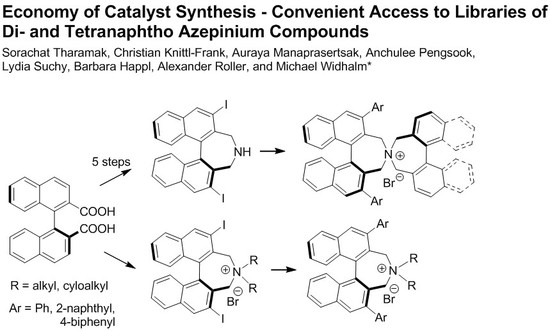
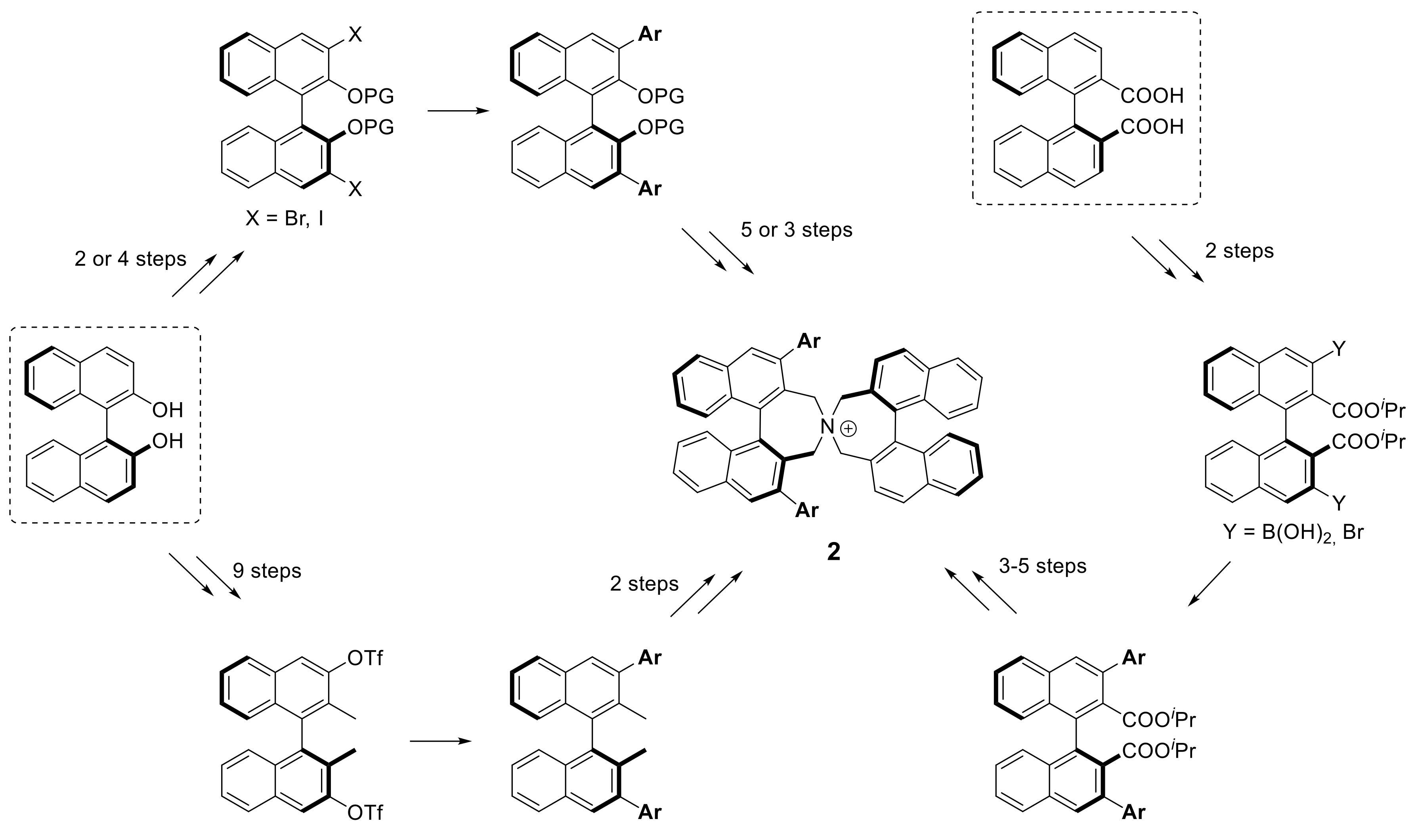
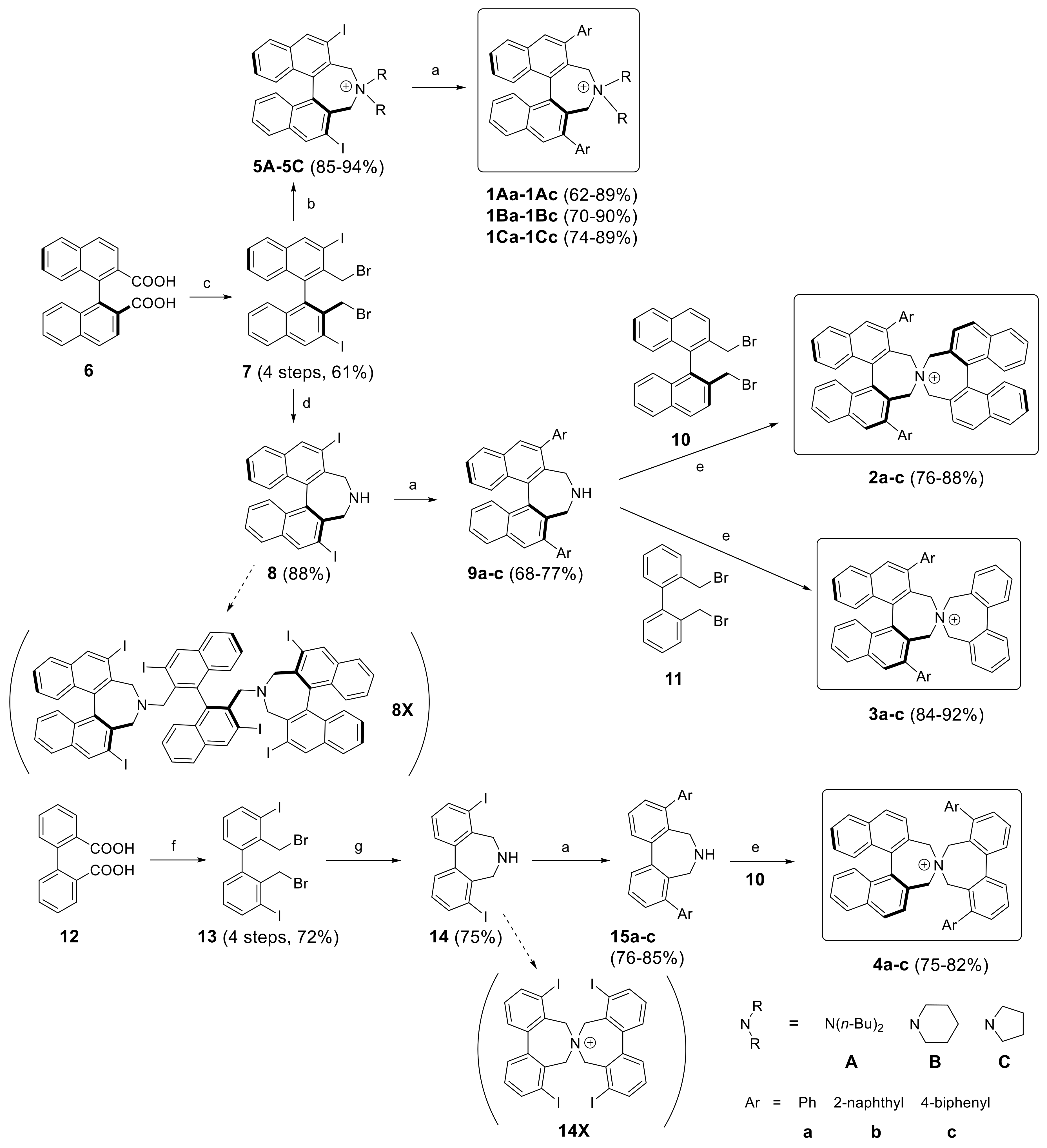
3.2. Synthesis
1Aa–1Cc (General Procedure A). To ammonium salt 5A–C (0.1 mmol) dissolved in toluene (3 mL) in a Schlenk tube was added Na2CO3 (1 mL of a 2 M solution) and arylboronic acid (0.4 mmol, dissolved in a minimum amount of EtOH). The mixture was degassed before adding Pd(PPh3)4 (20 mol%, 23 mg) and stirred at 80 °C (bath) under Ar atmosphere. After 4–20 h (TLC) DCM (10 mL) and water (10 mL) was added and the aqueous phase was extracted with DCM (10 mL). The combined organic phases were washed with KOH (20% in water, 10 mL) and sat. KBr solution (2 × 5 mL) and the solvents were evaporated. The residue was subjected to MPLC (aminopropyl-SiO2, MeOH(0→8%)/DCM) to give products 1Aa–1Cc as pale yellow solids.
