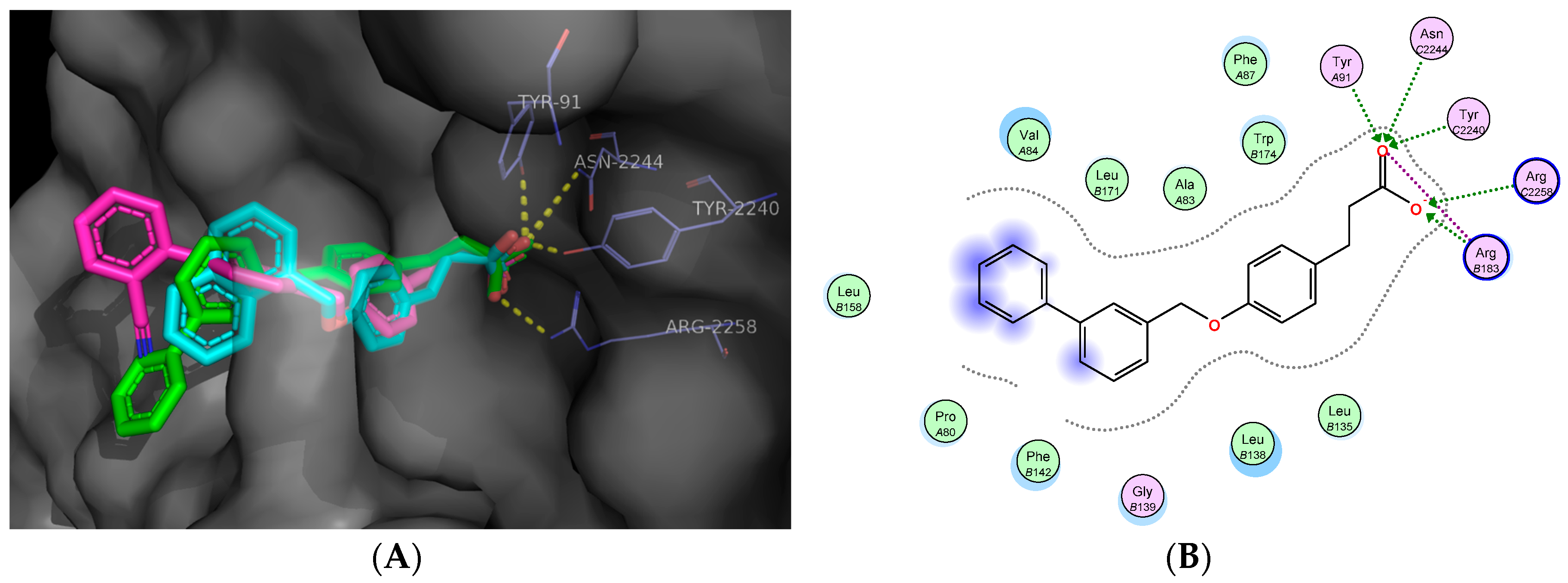
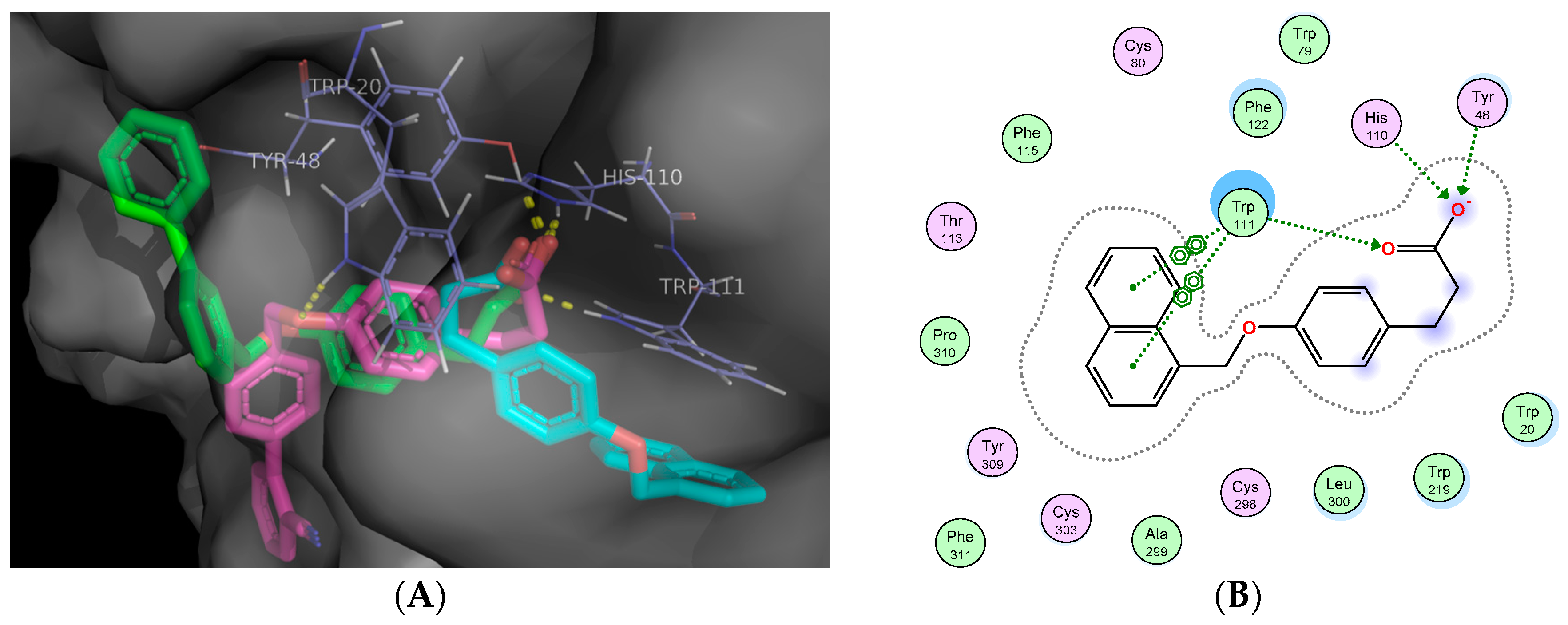
2.5. Molecular Docking Studies
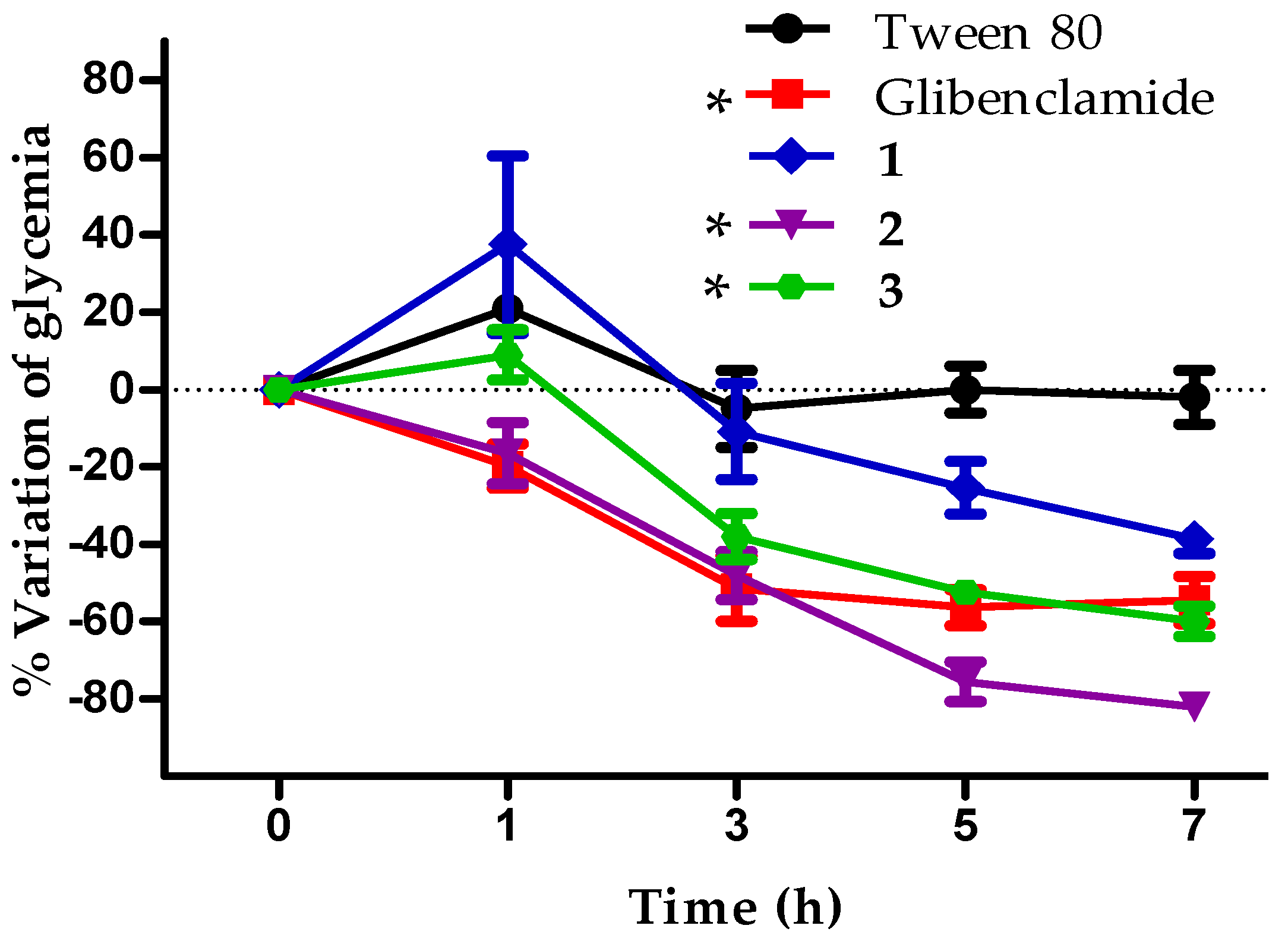
2.6. In Vivo Antidiabetic Effect of Compounds 1–3
2.7. Off-Target Toxicity Predictions
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for the Synthesis of Compounds 1–4
3.3. Synthesis of 3-{4-[2-(1,3-Benzothiazol-2-ylamino)-2 oxoethoxy]phenyl}propanoic Acid (5)
3.4. Synthesis of (2E)-3-(4-Hydroxyphenyl)acrylic Acid (8)
3.5. Synthesis of 3-(4-Hydroxyphenyl)propanoic Acid (9)
3.6. Synthesis of Ethyl 3-(4-hydroxyphenyl)propanoate (10)
3.7. General Method for the Preparation of Compounds 11–14
3.8. Synthesis of N-1,3-Benzothiazol-2-yl-2-chloroacetamide (15)
3.9. Biological Assays
3.9.1. GPR40 Agonistic Activities of Compounds 1–5
3.9.2. Aldose Reductase (AKR1B1) Inhibition Assay
3.9.3. In Vitro PPARγ and GLUT-4 Assay
3.10. In Vivo Assay
3.11. In Silico Docking Studies
Docking Validation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.; Gavin, J.R., 3rd; Aguilar, R.B. The time is right for a new classification system for diabetes: Rationale and implications of the β-cell-centric classification schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Guo, B.; Chu, W.J.; Xie, X.; Yang, Y.S.; Zhou, X.L. Design, synthesis and evaluation of potent G-protein coupled receptor 40 agonists. Chin. Chem. Lett. 2016, 27, 159–162. [Google Scholar] [CrossRef]
- Edfalk, S.; Steneberg, P.; Edlund, H. GPR40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008, 57, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.D.; Poitout, V. The fatty acid receptor FFA1/GPR40 a decade later: How much do we know? Trends Endocrinol. Metab. 2013, 24, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.G.; Dhayal, S. G-protein coupled receptors mediating long chain fatty acid signalling in the pancreatic beta-cell. Biochem. Pharmacol. 