Radical Polymerization of Alkyl 2-Cyanoacrylates



Abstract
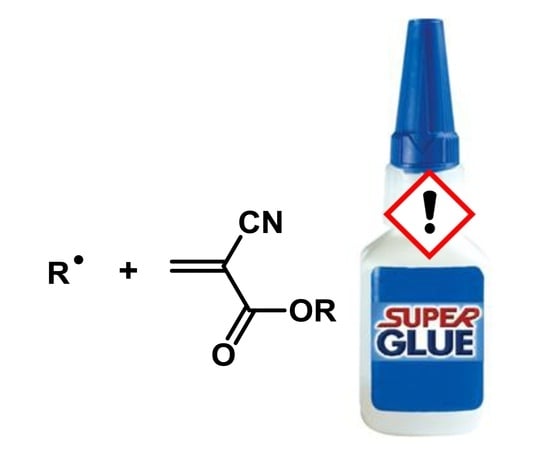
:
1. Introduction
2. General Reactivity and Properties
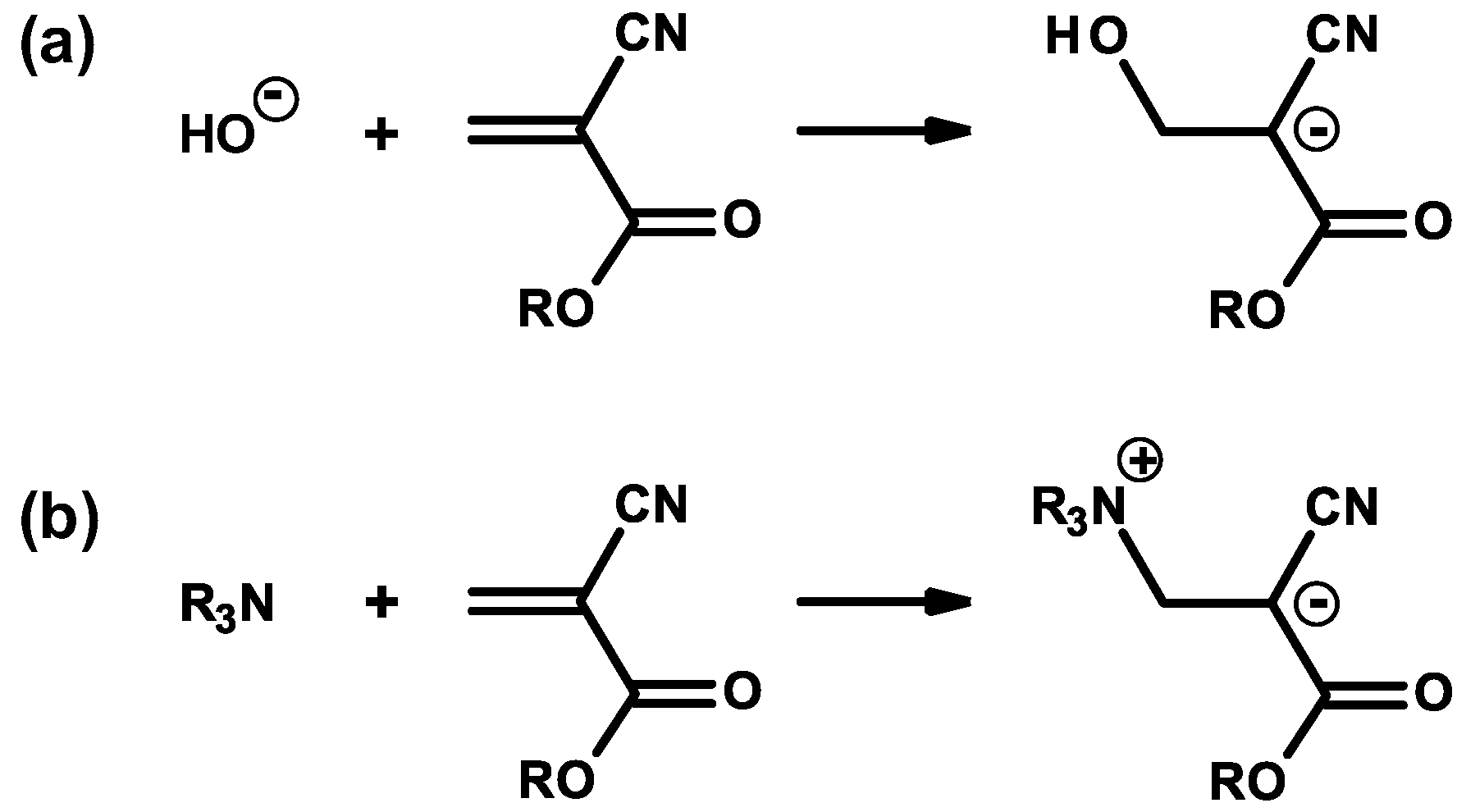
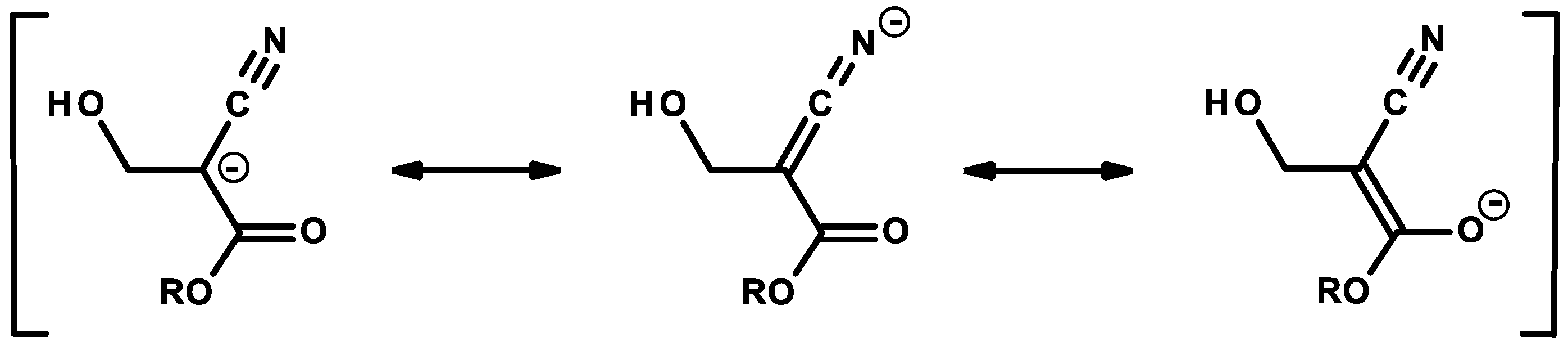
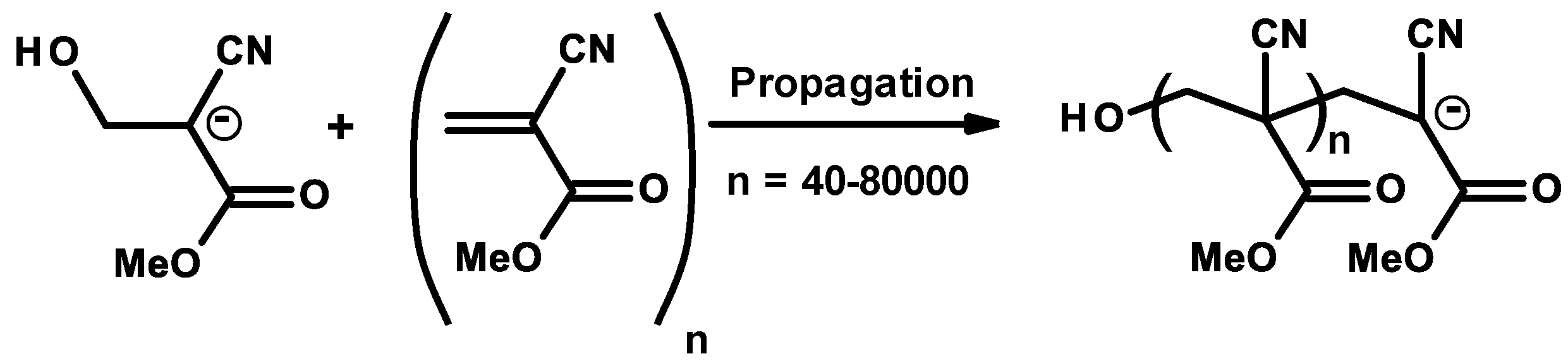
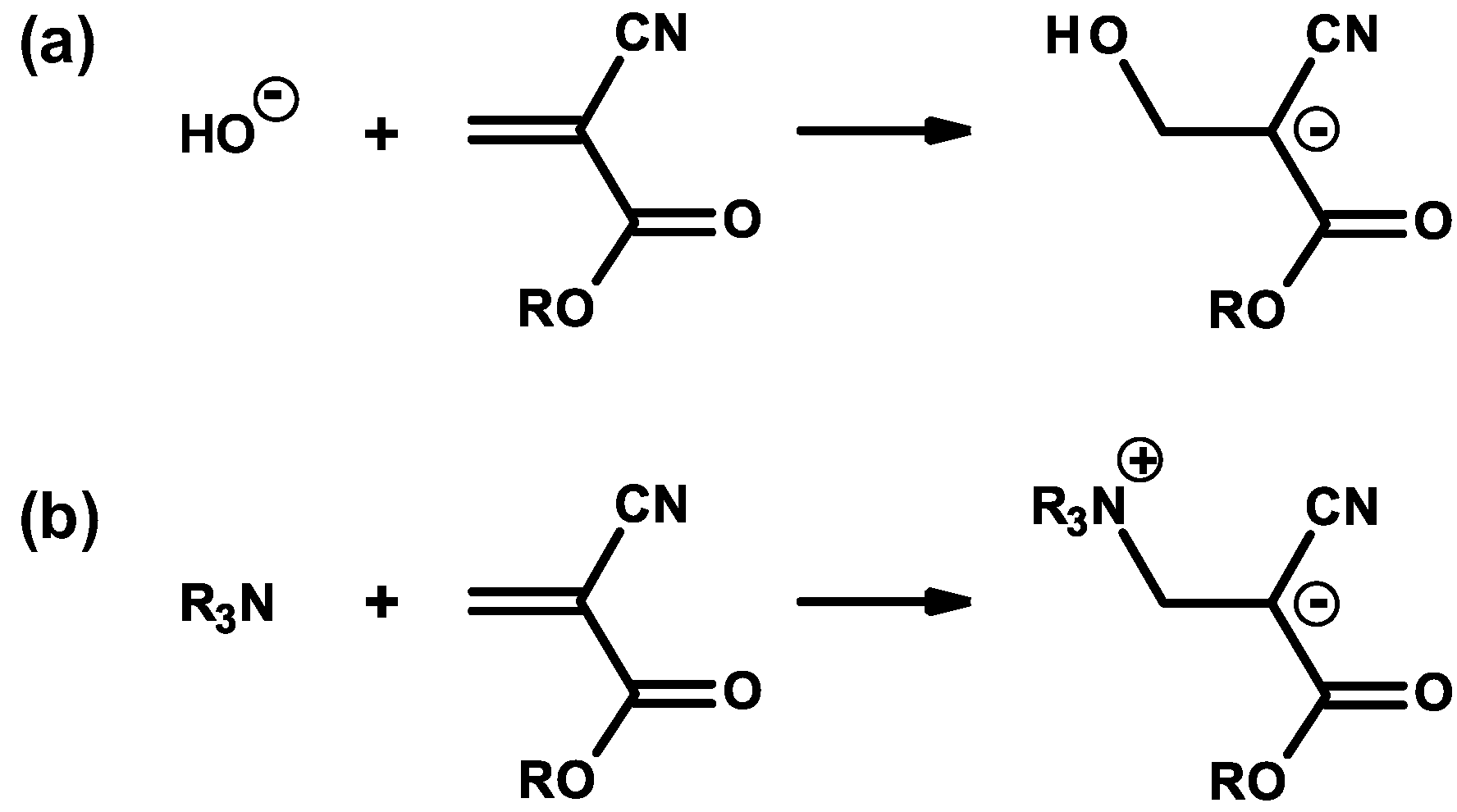
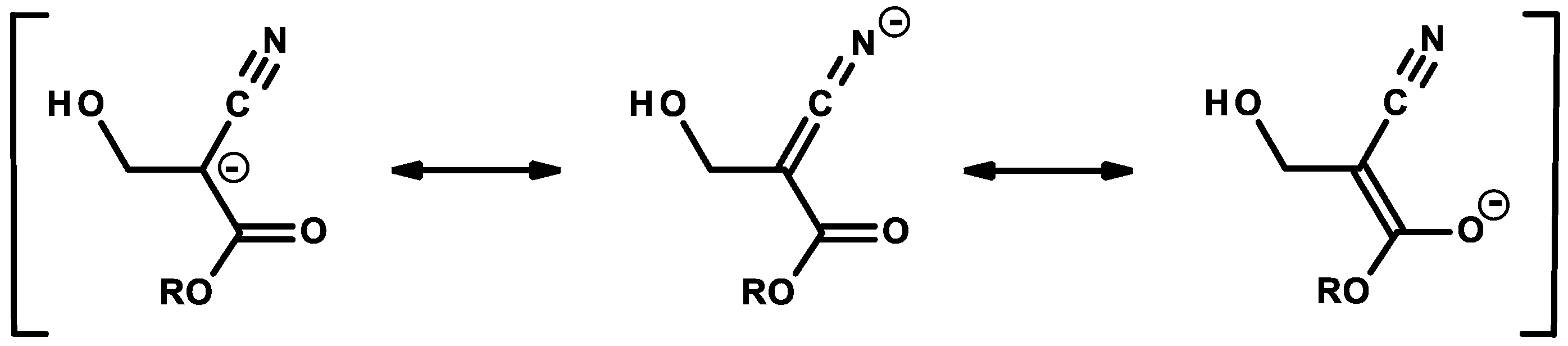
2.1. Anionic-Type Polymerizations
2.2. Polymer Properties and Stability
3. Conventional (Non-Living) Radical Polymerizations
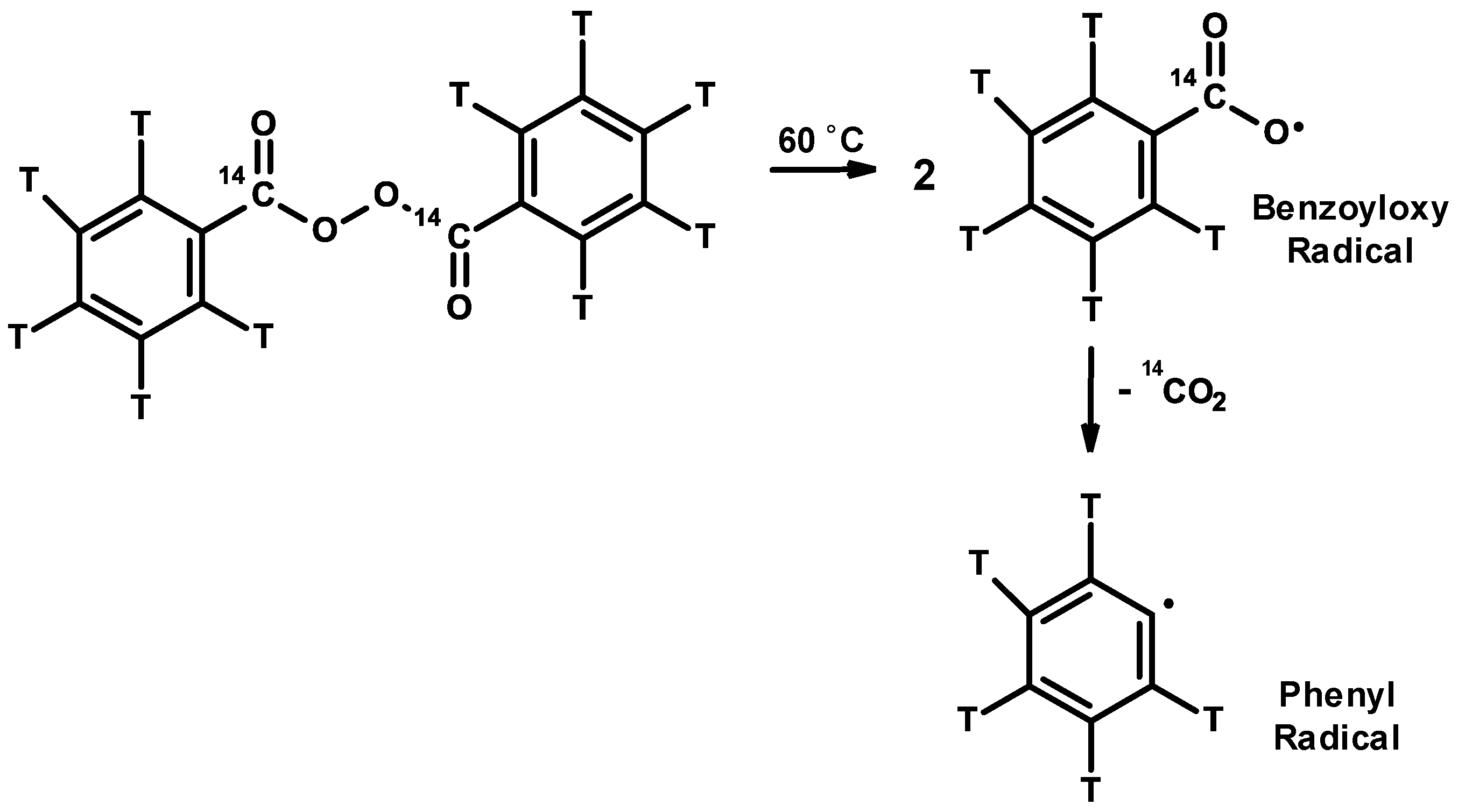
3.1. Inhibitors and Precautions
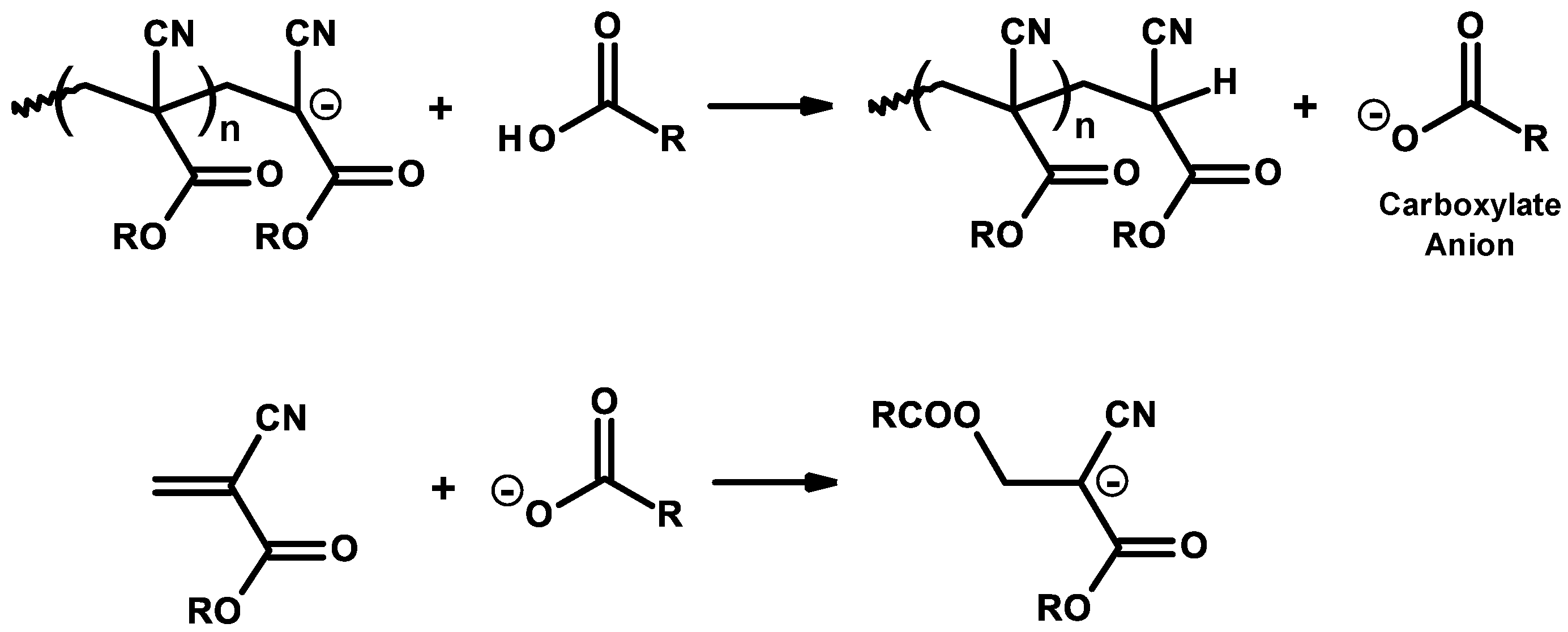
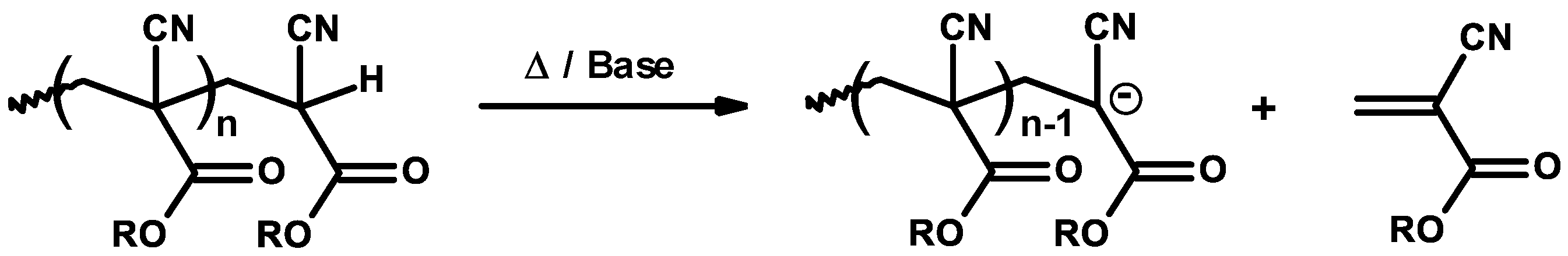
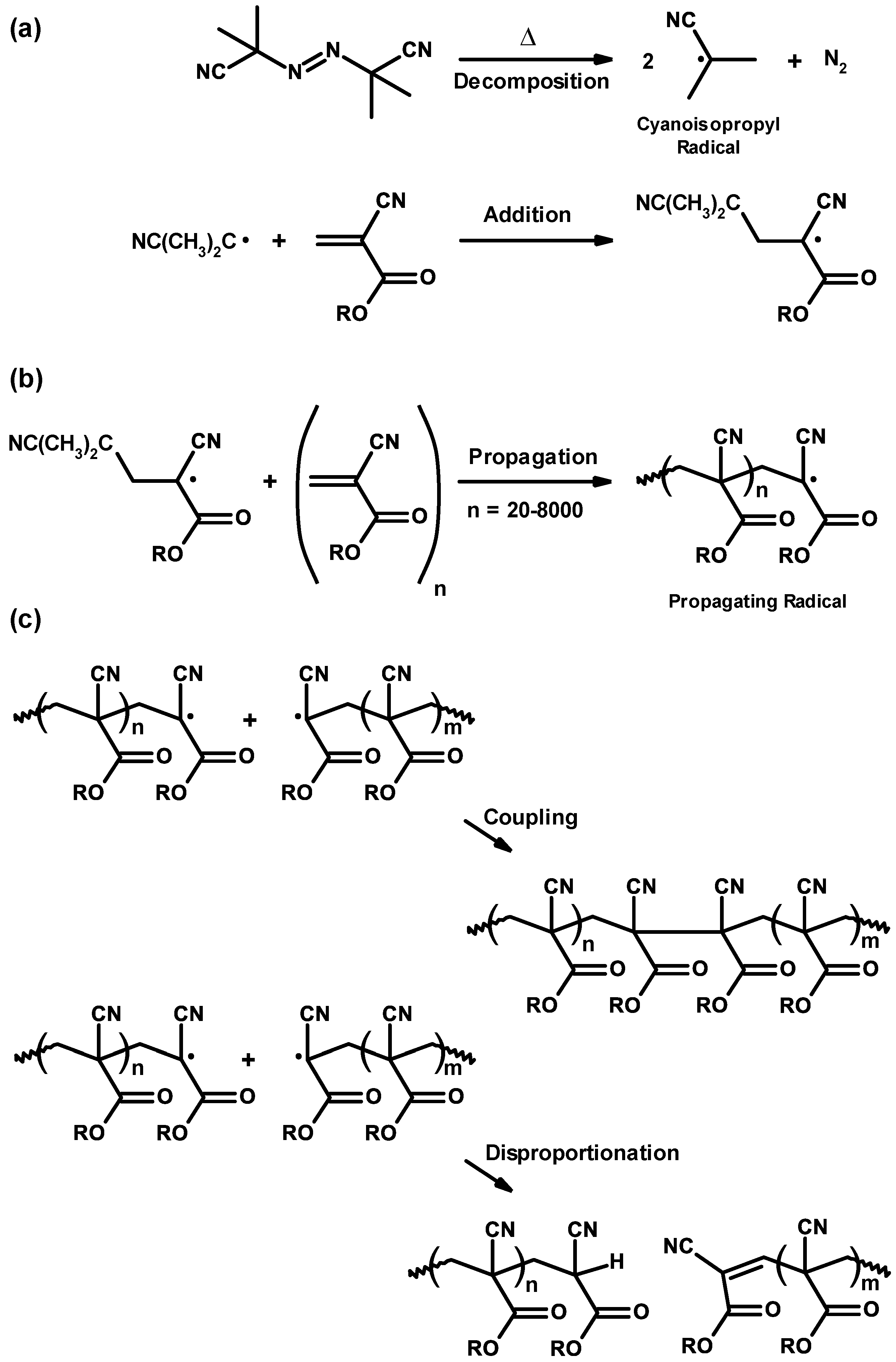
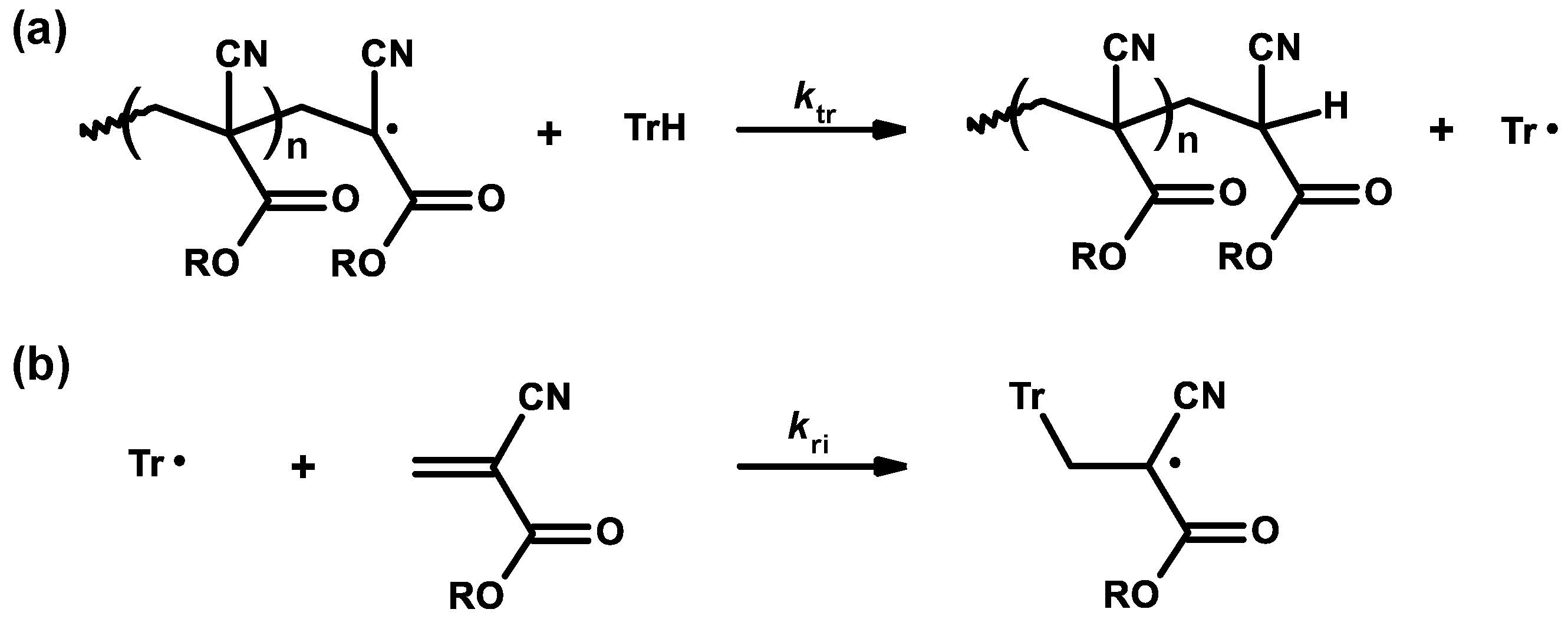
3.2. General Mechanism
3.3. Mechanism and Kinetics of Radical Homopolymerization
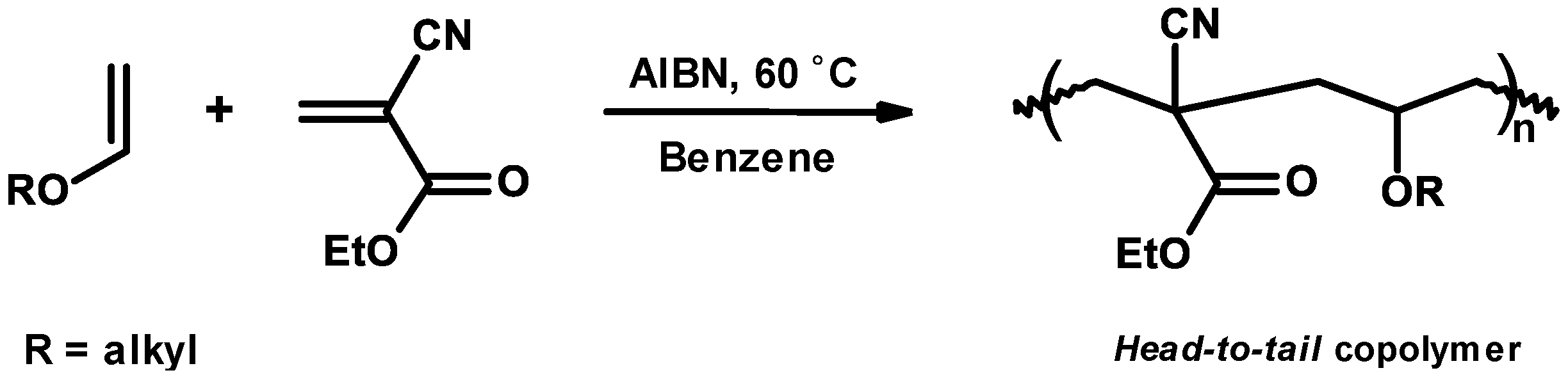
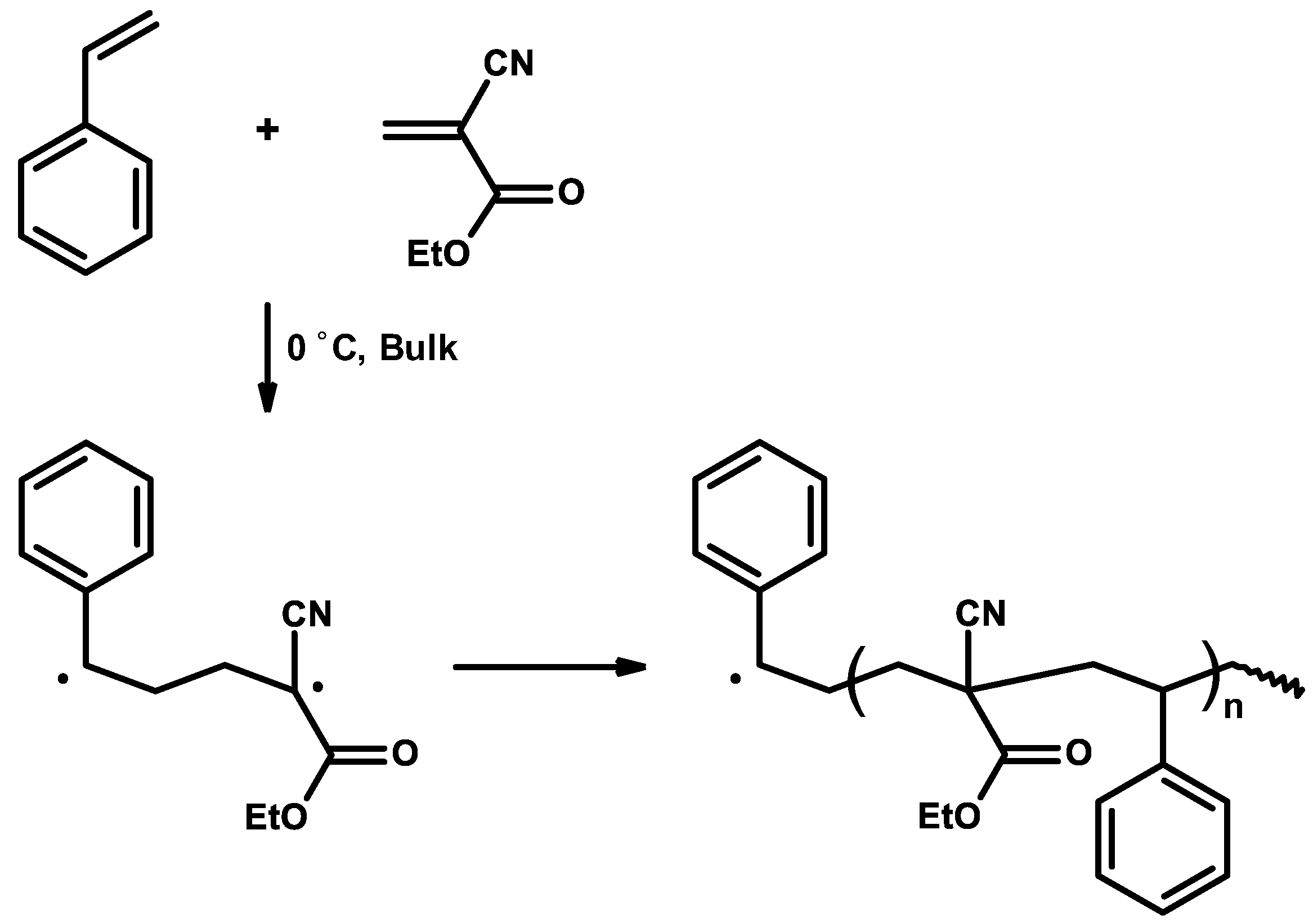
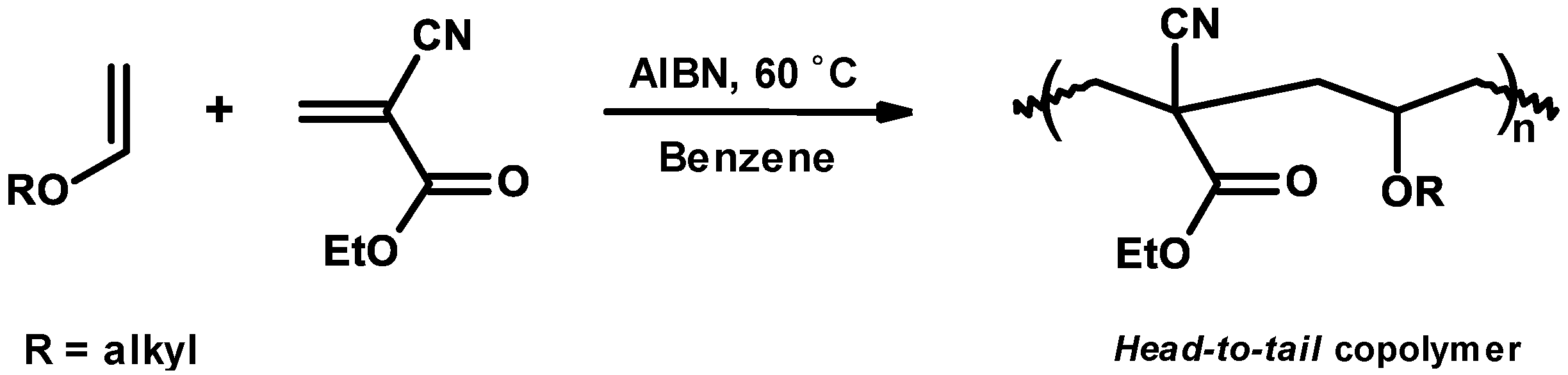
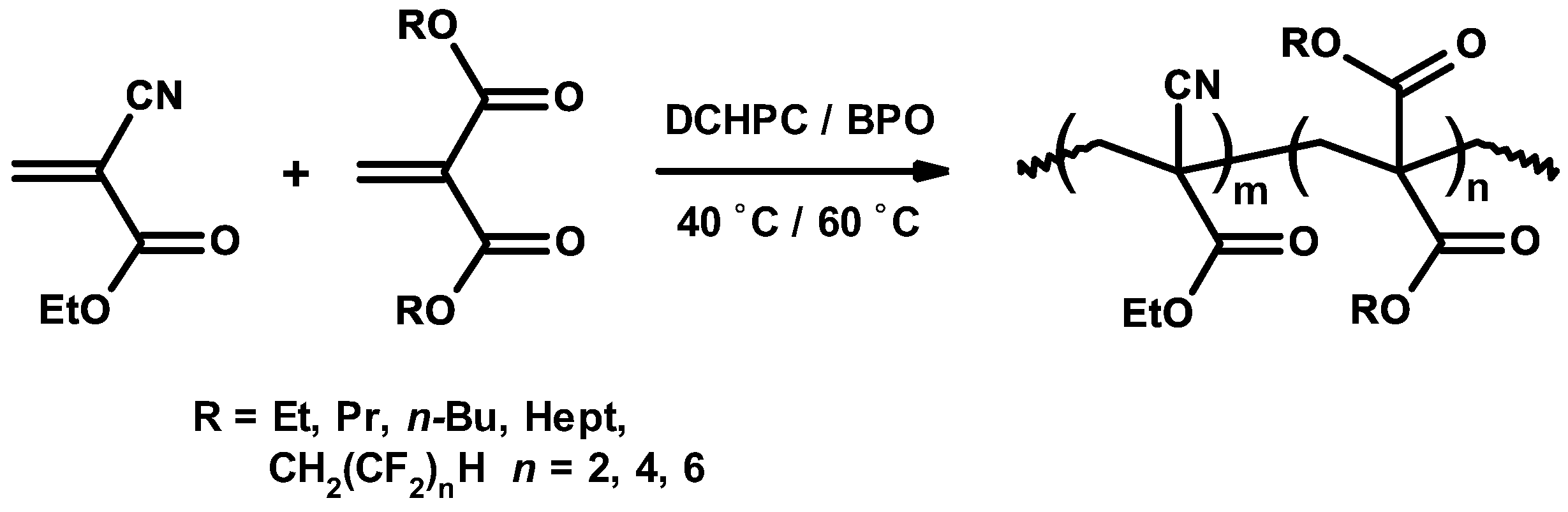
3.4. Mechanism and Kinetics of Radical Copolymerization
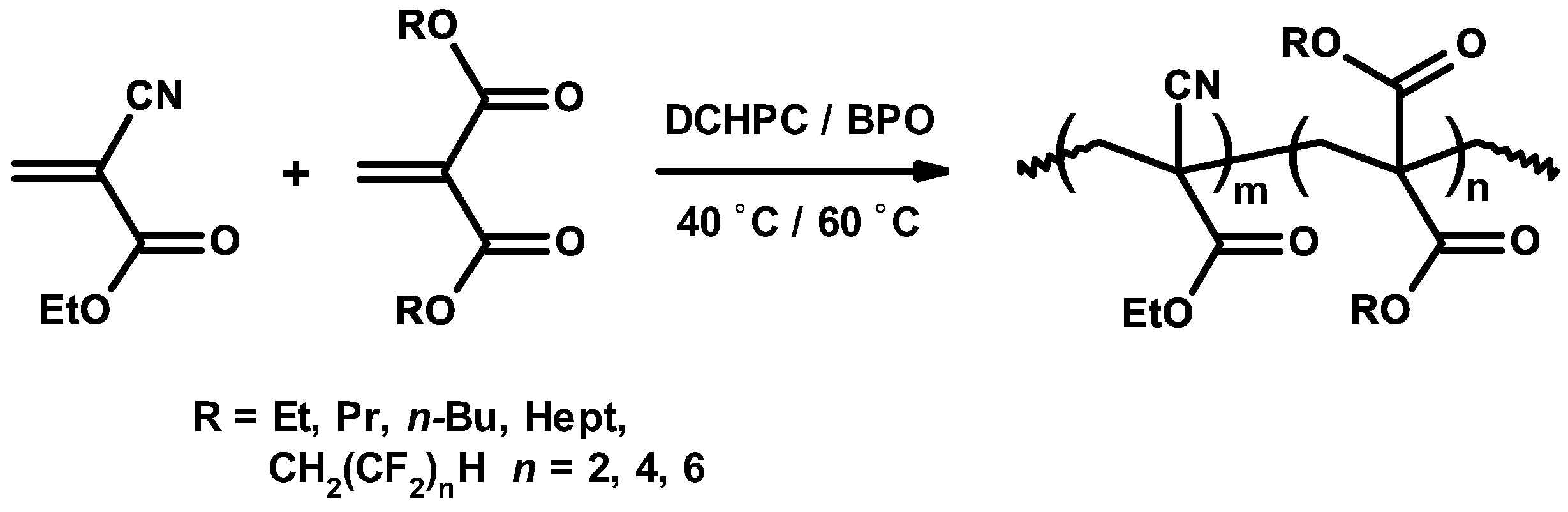
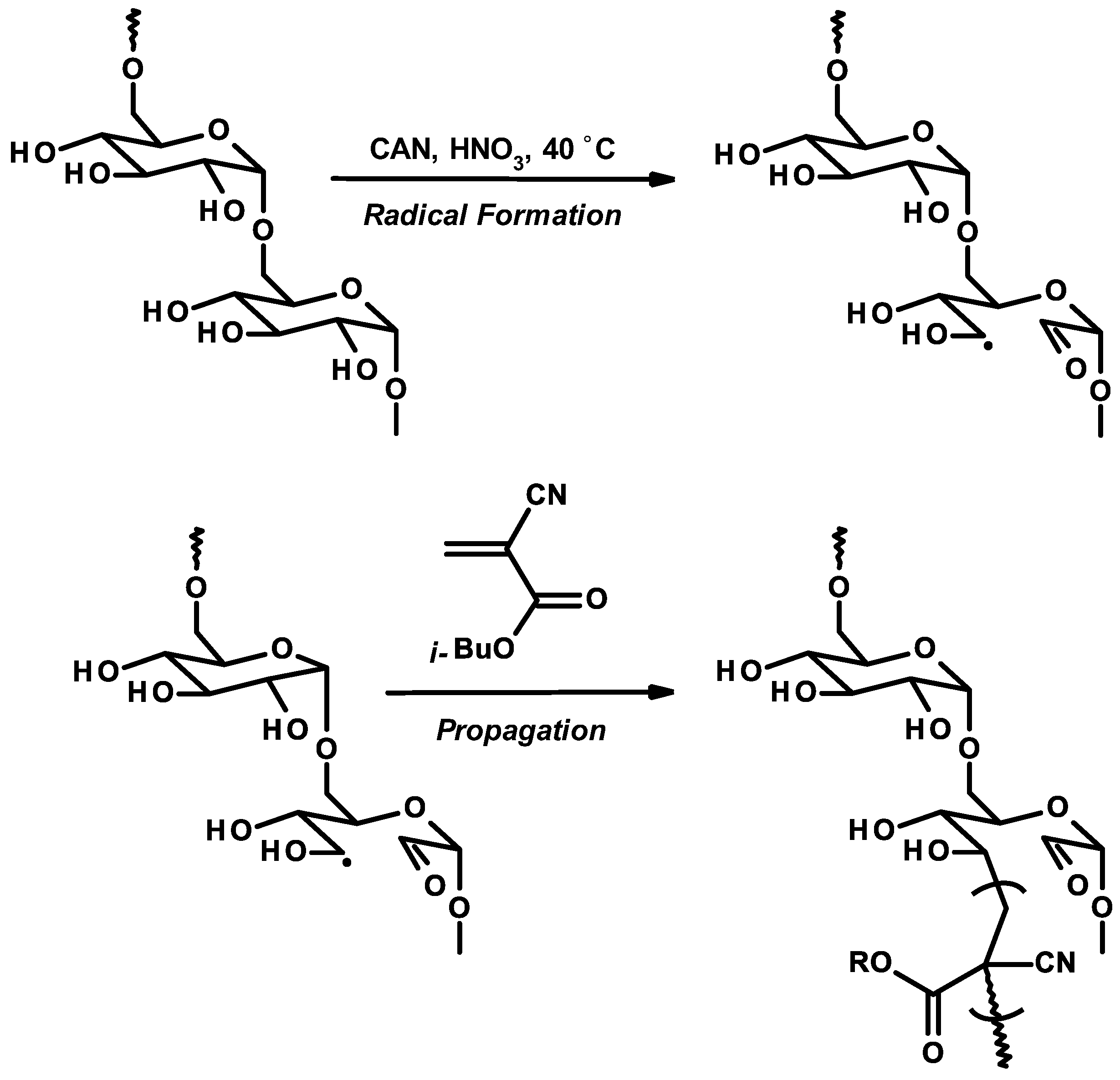
3.5. Synthetic Copolymerization Studies
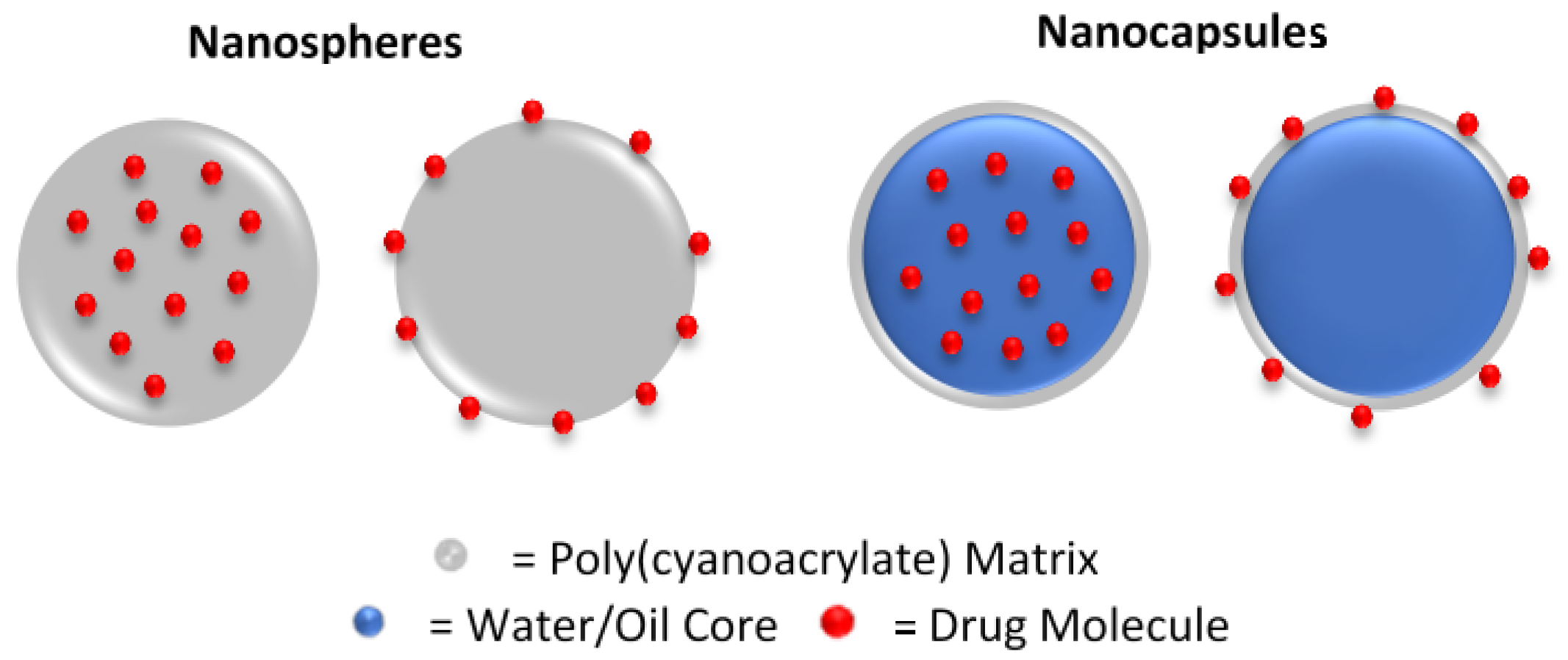
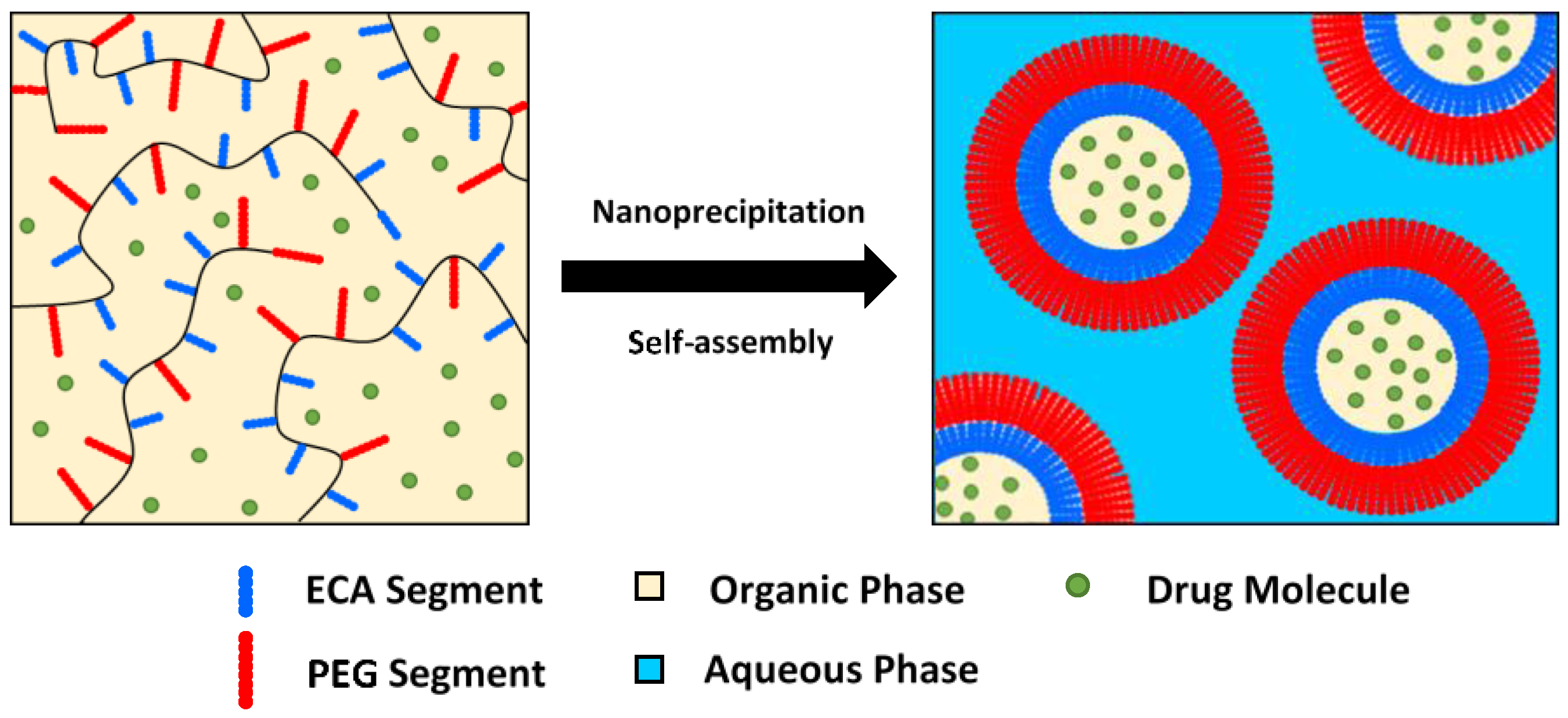
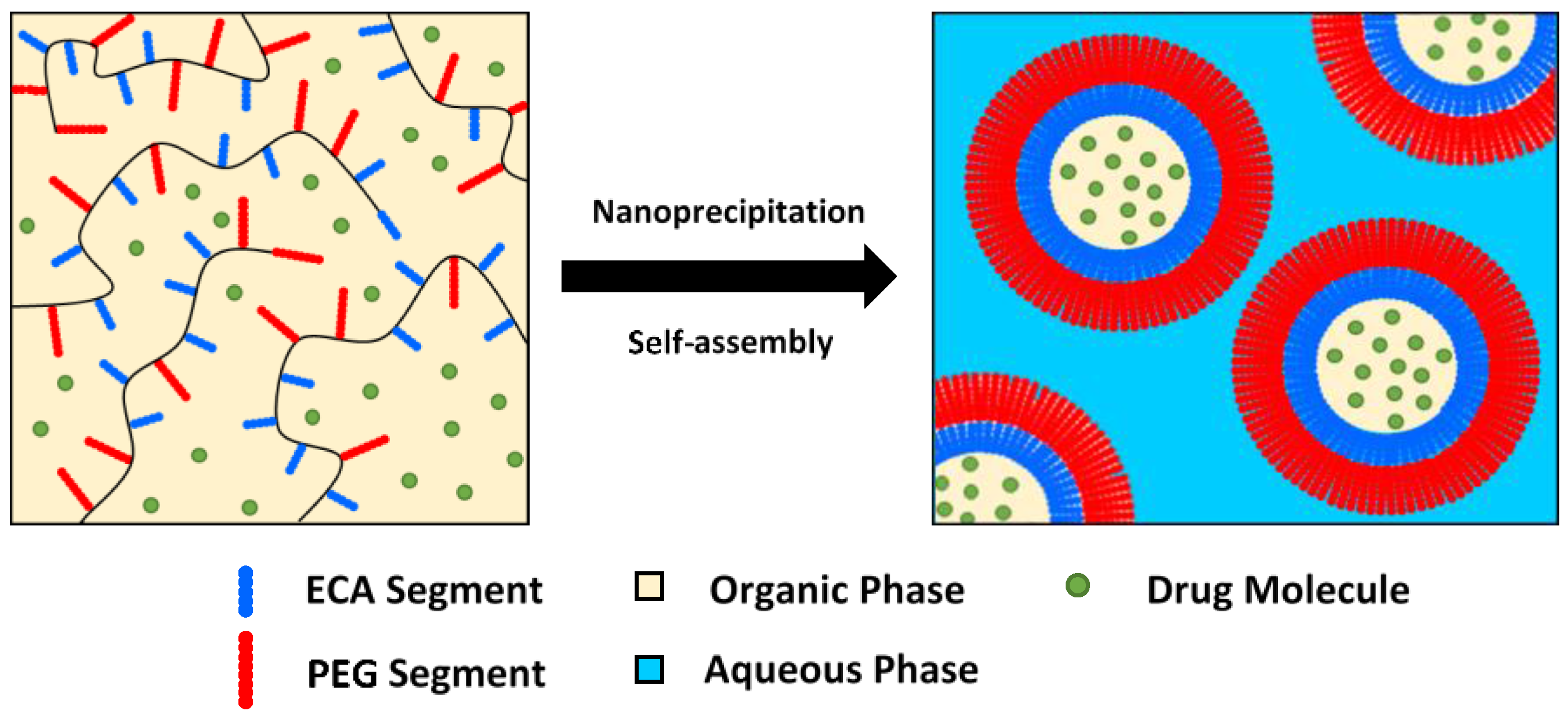
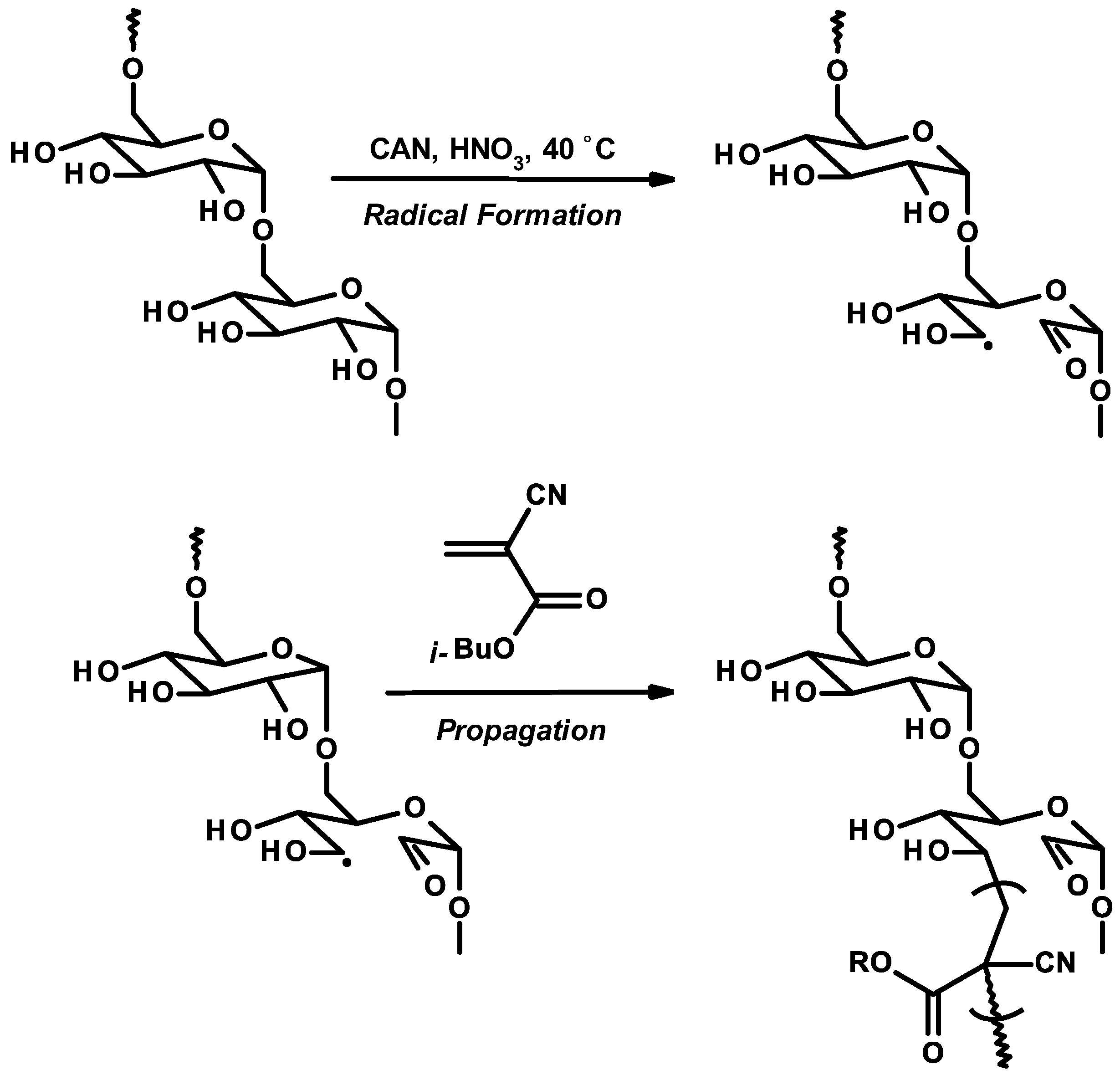
3.6. Nanoparticles
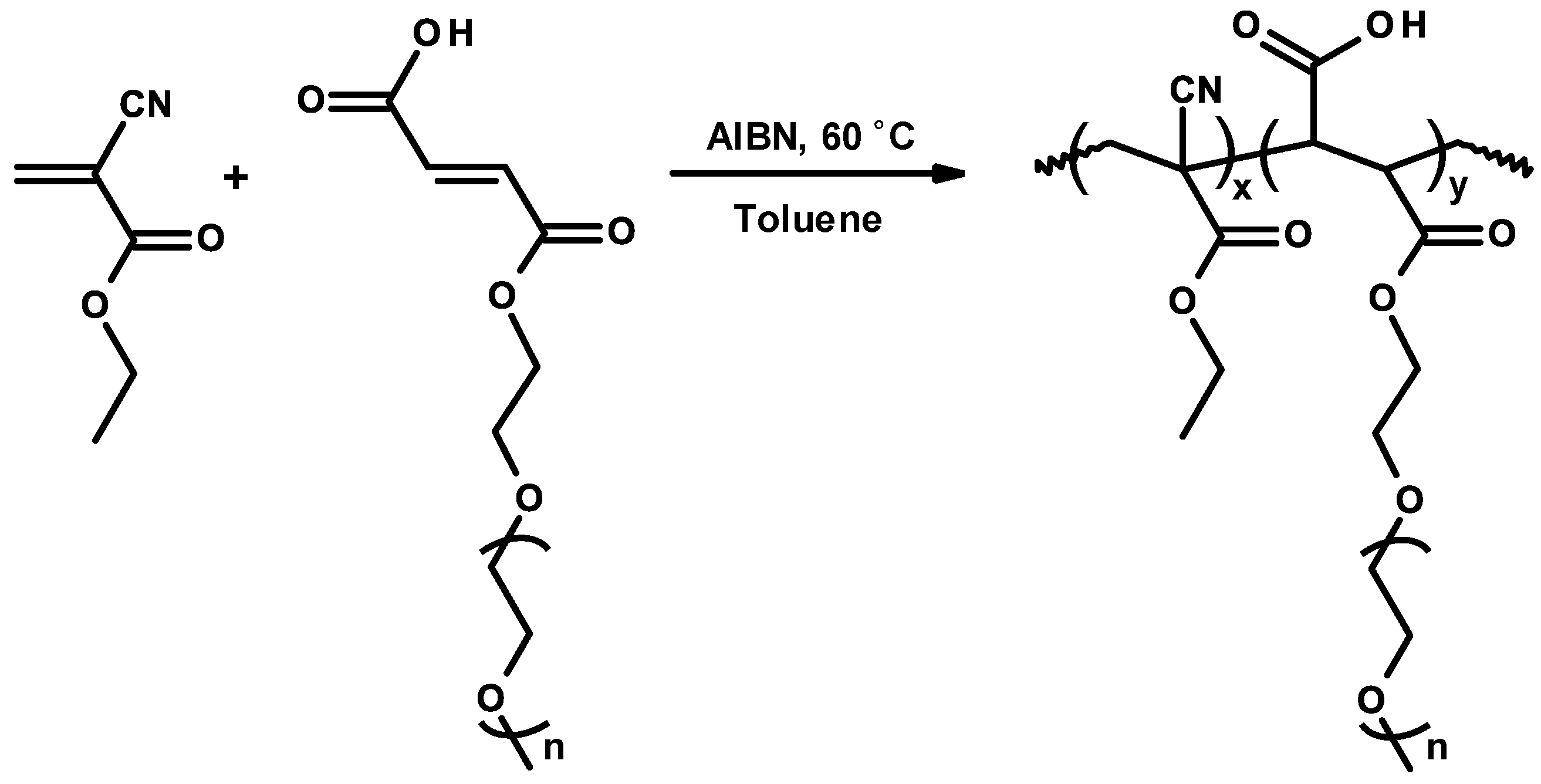
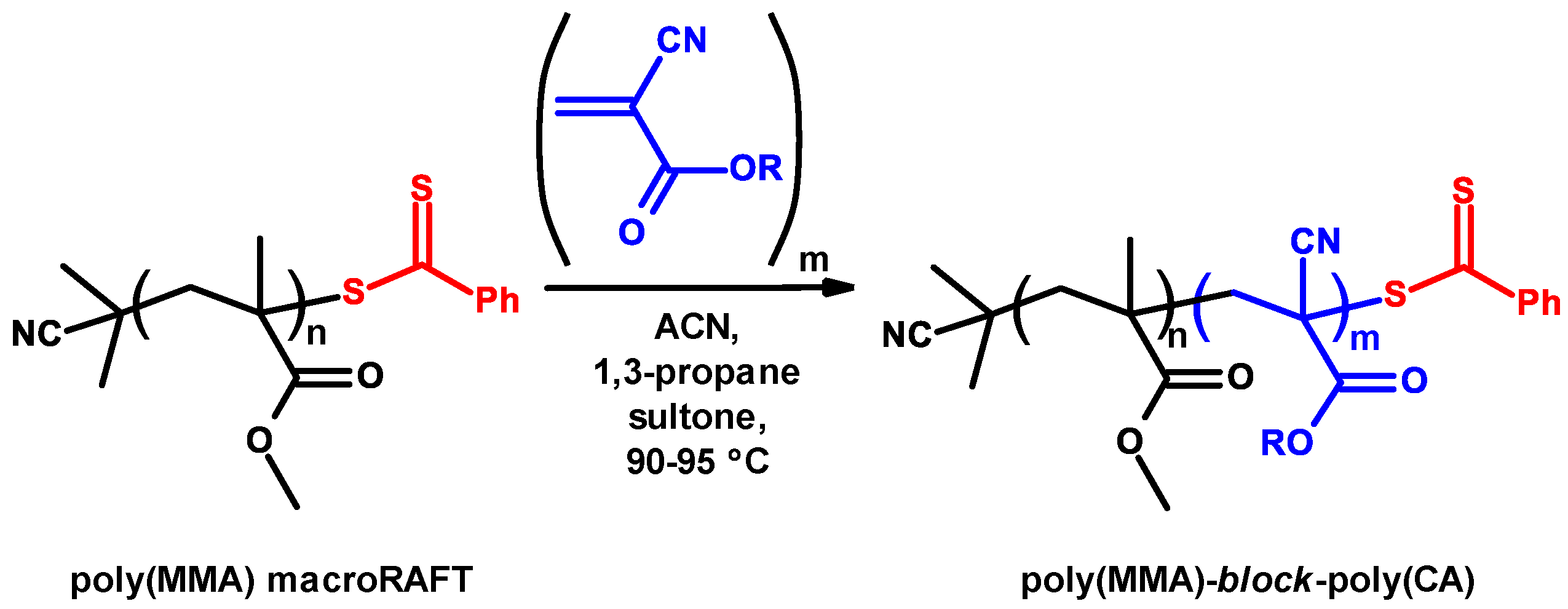
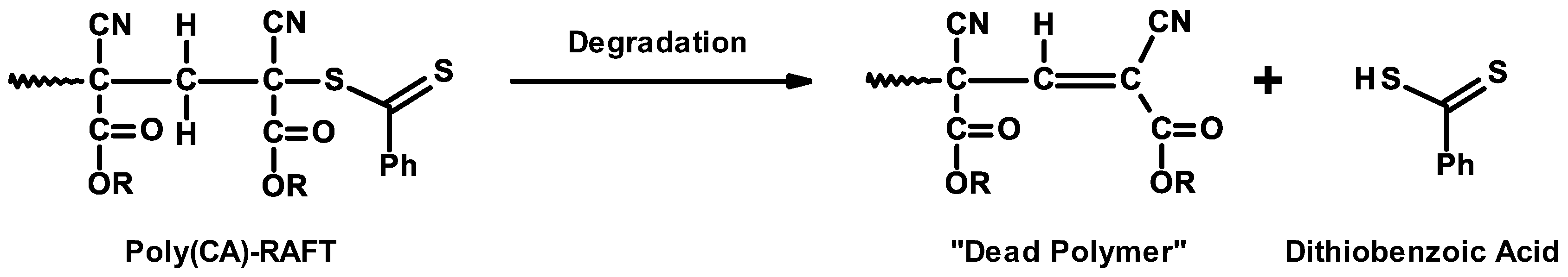
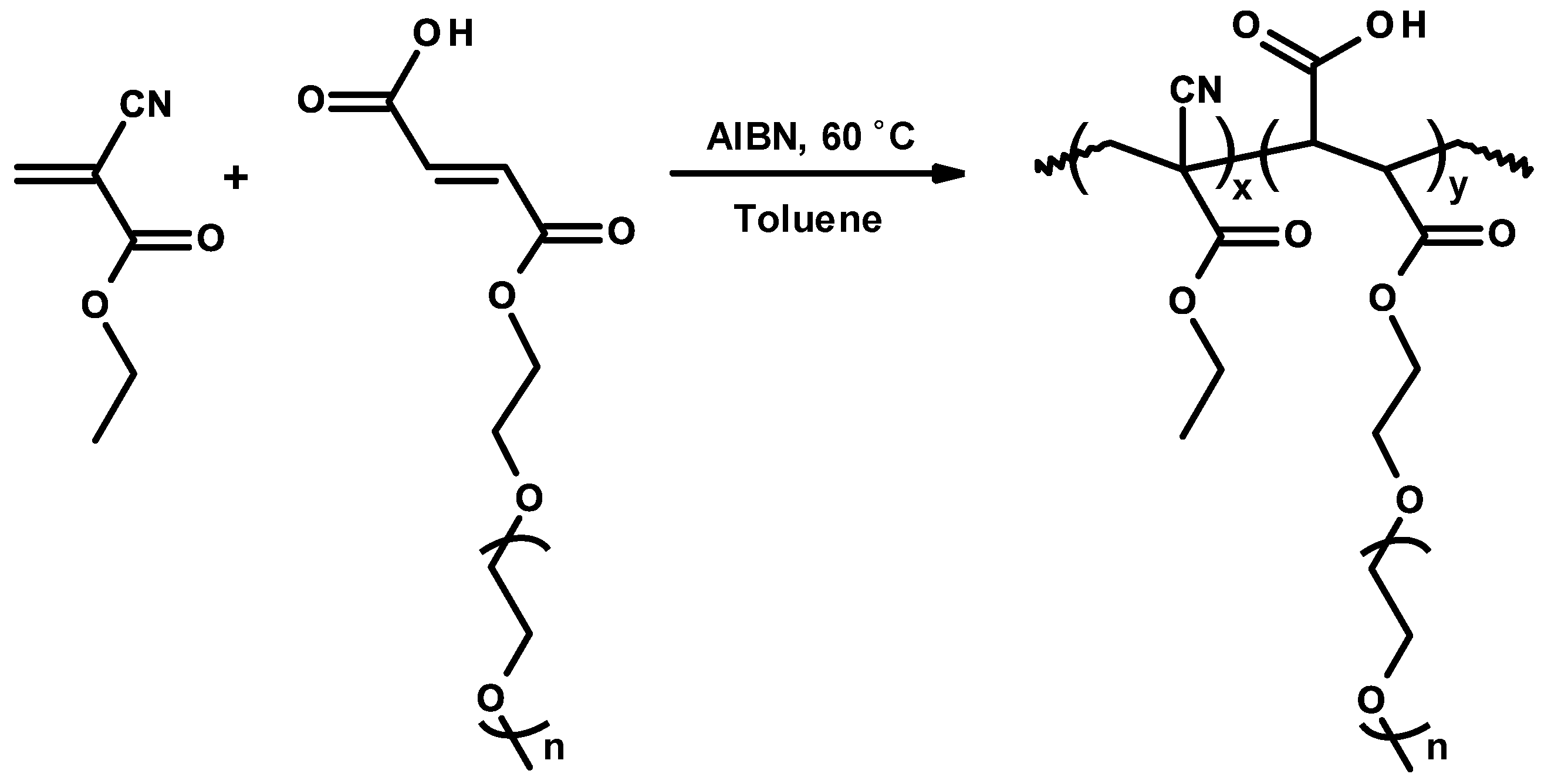
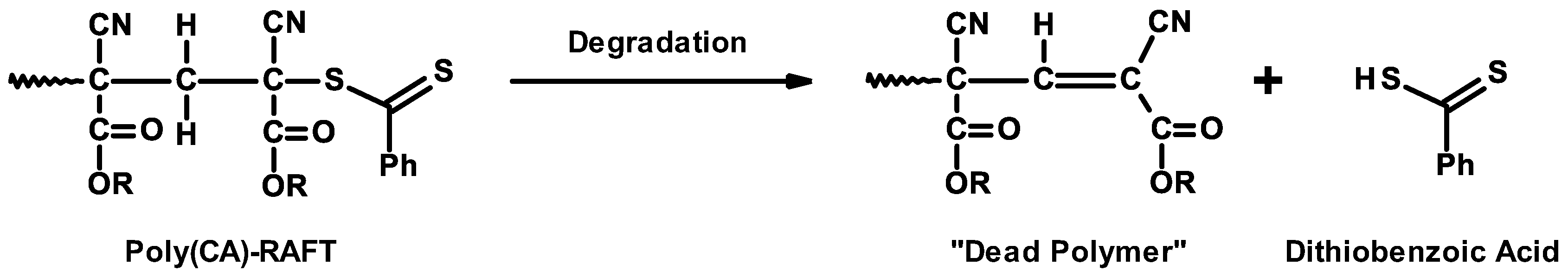
3.7. Controlled/Living Radical Polymerizations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klemarczyk, P.; Guthrie, J. Advances in anaerobic and cyanoacrylate adhesives. In Advances in Structural Adhesive Bonding, 1st ed.; Dillard, D., Ed.; Woodhead Publishing Limited: Cambridge, UK, 2010; pp. 96–131. ISBN 978-1-84569-435-7. [Google Scholar]
- Shantha, K.L.; Thennarasu, S.; Krishnamurti, N. Developments and applications of cyanoacrylate adhesives. J. Adhes. Sci. Technol. 1989, 3, 237–260. [Google Scholar] [CrossRef]
- Fink, J.K. Chapter 13—Cyanoacrylates. In Reactive Polymers Fundamentals and Applications, 2nd ed.; Fink, J.K., Ed.; Elsevier: London, UK, 2013; pp. 317–330. ISBN 978-1-4557-3149-7. [Google Scholar]
- Burns, B. Polycyanoacrylates. In Encyclopedia of Polymer Science and Technology, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; Volume 4, pp. 1–27. ISBN 9780471440260. [Google Scholar] [CrossRef]
- O’Connor, J.T. Acrylic ester polymers, 2-cyanoacrylic ester polymers. In Kirk-Othmer Encyclopedia of Chemical Technology, 4th ed.; Kroschwitz, J.I., Howe-Grant, M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; pp. 1–7. ISBN 9780471238966. [Google Scholar] [CrossRef]
- Woods, J. Polycyanoacrylates. In Encyclopedia of Polymer Science and Technology, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; pp. 643–652. ISBN 9780471288244. [Google Scholar] [CrossRef]