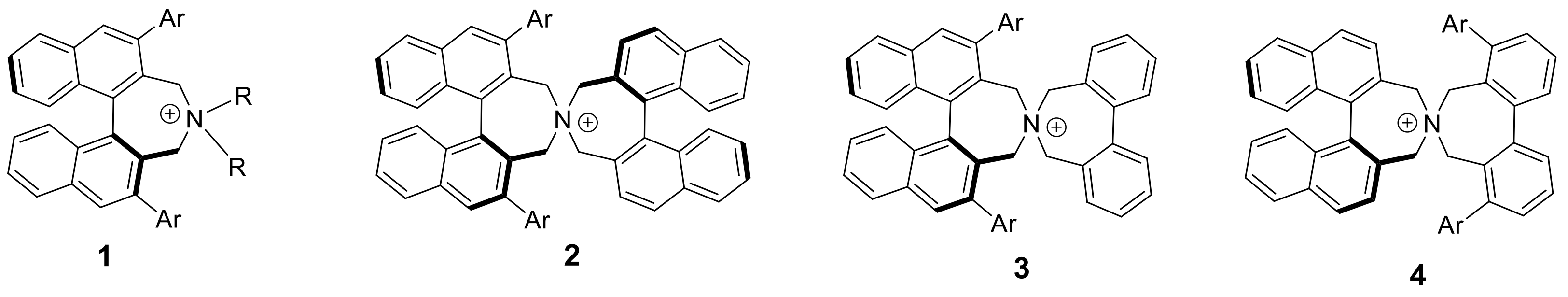
4,4-Dibutyl-2,6-diphenyl-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepin-4-ium bromide (
1Aa) [
27]: Yield: 62%; m.p.: 182–184 °C.
1H-NMR (CDCl
3) δ: 8.09 (s, 2H); 8.06 (br.d,
J = 8.3 Hz, 2H); 7.64 (ddd,
J = 8.0, 5.8, 2.2 Hz, 2H); 7.52–7.65 (br.m, ~8H); 7.46–7.52 (br.m, 2H); 7.35–7.41 (m, 4H); 5.11 (d,
J = 14.0 Hz, 2H); 3.65 (br.dd,
J = 14.0, 1.4 Hz, 2H); 3.17 (br.t,
J = 13.1 Hz, 2H); 2.56 (br.td,
J = 12.3, 4.7 Hz, 2H); 0.80–1.05 (m, 6H); 0.62 (t,
J = 7.0 Hz, 6H); 0.12 (m, 2H) ppm.
13C-NMR (CDCl
3) δ: 140.17 (C); 138.69 (C); 138.41 (C); 133.95 (C); 131.11 (CH); 130.72 (C); 129.65 (2CH); 128.53 (CH); 128.32 (CH); 128.25 (CH); 127.61 (CH); 127.40 (CH); 123.92 (C); 57.50 (CH
2); 57.21 (CH
2); 24.14 (CH
2); 19.53 (CH
2); 13.30 (CH
3) ppm. HRMS (ESI)
m/
z: calcd. for C
42H
42N [M − Br]
+: 560.3312, found: 560.3313.
4,4-Dibutyl-2,6-di(naphthalen-2-yl)-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepin-4-ium bromide (1Ab): Yield: 89%; m.p.: 255–260 °C. 1H-NMR (CDCl3) δ: 8.21 (br.s, 2H); 8.03 (br.d, J = 8.3 Hz, 2H); 7.67 (ddd, J = 8.1, 5.2, 2.7 Hz, 2H); 7.50–7.61 (m, 4H); 7.37–7.48 (m, 4H); 5.10 (br.m, 2H); 3.75 (d, J = 13.6 Hz, 2H); 3.05 (br.t, J = 12.6 Hz, 2H); 2.61 (m, 2H); −0.4–0.9 (br.m, ~14H) ppm. In addition a broad band was observed (7.6–8.2 ppm) corresponding to ~10H. 13C-NMR (CDCl3) δ: 140.15 (C); 138.46 (C); 134.03 (C); 132.62 (C); 131.43 (br.CH); 130.81 (C); 129.41 (br.CH); 128.57 (CH); 128.32 (CH); 127.71 (CH); 127.46 (CH); 127.18 (CH); 127.01 (CH); 124.15 (br.C); 57.50 (br.2CH2); 24.19 (CH2); 19.42 (CH2); ~12.5 (br.CH3) ppm (4CH and 2C not observed). HRMS (ESI) m/z: calcd. for C50H46N [M − Br]+: 660.3625, found 660.3626.
2,6-Di([1,1′-biphenyl]-4-yl)-4,4-dibutyl-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepin-4-ium bromide (
1Ac) [
27]: Yield: 82%; m.p.: 184–189 °C.
1H-NMR (CDCl
3) δ: 8.13 (s, 2H); 8.05 (d,
J = 8.3 Hz, 2H); 7.83 (br.d,
J = 7.6 Hz, 4H); 7.61–7.73 (m, 10H); 7.42–7.49 (m, 4H); 7.34–7.42 (m, 6H); 5.19 (br.d,
J = 13.3 Hz, 2H); 3.70 (br.d,
J = 13.4 Hz, 2H); 3.20 (br.m, 2H); 2.61 (br.m, 2H); 0.8–1.2 (br.m, 6H); 0.55 (t,
J = 6.6 Hz, 6H); 0.29 (br.m, 2H) ppm.
13C-NMR (CDCl
3) δ: 141.08 (C); 139.73 (C); 139.68 (C); 138.44 (C); 137.45 (C); 133.94 (C); 131.10 (CH); 130.68 (C); 128.91 (CH); 128.51 (CH); 128.28 (CH); 128.16 (CH); 127.78 (CH); 127.62 (CH); 127.43 (CH); 127.07 (CH); 123.84 (C); 57.41 (CH
2); 57.10 (CH
2); 24.36 (CH
2); 19.60 (CH
2); 13.39 (CH
3) ppm. HRMS (ESI)
m/
z: calcd. for C
54H
50N [M − Br]
+: 712.3938, found 712.3936.
2,6-Diphenyl-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-piperidin]-4-ium bromide (1Ba): Yield: 88%; m.p.: 246–250 °C. 1H-NMR (CDCl3) δ: 8.06 (s, 2H); 8.02 (d, J = 8.5 Hz, 2H); 7.63 (ddd, J = 8.1, 6.5, 1.4 Hz, 2H); 7.56 (m, 2H); 7.55 (br.d, J = 7.0 Hz, 2H); 7.44–7.51 (m, 8H); 7.39 (ddd, J = 8.6, 6.5, 1.3 Hz, 2H); 5.06 (d, J = 13.3 Hz); 3.61 (d, J = 13.3 Hz, 2H); 3.41 (m, 2H); 2.72 (m, 2H); 1.34 (br.m, 4H); 0.73 (br.m, 2H) ppm. 13C-NMR (CDCl3) δ: 140.20 (C); 138.73 (C); 138.11 (C); 133.90 (C); 130.88 (CH); 130.61 (C); 130.15 (CH); 129.51 (CH); 128.56 (CH); 128.41 (CH); 128.22 (CH); 127.51 (CH); 127.38 (CH); 123.94 (C); 59.12 (CH2); 57.85 (CH2); 20.27 (CH2); 19.76 (CH2) ppm. HRMS (ESI) m/z: calcd. for C39H34N [M − Br]+: 516.2686, found: 516.2699.
2,6-Di(naphthalen-2-yl)-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-piperidin]-4-ium bromide (1Bb): Yield: 90%; m.p.: 250–256 °C (dec.). 1H-NMR (CDCl3) δ: 8.16 (s, 2H); 7.86–8.14 (br.m, 10H); 7.36–7.75 (br.m, 12H); 5.16 (d, J = 13.1 Hz, 2H); 3.78 (d, J = 13.1 Hz, 2H); 3.39 (m, 2H); 2.69 (m, 2H); 1.19 (m, 2H); 1.04 (m, 2H); 0.57 (m, 2H) ppm. 13C-NMR (CDCl3) δ: 140.18 (C); 138.24 (C); 136.20 (br.C); 134.00 (C); 133.42 (br.C); 132.61 (C); 131.34 (CH); 130.73 (C); 129.38 (br.CH); 129.34 (CH); 128.63 (CH); 128.37 (br.CH); 128.31 (CH); 127.87 (CH); 127.67 (CH); 127.62 (CH); 127.47 (CH); 127.10 (CH); 126.95 (CH); 124.14 (C); 59.40 (CH2); 58.37 (br.CH2); 20.21 (CH2); 19.74 (CH2) ppm. HRMS (ESI) m/z: calcd. for C47H38N [M − Br]+: 616.2999, found: 616.2994.
