Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies
by
 ,
,
David Von der Ahe
1,†, 
Petra Huehnchen
1,2,3,†,
Mustafa Balkaya
4,
Sarah Peruzzaro
5,
Matthias Endres
1,2,3,6,7,8,*,† and
Wolfgang Boehmerle
1,2,3,† 1
Charité—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Klinik und Hochschulambulanz für Neurologie, Chariteplatz 1, 10117 Berlin, Germany
2
Charité—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Cluster of Excellence NeuroCure, 10117 Berlin, Germany
3
Berlin Institute of Health, Anna-Louisa-Karsch 2, 10178 Berlin, Germany
4
Burke-Cornell Medical Research Institute, White Plains, NY 10605, USA
5
Field Neurosciences Institute Laboratory for Restorative Neurology, Central Michigan University, Mt. Pleasant, MI 48859, USA
6
Charité—Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Center for Stroke Resarch Berlin, 10117 Berlin, Germany
7
German Center for Neurodegenerative Diseases (DZNE), 10117 Berlin, Germany
8
German Center for Cardiovascular Research (DZHK), Partner Site Berlin, 10117 Berlin, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2018, 23(2), 346; https://doi.org/10.3390/molecules23020346
Submission received: 15 December 2017
/
Revised: 23 January 2018
/
Accepted: 3 February 2018
/
Published: 7 February 2018
(This article belongs to the Special Issue Neuroprotective Agents)
Abstract
:Suramin is a trypan blue analogon originally developed to treat protozoan infections, which was found to have diverse antitumor effects. One of the most severe side effects in clinical trials was the development of a peripheral sensory-motor polyneuropathy. In this study, we aimed to investigate suramin-induced neuropathy with a focus on calcium (Ca2+) homeostasis as a potential pathomechanism. Adult C57Bl/6 mice treated with a single injection of 250 mg/kg bodyweight suramin developed locomotor and sensory deficits, which were confirmed by electrophysiological measurements showing a predominantly sensory axonal-demyelinating polyneuropathy. In a next step, we used cultured dorsal root ganglia neurons (DRGN) as an in vitro cell model to further investigate underlying pathomechanisms. Cell viability of DRGN was significantly decreased after 24-hour suramin treatment with a calculated IC50 of 283 µM. We detected a suramin-induced Ca2+ influx into DRGN from the extracellular space, which could be reduced with the voltage-gated calcium channel (VGCC) inhibitor nimodipine. Co-incubation of suramin and nimodipine partially improved cell viability of DRGN after suramin exposure. In summary, we describe suramin-induced neurotoxic effects on DRGN as well as potentially neuroprotective agents targeting intracellular Ca2+ dyshomeostasis.
1. Introduction
Suramin is a water-soluble polysulfonated naphtylurea. It was first synthesized in 1916 as a colorless derivate of trypan dyes for the treatment of African trypanosomiasis (reviewed by [1]). Later on it was found that suramin had an effect against the Human-Immunodeficiency Virus [2] and that non-Hodgkin lymphomas associated with this virus were sensitive to suramin treatment [3]. Further investigation revealed broad antitumor activity of suramin and a wide variety of mechanisms of action were described. Among others, it was shown that suramin inhibits several different enzymes and growth factors, which may account for the antiretroviral, antiprotozoal and antitumor activity. In different tumor entities suramin was proposed to bind to a range of growth factors such as the vascular endothelial growth factor or the epidermal growth factor [1]. Subsequently, suramin was tested in clinical trials of different metastatic tumor entities with emphasis on metastatic prostate cancer [4,5,6,7]. In line with the seemingly unspecific mode of action of suramin, the clinical application revealed several side effects such as coagulopathy, adrenal insufficiency, nephrotoxicity or hepatotoxicity [1,6]. One of the most frequently observed dose limiting side effects, however, is a toxic peripheral polyneuropathy, which occurs in 40–79% of suramin treated patients [6,8,9]. As suramin is a charged molecule and does not cross the blood brain barrier it exerts neurotoxic effects in the peripheral nervous system only [10]. Due to the potentially severe side effects, clinical use of suramin at the time of writing is limited to treatment of African trypanosomiasis and as an orphan drug for (metastatic) hormone refractory prostate cancer. Suramin-induced peripheral neuropathy most frequently shows as sensory-motor axonal neuropathy with sensory symptoms such as hypoesthesia, paresthesia, dysesthesia, hypoalgesia or pallhypesthesia [9,11]. The incidence of a demyelinating Guillian-Barré like syndrome under suramin treatment was reported less frequently [12]. Incidence and severity of suramin-induced neuropathy are influenced by plasma levels exceeding 350 µg/mL, the cumulative suramin dose and the duration of chemotherapy [12,13]. A variety of different pathomechanisms was described: The inhibition of lysosomal enzymes by suramin was proposed to cause an experimental form of gangliosidosis with intracellular lamellar inclusion bodies in neurons and Schwann cells [14,15]. Different experimental studies suggested the involvement of nerve growth factor and a binding affinity of suramin to the nerve growth factor receptor to account for the neurotoxic effect [16,17]. Furthermore, Sun et al. described that suramin leads to a disturbance of the intracellular calcium (Ca2+) homoeostasis in dorsal root ganglia neurons (DRGN) [18]. Intriguingly, other chemotherapeutic agents such as paclitaxel and salinomycin were also found to cause a peripheral neuropathy via a disturbance of intracellular Ca2+ levels [19,20]. Thus, it appears that the disruption of the intracellular Ca2+ homeostasis might be a common pathomechanism in chemotherapy-induced polyneuropathies. The aim of this study was thus to establish preclinical models of suramin-induced neurotoxicity in order to further elucidate the underlying pathomechanism with an emphasis on Ca2+ signaling and to identify potential molecular targets suitable for neuroprotection. We observed that suramin induces neurotoxicity in vitro and in vivo. Calcium influx into DRGN from the extracellular space seems to partially mediate suramin-induced neurotoxicity, as Nimodipine, an L-Type voltage-gated calcium channel (VGCC) inhibitor, showed limited neuroprotective effects.