- Singer, A.J.; Thode, H.C., Jr. A review of the literature on octylcyanoacrylate tissue adhesive. Am. J. Surg. 2004, 187, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Quinn, J.V.; Hollander, J.E. The cyanoacrylate topical skin adhesives. Am. J. Emerg. Med. 2008, 26, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Shivamurthy, D.M.; Singh, S.; Reddy, S. Comparison of octyl-2-cyanoacrylate and conventional sutures in facial skin closure. Natl. J. Maxillofac. Surg. 2010, 1, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, G.D.; Ballester, A.M.; Aliena-Valero, A.; Carabén-Redaño, A.; Lloris, J.M. Use of cyanoacrylate adhesives in general surgery. Surg. Today 2015, 45, 939–956. [Google Scholar] [CrossRef] [PubMed]
- Hynes, S.J. Biomedical applications of 2-cyanoacrylates. Irish Chem. News 2009, 25, 24–28. [Google Scholar]
- Nicolas, J.; Couvreur, P. Synthesis of poly(alkyl cyanoacrylate)-based colloidal nanomedicines. Wiley Interdiscipl. Rev. Nanomed. Nanobiotechnol. 2009, 1, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Dubernet, C.; Fattal, E.; Pinto-Alphandary, H.; Couvreur, P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv. Drug Deliv. Rev. 2003, 55, 519–548. [Google Scholar] [CrossRef]
- Graf, A.; McDowell, A.; Rades, T. Poly(alkycyanoacrylate) nanoparticles for enhanced delivery of therapeutics—Is there real potential? Expert Opin. Drug Deliv. 2009, 6, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Yordanov, G. Poly(alkyl cyanoacrylate) nanoparticles as drug carriers: 33 years later. Bulg. J. Chem. 2012, 1, 61–73. [Google Scholar]
- Arias, J.L.; Gallardo, V.; Ruiz, M.A. Chapter 4—Multifunctional anticancer nanomedicine based on a magnetically responsive cyanoacrylate polymer. Methods in Enzymol. 2012, 508, 61–88. [Google Scholar] [CrossRef]
- Bumbrah, G.S. Cyanoacrylate fuming method for detection of latent fingermarks: a review. Egypt J. Forensic Sci. 2017, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, M.; Kalman, J.R.; Reedy, B.J. Synthesis and characterization of four alkyl 2-cyanoacrylate monomers and their precursors for use in latent fingerprint detection. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 257–277. [Google Scholar] [CrossRef]
- McArdle, C.; Xiao, E.S.; Van Wijk, K.; Zhao, L.; Schneider, A.; Hansjoerg, A.; Petrick, P.; Domanski, R.; Schafer, V.; Schafer, S. An Article Comprising a Film on a Carrier or Release Substrate. Patent WO2013174430A1, 2013. [Google Scholar]
- Gololobov, Y.G.; Krylova, T.O. 2-Cyanoacrylates as reagents in heteroatomic Synthesis (a review). Heteroatom Chem. 1995, 6, 271–280. [Google Scholar] [CrossRef]
- Leggat, P.A.; Kedjarune, U.; Smith, D.R. Toxicity of cyanoacrylate adhesives and their occupational impacts for dental staff. Ind. Health 2004, 42, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Leggat, P.A.; Smith, D.R.; Kedjarune, U. Surgical applications of cyanoacrylate adhesives: A review of toxicity. ANZ J. Surg. 2007, 77, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, E.F.; Johnston, D.S.; Pepper, D.C.; Dunn, D.J. Ionic and zwitterionic polymerization of n-alkyl 2-cyanoacrylates. Polym. Lett. Ed. 1977, 15, 399–405. [Google Scholar] [CrossRef]
- Pepper, D.C.; Ryan, B. Initiation processes in polymerizations of alkyl cyanoacrylates by tertiary amines: Inhibition by strong acids. Makromol. Chem. 1983, 184, 383–394. [Google Scholar] [CrossRef]
- Pepper, D.C.; Ryan, B. Kinetics of polymerization of alkyl cyanoacrylates by tertiary amines and phosphines. Makromol. Chem. 1983, 184, 395–410. [Google Scholar] [CrossRef]