2,6-Di([1,1′-biphenyl]-4-yl)-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-piperidin]-4-ium bromide (1Bc): Yield: 70%; m.p.: 230–240 °C (slow dec. > 210 °C). 1H-NMR (CDCl3) δ: 8.12 (s, 2H); 8.05 (d, J = 8.2 Hz, 2H); 7.82 (d, J = 8.5 Hz, 2H); 7.82 (br.d, J = 6.5 Hz, 2H); 7.68–7.71 (m, 4H); 7.66 (ddd, J = 8.1, 6.6, 1.3 Hz, 2H); 7.59 (d, J = 8.2 Hz, 2H); 7.59 (m, 2H); 7.44–7.50 (m, 6H); 7.42 (ddd, J = 8.6, 6.7, 1.3 Hz, 2H); 7.37 (ddt, J = 7.8, 7.0, 1.3 Hz, 2H); 5.17 (d, J = 13.5 Hz, 2H); 3.69 (d, J = 13.4 Hz, 2H); 3.49 (m, 2H); 2.83 (m, 2H); 1.42 (m, 2H); 1.34 (m, 2H); 0.84 (m, 2H) ppm. 13C-NMR (CDCl3) δ: 141.21 (C); 139.92 (C); 138.25 (C); 137.61 (C); 133.99 (C); 130.95 (CH); 130.70 (C); 130.66 (CH); 128.91 (CH); 128.62 (CH); 129.29 (CH); 128.15 (CH); 127.78 (CH); 127.59 (CH); 127.45 (CH); 127.22 (CH); 124.08 (C); 59.25 (CH2); 58.00 (br.CH2); 20.33 (CH2); 19.85 (CH2) ppm. HRMS (ESI) m/z: calcd. for C51H42N [M − Br]+: 668.3312, found 668.3307.
2,6-Diphenyl-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-pyrrolidin]-4-ium bromide (1Ca): Yield: 74%; m.p.: 224–228 °C. 1H-NMR (CDCl3) δ: 8.08 (s, 2H); 8.04 (d, J = 8.2 Hz, 2H); 7.65 (ddd, J = 8.1, 6.7, 1.4 Hz, 2H); 7.56 (m, 2H); 7.54 (br.d, J = 6.8 Hz, 2H); 7.44–7.50 (m, 8H); 7.41 (ddd, J = 8.5, 6.6, 1.2 Hz, 2H); 4.85 (d, J = 13.5 Hz, 2H); 3.81 (d, J = 13.5 Hz, 2H); 3.46 (m, 2H); 2.96 (m, 2H); 1.81 (m, 2H); 1.24 (m, 2H) ppm. 13C-NMR (CDCl3) δ: 139.67 (C); 138.61 (C); 137.75 (C); 133.90 (C); 130.87 (CH); 130.68 (C); 130.10 (CH); 129.31 (CH); 128.57 (CH); 128.37 (CH); 128.13 (CH); 127.47 (CH); 127.33 (CH); 124.83 (C); 61.84 (CH2); 57.30 (CH2); 20.51 (CH2) ppm. HRMS (ESI) m/z: calcd. for C38H32N [M − Br]+: 502.2529, found: 502.2543.
2,6-Di(naphthalen-2-yl)-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-pyrrolidin]-4-ium bromide (1Cb): Yield: 81%; m.p.: 275–280 °C (dec.). 1H-NMR (CDCl3) δ: 8.17 (s, 2H); 8.07 (d, J = 8.4 Hz, 2H); 8.02 (d, J = 8.8 Hz, 2H); 8.01 (s, 2H); 7.96 (m, 2H); 7.90 (m, 2H); 7.67 (ddd, J = 8.1, 6.7, 1.2 Hz, 2H); 7.60 (br.d, J = 6.4 Hz, 2H); 7.51–7.57 (m, 6H); 7.45 (ddd, J = 8.4, 6.7, 1.2 Hz, 2H); 4.96 (d, J = 13.5 Hz, 2H); 3.95 (d, J = 13.4 Hz, 2H); 3.47 (m, 2H); 2.92 (m, 2H); 1.61 (m, 2H); 0.97 (m, 2H) ppm. 13C-NMR (CDCl3) δ: 139.68 (C); 137.93 (C); 136.07 (br.C); 134.03 (C); 133.26 (br.C); 132.61 (C); 131.33 (CH); 130.81 (C); 129.34 (CH); 129.24 (CH); 128.68 (CH); 128.32 (CH); 128.25 (CH); 127.87 (CH); 127.64 (CH); 127.59 (CH); 127.47 (CH); 127.14 (CH); 126.98 (CH); 125.05 (C); 61.93 (CH2); 57.83 (CH2); 20.56 (CH2) ppm. HRMS (ESI) m/z: calcd. for C46H36N [M − Br]+: 602.2842, found 602.2835.
2,6-Di([1,1′-biphenyl]-4-yl)-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-pyrrolidin]-4-ium bromide (1Cc): Yield: 89%; glassy material. 1H-NMR (600 MHz, CDCl3) δ: 8.13 (s, 2H); 8.06 (d, J = 8.3 Hz, 2H); 7.80 (d, J = 8.2 Hz, 4H); 7.68 (br.d, J = 7.3 Hz, 4H); 7.66 (m, 2H); 7.57 (br.d, J = 7.9 Hz, 4H); 7.50 (d, J = 8.4 Hz, 2H); 7.44–7.47 (m, 4H); 7.43 (m, 2H); 7.37 (m, 2H); 4.96 (d, J = 13.5 Hz, 2H); 3.87 (d, J = 13.5 Hz, 2H); 3.57 (m, 2H); 3.00 (m, 2H); 1.84 (m, 2H); 1.27 (m, 2H) ppm. 13C-NMR (151 MHz, CDCl3) δ: 141.14 (C); 139.83 (C); 139.28 (C); 137.86 (C); 137.39 (C); 133.97 (C); 130.97 (CH); 130.69 (C); 130.62 (CH); 128.88 (CH); 128.64 (CH); 128.24 (CH); 128.01 (CH); 127.76 (CH); 127.54 (CH); 127.45 (CH); 127.19 (CH); 124.84 (C); 61.97 (CH2); 57.48 (CH2); 20.60 (CH2) ppm. HRMS (ESI) m/z: calcd. for C50H40N [M − Br]+: 654.3155, found: 654.3134.
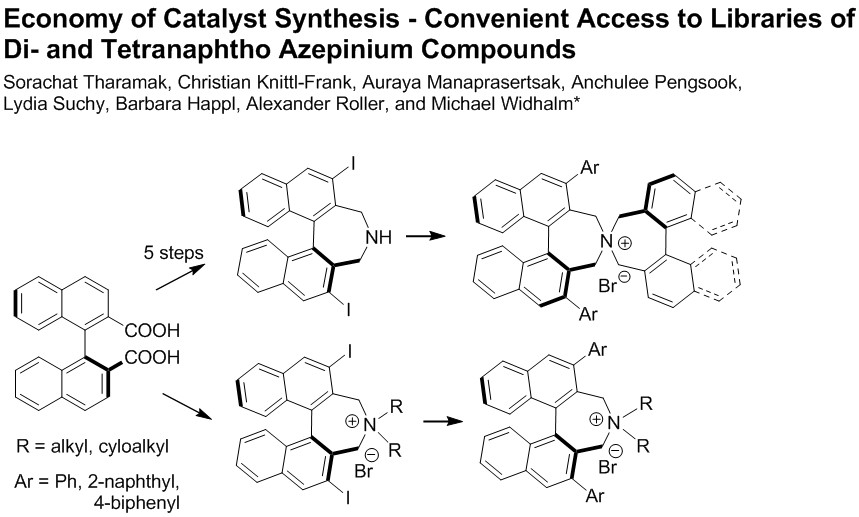
Spiro cyclization of (S)-9a–c with (S)-2,2′-bisbromomethyl-1,1′-binaphthyl 10 yielding (S,S)-2a–c, respectively. (General Procedure B): A Schlenk tube, equipped with magnetic stirring bar and glass stopper, was charged with a solution of substrate (0.3 mmol) in MeCN (6 mL) and K2CO3 (83 mg, 2 eq) followed by (S)-2,2′-bis(bromomethyl)-1,1′-binaphthyl 10 (132 mg, 0.3 mmol) and the mixture was degassed. The reaction was left stirring at 85–90 °C (bath) for 24 h. After cooling to r.t., DCM (30 mL) and H2O (30 mL) were added and the phases were separated. The aqueous one was extracted with DCM (3 × 15 mL) and the combined organic extracts were evaporated under reduced pressure. The crude material was purified by MPLC using a solvent gradient (MeOH(0→8%)/DCM).
(S,S)-2,6-Diphenyl-3,3′,5,5′-tetrahydro-4,4′-spirobi[dinaphtho[2,1-c:1′,2′-e]azepin]-4-ium bromide (
2a) [
8]: Yield: 76%.