2009, 78, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, H.; Long, Y.Q. GPR40 agonists for the treatment of type 2 diabetes mellitus: The biological characteristics and the chemical space. Bioorg. Med. Chem. Lett. 2016, 26, 5603–5612. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Vázquez, G.; Torres-Gómez, H.; Hidalgo-Figueroa, S.; Ramírez- Espinosa, J.J.; Estrada-Soto, S.; Medina-Franco, J.L.; León-Rivera, I.; Alarcón-Aguilar, F.J.; Almanza-Pérez, J.C. Synthesis, in vitro and in silico studies of a PPARγ and GLUT-4 modulator with hypoglycemic effect. Bioorg. Med. Chem. Lett. 2014, 24, 4575–4579. [Google Scholar] [CrossRef] [PubMed]
- Meinke, P.T.; Wood, H.B.; Szewczyk, J.W. Nuclear hormone receptor modulators for the treatment of diabetes and dyslipidemia. Annu. Rep. Med. Chem. 2006, 41, 99–126. [Google Scholar]
- Nevin, D.K.; Lloyd, D.G.; Fayne, D. Rational targeting of peroxisome proliferating activated receptor subtypes. Curr. Med. Chem. 2011, 18, 5598–5623. [Google Scholar] [CrossRef] [PubMed]
- Sternbach, D.D. Modulators of the peroxisome proliferator-activated receptors (PPARs). Annu. Rep. Med. Chem. 2003, 38, 71–80. [Google Scholar]
- Yasmin, S.; Jayaprakash, V. Thiazolidinediones and PPAR orchestra as antidiabetic agents: From past to present. Eur. J. Med. Chem. 2016, 126, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Abate, M.; Del Corso, A.; Mura, U. Modulation of aldose reductase activity by aldose hemiacetals. Biochim. Biophys. Acta 2015, 1850, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Del Corso, A.; Cappiello, M.; Mura, U. From a dull enzyme to something else: Facts and perspectives regarding aldose reductase. Curr. Med. Chem. 2008, 15, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chung, S.S. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999, 13, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Araki, N.; Koh, N.; Nakamura, J.; Horiuchi, S.; Hotta, N. Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem. Biophys. Res. Commun. 1996, 228, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Peat, A.J.; McKeown, S.C.; Corbett, D.F.; Goetz, A.S.; Littleton, T.R.; McCoy, D.C.; Kenakin, T.P.; Andrews, J.L.; Ammala, C.; et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: Identification of agonist and antagonist small molecules. Br. J. Pharmacol. 2006, 148, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Román-Ramos, R.; Alarcón-Aguilar, J.F.; Hernández-Rosado, J.V.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Navarrete-Vázquez, G. Discovery of thiazolidine-2,4-dione/biphenylcarbonitrile hybrid as dual PPAR α/γ modulator with antidiabetic effect: In vitro, in silico and in vivo approaches. Chem. Biol. Drug Des. 2013, 81, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Awad, K.S.; Elinoff, J.M.; Dougherty, E.J.; Ferreyra, G.A.; Wang, J.Y.; Cai, R.; Sun, J.; Ptasinska, A.; Danner, R.L. G protein-coupled receptor 40 (GPR40) and peroxisome proliferator-activated receptor γ (PPARγ): An integrated two-receptor signaling pathway. J. Biol. Chem. 2015, 290, 19544–19557. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.N.; Ravinder, M.; Bagul, P.; Ravikanti, K.; Bagul, C.; Nanubolu, J.B.; Srinivas, K.; Banerjee, S.K.; Rao, V.J. Synthesis and biological evaluation of new epalrestat analogues as aldose reductase inhibitors (ARIs). Eur. J. Med. Chem. 2014, 71, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiang, X.; Luo, L.; Yang, X.; Xiao, G.; Zheng, Y.; Cao, Z.; Sang, Z.; Su, F.; Deng, Y. Multitarget drug design strategy against Alzheimer’s disease: Homoisoflavonoid Mannich base derivatives serve as acetylcholinesterase and monoamine oxidase B dual inhibitors with multifunctional properties. Bioorg. Med. Chem. 2017, 25, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Kitamura, S.; Negoro, N.; Suzuki, M.; Tsujihata, Y.; Suzuki, N.; Santou, T.; Kanzaki, N.; Harada, M.; Tanaka, Y.; et