- Cronin, J.P.; Pepper, D.C. Zwitterionic polymerization of butyl cyanoacrylate by triphenylphosphine and pyridine. Makromol. Chem. 1988, 189, 85–102. [Google Scholar] [CrossRef]
- Johnston, D.S.; Pepper, D.C. Polymerisation via macrozwitterions, 1. Ethyl and butyl cyanoacrylates polymerised by triethyl and triphenylphosphines. Makromol. Chem. 1981, 182, 393–406. [Google Scholar] [CrossRef]
- Eromosele, I.C.; Pepper, D.C. Anionic polymerizations of butyl cyanoacrylate by tetrabutylammonium salts. Makromol. Chem. 1989, 190, 3095–3103. [Google Scholar] [CrossRef]
- Millet, G.H. Cyanoacrylate adhesive. In Structural Adhesives, Chemistry and Technology, 1st ed.; Hartshorn, S.R., Ed.; Plenum Press: New York, NY, USA, 1986; pp. 249–307. ISBN 978-1-4684-7781-8. [Google Scholar]
- Dossi, M.; Storti, G.; Moscatelli, D. Synthesis of poly(alkyl cyanoacrylates) as biodegradable polymers for drug delivery applications. Macromol. Symp. 2010, 289, 124–128. [Google Scholar] [CrossRef]
- Baskaran, D.; Müller, A.H.E. Kinetic investigation on metal free anionic polymerization of methyl methacrylate using tetraphenylphosphonium as the counterion in tetrahydrofuran. Macromolecules 1997, 30, 1869–1874. [Google Scholar] [CrossRef]
- Pepper, D.C. Transfer by weak acids in the slow-initiation-no-termination (SINT) polymerization of butyl cyanoacrylate. Makromol. Chem. 1987, 188, 527–536. [Google Scholar] [CrossRef]
- Geacintov, C.; Smid, J.; Szwarc, M. Kinetics of anionic polymerization of styrene in tetrahydrofuran. J. Am. Chem. Soc. 1962, 84, 2508–2514. [Google Scholar] [CrossRef]
- Georgopanos, P.; Lo, T.Y.; Ho, R.M.; Avgeropoulos, A. Synthesis, molecular characterization and self-assembly of (PS-b-PDMS)n type linear (n = 1, 2) and star (n = 3, 4) block copolymers. Polym. Chem. 2017, 8, 843–850. [Google Scholar] [CrossRef]
- Polymeropoulos, G.; Zapsas, G.; Ntetsikas, K.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. 50th Anniversary Perspective: Polymers with Complex Architectures. Macromolecules 2017, 50, 1253–1290. [Google Scholar] [CrossRef]
- Kulkarni, R.K.; Porter, H.J.; Leonard, F. Glass transition temperatures of poly(alkyl α-cyanoacrylates). J. Appl. Polym. Sci. 1973, 17, 3509–3514. [Google Scholar] [CrossRef]
- Cheung, K.H.; Guthrie, J.; Otterburn, M.S.; Rooney, J.M. The dynamic mechanical properties of poly(alkyl 2-cyanoacrylates). Makromol. Chem. 1987, 188, 3041–3046. [Google Scholar] [CrossRef]
- Bykova, T.A.; Kiparisova, Y.G.; Lebedev, B.V.; Mager, K.A.; Gololobov, Y.G. A calorimetric study of ethyl-α-cyanoacrylate and its polymerization and a study of polyethyl-α-cyanoacrylate at 13–450 K and normal pressure. Polym. Sci. U.S.S.R. 1991, 33, 537–543. [Google Scholar] [CrossRef]
- Chorbadjiev, K.G.; Novakov, P.C. Study of the thermal degradation of poly(alkyl α-cyanoacrylate)s. Eur. Polym. J. 1991, 27, 1009–1015. [Google Scholar] [CrossRef]
- Hickey, A.; Leahy, J.J.; Birkinshaw, C. End-group identity and its effect on the thermal degradation of poly(butyl cyanoacrylate). Macromol. Rapid Commun. 2001, 22, 1158–1162. [Google Scholar] [CrossRef]