2. Results
2.1. Suramin Induces a Sensory Axonal-Demyelinating Polyneuropathy in C57Bl/6 Mice
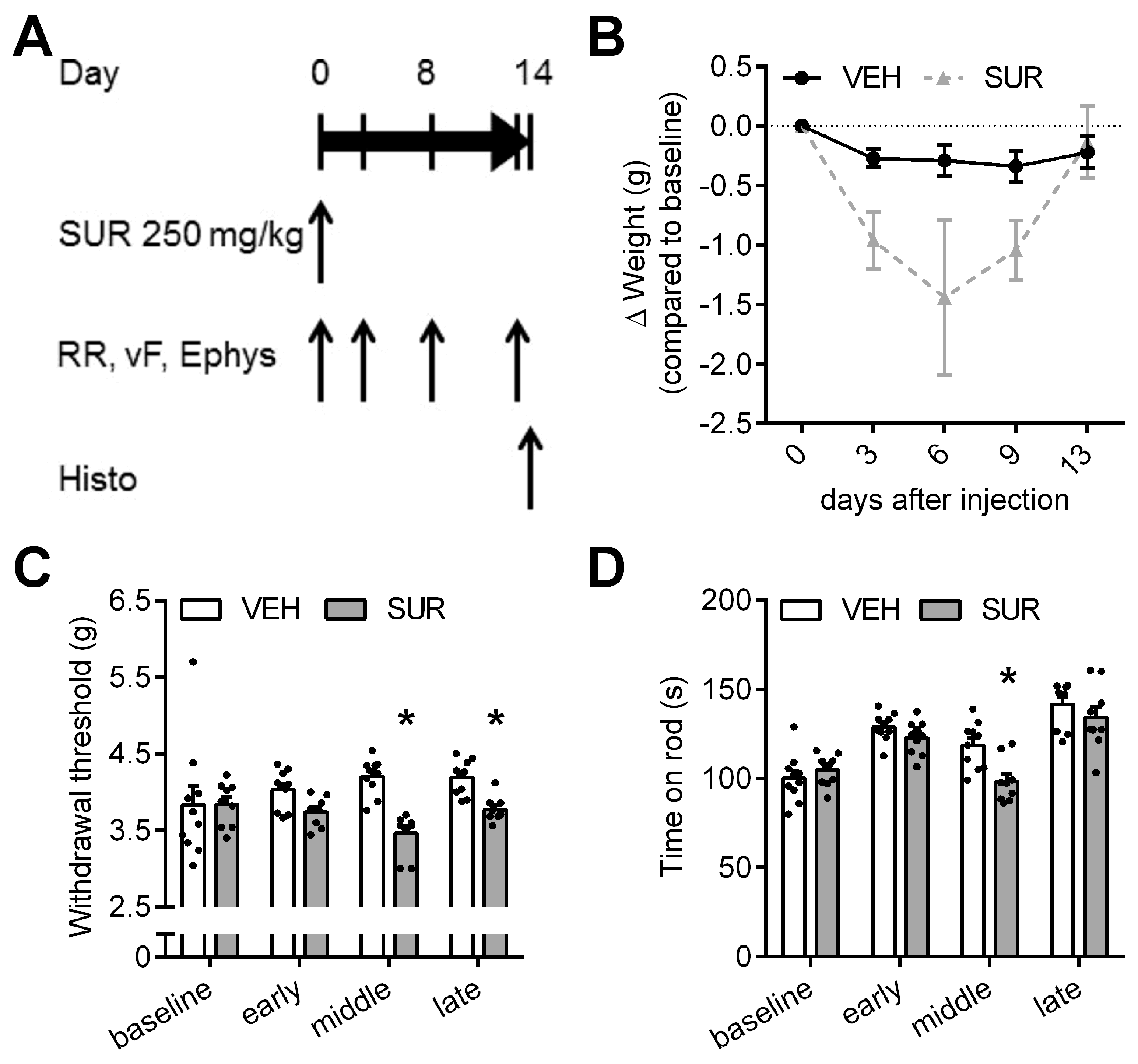
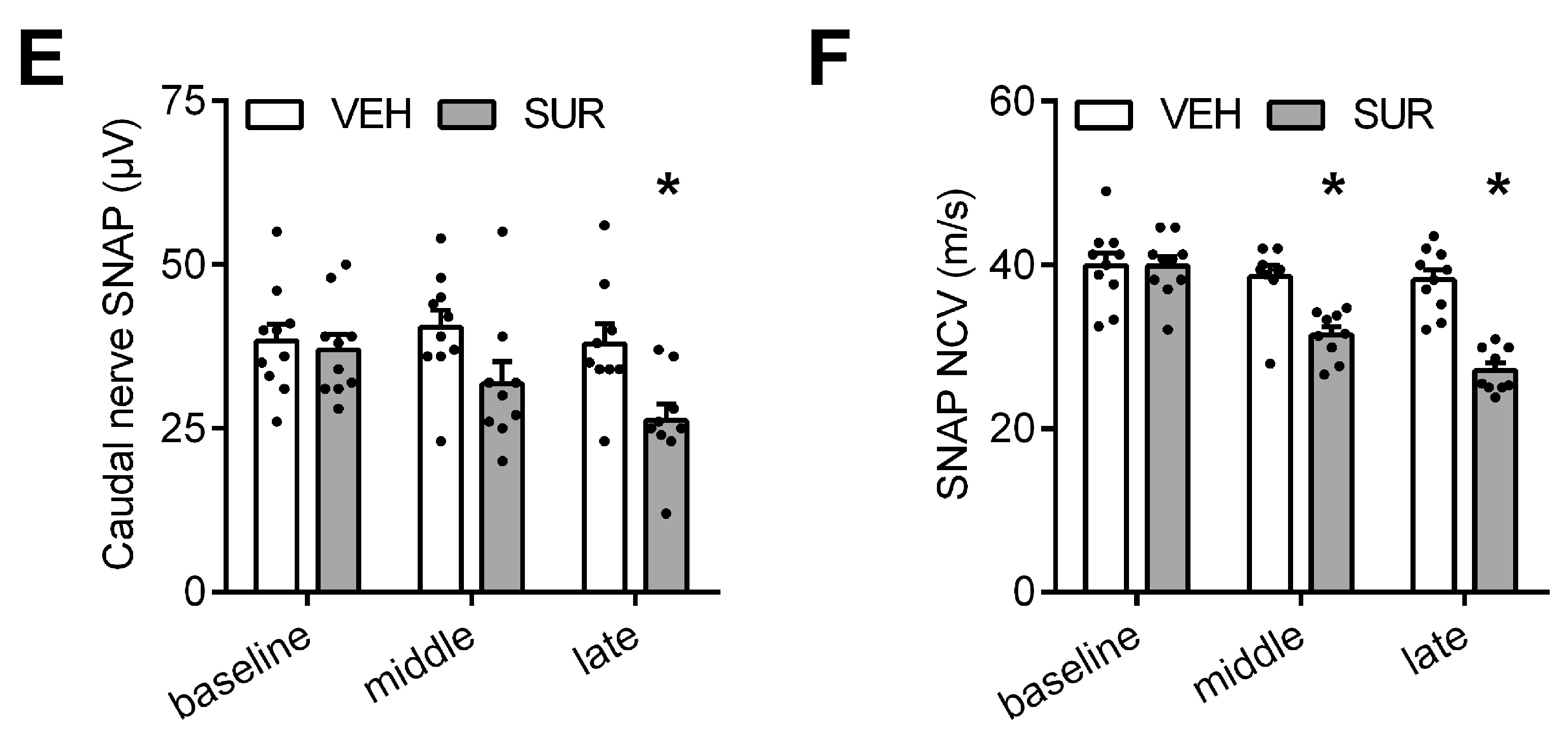
We previously characterized the behavioral and histological phenotype of chemotherapy-induced polyneuropathies in C57Bl/6 mice after application of paclitaxel, vincristine, cisplatin and bortezomib, respectively [21]. In order to establish a mouse model of a suramin-induced polyneuropathy, we adapted a previously published protocol. Russell et al. had shown that a single intraperitoneal injection of 500 mg/kg bodyweight suramin can induce chemotherapy-induced polyneuropathy in rats [15]. In mice, the published LD50 of intraperitoneal suramin application is 750 mg/kg bodyweight [22]. To avoid unspecific general side effects, we lowered the dose to a single intraperitoneal injection of 250 mg/kg bodyweight suramin. Mice were tested for sensory and motor deficits on days 3, 8 and 13 after suramin application (Figure 1A). Suramin treatment resulted in a steady decline in bodyweight, which reached its maximum on day 6 with −1.44 g ± 0.65 g compared to baseline (not significant, Kruskal-Wallis test; Figure 1B). Afterwards, mice regained bodyweight and reached control levels by the end of the experiment. One animal, which had lost >20% bodyweight compared to baseline, had to be sacrificed on day 6 according to predefined endpoints. As allodynia is a frequent symptom of chemotherapy-induced polyneuropathy, the mechanical withdrawal threshold was measured at various time points after suramin application. Suramin treated mice showed a significant decrease in the mechanical withdrawal threshold compared to vehicle-injected mice (Kruskal-Wallis test, p < 0.0001 and p = 0.0296 on day 8 and 13, respectively; Figure 1C), indicative of increased sensitivity to mechanical stimuli due to suramin treatment. In the rotarod test we also observed a mild deficit in locomotor function in suramin treated mice compared to control animals, which reached significance on day 8 (repeated measures 2-way ANOVA, p = 0.0022; Figure 1D). As the rotarod test measures a complex behavior which is influenced by motor performance but also coordination and sensory function, the observed decline in performance could be mediated by sensory neuropathy as well as motor neuropathy. Given the normal behavior of suramin treated animals in their home cage, the small effect size and our observations regarding the sensory nervous system, the effect of suramin on rotarod performance is likely dominated by sensory neuropathy. Electrophysiological assessment showed that suramin application led to a significant decline in the sensory nerve action potential amplitude (repeated measures 2-way ANOVA, p = 0.0158; Figure 1E) as well as the sensory nerve conduction velocity (Kruskal-Wallis, p = 0.0329 and p = 0.0008 on days 8 and 13, respectively; Figure 1F). In conclusion, our data provide evidence that a single injection of 250 mg/kg bodyweight suramin in C57Bl/6 mice is sufficient to induce a predominantly sensory axonal-demyelinating polyneuropathy.
