Separation and Purification of Fructo-Oligosaccharide by High-Speed Counter-Current Chromatography Coupled with Precolumn Derivatization


Abstract
:1. Introduction
2. Results and Discussion
2.1. Strategy for Separation and Purification of FOSs
2.2. Optimization of HPLC for FOSs
2.3. Selection of Precolumn Derivatization Method
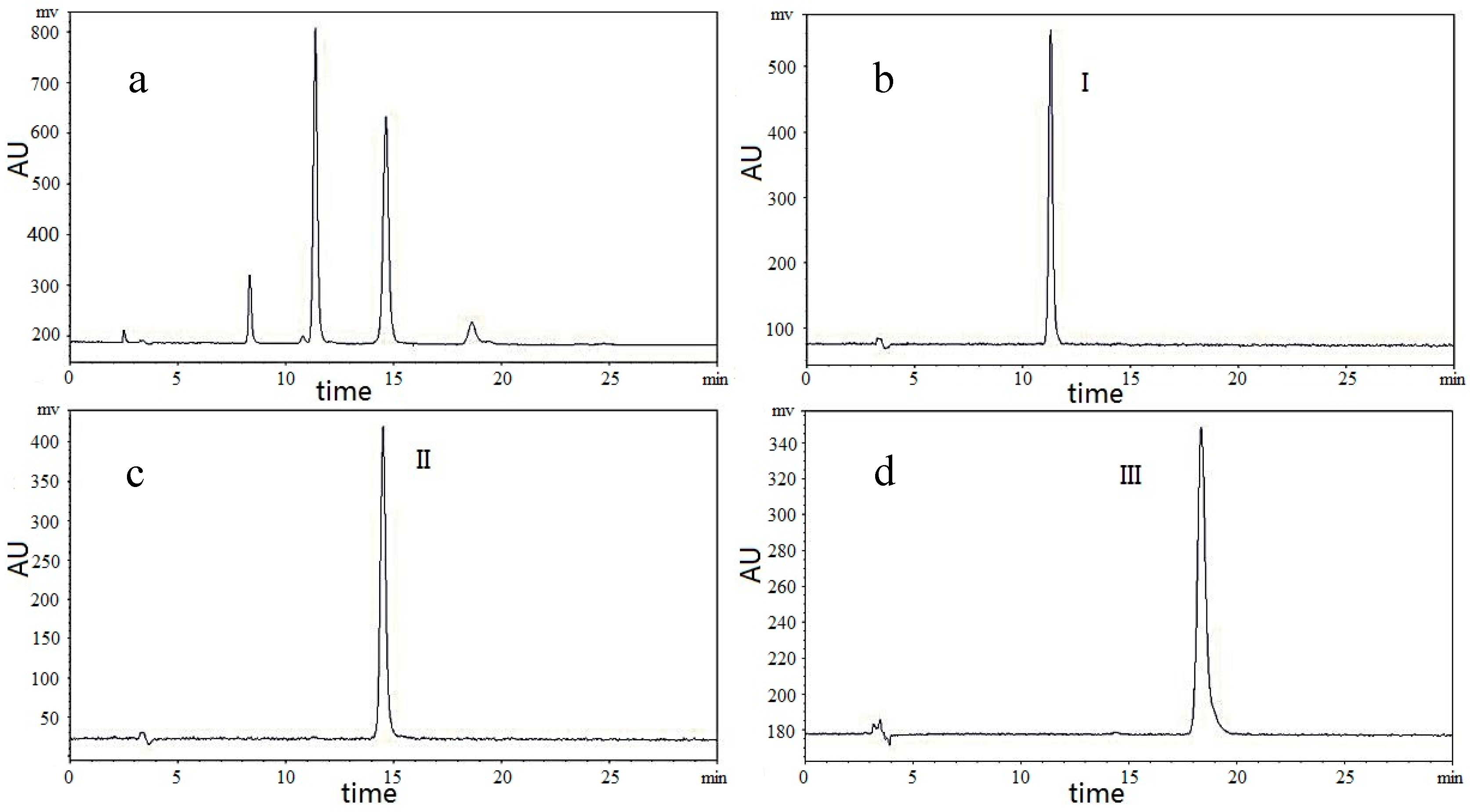
2.4. Selection of Two-Phase Solvent System and Other Conditions of HSCCC
3. Materials and Methods
3.1. Reagents and Materials
3.2. Precolumn Derivatization
3.3. HSCCC Separation
3.3.1. Apparatus
3.3.2. Measurement of Partition Coefficient
3.3.3. Preparation of Sample Solution and Two-Phase Solvent
3.3.4. Separation Procedure
3.4. Reduction Reaction
3.5. HPLC-ELSD Analysis
3.6. Structural Identification
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bindels, L.B.; Delzenne, N.M.; Cani, P.D.; Walter, J. Towards a more comprehensive concept for prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Catry, E.; Bindels, B.L.; Tailleux, A.; Lestave, S.; Neyrinck, M.A.; Goossens, J.F.; Lobysheva, I.; Plovier, H.; Essaghir, A.; Demoulin, J.B.; et al. Targeting the gut microbiota with inulin-type fructans: Preclinical demonstration of a novel approach in the management of endothelial dysfunction. Gut 2018, 67, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Fukuda, S.; Fujiwara, A.; Suda, W.; Hattori, M.; Kikuchi, J.; Ohno, H. Multiple omics uncovers host-gut microbial mutualism during prebiotic fructooligosaccharide supplementation. DNA Res. 2014, 21, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Weinborn, V.; Valenzuela, C.; Olivares, M.; Arredondo, M.; Weill, R.; Pizarro, F. Prebiotics increase heme iron bioavailability and do not affect non heme iron bioavailability in humans. Food Funct. 2017, 8, 1994–1999. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.Y.; Abdullah, M.; Määttänen, P.; Pilar, A.V.C.; Scruten, E.; Johnson-Henry, K.C.; Napper, S.; O’Brien, C.; Jones, N.L.; Sherman, P.M. Protein kinase C δ signaling is required for dietary prebioticinduced strengthening of intestinal epithelial barrier function. Sci. Rep. 2017, 1, 1–10. [Google Scholar]
- Zhou, S.; Liu, X.; Guo, Y.; Wang, Q.; Peng, D.; Cao, L. Comparison of the immunological activities of arabinoxylans from wheat bran with alkali and xylanase-aided extraction. Carbohydr. Polym. 2010, 81, 784–789. [Google Scholar] [CrossRef]
- Yen, C.H.; Kuo, Y.W.; Tseng, Y.H.; Lee, H.L.; Chen, H.L. Beneficial effects of fructo-oligosaccharides upplementation on fecal bifidobacteria and index of peroxidation status in constipated nursing-home residents—A placebocontrolled, diet-controlled trial. Nutrition 2011, 27, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Roberfroid, M.B. Fructo-oligosaccharide malabsorption: Benefit for gastrointestinal functions. Curr. Opin. Gastroenterol. 2000, 16, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Agheli, N.; Kabir, M.; Berni-Canani, S.; Petitjean, E.; Boussairi, A.; Luo, J.; Bornet, F.; Slama, G.; Rizkalla, S.W. Plasma lipids and fatty acid synthase activity are regulated by short-chain fructo-oligosaccharides in sucrose-fed insulin-resistant rats. J. Nutr. 1998, 128, 1283–1288. [Google Scholar] [CrossRef]