1H-NMR (CDCl
3) δ: 8.33 (s, 2H); 8.10 (br.d,
J = 8.2 Hz, 2H); 7.83 (d,
J = 8.1 Hz, 2H); 7.74 (m, 2H); 7.62 (ddd,
J = 7.9, 6.5, 1.1 Hz, 2H); 7.48 (ddd,
J = 7.9, 6.6, 1.1 Hz, 2H); 7.32 (ddd,
J = 8.3, 6.8, 1.1 Hz, 2H); 7.32 (d,
J = 8.5 Hz, 2H); 7.18 (ddd,
J = 8.1, 6.8, 1.2 Hz, 2H); 7.12 (d,
J = 8.6 Hz, 2H); 7.09 (d,
J = 8.4 Hz, 2H); 6.32 (d,
J = 8.4 Hz, 2H); 5.00 (d,
J = 13.8 Hz, 2H); 4.38 (d,
J = 13.7 Hz, 2H); 4.21 (d,
J = 13.4 Hz, 2H); 3.69 (d,
J = 13.3 Hz) ppm. In addition a broad band was observed (7.4–8.2 ppm) corresponding to ~8H.
13C-NMR (CDCl
3) δ: 139.27 (C); 139.13 (C); 136.15 (C); 133.95 (C); 133.85 (C); 132.69 (CH); 131.02 (C); 130.96 (C); 130.89 (br. CH); 130.15 (br. CH); 129.04 (CH); 128.74 (CH); 128.52 (CH); 128.33 (CH); 128.18 (CH); 127.51 (2CH); 127.42 (CH); 127.34 (CH); 126.87 (CH); 126.70 (CH); 124.81 (C); 122.36 (C); 62.33 (CH
2); 57.41 (CH
2) ppm (1C not observed). HRMS (ESI) calcd. for C
56H
40N [M − Br]
+: 726.3161, found: 726.3171.
(S,S)-2,6-Di(naphthalen-2-yl)-3,3′,5,5′-tetrahydro-4,4′-spirobi[dinaphtho[2,1-c:1′,2′-e]azepin]-4-ium bromide (
2b) [
8]: Yield: 88%; m.p.: 255–258 °C (dec.);
= +55 (c: 0.70, DCM).
1H-NMR (CDCl
3) δ: 8.47 (s, 2H); 8.15 (d,
J = 8.1 Hz, 2H); 8.13 (br.m, 2H); 7.77 (br.m, 2H); 7.65 (ddd,
J = 8.1, 6.8, 1.1 Hz, 2H); 7.30–7.42 (m, 6H); 7.19 (br.d,
J = 8.7 Hz, 2H); 7.07 (ddd,
J = 8.3, 6.7, 1.4 Hz, 2H); 6.94 (d,
J = 8.7 Hz, 2H); 6.06 (br.m, 4H); 5.06 (br.m, 2H); 4.49 (d,
J = 13.8 Hz, 2H); 4.20 (d,
J = 13.2 Hz, 2H); 3.65 (d,
J = 13.2 Hz, 2H) ppm. In addition a broad band was observed (7.5–9.1 ppm); integration corresponding to ~10H.
13C-NMR (CDCl
3) δ: 139.43 (C); 139.28 (C); 135.92 (C); 134.08 (C); 133.68 (C); 133.19 (C); 132.79 (br.CH); 131.27 (C); 130.75 (C); 129.97 (br.CH); 128.86 (br.CH); 128.57 (2CH); 128.40 (CH); 128.31 (br.CH); 127.95 (CH); 127.75 (br.CH); 127.59 (CH); 127.56 (CH); 127.30 (3CH); 127.19 (CH); 126.78 (br.CH); 126.56 (CH); 124.78 (C); 122.84 (C); 62.40 (CH
2); 57.61 (br.CH
2) ppm (1CH and 2C not observed). HRMS (ESI) calcd. for C
64H
44N [M − Br]
+: 826.3474, found: 826.3471.
(S,S)-2,6-Di([1,1′-biphenyl]-4-yl)-3,3′,5,5′-tetrahydro-4,4′-spirobi[dinaph-tho[2,1-c:1′,2′-e]azepin]-4-ium bromide (2c): Yield: 81%; m.p.: 252–255 °C (dec.); = +140 (c: 0.81, DCM). 1H-NMR (CDCl3) δ: 8.40 (s, 2H); 8.12 (d, J = 7.9 Hz, 2H); 7.84 (m, 4H); 7.57–7.67 (m, 8H); 7.53 (m, 2H); 7.43 (ddd, J = 8.1, 6.7, 1.1 Hz, 2H); 7.32 (ddd, J = 8.2, 6.8, 1.1 Hz, 2H); 7.13–7.20 (m, 6H); 7.08 (d, J = 8.4 Hz, 2H); 6.41 (d, J = 8.4 Hz, 2H); 5.08 (d, J = 13.8 Hz, 2H); 4.47 (d, J = 13.8 Hz, 2H); 4.27 (d, J = 13.4 Hz, 2H); 3.76 (d, J = 13.4 Hz, 2H) ppm; in addition a broad band was observed (7.4–8.5 ppm); integration corresponding to ~8H. 13C-NMR (CDCl3) δ: 141.76 (C); 140.18 (C); 139.40 (C); 138.77 (C); 138.18 (C); 136.24 (C); 134.05 (C); 133.84 (C); 132.42 (CH); 131.48 (br. CH); 131.22 (C); 131.03 (C); 129.27 (CH); 128.82 (CH); 128.77 (CH); 128.58 (CH); 128.34 (CH); 128.11 (CH); 128.00 (CH); 127.54 (2CH); 127.41 (CH); 127.32 (CH); 126.83 (CH); 126.71 (CH); 125.05 (C); 122.67 (C); 62.51 (CH2); 57.43 (CH2) ppm (1CH not observed). HRMS (ESI) calcd. for C68H48N [M − Br]+: 878.3781, found: 878.3781.
Spiro cyclization of (S)-9a–c with 2,2′-bisbromomethyl-1,1′-biphenyl 10 yielding (S)-3a–c; a similar procedure as for the synthesis of (S,S)-2a–c was applied.
(S)-2′,6′-Diphenyl-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]azepine-6,4′-dinaphtho[2,1-c:1′,2′-e]azepin]-6-ium bromide (3a): Yield: 89%; m.p.: 275–286 °C (dec.); = +12 (c: 0.51, DCM). 1H-NMR (500 MHz, DMSO-d6, 378 K) δ: 8.28 (s, 2H); 8.25 (d, J = 8.1 Hz, 2H); 7.72 (br.ddd, J = 8.0, 6.7, 1.0 Hz, 2H); 7.1–7.6 (br.m, 22H); 4.66 (br.d, J = 13.2 Hz, 2H); 4.52 (d, J = 13.3 Hz, 2H); 4.16 (br.s, 2H); 2.87 (br.s, 2H) ppm. 13C-NMR (125.8 MHz, DMSO-d6, 378 K) δ: 139.67 (C); 138.96 (C); 138.41 (C); 137.58 (C); 133.32 (C); 130.44 (CH); 130.31 (C); 130.20 (CH); 129.57 (CH); 128.25 (CH); 128.08 (CH); 127.83 (CH); 127.49 (CH); 127.39 (CH); 127.31 (CH); 126.61 (CH); 126.57 (CH); 126.27 (C); 123.85 (br.C); 61.30 (CH2); 57.74 (CH2) ppm. HRMS (ESI) calcd. for C48H36N [M − Br]+: 626.2842, found: 626.2839.
(S)-2′,6′-Di(naphthalen-2-yl)-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]azepine-6,4′-dinaphtho[2,1-c:1′,2′-e]-azepin]-6-ium bromide (3b): Yield: 92%; m.p.: 264–267 °C (dec.); = −21 (c: 0.35, DCM). 1H-NMR (500 MHz, DMSO-d6, 378 K) δ: 8.46 (s, 2H); 8.30 (d, J = 8.3 Hz, 2H); 8.18 (br.s, 2H); 8.00 (br.d, J = 8.2 Hz, 2H); 7.96 (br.d, J = 7.5 Hz, 2H); 7.92 (br.d, J = 7.3 Hz, 2H); 7.75 (ddd, J = 8.0, 7.0, 1.0 Hz, 2H); 7.73 (br.s, 2H); 7.58 (m, 4H); 7.51 (ddd, J = 8.2, 7.0, 1.1 Hz, 2H); 7.36 (br.d, J = 8.3 Hz, 2H); 7.23 (m, 4H); 6.76 (br.s, 2H); 6.57 (br.s, 2H); 4.85 (d, J = 13.9 Hz, 2H); 4.53 (d, J = 13.8 Hz, 2H); 3.97 (br.s, 2H); 3.13 (br.s, 2H) ppm. 13C-NMR (125.8 MHz, DMSO-d6, 378 K) δ: 139.48 (C); 138.86 (C); 137.88 (br.C); 135.79 (C); 133.34 (C); 132.46 (br.C); 131.82 (C); 130.81 (CH); 130.37 (C); 130.14 (CH); 128.92 (CH); 128.24 (CH); 127.99 (CH); 127.50 (CH); 127.42 (CH); 127.35 (CH); 126.96 (CH); 126.74 (CH); 126.59 (CH); 126.24 (2CH); 125.98 (C); 123.70 (br.C); 61.10 (CH2); 57.56 (CH2) ppm (3CH not observed). HRMS (ESI) calcd. for C56H40N [M − Br]+: 726.3155, found: 726.3145.