- Barkan, Y.; Levinman, M.; Veprinsky-Zuzuliya, I.; Tsach, T.; Merqioul, E.; Blum, G.; Domb, A.J.; Basu, A. Comparative evaluation of polycyanoacrylates. Acta Biomat. 2017, 48, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, J.; Otterburn, M.S.; Rooney, J.M.; Tsang, C.N. The effect of heat on the molecular weight of poly(ethyl 2-cyanoacrylate) adhesive. J. Appl. Polym. Sci. 1985, 30, 2863–2867. [Google Scholar] [CrossRef]
- Birkinshaw, C.; Pepper, D.C. The thermal degradation of polymers of n-butylcyanoacrylate prepared using tertiary phosphine and amine initiators. Polym. Degrad. Stab. 1986, 16, 241–259. [Google Scholar] [CrossRef]
- Leonard, F.; Kulkarni, R.K.; Brandes, G.; Nelson, J.; Cameron, J.J. Synthesis and degradation of poly(alkyl α-cyanoacrylates). J. Appl. Polym. Sci. 1966, 10, 259–272. [Google Scholar] [CrossRef]
- Ryan, B.; McCann, G. Novel sub-ceiling temperature rapid depolymerization-repolymerization reactions of cyanoacrylate polymers. Macromol. Rapid Commun. 1996, 17, 217–227. [Google Scholar] [CrossRef]
- Robello, D.R.; Eldridge, T.D.; Swanson, M.T. Degradation and stabilization of polycyanoacrylates. J. Polym. Sci. A Polym. Chem. 1999, 37, 4570–4581. [Google Scholar] [CrossRef]
- Han, G.H.; Kim, S. Controlled degradation of poly(ethyl cyanoacrylate-co-methyl methacrylate)(PECA-co-PMMA) copolymers. Polymer 2009, 50, 1270–1280. [Google Scholar] [CrossRef]
- Canale, A.J.; Goode, W.E.; Kinsinger, J.B.; Panchak, J.R.; Kelso, R.L.; Graham, R.K. Methyl α-cyanoacrylate. I. Free-radical homopolymerization. J. Appl. Polym. Sci. 1960, 4, 231–236. [Google Scholar] [CrossRef]
- Busfield, W.K. Ceiling Temperatures. In Aspects of Degradation and Stabilization of Polymers, 1st ed.; Jellinek, H.H.G., Ed.; Elsevier: Amsterdam, The Netherlands, 1978; pp. 39–78. ISBN 978-0444415639. [Google Scholar]
- Otsu, T.; Yamada, B. Determination of Q, e parameters for methyl α-cyanoacrylate. Makromol. Chem. 1967, 110, 297–299. [Google Scholar] [CrossRef]
- Yamada, B.; Yoshioka, M.; Otsu, T. Determination of absolute rate constants for radical polymerization and copolymerization of ethyl α-cyanoacrylate in the presence of effective inhibitors against anionic polymerization. Makromol. Chem. 1983, 184, 1025–1033. [Google Scholar] [CrossRef]
- Yamada, B.; Hayashi, T.; Otsu, T. Determination of absolute rate constants for radical polymerization and copolymerization of ethyl α-chloroacrylate: Effects of substituents on reaction rates of monomer and polymer radical. J. Macromol. Sci. Part A Chem. 1983, 7, 1023–1039. [Google Scholar] [CrossRef]
- Yamada, B.; Kontani, T.; Yoshioka, M.; Otsu, T. Determination of absolute rate constants for free radical polymerization of ethyl α-fluoroacrylate and characterization of the polymer. J. Polym. Sci. A Polym. Chem. 1984, 22, 2381–2393. [Google Scholar] [CrossRef]
- Rooney, T.R.; Mavroudakis, E.; Lacík, I.; Hutchinson, R.A.; Moscatelli, D. Pulsed-laser and quantum mechanics study of n-butyl cyanoacrylate and methyl methacrylate free-radical copolymerization. Polym. Chem. 2015, 6, 1594–1603. [Google Scholar] [CrossRef]
- Tang, H.; Tsarevsky, N.V. Preparation and Functionalization of Linear and Reductively Degradable Highly Branched Cyanoacrylate-Based Polymers. J. Polym. Sci. A Polym. Chem. 2016, 54, 3683–3693. [Google Scholar] [CrossRef]