al. Design, synthesis, and biological activity of potent and orally available G protein-coupled receptor 40 agonists. J. Med. Chem. 2011, 54, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, E.; Due-Hansen, M.E.; Urban, C.; Merten, N.; Pfleiderer, M.; Karlsen, K.K.; Rasmussen, S.S.; Steensgaard, M.; Hamacher, A.; Schmidt, J.; et al. Structure-activity study of dihydrocinnamic acids and discovery of the potent FFA1 (GPR40) agonist TUG-469. ACS Med. Chem. Lett. 2010, 1, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Negoro, N.; Sasaki, S.; Mikami, S.; Ito, M.; Tsujihata, Y.; Ito, R.; Suzuki, M.; Takeuchi, K.; Suzuki, N.; Miyazaki, J.; et al. Optimization of (2,3-dihydro-1-benzofuran-3-yl)acetic acids: Discovery of a non-free fatty acid-like, highly bioavailable G protein-coupled receptor 40/free fatty acid receptor 1 agonist as a glucose-dependent insulinotropic agent. J. Med. Chem. 2012, 55, 3960–3974. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del Corso, A. L-Idose: An attractive substrate alternative to D-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Rotondo, R.; Moschini, R.; Pellegrino, M.; Cappiello, M.; Barracco, V.; Misuri, L.; Sorce, C.; Andreucci, A.; Del-Corso, A.; et al. Zolfino landrace (Phaseolus vulgaris L.) from Pratomagno: General and specific features of a functional food. Food. Nutr. Res. 2016, 60, 31792. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Li, J.; Chang, K.S.; Stenbit, A.E.; Galuska, D.; Anderson, J.E.; Zierath, J.R.; McCarter, R.J.; Charron, M.J. Metabolic adaptations in skeletal muscle overexpressing GLUT4: Effects on muscle and physical activity. FASEB J. 2001, 15, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Dia-DB, A Web Server for the Prediction of Diabetes Drugs. Available online: http://bio-hpc.ucam.edu/dia-db/ (accessed on 5 January 2017).