(S)-2′,6′-Di([1,1′-biphenyl]-4-yl)-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]aze-pine-6,4′-dinaphtho[2,1-c:1′,2′-e]azepin]-6-ium bromide (3c): Yield: 84%; glassy material. = +9 (c: 0.61, DCM). 1H-NMR (500 MHz, DMSO-d6, 378 K) δ: 8.37 (s, 2H); 8.28 (d, J = 8.2 Hz, 2H); 7.55–7.80 (m, 14H); 7.31–7.54 (m, 14H); 7.17 (br.s, 4H); 4.79 (br.d, J = 13.0 Hz, 2H); 4.51 (d, J = 13.8 Hz, 2H); 4.10 (br.s, 2H); 3.08 (br.s, 2H) ppm. 13C-NMR (125.8 MHz, DMSO-d6, 378 K) δ: 139.75 (C); 139.73 (C); 138.89 (C); 138.53 (C); 137.72 (C); 137.38 (C); 133.33 (C); 130.34 (C); 130.32 (CH); 130.27 (CH); 128.39 (CH); 128.25 (CH); 127.59 (br.CH); 127.51 (CH); 127.36 (CH); 127.17 (CH); 126.67 (CH); 126.57 (CH); 126.48 (CH); 126.29 (C); 126.11 (2CH); 123.79 (br.C); 61.21 (CH2); 57.49 (CH2) ppm (1CH not observed). HRMS (ESI) calcd for C60H44N [M − Br]+: 778.3468, found: 778.3441.
(S)-4,8-Diphenyl-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]azepine-6,4′-dinaphtho[2,1-c:1′,2′-e]azepin]-6-ium bromide (
4a) [
14]: Yield: 82%; m.p.: 225–228 °C;
= +225 (c: 0.49, DCM).
1H-NMR (CDCl
3) δ: 6.66–8.10 (several broad m, ~24H), 7.92 (br.d,
J = 8.3 Hz, ~2H); 7.18 (br.m,
J = 8.8 Hz, ~2H); 4.60–4.75 (m, 4H); 4.43 (d,
J = 13.3 Hz, 2H); 2.46–3.10 (br.m, 2H) ppm.
13C-NMR (CDCl
3) δ: 143.52 (C); 142.57 (br.C); 138.92 (C); 136.27 (C); 134.00 (C); 130.95 (CH); 130.65 (C); 129.60 (br.CH); 129.53 (CH); 129.11 (CH); 128.33 (CH), 127.57 (br.CH); 127.21 (CH); 127.11 (2CH); 126.74 (CH); 126.09 (br.C); 124.16 (br.C); 63.17 (CH
2); 57.86 (CH
2) ppm; (2CH not observed). HRMS (ESI) calcd. for C
48H
36N [M − Br]
+: 626.2842, found: 626.2828.
(S)-4,8-Di(naphthalen-2-yl)-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]azepine-6,4′-dinaphtho[2,1-c:1′,2′-e]azepin]-6-ium bromide (
4b) [
14]: Yield: 80%; m.p.: 238–241 °C (dec.);
= +228 (c: 0.54, DCM).
1H-NMR (CDCl
3) δ: 7.29–8.23 (several broad m, ~26H), 7.35 (m, 2H); 7.05 (br.ddd,
J = 8.5, 6.9, 1.0 Hz, 2H); 6.90 (d,
J = 8.5 Hz, 2H); 4.76–4.92 (br.m, 2H); 4.48–4.65 (br.m, 4H); 2.73–3.25 (br.m, 2H) ppm.
13C-NMR (CDCl
3) δ: 143.52 (C); 142.87 (br.C); 136.31 (br.C); 135.95 (C); 133.62 (C); 132.96 (br.C); 132.37 (br.C); 131.05 (CH); 130.40 (C); 129.39 (CH); 128.95 (several br.CH); 128.07 (CH); 127.64 (br.CH); 127.35 (CH); 126.97 (CH); 126.88 (CH); 126.78 (CH); 126.70 (CH); 126.59 (CH); 126.48 (CH); 125.48 (br.C); 124.11 (br.C); 62.96 (CH
2); 57.55 (br.CH
2) ppm. HRMS (ESI) calcd. for C
56H
40N [M − Br]
+: 726.3155, found: 726.3145.
(S)-4,8-Di([1,1′-biphenyl]-4-yl)-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]azepine-6,4′-dinaphtho[2,1-c:1′,2′-e]azepin]-6-ium bromide (4c): Yield: 75%; m.p.: 236–241 °C; = +211 (c: 0.62, DCM). 1H-NMR (500 MHz, DMSO-d6, 378 K) δ: 7.98 (br.m, 2H); 7.93 (br.d, J = 8.3 Hz, 2H); 7.88 (s, 2H); 7.85–7.90 (m, 2H); 7.62–7.66 (m, 2H); 7.58 (br.m, 2H); 7.46 (ddd, J = 8.0, 6.8, 1.0 Hz, 2H); 7.23–7.45 (br.m, ~14H); 7.21 (ddd, J = 8.2, 6.8, 1.1 Hz, 2H); 7.06 (d, J = 8.5 Hz, 2H); 7.04 (br.m, 4H); 4.49 (d, J = 13.3 Hz, 2H); 4.48 (br.m, 2H); 4.35 (d, J = 13.3 Hz, 2H); 2.65 (br.m, 2H) ppm. 13C-NMR (125.8 MHz, DMSO-d6, 378 K) δ: 142.60 (C); 141.81 (C); 139.77 (C); 138.81 (C); 137.08 (C); 135.38 (C); 133.17 (C); 130.31 (CH); 129.75 (br.CH); 129.64 (C); 129.43 (CH); 128.64 (CH); 128.37 (CH); 128.03 (CH); 127.70 (CH); 126.83 (CH); 126.70 (CH); 126.44 (CH); 126.24 (CH); 125.97 (2CH); 125.87 (C); 125.69 (CH); 124.33 (C); 62.16 (CH2); 56.78 (CH2) ppm. HRMS (ESI) calcd for C60H44N [M − Br]+: 778.3468, found: 778.3451.
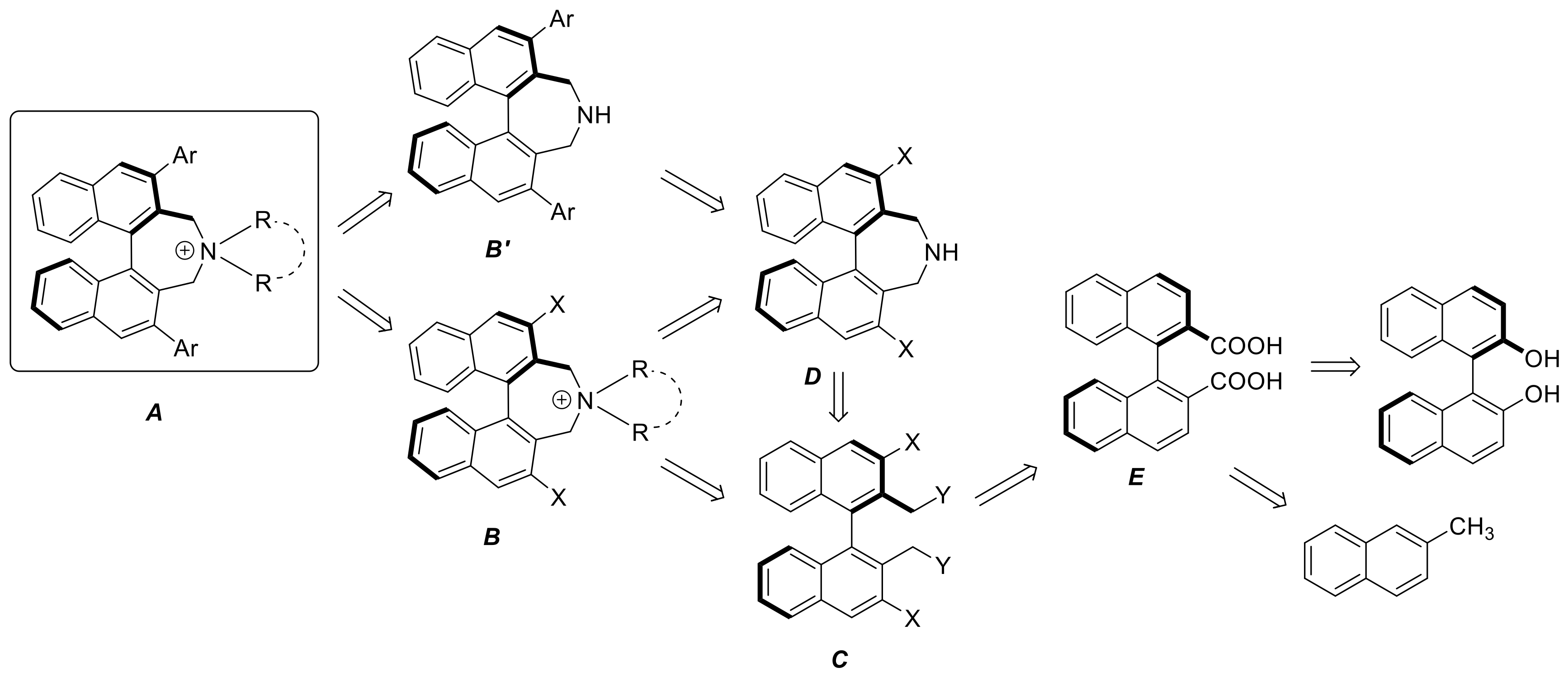
5A–C (General Procedure C) A round-bottomed flask (10 mL) was charged with racemic dibromide 7 (0.2 mmol) and secondary amine (0.8 mmol for 5A and 0.4 mmol for 5B and 5C, respectively) in MeCN (4 mL). To this was added K2CO3 (110 mg, 0.8 mmol) and the reaction was stirred for 22–24 h at 80 °C. Volatiles were removed under vaccum and the residue was suspended in water (2 mL). The precipitate was separated, washed with water (2 mL), Et2O (2 × 2 mL) and air-dried to give a pale yellow solid which was found to be pure by 1H-NMR.