2.2. Effects of Suramin on Cell Viability and Calcium Homeostasis in Dorsal Root Ganglia Neurons
We have previously shown that some cytostatic agents cause alterations in intracellular calcium (Ca2+) homeostasis in dorsal root ganglia neurons (DRGN) and thus contribute to the development of chemotherapy-induced polyneuropathy [19,20,23,24]. In a first step, we were interested in the toxicity of suramin towards DRGN. We measured cell viability of DRGN in response to increasing concentrations of suramin and observed marked toxicity above 300 µM suramin concentrations. The calculated half maximal inhibitory concentration (IC50) of 22–24 h (hour) suramin treatment was 283 µM (non-linear regression analysis, 95% confidence interval 226–355 µM) in DRGN (Figure 2A). For the following experiments studying cell viability we used a dose of 400 µM. Next, we were interested in the effects suramin might exhibit on intracellular Ca2+ homeostasis. Sun and Windebank could previously show a calcium (Ca2+) influx into DRGN under “acute” suramin treatment [18]. In order to quantify and localize the impact of suramin on intraneuronal Ca2+ homeostasis, Ca2+ imaging experiments were performed using the fluorescent Ca2+ indicator dye fura-2. A higher dose of 1 mM suramin was chosen in order to ensure robust activation of potential receptors in the plasma membrane of DRGN despite of the short incubation time. First, we monitored Ca2+ levels in DRGN cultured in Ca2+ free medium, which did not result in any changes of intracellular Ca2+ levels (unpaired two-sided t-test; Figure 2B). However, when DRGN were cultured in Ca2+ containing medium, we observed a strong increase of intracellular Ca2+ levels (SUR 65.2 nM ± 7.4 nM, VEH 3.2 nM ± 0.6 nM), which is in line with previous findings (Mann-Whitney-U test, p < 0.0001; Figure 2C). These findings suggest that suramin exposure leads to an influx of Ca2+ from the extracellular space into the cytosol of DRGN via the plasma membrane. Intriguingly, the addition of suramin to DRGN cultures in Ca2+ containing imaging buffer caused different profiles of Ca2+ influx (Figure 2D): Most DRGN showed a rapid but transient Ca2+ influx followed by a slow Ca2+ increase (profile 1, 54%), others reacted with a slow but steady Ca2+ increase (profile 2, 23%) whereas some cells showed a short but transient Ca2+ increase only (profile 3, 18%). A smaller group of cells did not show any rise in intracellular Ca2+ levels after suramin treatment (profile 4, 7%) (Figure 2E).
2.3. Effects of Various Inhibitors of Plasmamembrane Channels on Suramin-Induced Neurotoxicity
Our data obtained from Ca2+ imaging indicated, that suramin treatment leads to an increase of intracellular Ca2+ levels via a Ca2+ influx across the plasma membrane. In a next step, we aimed to identify specific Ca2+ channels in the plasma membrane which are modulated by suramin. It was previously shown that nimodipine, an inhibitor of L-Type VGCC, has a protective effect on axonal outgrowth in suramin treated DRGN [18]. Based on our dose-response curve, we used suramin concentrations of 400 µM as a clinically relevant concentration that reproducibly led to a decrease of cell viability down to 52% ± 4% of vehicle treatment. We measured cell viability of suramin treated cells with and without co-incubation with various inhibitors of VGCC. Because of the complex chemical structure of suramin, we hypothesized that suramin could also induce Ca2+ influx into DRGN via transient-receptor-potential- (TRP-) channels. These are cation-conducting channels, which play an important role in sensory perception. A variety of TRP-channels subgroups are found in the plasma membrane of DRGN. At first, we screened for potential neuroprotective effects of different VGCC and TRP-channel inhibitors after co-incubation with 400 µM suramin using the MTT-assay. In case of neuroprotective effects on cell viability, calcium imaging was performed with those previously identified inhibitors. Table 1 summarizes the results of cell viability measurements of DRGN treated with 400 µM suramin in the presence of different VGCC- and TRP-inhibitors.
We observed a dose-dependent increase of cell viability, when DRGN exposed to 400 µM suramin were co-incubated with nimodipine (Figure 3A). This effect reached statistical significance for nimodipine concentrations of 100 µM and 150 µM, but could not restore impaired cell viability to control levels (1-way ANOVA, p < 0.0001; Figure 3B). We additionally measured intracellular Ca2+ levels of suramin treated DRGN under co-incubation with nimodipine. In line with the findings of the cell viability assays, we observed a significantly reduced Ca2+ influx in DRGN under co-incubation with suramin and nimodipine, compared to DRGN treated with suramin and the corresponding vehicle (Mann-Whitney-U test, p = 0.0229; n = 35 SUR/VEH, n = 34 SUR/Nimo; Figure 3C). Again, nimodipine was not able to prevent suramin-induced Ca2+ influx completely.
Efonidipine, another L-Type channel antagonist, did not exert protective effects on suramin-induced cell death. However, data on efonidipine is hard to interpret as increasing doses of efonidipine in the absence of suramin per se exert toxic effects on cultured DRGN with a significantly lower cell viability (1-way ANOVA, p = 0.0388 (10 µM) and p < 0.0001 (50 µM), respectively). The same problem was observed for the unselective VGCC and TRP inhibitor Ruthenium Red (1-way ANOVA, p = 0.0005; Supplementary Materials Table S1 and Supplementary Materials Figure S1). Additional VGCC antagonists did not show any effects on cell viability of DRGN after suramin treatment (Table 1).
Next, we investigated various TRP channel inhibitors regarding their neuroprotective effects under suramin exposure. We only observed a small but significant protective effect on cell viability when DRGN were co-incubated with concentrations of 10–100 nM of the selective TRPV4 antagonist HC067047 (1-way ANOVA, p = 0.0294 (10 nM) and p = 0.0403 (100 nM), respectively; Figure 3D,E). However, HC067047 did not change Ca2+ influx due to suramin treatment (Mann-Whitney-U test; Figure 3F). Other TRP antagonists had no effects on cell viability after suramin exposure and measurements of intracellular Ca2+ levels of suramin treated DRGN did not show any alterations when DRGN were co-incubated with TRP antagonists.