- Bevington, J.C.; Jemmett, J.A.L. Polymerization of methyl α-cyanoacrylate. Part 1.-Initiation by benzoyl peroxide. J. Chem. Soc. Faraday Trans. 1 1973, 69, 1866–1871. [Google Scholar] [CrossRef]
- Bevington, J.C.; Jemmett, J.A.L.; Onyon, P.F. Polymerization of methyl α-cyanoacrylate-II: Conditions for radical polymerization. Eur. Polym. J. 1976, 12, 255–257. [Google Scholar] [CrossRef]
- Duffy, C.; Phelan, M.; Zetterlund, P.B.; Aldabbagh, F. Reversible addition-fragmentation chain transfer polymerization of alkyl-2-cyanoacrylates: An assessment of livingness. J. Polym. Sci. A Polym. Chem. 2017, 55, 1397–1408. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 198–350. ISBN 978-0-471-27400-1. [Google Scholar]
- Chappelow, C.C.; Pinzino, C.S.; Byerley, T.J.; Eick, J.D. Tri-n-butylborane Oxide-Initiated Homopolymerization of Vinyl Monomers Containing Cyano or lsocyanato Groups. J. Appl. Polym. Sci. 1995, 58, 1147–1150. [Google Scholar] [CrossRef]
- Renaud, P.; Beauseigneur, A.; Brecht-Forster, A.; Becattini, B.; Darmency, V.; Kandhasamy, S.; Montermini, F.; Ollivier, C.; Panchaud, P.; Pozzi, D.; et al. Boron: A key element in radical reactions. Pure Appl. Chem. 2007, 79, 223–233. [Google Scholar] [CrossRef]
- Van Herk, A.M. Pulsed initiation polymerization as a means of obtaining propagation rate coefficients in free-radical polymerizations. J. Macromol. Sci. Polymer Rev. 1997, 37, 633–648. [Google Scholar] [CrossRef]
- Değirmenci, I.; Aviyente, V. DFT study on the propagation kinetics of free-radical polymerization of α-substituted acrylates. Macromolecules 2009, 42, 3033–3041. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M. Rate coefficients of free-radical polymerization deduced from pulsed laser experiments. Prog. Polym. Sci. 2002, 27, 191–254. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Beuermann, S.; Buback, M.; Castignolles, P.; Charleux, B.; Coote, M.L.; Hutchinson, R.A.; Junckers, T.; Lacik, I.; Russell, G.T.; et al. Critically-evaluated rate coefficients in radical polymerization—7. Secondary-radical propagation rate coefficients for methyl acrylate in the bulk. Polym. Chem. 2014, 5, 204–212. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Davis, T.P.; Gilbert, R.G.; Hutchinson, R.A.; Olaj, O.F.; Russell, G.T.; Schweer, J.; Van Herk, A.M. Critically evaluated rate coefficients for free-radical polymerization, 2. Propagation rate coefficients for methyl methacrylate. Macromol. Chem. Phys. 1997, 198, 1545–1560. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Davis, T.P.; Gilbert, R.G.; Hutchinson, R.A.; Kajiwara, A.; Klumperman, B.; Russell, G.T. Critically evaluated rate coefficients for free-radical polymerization, 3 Propagation rate coefficients for alkyl methacrylates. Macromol. Chem. Phys. 2000, 201, 1355–1364. [Google Scholar] [CrossRef]
- Buback, M.; Gilbert, R.G.; Hutchinson, R.A.; Klumperman, B.; Kuchta, F.D.; Manders, B.G.; O’Driscoll, K.F.; Russell, G.T.; Schweer, J. Critically evaluated rate coefficients for free-radical polymerization, I. Propagation rate coefficient for styrene. Macromol. Chem. Phys. 1995, 196, 3267–3280. [Google Scholar] [CrossRef]
- Cho, I. Copolymer sequence control by ring-opening polymerization of prestructured cyclic monomers. Makromol. Chem., Macromol. Symp. 1990, 33, 45–54. [Google Scholar] [CrossRef]