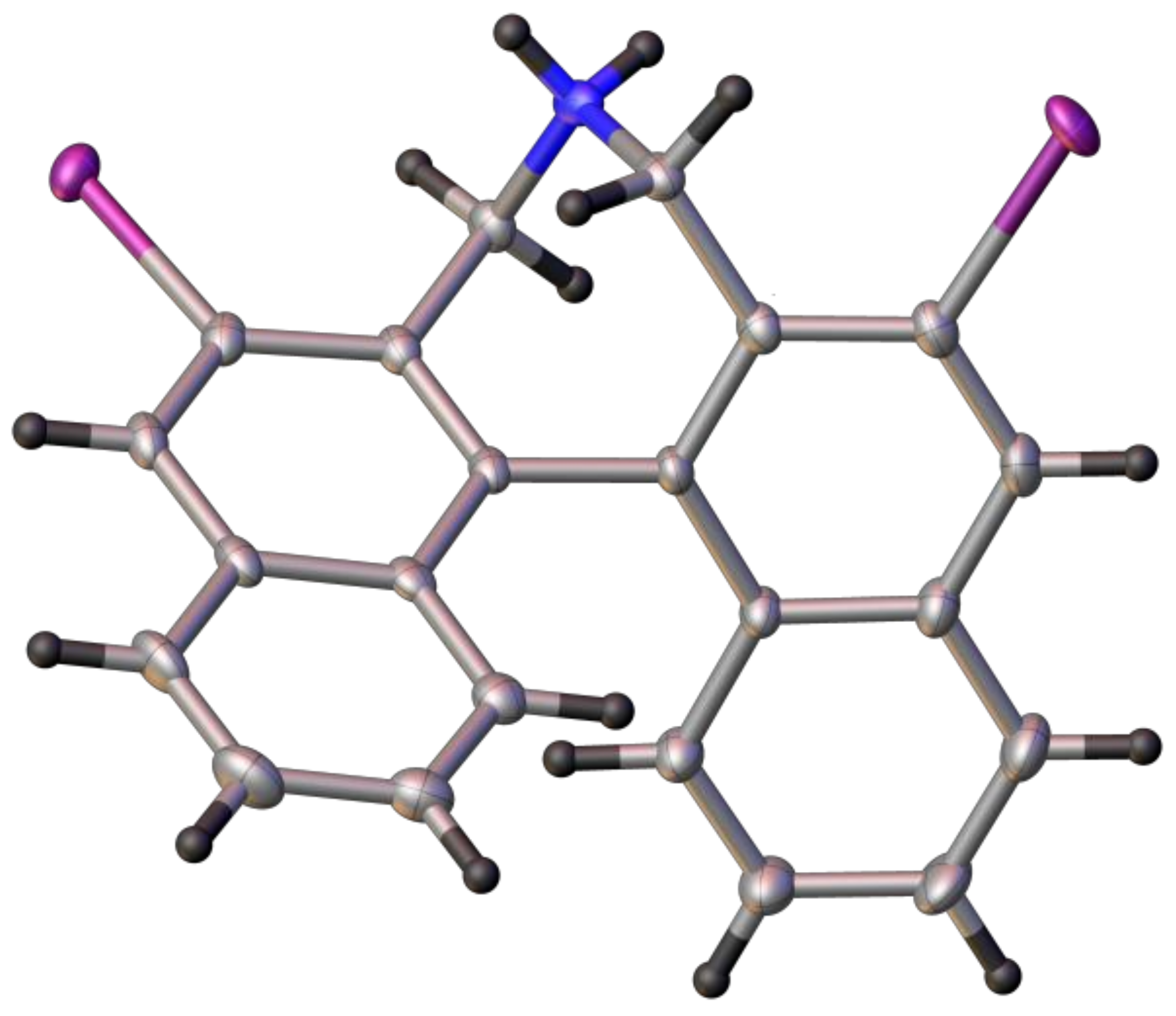
4,4-Dibutyl-2,6-diiodo-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepin-4-ium bromide (5A): Yield: 86%; m.p.: 220–225 °C. 1H-NMR (CDCl3) δ: 8.70 (s, 2H); 7.89 (d, J = 8.2 Hz, 2H); 7.59 (ddd, J = 8.0, 6.9, 1.0 Hz, 2H); 7.32 (ddd, J = 8.2, 6.9, 1.2 Hz, 2H); 7.09 (d, J = 8.7 Hz, 2H); 5.12 (d, J = 13.9 Hz, 2H); 3.97 (d, J = 13.7 Hz, 2H); 3.52 (m, 4H); 2.18 (m, 2H); 1.83 (m. 2H); 1.51 (m, 4H); 1.02 (t, J = 7.4 Hz, 6H) ppm. 13C-NMR (CDCl3) δ: 141.36 (CH); 137.89 (C); 135.31 (C); 130.88 (C); 128.87 (CH); 128.16 (CH); 127.71 (C); 127.47 (CH); 127.44 (CH); 97.56 (C); 66.01 (CH2); 59.60 (CH2); 25.44 (CH2); 19.97 (CH2); 13.69 (CH3) ppm. HRMS (ESI) m/z: Calcd. for C30H32I2N [M − Br]+: 660.0619, found: 660.0610.
2,6-Diiodo-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-piperidin]-4-ium bromide (5B): Yield: 85%; m.p.: ~290 °C (dec.). 1H-NMR (CDCl3) δ: 8.71 (s, 2H); 7.89 (d, J = 8.3 Hz, 2H); 7.60 (ddd, J = 8.0, 6.9, 1.1 Hz, 2H); 7.33 (ddd, J = 8.2, 6.9, 1.2 Hz, 2H); 7.10 (dm, J = 8.6 Hz, 2H); 5.42 (d, J = 13.9 Hz); 4.11 (m, 2H); 3.88 (m, 2H); 3.76 (d, J = 13.8 Hz, 2H); 2.38 (m, 2H); 2.10 (m, 2H); 2.02 (m, 2H) ppm. 13C-NMR (CDCl3) δ: 141.42 (CH); 137.97 (C); 135.27 (C); 130.69 (C); 128.84 (CH); 128.14 (CH); 128.06 (C); 127.40 (CH); 96.99 (C); 65.34 (CH2); 59.23 (CH2); 21.80 (CH2); 20.42 (CH2) ppm. HRMS (ESI) m/z: Calcd. for C27H24I2N [M − Br]+: 615.9993, found: 616.0016.
2,6-Diiodo-3,5-dihydrospiro[dinaphtho[2,1-c:1′,2′-e]azepine-4,1′-pyrrolidin]-4-ium bromide (5C): Yield: 94%; m.p.: 234–238 °C. 1H-NMR (CDCl3) δ: 8.71 (s, 2H); 7.90 (ddd, J = 8.0, 6.8, 1.0 Hz, 2H); 7.35 (ddd, J = 8.2, 6.9, 1.3 Hz, 2H); 7.14 (br.d, J = 8.5 Hz, 2H); 5.25 (d, J = 13.8 Hz, 2H); 4.24 (m, 2H); 4.09 (m, 2H); 3.92 (d, J = 13.7 Hz, 2H); 2.62 (m, 2H); 2.51 (m, 2H) ppm. 13C-NMR (CDCl3) δ: 141.46 (CH); 137.65 (C); 135.34 (C); 130.75 (C); 128.80 (CH); 128.66 (C); 128.17 (CH); 127.49 (CH); 127.39 (CH); 96.56 (C); 65.03 (CH2); 62.55 (CH2); 22.16 (CH2) ppm. HRMS (ESI) m/z: Calcd. for C26H22I2N [M − Br]+: 601.9836, found 601.9861.
(S)-2,2′-Bis(bromomethyl)-3,3′-diiodo-1,1′-binaphthalene (
7) [
21]: (
S)-2,2′-Bis(hydroxymethyl)-3,3′-diiodo-1,1′-binaphthalene (2.971 g, 5.247 mmol) was suspended in HBr (30% in HOAc, 50 mL) and heated to 90 °C for 1.5 h. The cooled reaction mixture was poured into water (500 mL) and sufficient DCM was added to dissolve the precipitate (usually 350–400 mL). The aqueous layer was extracted with DCM (2 × 50 mL) and the combined organic phases washed with water, sat. NaHCO
3, and sat. NaCl and dried (MgSO
4). Evaporation afforded crude
7 as a pale yellow crystalline precipitate; yield: 3.531 g (96%, 99% purity by NMR). The material was pure enough for the next step.