2.4. Downstream Apoptotic Pathways and Experimental Limitations
In order to investigate apoptotic pathways further downstream of Ca2+ influx, we measured caspase activity after suramin treatment of DRGN. Surprisingly, we did not detect any protective effects of caspase-3/7 or calpain inhibition on cell viability when DRGN were co-incubated with suramin and the respective inhibitors (Supplementary Materials Table S2). We also observed a significant decrease of caspase-3/7 activity after 0, 6 and 12 h suramin treatment (Supplementary Materials Figure S2A). It was previously described in the Jurkat cell line that suramin itself can inhibit caspase-3/7 activity [25]. However, we also observed a significant increase in caspase-3/7 activity after 24 h suramin incubation. This observation is difficult to reconcile with the initially observed decrease of the luminescenct signal and we hypothesized that the luminescent assay signal might be quenched in the presence of suramin. Indeed, when we added suramin directly before luminescent signal detection, we also observed a significant decrease in measured caspase activity (Supplementary Materials Figure S2B). We therefore suggest that suramin either binds under a chemical reaction directly to the luminophore and thereby alters photon emission or it potentially absorbs photons emitted by the luminophore. Nevertheless, the significantly increased caspase activity after 24 h of suramin incubation suggests caspase activation by suramin, even considering the experimental limitations of the assay. We additionally performed immunhistochemical staining in order to verify our hypothesis. During those experiments, suramin interaction with cell culture coated dishes was observed. DRGN did not adhere to cell culture dishes under suramin treatment when washed according to the staining protocol. All cell culture surfaces were coated with a well-established protocol for DRGN cultures using poly-l-lysin and laminin. Prighozinha et al. described Matrigel dissolvement by suramin, which is of interest as Matrigel consists of 61% laminin. It was hypothesized that negatively charged sulfate groups would account for laminin binding and Matrigel dissolvement [26]. In line with these findings suramin was previously described to act as a heparan-sulfate analogue and antimetastic tumor activity of suramin was linked to this mode of action. Furthermore, suramin inhibition of binding of extracellular tumorcell-glycocalyx to the extracellular matrixprotein laminin was proposed to prevent tumor cell migration [27,28,29]. The same mechanism could be responsible for reduced binding activity of DRGN to laminin-coated surfaces. This means that suramin could inhibit binding of extracellular glycocalyx of DRGN to laminin molecules of the coated cell culture dish.
3. Discussion
The objective of this study was to elucidate the pathomechanism underlying suramin-induced neurotoxicity and its potential mechanistic link to neuronal Ca2+ dyshomeostasis. Intriguingly, different chemotherapeutic agents were found to disturb intracellular Ca2+ levels in neurons resulting in neuronal apoptosis. One example is paclitaxel, a chemotherapeutic agent causing a peripheral sensory neuropathy, which induces a dyshomeostasis of intracellular Ca2+ in DRGN [20,23]. It was proposed that the phosphoinositide pathway is involved and causes Ca2+ release from the endoplasmic reticulum under treatment with paclitaxel. Salinomycin, another experimental chemotherapeutic agent, was found to cause increased intracellular Ca2+ levels by its ionophoric capacity [19]. Furthermore, caspases and Ca2+ activated proteases such as calpains were shown to beinvolved in Ca2+-induced apoptosis. Caspase 12, a cytosolic caspase bound to the endoplasmic reticulum, also plays a role in Ca2+ triggered apoptosis [30]. In the context of suramin, Sun and Windebank proposed that Ca2+ influx into the cytosol would be mediated by channels in the plasma membrane [18]. Our Ca2+ imaging experiments confirm this hypothesis. In our study, we observed that suramin induces an increase of intracellular Ca2+ in DRGN only in Ca2+ containing buffer, whereas intracellular Ca2+ levels were not affected when DRGN were cultured in Ca2+ free buffer. This implies that suramin activates Ca2+ conducting channels in the plasma membrane causing a Ca2+ influx via the plasma membrane into the cytosol. Furthermore, it was shown that the L-type VGCC-inhibitor nimodipine can restore neuronal survival and axonal outgrowth after suramin treatment [18]. In our experiments, we also observed that nimodipine improves cell viability compared to suramin/vehicle co-treatment. Our data however also suggest that nimodipine cannot fully reverse suramin’s neurotoxic effects. In line with these findings, suramin-induced Ca2+ influx was significantly reduced when DRGN were co-incubated with nimodipine, but intracellular Ca2+ levels still remained elevated compared to control levels. Although these observations suggest that, the effect of nimodipine depends at least partially on inhibition of L-type VGCC, we observed robust neuroprotective effects only at comparatively high nimodipine concentrations above 100 µM. This concentration is at least an order of magnitude higher compared to the concentration of 1–10 µM typically used in in vitro studies to block L-type VGCC (e.g., [31,32]), suggesting the possibility of additional molecular mechanisms. Among others, nimodipine was shown to interact with multidrug resistance transporters such as P-glycoprotein [33] and at higher concentrations with voltage dependent potassium channels [34]. To further investigate this aspect we used efonidipine as another specific inhibitor of L-type VGCC and ruthenium red as a broad range inhibitor of Ca2+ channels in the plasma membrane (including VGCC and TRP). Unfortunately, these substances were themselves toxic, making the results hard to interpret.
Another possible source for the Ca2+ influx across the plasma membrane are TRP channels. We tested a number of TRP channel inhibitors targeting TRP channel 3 (TRPC3), TRP channel subfamily A (TRPA1), subfamily V (TRPV4) and subfamily M inhibitors, of which only the TRPV4 inhibitor yielded a slightly positive effect on suramin-induced cell death. This is an interesting finding, as TRPV4 has been shown to play an important role in neuropathic pain caused by paclitaxel [35]. Given the diversity of TRP channels alone however, one limitation of the present work is that only a small subset of potentially relevant ion channels was studied. A logical next step for future experiments would be to repeat our experimental paradigm combined with whole cell patch clamp experiments to further characterize the mechanisms underlying Ca2+ influx. Subsequently additional inhibitors should be evaluated.
DRGN exhibited different responses to suramin in terms of their intracellular Ca2+ levels. We hypothesize that suramin might stimulate different ion channels or receptors in the plasma membrane by its unspecific binding properties. It should also be noted that DRGN differ in receptor and ion channel status. VGCC expression patterns vary especially between small, medium and large diameter neurons [36,37,38]. Also TRP-channel subtype expression was found to be variable in different DRGN [39]. Therefore, the unspecific binding properties of suramin on the one hand and varying receptor/ion channel status on DRGN on the other hand could lead to different reaction patterns of Ca2+ influx in DRGN after suramin treatment. This observation might explain the small effect size and heterogenous results observed in case of TRPV4 inhibition and the lack of changes observed in Ca2+ imaging experiments. This experimental hurdle could be overcome by studying for instance only TRPV4 positive DRGN.
Increasing intracellular concentrations of Ca2+ in the cytosol can trigger apoptotic processes by initiating different signaling pathways. Most frequently caspases and calpains are involved. Unexpectedly, we did not see any positive effects of caspase or calpain inhibition regarding cell viability after suramin treatment, which is in contrast to previous results of Sun and Windebank, who observed protective effects of calpain inhibition. Future studies should expand on this aspect as it suggests a more complex mechanism of cell death.
Taken together, our data suggest a link between suramin-induced neurotoxicity and Ca2+ dyshomeostasis in DRGN and we could demonstrate that suramin treatment leads to a predominantly sensory axonal-demyelinating neuropathy in mice. However, it has also become clear in our experiments that suramin is a complex molecule and a whole array of cellular effects induced by suramin were described in the past (reviewed by [1]) making it a “dirty” drug. A number of mechanisms may thus contribute to suramin-induced neurotoxicity in the peripheral nervous system including but not limited to Ca2+ dyshomeostasis, interference with nerve growth factor signaling [16,17], glycolipid homeostasis [40], inhibition of P2X/P2Y purinoreceptors (reviewed by [41]) and mitochondrial toxicity [42]. As suramin acts as inhibitor of calcium conducting P2X purinoreceptors it seems that this target does not play a role in suramin-induced calcium influx in DRGN [43]. L-type Ca2+ channels seem to account only for a partial increase in intracellular Ca2+ levels after suramin treatment. Nevertheless, they present a potentially interesting molecular target as VGCC inhibitors such as nimodipine have already been approved for clinical use and could theoretically be easily translated to clinical application. Additional animal studies are however needed to prove a beneficial effect of VGCC inhibition in chemotherapy-induced polyneuropathy. Further ion channels in the plasma membrane also seem to mediate suramin-induced Ca2+ influx. One of those targets could be TRP channels, which have already been linked to chemotherapy-induced polyneuropathy in other antineoplastic substances such as paclitaxel.
In summary, given suramin’s complex biological and chemical characteristics and proven neurotoxicity, developing a neuroprotective co-treatment may help to overcome suramin’s translational challenge and enable a broader clinical use of this drug.