- Lee, J.; Cho, I. Synthesis and ring-opening polymerization of 3-methoxy-4-cyano-2,9-dioxabicyclo[4.3.0]non-3-ene: Preparation of alternating head-to-head copolymer of methyl α-cyanoacrylate and 2,3-dihydrofuran. J. Polym. Sci. C Polym. Lett. 1989, 27, 85–91. [Google Scholar] [CrossRef]
- Alfrey, T., Jr.; Price, C.C. Relative reactivities in vinyl copolymerization. J. Polym. Sci. 1947, 2, 101–106. [Google Scholar] [CrossRef]
- Coote, M.L.; Davis, T.P. The mechanism of the propagation step in free-radical copolymerization. Prog. Polym. Sci. 1999, 24, 1217–1251. [Google Scholar] [CrossRef]
- Jenkins, A. Interpretation of reactivity in radical polymerization-Radicals, monomers, and transfer agents: Beyond the Q-e scheme. J. Polym. Sci. A: Polym. Chem. 1999, 37, 113–126. [Google Scholar] [CrossRef]
- McFarlane, R.C.; Reilly, P.M.; O’Driscoll, K.F. Comparison of the precision of estimation of copolymerization reactivity ratios by current methods. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 251–257. [Google Scholar] [CrossRef]
- Greenley, R.Z. Q and e Values for free radical copolymerizations of vinyl monomers and telogens. In Polymer Handbook, 4th ed.; Brandup, J., Immergut, E.H., Grulke, E.A., Eds.; John Wiley and Sons: New York, NY, 1999; Chapter II; pp. 309–320. ISBN 9780471479369. [Google Scholar]
- Kinsinger, J.B.; Panchak, J.R.; Kelso, R.L.; Bartlett, J.S.; Graham, R.K. Methyl α-cyanoacrylate. II. Copolymerization studies. J. Appl. Polym. Sci. 1965, 9, 429–437. [Google Scholar] [CrossRef]
- Polyakova, A.M.; Suchkova, M.D.; Mager, K.A.; Korshak, V.V. Copolymerization of the ethyl ester of α-cyanoacrylic acid with di(alkyl) and di(fluoroalkyl) methylene malonates. Polym. Sci. U.S.S.R. 1984, 26, 77–82. [Google Scholar] [CrossRef]
- Sperlich, B.; Eisenbach, C.D. Copolymerization of ethyl cyanoacrylate and ethylene in the presence of zinc chloride or trifluoroacetic acid as complexing agent. Acta Polym. 1996, 47, 280–284. [Google Scholar] [CrossRef]
- Dikov, V.K.; Kotzev, D.L.; Kabaivanov, V.S. Polymerization of ethyl 2-cyanoacrylate in the presence of poly (butadiene-co-acrylonitrile). Polym. Int. 1988, 20, 9–12. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Padias, A.B.; Chu, G.; Lee, H.Y.; Kalinin, I.; Sansone, M. Breckenridge, G. Novel cyano-containing copolymers of vinyl esters for piezoelectric materials. J. Polym. Sci. A Polym. Chem. 1992, 30, 2341–2347. [Google Scholar] [CrossRef]
- Rasoul, H.A.A.; Hall, H.K., Jr. Cycloaddition and polymerization reactions of methyl α-cyanoacrylate with electron-rich olefins. J. Org. Chem. 1982, 47, 2080–2083. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Padias, A.B. A paradigm for the mechanisms and products of spontaneous polymerizations. J. Polym. Sci. A Polym. Chem. 2009, 47, 6735–6749. [Google Scholar] [CrossRef]
- Elsabahy, M.; Heo, G.S.; Lim, S.M.; Sun, G.; Wooley, K.L. Polymeric nanostructures for imaging and therapy. Chem. Rev. 2015, 115, 10967–11011. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Kante, B.; Roland, M.; Guiot, P.; Bauduin, P.; Speiser, P. Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: preparation, morphological and sorptive properties. J. Pharm. Pharmacol. 1979, 31, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.J.; Illum, L.; Davis, S.S.; Kreuter, J. Particle-size and size distribution of poly(butyl-2-cyanoacrylate) nanoparticles. 1. Influence of physicochemical factors. J. Colloid Interface Sci. 1984, 101, 149–158. [Google Scholar] [CrossRef]