- Li, J.; Cheong, K.L.; Zhao, J.; Hu, D.J.; Chen, X.Q.; Qiao, C.F.; Zhang, Q.W.; Chen, Y.W.; Li, S.P. Preparation of inulin-type fructooligosaccharides using fast protein liquid chromatography coupled with refractive index detection. J. Chromatogr. A 2013, 1308, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.M.; Hu, J.; Zhao, M.Y. Isolation and quantitative determination of inulin-type oligosaccharides in roots of Morinda officinalis. Carbohydr. Polym. 2011, 83, 1997–2004. [Google Scholar] [CrossRef]
- Ronkart, S.N.; Blecker, C.S.; Fourmanoir, H.; Fougnies, C.; Deroanne, C.; Herck, J.C.V.; Paquot, M. Isolation and identification of inulooligosaccharides resulting from inulin hydrolysis. Anal. Chim. Acta 2007, 604, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.K.; Duan, W.J.; Liu, F.; Shi, X.G.; Geng, Y.L.; Wang, X. Preparative separation of polyphenols from the flowers of Paeonia lactiflora Pall. by high-speed counter-current chromatography. J. Chromatogr. B 2014, 947–948, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Q.; Zhao, X.; Li, Y.; Yang, D.; Zhou, T.T.; Fan, G.R.J. Impurities preparation of sodium tanshinone IIA sulfonate by high-speed counter-current chromatography and identification by liquid chromatography/multistage tandem mass spectrometry. J. Chromatogr. B 2013, 1288, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Adhami, H.R.; Zehl, M.; Dangl, C.; Dorfmeister, D.; Stadler, M.; Urban, E. Preparative isolation of oleocanthal, tyrosol, and hydroxytyrosol from olive oil by HPCCC. Food Chem. 2015, 170, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Shu, X.K.; Wang, X.; Yu, J.Q.; Jing, F. Preparative isolation of seven diterpenoid alkaloids from Aconitum coreanum by pH-zone-refining counter-current chromatograph. Molecules 2014, 19, 12619–12629. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, Z.J.; Guo, X.F.; Yuan, J.P.; Zheng, C.C. Preparative separation of gingerols from Zingiber officinale by high speed counter-current chromatography using stepwise elution. Food Chem. 2011, 125, 1476–1480. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, Z.Y.; Duan, W.J.; Lei, F.; Xu, S.S.; Wang, X. Isolation and purification of seven lignans from Magnolia sparengeri by high-speed-counter-current chromatography. J. Chromatogr. B 2011, 879, 3775–3779. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Betz, P.; Sun, Y.C.; Gorb, S.N.; Lindhorst, T.K.; Krueger, A. Saccharide-modified nanodiamond conjugates for the efficient detection and removal of pathogenic bacteria. Chemistry 2012, 18, 6485–6492. [Google Scholar] [CrossRef] [PubMed]