- Sánchez-Pérez, A.; Muñoz, A.; Peña-García, J.; den-Haan, H.; Bekas, N.; Katsikoudi, A.; Tzakos, A.G.; Pérez-Sánchez, H. DIA-DB: A Web-Accessible Database for the Prediction of Diabetes Drugs. Bioinform. Biomed. Eng. 2015, 9044, 655–663. [Google Scholar]
- Guasch, L.; Sala, E.; Valls, C.; Blay, M.; Mulero, M.; Arola, L.; Pujadas, G.; Garcia-Vallvé, S. Structural insights for the design of new PPARgamma partial agonists with high binding affinity and low transactivation activity. J. Comput. Aided Mol. Des. 2011, 25, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci. Rep. 2016, 6, 34792. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, Y.; Pan, Y.; Skiles, G.L.; Shou, M. Prediction of human drug-drug interactions from time-dependent inactivation of CYP3A4 in primary hepatocytes using a population-based simulator. Drug Metabol. Dispos. 2009, 37, 2330–2339. [Google Scholar] [CrossRef] [PubMed]
- Taboureau, O.; Jørgensen, F.S. In silico predictions of hERG channel blockers in drug discovery: From ligand-based and target-based approaches to systems chemical biology. Comb. Chem. High Throughput Screen. 2011, 14, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-U. Polypharmacology—Foe or Friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Lucio, O.; Naveja, J.J.; Vite-Caritino, H.; Prieto-Martínez, F.D.; Medina-Franco, J.L. One Drug for Multiple Targets: A Computational Perspective. J. Mex. Chem. Soc. 2016, 60, 168–181. [Google Scholar] [CrossRef]
- García-Macedo, R.; Sánchez-Muñoz, F.; Almanza-Pérez, J.C.; Duran-Reyes, G.; Alarcón-Aguilar, F.; Cruz, M. Glycine increases mRNA adiponectin and diminishes pro-inflammatory adipokines expression in 3T3-L1 cells. Eur. J. Pharmacol. 2008, 587, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Almanza-Pérez, J.C.; Alarcón-Aguilar, F.J.; Blancas-Flores, G.; Campos-Sepúlveda, A.E.; Román-Ramos, R.; García-Macedo, R.; Cruz, M. Glycine regulates inflammatory markers modifying the energetic balance through PPAR and UCP-2. Biomed. Pharmacother. 2010, 64, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Figueroa, S.; Navarrete-Vázquez, G.; Estrada-Soto, S.; Giles-Rivas, D.; Alarcón-Aguilar, F.J.; León-Rivera, I.; Giacoman-Martínez, A.; Miranda-Pérez, E.; Almanza-Pérez, J.C. Discovery of new dual PPARγ-GPR40 agonists with robust antidiabetic activity: Design, synthesis and in combo drug evaluation. Biomed. Pharmacother. 2017, 90, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Verspohl, E.J. Recommended testing in diabetes research. Planta Medica 2002, 68, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.N.; Reddy, N.M.; Patil, K.R.; Nakhate, K.T.; Ojha, S.; Patil, C.R.; Agrawal, Y.O. Challenges and issues with streptozotocin-induced diabetes—A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem. Biol. Interact. 2016, 244, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Kojima, R.; Ito, M. Strain differences in the diabetogenic activity of streptozotocin in mice. Biol. Pharm. Bull. 2006, 6, 1110–1119. [Google Scholar] [CrossRef]
- Radenkovíc, M.; Stojanović, M.; Prostran, M. Experimental diabetes induced by alloxan and streptozotocin: The current state of the art. J. Pharm. Toxicol. Methods 2016, 78, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Andrade, R.; Rodríguez-López, V.; Garduño-Ramírez, M.; Castillo-España, P.; Estrada-Soto, S. Anti-diabetic effect on alloxanized and normoglycemic rats and some pharmacological evaluations of Tournefortia hartwegiana. J. Ethnopharmacol. 2005, 101, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Lara, E.; Martínez-Conde, C.; Rosales-Ortega, E.; Ramírez-Espinosa, J.J.; Rivera-Leyva, J.C.; Centurión, D.; Carvajal, K.; Ortega-Cuellar, D.; Estrada-Soto, S.; Navarrete-Vázquez, G. Synthesis and in vitro AMPK activation of cycloalkyl/alkarylbiguanides with robust in vivo antihyperglycemic action. J. Chem. 2017, 2017, 1212609. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2016.08; Chemical Computing group ULC, 1010 Sherbooke St. West, Site #910, Montreal, QC, Canada, H3A 2R7, 2018. Available online: http://www.chemcomp.com (accessed on 30 September 2017).