4. Materials and Methods
4.1. In Vivo
4.1.1. Animal Housing, Sample Sizes and Methods of Randomization and Blinding
We used 20 nine-week old male C57Bl/6 mice from Charles River (Sulzfeld, Germany) for the experiment. All procedures had been previously approved by the State Office for Health and Social Affairs (Landesamt für Gesundheit und Soziales (LaGeSo), Berlin, Germany, Reg.-Nr: G0092-10) and conformed to animal welfare guidelines. Mice were housed in groups of five in an enriched environment and a 12-hour light/dark cycle (7 a.m.–7 p.m.) with unlimited access to food and water. The a priori calculated sample size was 10 mice/group, based on previously observed effect sizes of electrophysiological parameters [21], a power of 0.8 and an alpha-error of 0.05. Sample size calculation was done using G*Power statistical software (Heinrich Heine Universität Düsseldorf, Düsseldorf, Germany) [44]. Mice were randomized to one of two groups (SUR vs. VEH) using an online randomization tool (GraphPad Software, La Jolla, CA, USA; https://www.graphpad.com/quickcalcs/randomize1.cfm). Behavior experiments were conducted by a blind investigator.
4.1.2. Drug Preparation and Injection
Suramin (Tocris, Bioscience, Bristol, UK) was dissolved in Aqua dest. to a concentration of 50 mg/mL and diluted in sterile 0.9% saline to a final concentration of 25 mg/mL and injected once intraperitoneally at 10 µL/g bodyweight. As vehicle treatment, Aqua dest. was diluted 1:1 in 0.9% sterile saline and injected once intraperitoneally at 10 µL/g bodyweight.
4.1.3. Behavior Analysis
Mice were handled for five consecutive days before behavior experiments to ensure familiarization to the investigator and to minimize stress and anxiety. All behavior experiments were done in a dedicated laboratory with soundproof chambers during 10 a.m. and 4 p.m. The general wellbeing of the mice was checked daily and their weight recorded. The mechanical withdrawal threshold was obtained using the von Frey filament test as previously described [21,45]. In brief: a hand-held force transducer was fitted with a 0.5 mm2 polypropylene tip. Mice were placed in a clear inverted plastic cage on a wire-mesh floor. Trained investigators applied increasing pressure to the center of the hind paws until a clear withdrawal response was evoked. If the animal did not exhibit a clear withdrawal response within 5 s the trial was deemed invalid and was repeated. The withdrawal threshold in grams (g) was automatically measured by the device (IITC, Woodland Hills, CA, USA). The maximum applied force was 10 g. For each time point, 5 trials were averaged. Locomotor function was measured with the rotarod test as previously described [21,45]. Mice were placed on a rotating rod (TSE Systems, Bad Homburg, Germany), which gradually increased in speed from 4 to 40 rpm over the course of 300 s. The latency for the mice to fall off the rod was automatically measured by a floor sensor. Mice were trained in the task for four consecutive days prior to baseline measurement. For each time point, three trials for each animal were averaged.
4.1.4. Nerve Conduction Studies
Mice were anaesthetized with isoflurane 1.5%/50% O2 (volume/volume) and placed on a heating pad with a constant temperature of 37.5 °C (Harvard Apparatus, Holliston, MA, USA). Stimulating needle electrodes were placed at the base of the tail with the recording electrodes approx. 5 cm distal. A ground electrode was place in the middle. Single stimuli were applied to determine supramaximal level and afterwards 50 serial supramaximal stimuli were averaged to obtain the sensory nerve action potential (SNAP) amplitudes and NCV [21,45].
4.2. In Vitro
4.2.1. DRGN Cell Culture
DRGN cultures were obtained from P 0–3 Wistar rat neonates as described before [23]. Cells were plated on poly-l-lysin and laminin coated 96-well plates for cell viability measurements and on cover slips with the same coating for Ca2+ imaging experiments. DRGN cultures were incubated overnight at 37 °C in a 95% air/5% CO2 humidified atmosphere before experimental use. Neurobasal A medium supplemented with B-27 (Life Technologies GmbH, Darmstadt, Germany), 0.5 mM glutamine, fresh nerve growth factor (10 ng/mL) was used for cell maintenance.
4.2.2. Calcium Imaging
Cells were mounted with Fura-2 AM 5 µM (Life Technologies GmbH, Darmstadt, Germany) for 30 min at 37 °C in a standard HEPES buffered solution: 130 mM NaCl/4.7 mM KCl/1 mM MgSO4/1.2 mM KH2PO4/1.3 mM CaCl2/20 mM Hepes/5 mM glucose (pH 7.4)/0.02% pluronic F-127 (Life Technologies GmbH, Darmstadt, Deutschland). After incubation cell cultures were washed with standard HEPES buffered solution and coverslips were placed in a Olympus IX 81 microscope equipped with a Uplan FLN oil objective 40×/1.13 (Olympus Corporation, Tokyo, Japan). For calcium imaging experiments in Ca2+ free extracellular medium a standard HEPES solution without Ca2+ was used: 130 mM NaCl/4.7 mM KCl/2.3 mM MgSO4/1.2 mM KH2PO4/10 mM EDTA/20 mM Hepes/5 mM glucose, pH 7.4. For experiments with ion channel inhibitors DRGN cultures were preincubated in standard HEPES buffered solution with the respective channel blocker for 5 min before Ca2+ levels were measured. Fluorescence signals were detected by a cooled CCD-camera and data was processed on a computer using Olympus xcellence imaging software. Intracellular Ca2+ concentrations [(Ca2+) int] (nM) were calculated using ratios of F340/F380 after background subtraction with the following equation: [Ca2+]int (nM) = Kd*Q*(R − Rmin)/(Rmax − R), where Kd is the dissociation constant of Fura-2 for Ca2+ at room temperature (225 nM); Q is the fluorescence ratio of the emission intensity excited by 380 nm in the absence of Ca2+ to that during the presence of saturating Ca2+; and Rmin and Rmax are the minimal or maximal fluorescence ratios, respectively. Values for Rmin and Rmax were derived from in situ calibration using Ca2+ free HEPES buffered standard solution with 10 µM ionomycin for Rmin values and HEPES buffered standard solution with CaCl2 for Rmax values. Intracellular free Ca2+ concentrations were measured over 4.37 min adding 1 µM suramin or VEH (Aq. dest.) after 20 s and 10 µM ionomycin after 4.15 min (internal control). Cells with an increase of <200 nM [Ca2+ int.] (Δ) in HEPES buffered solution with Ca2+ or <50 nM [Ca2+ int.] (Δ) in HEPES buffered solution without Ca2+ were excluded from the data analysis.
4.2.3. MTT-Assay
DRGN cultures plated in 96 well plates were incubated with suramin and ion channel inhibitors (VGCC and TRP-channels) and respective controls for 24 h at 37 °C in a 95% air/5% CO2 humidified atmosphere. MTT was added to each well in a ratio of 1:10 and cell cultures were reincubated for 30 min at 37 °C in a 95% air/5% CO2 humidified atmosphere. Reaction was stopped using SDS in a ratio of 1:1. After cell dissolution overnight, absorption values were gained at a wavelength of 550 nm, lamp energy of 10,000 during 0.2 s in a microplate reader (Tristar LB941 Multimode Microplate Reader Berthold Technologies GmbH and Co. KG, Bad Wildbad, Germany). Values were background subtracted and standardized to percentage of control. For coincubation with suramin and ion channel inhibitors a second standardization was used and the mean difference suramin plus inhibitor to suramin plus vehicle treatment (ΔSUR/VEH) was calculated.
4.2.4. Caspase Assay
Caspase activity was assessed using the Promega Caspase-Glo® 3/7 Assay kit (Promega, Mannheim, Germany) according to the manufacturer’s protocol as described [46].