(S)-2,6-Diiodo-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepine (8): In a pressure tube with stirring bar dibromide 7 (1 mmol, 692 mg) was suspended in aqueous NH3 (25%, 15 mL) and acetonitrile (25 mL) and the mixture was heated to 60 °C (oil bath) with stirring for 24 h. The pressure tube was opened at r.t. and the slurry was transferred to a 250 mL flask and ammonia and in part acetonitrile was evaporated. To the residue was added KOH (50 mL, 5% in water) and DCM (50 mL) to obtain a clear 2-phase system. The alkaline phase was extracted with DCM (2 × 20 mL) and the combined organic phase was washed neutral, dried (K2CO3) and evaporated to yield 495–520 mg (86–90%) of crude 8 (95% purity by NMR) which was pure enough for subsequent reactions. Further purification by chromatography (EtOAc/heptane, 50:50) yielded a crystalline product, m.p.: 239–241 °C; = +231 (c: 1.0, EtOH). 1H-NMR (CDCl3) δ: 8.58 (s, 2H); 7.81 (d, J = 8.3 Hz, 2H); 7.45 (m, 2H); 7.26 (m, 4H); 4.35 (d, J = 12.8 Hz, 2H); 3.34 (d, J = 12.8 Hz, 2H); ~2.10 (br.s, 1H) ppm. 13C-NMR (CDCl3) δ: 139.78 (CH); 136.05 (C); 135.63 (C); 134.17 (C); 130.80 (C); 127.38 (CH); 127.14 (CH); 126.61 (CH); 126.42 (CH); 97.74 (C); 52.57 (CH2) ppm. HRMS (ESI) calcd. for C22H16I2N [M + H]+: 547.9372, found: 547.9367.
(11bS,11b′S)-4,4′-(((S)-3,3′-Diiodo-[1,1′-binaphthalene]-2,2′-diyl)bis(methylene))bis(2,6-diiodo-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepine) (8X) (by-product). m.p.: 233–236 °C (dec.). 1H-NMR (CDCl3, 600 MHz) δ: 8.54 (s, 2H); 8.43 (s, 4H); 7.74 (d, J = 8.2 Hz, 4H); 7.49 (m, 2H); 7.37–7.42 (m, 6H); 7.16 (m, 4H); 7.01–7.10 (m, 8H); 4.37 (d, J = 13.7 Hz, 2H); 3.79 (d, J = 13.6 Hz) ppm; In addition a broad band was observed (2.3–3.5 ppm); integration corresponding to ~4H. 13C-NMR (CDCl3, 151 MHz) δ: 139.81 (C); 139.00 (CH); 138.92 (CH); 136.22 (C); 134.06 (C); 134.02 (C); 133.88 (C); 130.73 (C); 129.16 (CH); 127.30 (CH); 127.03 (CH); 126.28 (CH); 126.10 (2CH); 125.76 (CH); 125.36 (CH); 99.50 (C); 98.74 (C); 62.68 (CH2) ppm (1CH2 not observed). HRMS (ESI) calcd. for C66H43I6N2 [M + H]+: 1624.7694, found: 1624.7684.
Suzuki-Miyaura coupling of (S)-8 yielding (S)-9a–9c (General Procedure D): A Schlenk tube, equipped with magnetic stirring bar and glass stopper, was charged with a solution of diiodoazepine (S)-8 (274 mg, 0.50 mmol) in toluene (10 mL) and a Na2CO3-solution (2 M in H2O, 5.0 mL). Then arylboronic acid (2.0 mmol, 4 eq) in a minimum amount of EtOH (~2 mL) was added and the mixture was degassed. After the addition of Pd(PPh3)4 (115 mg, 20 mol %), the reaction was left stirring at 80 °C for 48 h. The reaction was monitored by TLC (EtOAc/heptane, 30/70). After cooling to r.t., DCM (50 mL) and H2O (30 mL) were added and the phases were separated. The aqueous phase was extracted with DCM (3 × 20 mL). The combined organic phases were washed with KOH solution (10%, 20 mL) and sat. NaCl solution and dried (K2CO3). After evaporation of solvents the crude material was purified by MPLC using a solvent gradient (EtOAc + 5% triethylamine(0→30%)/heptane).
(S)-2,6-Diphenyl-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepine (
9a) [
33]: Yield: 77%; glassy material;
= +294 (c: 0.56, DCM).
1H-NMR (CDCl
3) δ: 7.95 (s, 2H); 7.94 (br.d,
J = 8.2 Hz, 2H); 7.60 (m, 4H); 7.38–7.50 (m, 10H); 7.27 (ddd,
J = 8.5, 6.7, 1.2 Hz, 2H); 4.00 (d,
J = 12.5 Hz, 2H); 3.36 (d,
J = 12.5 Hz, 2H) ppm.
13C-NMR (CDCl
3) δ: 141.35 (C); 139.75 (C); 136.00 (C); 133.37 (C); 132.44 (C); 130.80(C); 129.65 (CH); 129.60 (CH); 128.26 (CH); 128.19 (CH); 127.53 (CH); 127.16 (CH); 125.76 (CH); 125.69 (CH); 44.58 (CH
2) ppm. HRMS (ESI) calcd for C
34H
26N [M + H]
+: 448.2065, found: 448.2054.
(S)-2,6-Di(naphthalen-2-yl)-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepine (9b): Yield: 68%; glassy material; = +221 (c: 0.48, DCM). 1H-NMR (CDCl3) δ: 8.08 (br.d, J = 1.2 Hz, 2H); 8.06 (s, 2H); 7.89–8.00 (m, 8H); 7.75 (dd, J = 8.3, 1.5 Hz, 2H); 7.48–7.56 (m, 8H); 7.31 (m, 2H); 4.08 (d, J = 12.6 Hz, 2H); 3.44 (d, J = 12.6 Hz, 2H) ppm. 13C-NMR (CDCl3) δ: 139.70 (C); 138.93 (C); 136.10 (C); 133.53 (C); 133.35 (C); 132.53 (C); 132.50 (C); 130.93 (C); 129.92 (CH); 128.32 (2CH); 128.15 (CH); 128.09 (CH); 127.72 (CH); 127.62 (CH); 127.57 (CH); 126.32 (CH); 126.01 (CH); 125.83 (CH); 125.79 (CH); 44.74 (CH2) ppm. HRMS (ESI) calcd. for C42H30N [M + H]+: 548.2378, found: 548.2383.
(S)-2,6-Di([1,1′-biphenyl]-4-yl)-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepine (9c): Yield: 68%; m.p.: 185–190 °C; = +274 (c: 0.49, DCM). 1H-NMR (CDCl3) δ: 8.01 (s, 2H); 7.97 (br.d, J = 8.2 Hz, 2H); 7.66–7.73 (m, 12H); 7.45–7.52 (m, 8H); 7.38 (m, 2H); 7.29 (ddd, J = 8.6, 6.8, 1.4 Hz, 2H); 4.10 (d, J = 12.5 Hz, 2H); 3.41 (d, J = 12.5 Hz, 2H) ppm. 13C-NMR (CDCl3) δ: 140.77 (C); 140.30 (C); 140.03 (C); 139.32 (C); 136.06 (C); 133.33 (C); 132.46 (C); 130.82 (C); 130.10 (CH); 129.65 (CH); 128.83 (CH); 128.29 (CH); 127.54 (CH); 127.35 (CH); 127.12 (CH); 126.94 (CH); 125.81 (CH); 125.76 (CH); 44.65 (CH2) ppm. HRMS (ESI) calcd. for C46H34N [M + H]+: 600.2686, found: 600.2704.
3,3′-Bis(trimethylsilyl)-[1,1′-biphenyl]-2,2′-dicarboxylic acid: A solution of 2,2,6,6-teramethylpiperidine (1.730 g, 12 mmol, 2.08 mL) in THF (20 mL) was degassed and cooled to 0 °C. To this was added n-BuLi (4.80 mL of a 2.5 molar solution, 12 mmol) and stirring was continued for 20 min. The reaction was cooled to −78 °C and Me3SiCl (2.53 mL, 20 mmol) was added followed by dropwise addition of diphenic acid (484 mg, 2 mmol) in degassed THF (10 mL) during 30 min. The mixture was allowed to reach r.t. overnight. For work-up HCl (4 M, 20 mL) was carefully added with stirring followed by Et2O (40 mL). The aqueous phase was extracted with Et2O (20 mL) and the combined organic phases stirred with NaOH (1 molar, 40 mL) for 15 min. The organic phases was washed another time with NaOH and the alkaline extracts washed with ether (20 mL) and acidified (HCl, 6 M). The mixture was extracted with Et2O (2 × 100 mL) and the combined organic phase was washed with brine and dried (MgSO4). Evaporation of solvent left 704 mg (90%) of 3,3′-bis(trimethylsilyl)-[1,1′-biphenyl]-2,2′-dicarboxylic acid as an off-white powder which was pure enough for the next step (99% by NMR). M.p.: 199–202 °C. 1H-NMR (CDCl3) δ: 7.62 (dd, J = 7.5, 1.2 Hz, 2H); 7.39 (t, J = 7.5 Hz, 2H); 7.20 (dd, J = 7.6, 1.2 Hz, 2H); 0.32 (s, 18H) ppm. 13C-NMR (CDCl3) δ: 173.71 (C); 138.73 (C); 138.08 (C); 137.47 (C); 134.42 (CH); 130.15 (CH); 129.22 (CH); 0.24 (CH3) ppm. HRMS (ESI) calcd. for C20H25O4Si2 [M − H]−: 385.1297, found 385.1298.