- Douglas, S.J.; Illum, L.; Davis, S.S. Particle-size and size distribution of poly(butyl 2-cyanoacrylate) nanoparticles. 2. Influence of stabilizers. J. Colloid Interface Sci. 1985, 103, 154–163. [Google Scholar] [CrossRef]
- Behan, N.; Birkinshaw, C.; Clarke, N. Poly n-butyl cyanoacrylate nanoparticles: a mechanistic study of polymerisation and particle formation. Biomaterials 2001, 22, 1335–1344. [Google Scholar] [CrossRef]
- Hansali, F.; Poisson, G.; Wu, M.; Bendedouch, D.; Marie, E. Miniemulsion polymerizations of n-butyl cyanoacrylate via two routes: Towards a control of particle degradation. Colloids and Surf. B Biointerfaces 2011, 88, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Frochot, C.; Dellacherie, E.; Marie, E. Well-defined poly(butyl cyanoacrylate) nanoparticles via miniemulsion polymerization. Macromol. Symp. 2009, 281, 39–46. [Google Scholar] [CrossRef]
- Chauvierre, C.; Labarre, D.; Couvreur, P.; Vauthier, C. Novel polysaccharide-decorated poly(isobutyl cyanoacrylate) nanoparticles. Pharm. Res. 2003, 20, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Osuna, I.; Ponchel, G.; Vauthier, C. Tuning of shell and core characteristics of chitosan-decorated acrylic nanoparticles. Eur. J. Pharm. Sci. 2007, 30, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Bertholon, I.; Lesieur, S.; Labarre, D.; Besnard, M.; Vauthier, C. Characterization of dextran-poly(isobutylcyanoacrylate) copolymers obtained by redox radical and anionic emulsion polymerization. Macromolecules 2006, 39, 3559–3567. [Google Scholar] [CrossRef]
- Fallouh, N.A.; Roblot-Treupel, L.; Fessi, H.; Devissaguet, J.P.; Puisieux, F. Development of a new process for the manufacture of polyisobutylcyanoacrylate nanocapsules. Int. J. Pharm. 1986, 28, 125–132. [Google Scholar] [CrossRef]
- Lambert, G.; Fattal, E.; Pinto-Alphandary, H.; Gulik, A.; Couvreur, P. Polyisobutylcyanoacrylate nanocapsules containing an aqueous core as a novel colloidal carrier for the delivery of oligonucleotides. Pharm. Res. 2000, 17, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Thioune, O.; Fessi, H.; Devissaguet, J.; Puisieux, F. Preparation of pseudolatex by nanoprecipitation: Influence of the solvent nature on intrinsic viscosity and interaction constant. Int. J. Pharm. 1997, 146, 233–238. [Google Scholar] [CrossRef]
- Xing, J.; Deng, L.; Li, J.; Dong, A. Amphiphilic poly{[α-maleic anhydride-ω-methoxypoly(ethylene glycol)]-co-(ethyl cyanoacrylate)} graft copolymer nanoparticles as carriers for transdermal drug delivery. Int. J. Nanomed. 2009, 4, 227–232. [Google Scholar] [CrossRef]
- Deng, L.; Yao, C.; Li, A.; Dong, A. Preparation and characterization of poly{[α-maleic anhydride- ω-methoxy-poly(ethylene glycol)]-co-(ethyl cyanoacrylate)} copolymer nanoparticles. Polym. Int. 2005, 54, 1007–1013. [Google Scholar] [CrossRef]
- Stolnik, S.; Illum, L.; Davis, S.S. Long circulating microparticulate drug carriers. Adv. Drug Deliv. Rev. 1995, 16, 195–214. [Google Scholar] [CrossRef]