4.3. Statistical Analysis and Exclusion Criteria
The manuscript was prepared in accordance with ARRIVE guidelines [47]. Data is presented as mean ± SEM with individual values depicted for in vivo studies. For MTT assay experiments, data was obtained from n = 3–6 individual experiments (biological replicates) with n = 3 technical replicates. For Ca2+ imaging experiments (n/N) describes the number of cells studied (n) in (N) independent cultures. Statistical analysis was performed using Prism v6.0 (GraphPad Software, La Jolla, CA, USA). Gaussian distribution of data was checked prior to statistical analysis using Shapiro-Wilk normality test. Normally distributed data was analyzed using unpaired two-sided t-tests (two groups), one-way ANOVA with Sidak post hoc test (≥three groups) or (repeated measures) 2-way ANOVA (two variables). Not normally distributed data was analyzed with Mann-Whitney-U test (2 groups) or Kruskal-Wallis test with Dunn’s method (≥3 groups). p < 0.05 was considered statistically significant and is depicted by asterisk. Only statistical outliers that conformed with Peirce criterion were excluded from the dataset [48,49].
Supplementary Materials
The Supplementary Materials are available online.
Acknowledgments
We would like to thank Petra Loge for excellent technical assistance. The researchers have received funding from the Federal Ministry of Education and Research via the grant Center for Stroke Research Berlin (01 EO 0801) and from the Deutsche Forschungsgemeinschaft (EXC 257 NeuroCure). PH receives funding from the Charité Clinician Scientist program funded by the Charité Universitätsmedizin Berlin and the Berlin Institute of Health.
Author Contributions
W.B. and M.E. conceived and designed the experiments. D.v.d.A. performed the in vitro experiments. P.H., M.B. and S.P. performed the in vivo experiments. D.v.d.A., P.H. and W.B. analyzed the data and compiled the figures. D.v.d.A., P.H. and W.B. wrote the manuscript. All authors reviewed the manuscript. M.E. provided funding for the study.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Ca2+ | calcium |
| DRGN | dorsal root ganglia neurons |
| VGCC | voltage-gated calcium channels |
| TRP | transient-receptor-potential |
References
- McGeary, R.P.; Bennett, A.J.; Tran, Q.B.; Cosgrove, K.L.; Ross, B.P. Suramin: Clinical uses and structure-activity relationships. Mini Rev. Med. Chem. 2008, 8, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, H.; Popovic, M.; Yarchoan, R.; Matsushita, S.; Gallo, R.C.; Broder, S. Suramin protection of T cells in vitro against infectivity and cytopathic effect of HTLV-III. Science 1984, 226, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Levine, A.M.; Mildvan, D.; Kaplan, L.D.; Wolfe, P.; Rios, A.; Groopman, J.E.; Gill, P.; Volberding, P.A.; Poiesz, B.J. Suramin therapy in AIDS and related disorders: Report of the US Suramin Working Group. JAMA 1987, 258, 1347–1351. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.J.; Ranson, M.; Margison, J.; Thomson, Y.; van der Water, L.; George, N.; Howell, A. Suramin for breast and prostate cancer: A pilot study of intermittent short infusions without adaptive control. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1994, 5, 597–600. [Google Scholar] [CrossRef]
- Small, E.J.; Meyer, M.; Marshall, M.E.; Reyno, L.M.; Meyers, F.J.; Natale, R.B.; Lenehan, P.F.; Chen, L.; Slichenmyer, W.J.; Eisenberger, M. Suramin therapy for patients with symptomatic hormone-refractory prostate cancer: Results of a randomized phase III trial comparing suramin plus hydrocortisone to placebo plus hydrocortisone. J. Clin. Oncol. 2000, 18, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, M.A.; Reyno, L.M.; Jodrell, D.I.; Sinibaldi, V.J.; Tkaczuk, K.H.; Sridhara, R.; Zuhowski, E.G.; Lowitt, M.H.; Jacobs, S.C.; Egorin, M.J. Suramin, an active drug for prostate cancer: Interim observations in a phase I trial. J. Natl. Cancer Inst. 1993, 85, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.A.; Figg, W.D.; Cooper, M.R.; Sartor, O.; Bergan, R.C.; Senderowicz, A.M.; Steinberg, S.M.; Tompkins, A.; Weinberger, B.; Sausville, E.A.; et al. Phase II trial of suramin, leuprolide, and flutamide in previously untreated metastatic prostate cancer. J. Clin. Oncol. 1997, 15, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.; Cooper, M.; Stein, C.; LaRocca, R.; Walther, M.M.; Weiss, G.; Choyke, P.; Dawson, N.; Steinberg, S.; Uhrich, M.M.; et al. Suramin: A novel growth factor antagonist with activity in hormone-refractory metastatic prostate cancer. J. Clin. Oncol. 1992, 10, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, V.; Eisenberger, M.A.; Sinibaldi, V.J.; Sheikh, K.; Griffin, J.W.; Cornblath, D.R. A prospective study of suramin-induced peripheral neuropathy. Brain J. Neurol. 1996, 119, 2039–2052. [Google Scholar] [CrossRef]
- Voogd, T.E.; Vansterkenburg, E.L.; Wilting, J.; Janssen, L.H. Recent research on the biological activity of suramin. Pharmacol. Rev. 1993, 45, 177–203. [Google Scholar] [PubMed]
- Peltier, A.C.; Russell, J.W. Recent advances in drug-induced neuropathies. Curr. Opin. Neurol. 2002, 15, 633–638. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, R.V.; Meer, J.; Gilliatt, R.W.; Stein, C.A.; Cassidy, J.; Myers, C.E.; Dalakas, M.C. Suramin-induced polyneuropathy. Neurology 1990, 40, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Bitton, R.J.; Figg, W.D.; Venzon, D.J.; Dalakas, M.C.; Bowden, C.; Headlee, D.; Reed, E.; Myers, C.E.; Cooper, M.R. Pharmacologic variables associated with the development of neurologic toxicity in patients treated with suramin. J. Clin. Oncol. 1995, 13, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Hobday, K.L.; Windebank, A.J. Mechanism of suramin toxicity in stable myelinating dorsal root ganglion cultures. Exp. Neurol. 1995, 133, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.W.; Gill, J.S.; Sorenson, E.J.; Schultz, D.A.; Windebank, A.J. Suramin-induced neuropathy in an animal model. J. Neurol. Sci. 2001, 192, 71–80. [Google Scholar] [CrossRef]
- Russell, J.W.; Windebank, A.J.; Podratz, J.L. Role of nerve growth factor in suramin neurotoxicity studied in vitro. Ann.Neurol. 1994, 36, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Connolly, D.C.; McManus, M.J.; Maihle, N.J.; Windebank, A.J. Suramin induces phosphorylation of the high-affinity nerve growth factor receptor in PC12 cells and dorsal root ganglion neurons. J. Neurochem. 1996, 66, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Windebank, A.J. Calcium in suramin-induced rat sensory neuron toxicity in vitro. Brain Res. 1996, 742, 149–156. [Google Scholar] [PubMed]
- Boehmerle, W.; Endres, M. Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis. 2011, 2, e168. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Splittgerber, U.; Lazarus, M.B.; McKenzie, K.M.; Johnston, D.G.; Austin, D.J.; Ehrlich, B.E. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 18356–18361. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Huehnchen, P.; Peruzzaro, S.; Balkaya, M.; Endres, M. Electrophysiological, behavioral and histological characterization of paclitaxel, cisplatin, vincristine and bortezomib-induced neuropathy in C57Bl/6 mice. Sci. Rep. 2014, 4, 6370. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Mitsuya, H.; De Clercq, E.; Broder, S. Aurintricarboxylic acid and evans blue represent two different classes of anionic compounds which selectively inhibit the cytopathogenicity of human T-cell lymphotropic virus type III/lymphadenopathy-associated virus. Biochem. Biophys. Res. Commun. 1986, 136, 64–71. [Google Scholar] [CrossRef]