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Maximal Response % (100 μM) | EC50 (μM) a |
|---|---|---|
| 1 | 98 ± 10 | 0.075 ± 0.02 |
| 2 | 103 ± 3 | 0.648 ± 0.10 |
| 3 | 105 ± 15 | 0.797 ± 0.17 |
| 4 | 84 | ≈100 |
| 5 | 9.9 | >100 |
| LA | 100 | 8.30 ± 1.7 |
| Comp. | IC50 (μM) a |
|---|---|
| 1 | 8.9 (6.5–12.1) |
| 2 | 17.4 (14.3–21.2) |
| 3 | 21.0 (12.8–34.3) |
| 4 | 31.7 (22.3–44.9) |
| 5 | 23.3 (19.0–25.5) |
| Sorbinil | 1.2 (0.9–1.5) |
| Compd | LD50 (mg/kg) | Probability of Inhibition (IC50 or Ki < 10 μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| Mouse | Rat | CYP450 Isoform | hERG | |||||
| i.p. | p.o. | i.p. | p.o. | 3A4 | 2D6 | 1A2 | ||
| 1 | 320 | 1400 | 560 | 3800 | 0.08 | 0.05 | 0.17 | 0.10 |
| 2 | 530 | 2000 | 460 | 2700 | 0.07 | 0.05 | 0.15 | 0.04 |
| 3 | 580 | 1990 | 1200 | 4000 | 0.07 | 0.02 | 0.04 | 0.13 |
| Pioglitazone | 440 | 1900 | 400 | 1100 | 0.22 | 0.03 | 0.08 | 0.21 |
| Sorbinil | 420 | 680 | 190 | 1600 | 0.02 | 0.02 | 0.01 | 0.10 |
| Glibenclamide | 980 | 1900 | 500 | 2400 | 0.20 | 0.08 | 0.01 | 0.60 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colín-Lozano, B.; Estrada-Soto, S.; Chávez-Silva, F.; Gutiérrez-Hernández, A.; Cerón-Romero, L.; Giacoman-Martínez, A.; Almanza-Pérez, J.C.; Hernández-Núñez, E.; Wang, Z.; Xie, X.; et al. Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids. Molecules 2018, 23, 340. https://doi.org/10.3390/molecules23020340
Colín-Lozano B, Estrada-Soto S, Chávez-Silva F, Gutiérrez-Hernández A, Cerón-Romero L, Giacoman-Martínez A, Almanza-Pérez JC, Hernández-Núñez E, Wang Z, Xie X, et al. Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids. Molecules. 2018; 23(2):340. https://doi.org/10.3390/molecules23020340
Chicago/Turabian StyleColín-Lozano, Blanca, Samuel Estrada-Soto, Fabiola Chávez-Silva, Abraham Gutiérrez-Hernández, Litzia Cerón-Romero, Abraham Giacoman-Martínez, Julio Cesar Almanza-Pérez, Emanuel Hernández-Núñez, Zhilong Wang, Xin Xie, and et al. 2018. "Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids" Molecules 23, no. 2: 340. https://doi.org/10.3390/molecules23020340
APA StyleColín-Lozano, B., Estrada-Soto, S., Chávez-Silva, F., Gutiérrez-Hernández, A., Cerón-Romero, L., Giacoman-Martínez, A., Almanza-Pérez, J. C., Hernández-Núñez, E., Wang, Z., Xie, X., Cappiello, M., Balestri, F., Mura, U., & Navarrete-Vazquez, G. (2018). Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids. Molecules, 23(2), 340. https://doi.org/10.3390/molecules23020340