- Storm, G.; Belliot, S.O.; Daemen, T.; Lasic, D.D. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv. Drug Deliv. Rev. 1995, 17, 31–48. [Google Scholar] [CrossRef]
- Soma, E.; Attali, P.; Merle, P. Chapter 11: A clinically relevant case study: The development of livatag for the treatment of advanced hepatocellular carcinoma. In Nanostructured Biomaterials for Overcoming Biological Barriers, 1st ed.; Alonso, M.J., Csaba, N.S., Eds.; Royal Society of Chemistry: Cambridge, UK, 2012; pp. 591–600. ISBN 978-1-84973-529-2. [Google Scholar]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT agent Design and synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
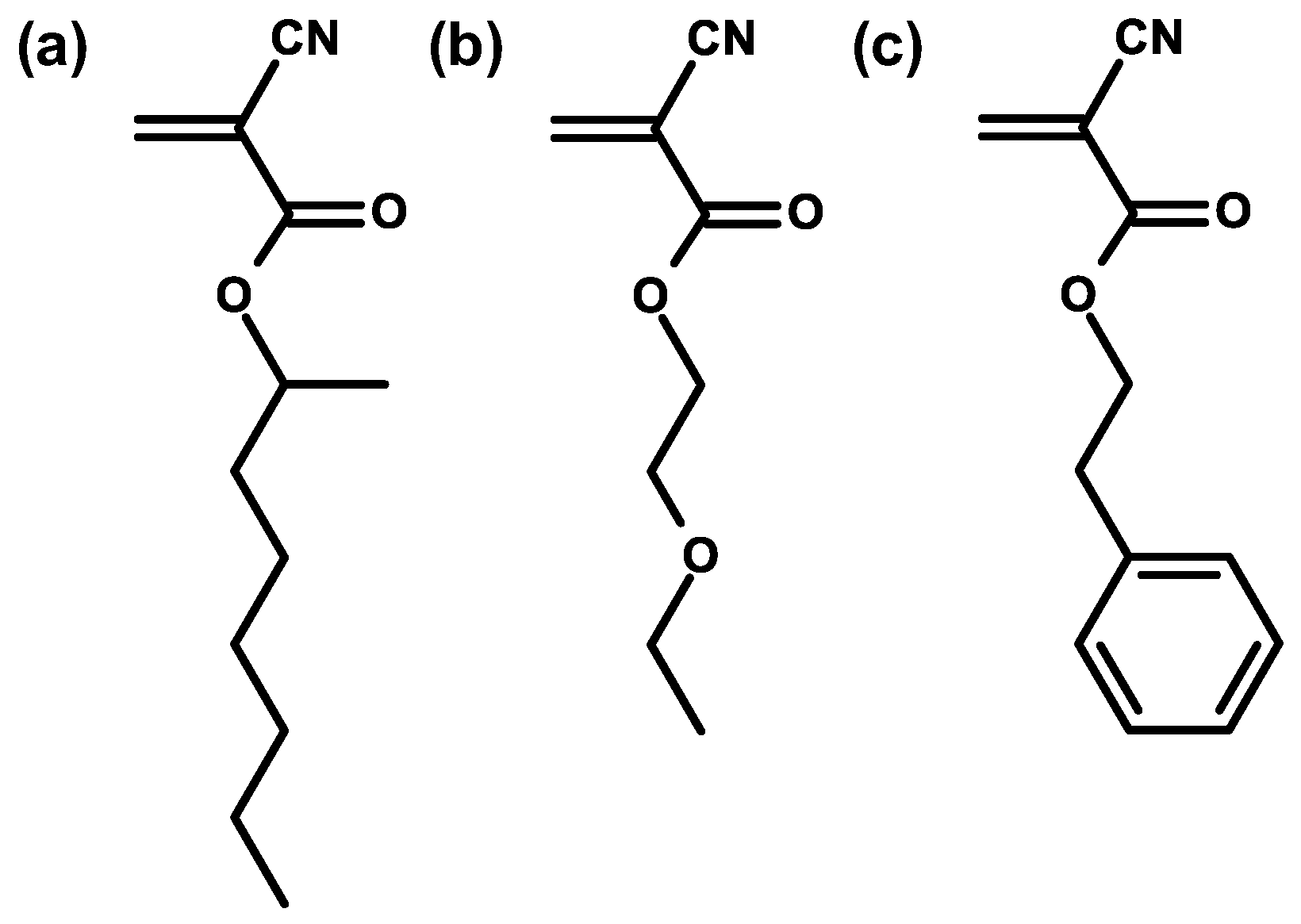
| Structure | R | Uses |
|---|---|---|
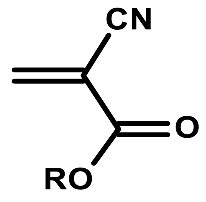 | Me | Loctite 496 super bonder |
| Et | Loctite 401 instant adhesive | |
| i-Propyl | Root canal sealant | |
| n-Butyl | Skin/surgical adhesive | |
| 2-Octyl | Skin surgical adhesive | |
| Allyl | High temperature adhesive | |
| β-Methoxyethyl | Low odor adhesive | |
| β-Ethoxyethyl | Low odor adhesive | |
| Phenylethyl | Adhesive tapes |
| Poly(CA) | Tg, °C | Ref. |
|---|---|---|
| MeCA | 160 | [36] |
| EtCA | 150 | [38] |
| n-BuCA (isobutylcyanoacrylate) | 130 | [37] |
| 2-OctylCA | 10 | [1] |
| AllylCA | 90 | [37] |
| β-MethoxyethylCA | 85 | [1] |
| Monomer | kp (L·mol−1·s−1) | Ref. |
|---|---|---|
| EtCA | 1610–1622 | [51] |
| Ethyl chloroacrylate | 1660 | [52] |
| Ethyl fluoroacrylate | 1120 | [53] |
| Monomer | kp (L·mol−1·s−1) | Ref. |
|---|---|---|
| n-BuCA a | 226 ± 32 | [54] |
| methyl methacrylate | 270 | [66] |
| n-BuMA | 310 | [67] |
| MA | 15,851 | [65] |
| St a | 107 | [68] |
| Monomer | Q | e |
|---|---|---|
| MeCA | 4.91 | 0.91 |
| Vinyl Acetate (VAc) | 0.026 | −0.88 |
| St | 1.00 | −0.80 |
| Isoprene | 1.99 | −0.55 |
| 1,3-Butadiene | 1.70 | −0.50 |
| MMA | 0.78 | 0.40 |
| Acrylamide | 0.23 | 0.54 |
| MA | 0.45 | 0.64 |
| Ethyl vinyl ether | 0.018 | −1.80 |
| Acrylonitrile (AN) | 0.48 | 1.23 |
| CA (M1) | Monomer (M2) | r 1 | r 2 | Medium | Ref. |
|---|---|---|---|---|---|
| MeCA | MA | 1.2 | 0.1 | Benzene | [76] |
| MeCA | MMA | 0.25 | 0.04 | Benzene | [76] |
| MeCA | St | 0.03 | 0.01 | Benzene | [76] |
| MeCA | α-Methyl St | 0.001 | 0.05 | Benzene | [76] |
| MeCA | VAc c | 0.5 | 0.005 | Benzene | [76] |
| MeCA | MMA | 0.13 | 0.10 | Bulk | [50] |
| EtCA | MMA | 0.85 | 0.41 | Bulk | [47] |
| EtCA | MMA | 0.16 | 0.08 a | Bulk | [51] |
| n-BuCA | MMA | 0.236 | 0.057 b | Bulk | [54] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duffy, C.; Zetterlund, P.B.; Aldabbagh, F. Radical Polymerization of Alkyl 2-Cyanoacrylates. Molecules 2018, 23, 465. https://doi.org/10.3390/molecules23020465
Duffy C, Zetterlund PB, Aldabbagh F. Radical Polymerization of Alkyl 2-Cyanoacrylates. Molecules. 2018; 23(2):465. https://doi.org/10.3390/molecules23020465
Chicago/Turabian StyleDuffy, Cormac, Per B. Zetterlund, and Fawaz Aldabbagh. 2018. "Radical Polymerization of Alkyl 2-Cyanoacrylates" Molecules 23, no. 2: 465. https://doi.org/10.3390/molecules23020465
APA StyleDuffy, C., Zetterlund, P. B., & Aldabbagh, F. (2018). Radical Polymerization of Alkyl 2-Cyanoacrylates. Molecules, 23(2), 465. https://doi.org/10.3390/molecules23020465