- Boehmerle, W.; Zhang, K.; Sivula, M.; Heidrich, F.M.; Lee, Y.; Jordt, S.E.; Ehrlich, B.E. Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain-mediated neuronal calcium sensor-1 degradation. Proc. Natl. Acad. Sci. USA 2007, 104, 11103–11108. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Muenzfeld, H.; Springer, A.; Huehnchen, P.; Endres, M. Specific targeting of neurotoxic side effects and pharmacological profile of the novel cancer stem cell drug salinomycin in mice. J. Mol. Med. 2014, 92, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, S.T.; Krueger, A.; Muerkoster, S.; Fas, S.C.; Golks, A.; Gruetzner, U.; Schubert, L.; Opelz, C.; Bilzer, M.; Gerbes, A.L.; et al. Suramin inhibits death receptor-induced apoptosis in vitro and fulminant apoptotic liver damage in mice. Nat. Med. 2004, 10, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Prigozhina, N.L.; Heisel, A.J.; Seldeen, J.R.; Cosford, N.D.; Price, J.H. Amphiphilic suramin dissolves Matrigel, causing an ‘inhibition’ artefact within in vitro angiogenesis assays. Int. J. Exp. Pathol. 2013, 94, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Zabrenetzky, V.S.; Kohn, E.C.; Roberts, D.D. Suramin inhibits laminin- and thrombospondin-mediated melanoma cell adhesion and migration and binding of these adhesive proteins to sulfatide. Cancer Res. 1990, 50, 5937–5942. [Google Scholar] [PubMed]
- Constantopoulos, G.; Rees, S.; Cragg, B.G.; Barranger, J.A.; Brady, R.O. Experimental animal model for mucopolysaccharidosis: Suramin-induced glycosaminoglycan and sphingolipid accumulation in the rat. Proc. Natl. Acad. Sci. USA 1980, 77, 3700–3704. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; DeChavigny, A.; Johnson, C.E.; Hamada, J.; Stein, C.A.; Nicolson, G.L. Suramin. A potent inhibitor of melanoma heparanase and invasion. J. Biol. Chem. 1991, 266, 9661–9666. [Google Scholar] [PubMed]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Liljelund, P.; Netzeband, J.G.; Gruol, D.L. L-Type calcium channels mediate calcium oscillations in early postnatal Purkinje neurons. J. Neurosci. 2000, 20, 7394–7403. [Google Scholar] [PubMed]
- Galvan, E.J.; Calixto, E.; Barrionuevo, G. Bidirectional Hebbian plasticity at hippocampal mossy fiber synapses on CA3 interneurons. J. Neurosci. 2008, 28, 14042–14055. [Google Scholar] [CrossRef] [PubMed]
- Pascaud, C.; Garrigos, M.; Orlowski, S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem. J. 1998, 333, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Gold, M.S. Dihydropyridine block of voltage-dependent K+ currents in rat dorsal root ganglion neurons. Neuroscience 2009, 161, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Alessandri-Haber, N.; Dina, O.A.; Yeh, J.J.; Parada, C.A.; Reichling, D.B.; Levine, J.D. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J. Neurosci. 2004, 24, 4444–4452. [Google Scholar] [CrossRef] [PubMed]
- Yusaf, S.P.; Goodman, J.; Pinnock, R.D.; Dixon, A.K.; Lee, K. Expression of voltage-gated calcium channel subunits in rat dorsal root ganglion neurons. Neurosci. Lett. 2001, 311, 137–141. [Google Scholar] [CrossRef]
- Scroggs, R.S.; Fox, A.P. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Physiol. 1992, 445, 639–658. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.G.; Lopez, H.S. delta opioid receptor modulation of several voltage-dependent Ca2+ currents in rat sensory neurons. J. Neurosci. 1999, 19, 8337–8348. [Google Scholar] [PubMed]
- Vandewauw, I.; Owsianik, G.; Voets, T. Systematic and quantitative mRNA expression analysis of TRP channel genes at the single trigeminal and dorsal root ganglion level in mouse. BMC Neurosci. 2013, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Windebank, A.J. Suramin induced ceramide accumulation leads to apoptotic cell death in dorsal root ganglion neurons. Cell Death Differ. 1998, 5, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C. P1- and P2-purinoceptor subtypes—An update. Arch. Int. Pharmacodyn. Ther. 1990, 303, 30–50. [Google Scholar] [PubMed]
- Rago, R.P.; Miles, J.M.; Sufit, R.L.; Spriggs, D.R.; Wilding, G. Suramin-induced weakness from hypophosphatemia and mitochondrial myopathy. Association of suramin with mitochondrial toxicity in humans. Cancer 1994, 73, 1954–1959. [Google Scholar] [CrossRef]
- Light, A.R.; Wu, Y.; Hughen, R.W.; Guthrie, P.B. Purinergic receptors activating rapid intracellular Ca increases in microglia. Neuron Glia Biol. 2006, 2, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Huehnchen, P.; Boehmerle, W.; Endres, M. Assessment of paclitaxel induced sensory polyneuropathy with "Catwalk" automated gait analysis in mice. PLoS ONE 2013, 8, e76772. [Google Scholar] [CrossRef] [PubMed]
- Huehnchen, P.; Boehmerle, W.; Springer, A.; Freyer, D.; Endres, M. A novel preventive therapy for paclitaxel-induced cognitive deficits: Preclinical evidence from C57BL/6 mice. Transl. Psychiatry 2017, 7, e1185. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Dardis, C. Peirce’s criterion for the rejection of non-normal outliers: Defining the range of applicability. J. Stat. Softw. 2004, 10, 1–8. [Google Scholar]
- Ross, S.M. Peirce’s criterion for the elimination of suspect experimental data. J. Eng. Technol. 2003, 20, 38–41. [Google Scholar]
Sample Availability: Samples of the compounds are available from commercial sources. |
Figure 1.
Mouse model of suramin-induced sensory-motor polyneuropathy. (A) Schematic outline of the experiment: Adult C57Bl/6 mice received a single intraperitoneal injection of 250 mg/kg bodyweight suramin (SUR) or vehicle (VEH) on day 0. Mice were tested on the rotarod (RR) and with the von Frey (vF) test for locomotor deficits and alterations of mechanical withdrawal threshold four times. Electropysiological (Ephys) measurements were obtained on day 0, 8 and 13; (B) SUR treated mice showed a decline in body weight, but recovered by day 13. One animal had to be sacrificed due to weight loss >20%; (C) SUR application resulted in a decrease of the mechanical withdrawal threshold and (D) locomotor function, which was most pronounced on day 8; (E) SUR treated mice showed a decline of the sensory nerve action potential (SNAP) amplitude as well as the (F) SNAP nerve conduction velocity (NCV), indicative of an axonal-demyelinating neuropathy. Group sizes: n = 10 (vehicle), n = 9–10 (suramin), * p < 0.05.
Figure 1.
Mouse model of suramin-induced sensory-motor polyneuropathy. (A) Schematic outline of the experiment: Adult C57Bl/6 mice received a single intraperitoneal injection of 250 mg/kg bodyweight suramin (SUR) or vehicle (VEH) on day 0. Mice were tested on the rotarod (RR) and with the von Frey (vF) test for locomotor deficits and alterations of mechanical withdrawal threshold four times. Electropysiological (Ephys) measurements were obtained on day 0, 8 and 13; (B) SUR treated mice showed a decline in body weight, but recovered by day 13. One animal had to be sacrificed due to weight loss >20%; (C) SUR application resulted in a decrease of the mechanical withdrawal threshold and (D) locomotor function, which was most pronounced on day 8; (E) SUR treated mice showed a decline of the sensory nerve action potential (SNAP) amplitude as well as the (F) SNAP nerve conduction velocity (NCV), indicative of an axonal-demyelinating neuropathy. Group sizes: n = 10 (vehicle), n = 9–10 (suramin), * p < 0.05.


Figure 2.