- Ciucanu, J.; Kerek, F.A. Simple and rapid method for the permethylation of carbohydrates. Carbohydr. Res. 1984, 131, 209–217. [Google Scholar] [CrossRef]
- Patankar, M.S.; Oehninger, S.; Barnett, T.R.; Williams, L.; Clark, G.F. A revised structure for fucoidan may explain some of its biological activities. J. Biol. Chem. 1993, 268, 21770–21776. [Google Scholar] [PubMed]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.K.; Jin, M.C.; He, C.H. Preparative separation of four majoralkaloids from medicinal plant of Tripterygium Wilfordii Hook F using high-speed counter-current chromatography. Sep. Purif. Technol. 2007, 56, 319–324. [Google Scholar] [CrossRef]
- Lin, Y.A.; Chalker, J.M.; Davis, B.G. Olefin Cross-Metathesis on Proteins: Investigation of Allylic Chalcogen Effects and Guiding Principles in Metathesis Partner Selection. J. Am. Chem. Soc. 2010, 132, 16805–16811. [Google Scholar] [CrossRef] [PubMed]
- Bruyn, A.D.; Loo, J.V. The identification by 1H- and 13C-n.m.r. spectroscopy of sucrose, 1-kestose, and neokestose in mixtures present in plant extracts. Carbohydr. Res. 1991, 211, 131–136. [Google Scholar] [CrossRef]
- Timmermans, J.W.; Waard, P.D.; Tournois, H.; Leeflang, B.R.; Vliegenthart, J.F.G. NMR spectroscopy of nystose. Carbohydr. Res. 1993, 243, 379–384. [Google Scholar] [CrossRef]
- Cui, C.B.; Yang, M.; Yao, Z.W.; Cai, B.; Luo, Z.P.; Xu, Y.K. Studies on proton and carbon-13 nuclear magnetic resonance of inulin-type oligosaccharides. Chin. J. Med. Chem. 1995, 5, 32–39. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Solvent Systems | KD Values | |||
|---|---|---|---|---|
| I | II | III | ||
| petroleum ether–ethyl acetate–methanol–water | 1:1:1:1 | 1.29 | 1.47 | 1.61 |
| 4:5:4:5 | 2.04 | 2.41 | 2.93 | |
| petroleum ether–n-butanol–methanol–water | 3:2:2:3 | 0.28 | 0.35 | 0.52 |
| 3:2:1:4 | 1.34 | 1.97 | 2.68 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, W.; Ji, W.; Wei, Y.; Zhao, R.; Chen, Z.; Geng, Y.; Jing, F.; Wang, X. Separation and Purification of Fructo-Oligosaccharide by High-Speed Counter-Current Chromatography Coupled with Precolumn Derivatization. Molecules 2018, 23, 381. https://doi.org/10.3390/molecules23020381
Duan W, Ji W, Wei Y, Zhao R, Chen Z, Geng Y, Jing F, Wang X. Separation and Purification of Fructo-Oligosaccharide by High-Speed Counter-Current Chromatography Coupled with Precolumn Derivatization. Molecules. 2018; 23(2):381. https://doi.org/10.3390/molecules23020381
Chicago/Turabian StyleDuan, Wenjuan, Wenhua Ji, Yuanan Wei, Ruixuan Zhao, Zijian Chen, Yanling Geng, Feng Jing, and Xiao Wang. 2018. "Separation and Purification of Fructo-Oligosaccharide by High-Speed Counter-Current Chromatography Coupled with Precolumn Derivatization" Molecules 23, no. 2: 381. https://doi.org/10.3390/molecules23020381