Effects of suramin on cell viability and intracellular calcium in dorsal root ganglia neurons. (A) Increasing suramin concentrations between 10 and 10,000 µM led to a dose-dependent decrease of cell viability with a calculated IC50 of 283 µM (non-linear regression analysis); (B) DRGN cultured in Ca2+ free buffer did not show alterations of intracellular Ca2+ levels after exposure with 1 mM suramin (24/9), whereas (C) increased intracellular Ca2+ levels could be observed when DRGN were incubated in Ca2+ containing medium and treated with 1 mM suramin (26/5); (D) Representative Ca2+ measurements of suramin treated DRGN: DRGN reacted to 1 mM suramin exposure with a transient Ca2+ increase followed by a slow increase (profile 1, black solid line), steady Ca2+ increase (profile 2, dark grey solid line) or a transient Ca2+ increase only (profile 3, light grey solid line). First arrow marks addition of 1 mM suramin, second arrow represents addition of 5 µM ionomycine (internal positive control); (E) Percentages of DRGN showing the different profiles of Ca2+ levels in response to 1 mM suramin exposure. * p < 0.05.
Figure 2.
Effects of suramin on cell viability and intracellular calcium in dorsal root ganglia neurons. (A) Increasing suramin concentrations between 10 and 10,000 µM led to a dose-dependent decrease of cell viability with a calculated IC50 of 283 µM (non-linear regression analysis); (B) DRGN cultured in Ca2+ free buffer did not show alterations of intracellular Ca2+ levels after exposure with 1 mM suramin (24/9), whereas (C) increased intracellular Ca2+ levels could be observed when DRGN were incubated in Ca2+ containing medium and treated with 1 mM suramin (26/5); (D) Representative Ca2+ measurements of suramin treated DRGN: DRGN reacted to 1 mM suramin exposure with a transient Ca2+ increase followed by a slow increase (profile 1, black solid line), steady Ca2+ increase (profile 2, dark grey solid line) or a transient Ca2+ increase only (profile 3, light grey solid line). First arrow marks addition of 1 mM suramin, second arrow represents addition of 5 µM ionomycine (internal positive control); (E) Percentages of DRGN showing the different profiles of Ca2+ levels in response to 1 mM suramin exposure. * p < 0.05.

Figure 3.
Effects of nimodipine and HC067047 on cell viability and intracellular Ca2+ in DRGN. (A) Treatment of DRGN for 24 h with suramin (400 µM) significantly decreased cell viability. Co-incubation with L-type VGCC nimodipine had a dose-dependent protective effect on cell viability; (B) Nimodipine (Nimo) concentrations of 100 and 150 µM significantly increased cell viability of DRGN in the presence of suramin; (C) Nimodipine (150 µM) reduced suramin-induced Ca2+ influx in DRGN, but could not fully prevent it; (D) Cell viability measured after treatment of DRGN with the increasing concentrations of the TRPV4 inhibitor HC067047 with and without suramin; (E) We observed a small but significant increase of cell viability when DRGN were co-incubated with HC067047 (10–100 nM), while (F) HC067047 (HC, 100 nM) did not improve suramin-induced Ca2+ influx into DRGN (18-42/3-7). * p < 0.05.
Figure 3.
Effects of nimodipine and HC067047 on cell viability and intracellular Ca2+ in DRGN. (A) Treatment of DRGN for 24 h with suramin (400 µM) significantly decreased cell viability. Co-incubation with L-type VGCC nimodipine had a dose-dependent protective effect on cell viability; (B) Nimodipine (Nimo) concentrations of 100 and 150 µM significantly increased cell viability of DRGN in the presence of suramin; (C) Nimodipine (150 µM) reduced suramin-induced Ca2+ influx in DRGN, but could not fully prevent it; (D) Cell viability measured after treatment of DRGN with the increasing concentrations of the TRPV4 inhibitor HC067047 with and without suramin; (E) We observed a small but significant increase of cell viability when DRGN were co-incubated with HC067047 (10–100 nM), while (F) HC067047 (HC, 100 nM) did not improve suramin-induced Ca2+ influx into DRGN (18-42/3-7). * p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of observed cell viability of DRGN after suramin treatment in the presence of different inhibitors.
Table 1.
Summary of observed cell viability of DRGN after suramin treatment in the presence of different inhibitors.
| Substance | Nimodipine (L-Type VGCC Inhibitor) | A 967079 (TRPA1-Inhibitor) | ||||||||
| SUR 400 µM + | 0.1 µM | 1 µM | 10 µM | 100 µM | 150 µM | 1 nM | 10 nM | 100 nM | 1 µM | 10 µM |
| Change attributable to intervention (Δ% of SUR/VEH) | +3.5 ±2.6 | +2.7 ±2.0 | +3.9 ±1.9 | +15.1 * ±2.9 | +18.7 * ±2.1 | +0.2 ±2.3 | +7.1 ±3.5 | +3.0 ±2.4 | +2.6 ±1.2 | +2.8 ±4.8 |
| Substance | Efonidipine (L-Type VGCC Inhibitor) | HC 067047 (TRPV4 Inhibitor) | ||||||||
| SUR 400 µM + | 0.1 µM | 1 µM | 10 µM | 50 µM | 1 nM | 10 nM | 100 nM | 1 µM | 10 µM | |
| Change attributable to intervention (Δ% of SUR/VEH) | +3.5 ±2.7 | +0.5 ±2.6 | +0.6 ±2.7 | −1.6 ±3.2 | −7.5 * ±3.0 | +7.2 * ±2.9 | +7.1 * ±2.0 | +1.1 ±1.2 | +2.9 ±2.3 | |
| Substance | Ruthenium Red (unselective including VGCC and TRP Inhibition) | Pyr 3 (TRPC3 Inhibitor) | ||||||||
| SUR 400 µM + | 10 nM | 100 nM | 1 µM | 10 µM | 1 nM | 10 nM | 100 nM | 1 µM | 10 µM | |
| Change attributable to intervention (Δ% of SUR/VEH) | +8.1 ±4.2 | +4.4 ±1.6 | +7.6 ±3.9 | −1.5 ±2.8 | +0.2 ±2.5 | −3.4 ±4.2 | −1.3 ±3.8 | −2.8 ±3.0 | −2.6 ±2.5 | |
| Substance | SNX 482 (R-Type VGCC Inhibitor) | Ononetin (TRPM3 Inhibitor) | ||||||||
| SUR 400 µM + | 2 nM | 20 nM | 200 nM | 3 nM | 30 nM | 300 nM | 3 µM | 30 µM | ||
| Change attributable to intervention (Δ% of SUR/VEH) | −3.2 ±1.8 | −2.2 ±3.0 | +2.1 ±2.3 | −5.9 ±2.6 | +2.5 ±2.1 | −2.2 ±2.4 | −2.7 ±3.3 | −1.9 ±2.7 | ||
| Substance | Ω-Conotoxin MVIIC (N-, P-, Q-Type VGCC Inh) | |||||||||
| SUR 400 µM + | 1 nM | 10 nM | 100 nM | 1 µM | ||||||
| Change attributable to intervention (Δ% of SUR/VEH) | −2.2 ±1.8 | −2.0 ±2.6 | +1.9 ±1.6 | +1.9 ±3.3 | ||||||
* Statistically significant (p < 0.05).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Von der Ahe, D.; Huehnchen, P.; Balkaya, M.; Peruzzaro, S.; Endres, M.; Boehmerle, W. Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies. Molecules 2018, 23, 346. https://doi.org/10.3390/molecules23020346
AMA Style
Von der Ahe D, Huehnchen P, Balkaya M, Peruzzaro S, Endres M, Boehmerle W. Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies. Molecules. 2018; 23(2):346. https://doi.org/10.3390/molecules23020346
Chicago/Turabian StyleVon der Ahe, David, Petra Huehnchen, Mustafa Balkaya, Sarah Peruzzaro, Matthias Endres, and Wolfgang Boehmerle. 2018. "Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies" Molecules 23, no. 2: 346. https://doi.org/10.3390/molecules23020346