(3,3′-Bis(trimethylsilyl)-[1,1′-biphenyl]-2,2′-diyl)dimethanol: To a degassed solution of 3,3′-bis(trimethylsilyl)-[1,1′-biphenyl]-2,2′-dicarboxylic acid (3.67 g, 9.50 mmol) in THF (180 mL) was added BH3·THF complex (38 mL of a 1 M solution in THF, 38 mmol, 4 eq.) by syringe and the reaction was refluxed under Ar for 20–24 h (TLC control). Diluted HCl (2 M) was carefully added at 0 °C to decompose excess of BH3. After removing bulk of THF the residue was partioned between HCl (2 M, 200 mL) and DCM (400 mL). The aqueous phase was extracted with more DCM (100 mL and 50 mL) and the combined organic phase was washed with water and brine and dried (MgSO4). Evaporation of solvents and drying under vacuum overnight left 3.299 g (95% purity by 1H-NMR, 92% yield) of (3,3′-bis(trimethylsilyl)-[1,1′-biphenyl]-2,2′-diyl)dimethanol which was pure enough for the next step. M.p.: 171–175 °C. 1H-NMR (CDCl3) δ: 7.59 (dd, J = 7.5, 1.5 Hz, 2H); 7.32 (t, J = 7.5 Hz, 2H); 7.17 (dd, J = 7.5, 1.4 Hz, 2H); 4.54 (d, J = 11.5 Hz, 2H); 4.50 (d, J = 11.5 Hz, 2H); 2.56 (br.s, 2H); 0.40 (s, 18H) ppm. 13C-NMR (CDCl3) δ: 143.43 (C); 141.77 (C); 141.00 (C); 134.44 (CH); 130.94 (CH); 126.83 (CH); 61.83 (CH2); 0.83 (CH3) ppm. HRMS (ESI) calcd. for C20H31O2Si2 [M + Na]+: 381.1682, found: 381.1677.
(3,3′-Diiodo-[1,1′-biphenyl]-2,2′-diyl)dimethanol: A solution of ICl (4.14 g, 25.5 mmol, 3 eq.) in DCM (100 mL) was dropwise added to (3,3′-bis(trimethylsilyl)-[1,1′-biphenyl]-2,2′-diyl)dimethanol (3.21 g, 8.5 mmol, 95% purity from the previous step) in DCM (140 mL) at −40 °C and the reaction was stirred at the same temperature for 2 h. A solution of NaHSO3 (100 mL, 10%) was added with vigorous stirring and after formation of a semifrozen slury the mixture was warmed up to r.t. The crystalline product was separated, washed with some water and Et2O and dried under vacuum to give 3.435 g of product. The organic phase was separated, washed with water and dried (MgSO4) and evaporated. The residue was treated with DCM/heptane (1:1, 10 mL) to leave a white precipitate which was pure product as well, giving a total yield of 3.763 g (95%) of (3,3′-diiodo-[1,1′-biphenyl]-2,2′-diyl)dimethanol. M.p.: 230–233 °C. 1H-NMR (CDCl3) δ: 7.94 (dd, J = 7.9, 1.3 Hz, 2H); 7.11 (dd, J = 7.6, 1.3 Hz, 2H); 7.02 (t, J = 7.7 Hz, 2H); 4.52 (d, J = 12.2 Hz, 2H); 4.36 (d, J = 12.3 Hz, 2H); 3.32 (br.s, 2H) ppm. 13C-NMR (CDCl3) δ: 141.84 (C); 140.62 (C); 139.88 (CH); 129.87 (CH); 129.29 (CH); 102.04 (C); 65.75 (CH2) ppm. HRMS (ESI) calcd. for C14H12I2NaO2 [M + Na]+: 488.8824, found 488.8821.
2,2′-Bis(bromomethyl)-3,3′-diiodo-1,1′-biphenyl (13): To a mixture of HBr (100 mL, 30% in HOAc) and HOAc (100 mL) was added (3,3′-diiodo-[1,1′-biphenyl]-2,2′-diyl)dimethanol (3.593 g, 7.71 mmol) and the mixture was refluxed for 2 h. Upon cooling the first crop of product crystallized which was separated and dried under vacuum (3.776 g, 83%). To the clear filtrate was added ice water (300 mL) and DCM (200 mL). The aqueous phase was separated and extracted with DCM (2 × 50 mL). The combined organic phase was washed with water, sat. NaHCO3 solution and brine and dried (MgSO4). Evaporation gave a slightly less pure second crop of product (>90% NMR, 412 mg, 8%); total yield of 13: 91%; m.p.: 173–175 °C. 1H-NMR (CDCl3) δ: 7.96 (dd, J = 8.0, 1.3 Hz, 2H); 7.26 (dd, J = 7.6, 1.2 Hz, 2H); 7.06 (t, J = 7.8 Hz, 2H); 4.43 (d, J = 10.2 Hz, 2H); 4.29 (d, J = 10.2 Hz, 2H) ppm. 13C-NMR (CDCl3) δ: 141.10 (C); 140.62 (CH); 137.51 (C); 130.08 (CH); 129.69 (CH); 102.02 (C); 37.64 (CH2) ppm. HRMS (EI) calcd. for C14H1079Br81BrI2: 591.7218, found: 591.7278.
4,8-Diiodo-6,7-dihydro-5H-dibenzo[c,e]azepine (14): A similar procedure as in the synthesis of 8 was applied with following modifications: The reaction was conducted at 50 °C for 48 h and the crude product was purified by MPLC in a solvent gradient (MeOH(0→10)/DCM) to yield 14 and minor amounts of N-spiro compound 14X.
14: Yield: 73–77%; m.p.: 164–167 °C. 1H-NMR (CDCl3) δ: 7.91 (dd, J = 7.9, 1.2 Hz, 2H); 7.39 (dd, J = 7.6, 1.2 Hz, 2H); 7.09 (t, J = 7.8 Hz, 2H); 3.79 (br.m, 4H) ppm. 13C-NMR (CDCl3) δ: 142.33 (C); 139.51 (CH); 138.94 (C); 129.21 (CH); 127.93 (CH); 100.56 (C); 53.20 (CH2) ppm. HRMS (ESI) calcd. for C14H12I2N [M + H]+: 447.9054, found: 447.9050.
14X: Yield: 8–13%; m.p.: 295–298 °C. 1H-NMR (CDCl3) δ: 8.13 (dd, J = 8.0, 1.1 Hz, 4H); 7.70 (dd, J = 7.8, 1.1Hz, 4H); 7.47 (t, J = 7.9 Hz, 4H); 4.88 (d, J = 13.7 Hz, 4H); 4.59 (d, J = 13.7 Hz, 4H) ppm. 13C-NMR (CDCl3) δ: 142.75 (C); 141.31 (CH); 133.56 (CH); 130.27 (CH); 128.90 (C); 103.79 (C); 67.36 (CH2) ppm. HRMS (ESI) calcd for C28H20I4N [M − Br]+: 877.7769, found: 877.7763.
Suzuki-Miyaura coupling of 14 yielding 15a–15c: General Procedure D was applied with exception of condition for the MPLC separations; a gradient EtOAc(20→50%)/heptane was used.
4,8-Diphenyl-6,7-dihydro-5H-dibenzo[c,e]azepine (15a): Yield: 78%; colorless foam. 1H-NMR (CDCl3) δ: 7.35–7.55 (m, 16H); 3.65 (br.m, 4H) ppm. 13C-NMR (CDCl3) δ: 142.55 (C); 142.02 (C); 141.28 (C); 134.24 (C); 129.74 (CH); 129.46 (CH); 128.17 (CH); 127.38 (CH); 127.18 (CH); 127.13 (CH); 45.20 (CH2) ppm. HRMS (ESI) calcd. for C26H22N [M + H]+: 348.1752, found 348.1740.
4,8-Di(naphthalen-2-yl)-6,7-dihydro-5H-dibenzo[c,e]azepine (15b): Yield: 76%; colorless foam. 1H-NMR (CDCl3) δ: 7.98 (br.s, 2H); 7.87–7.93 (m, 6H); 7.68 (dd, J = 8.1, 1.5 Hz, 2H); 7.59 (dd, J = 6.4, 2.6 Hz, 2H); 7.49–7.55 (m, 8H); 3.73 (br.m, 4H) ppm. 13C-NMR (CDCl3) δ: 142.61 (C); 141.94 (C); 138.83 (C); 134.45 (C); 133.29 (C); 132.47 (C); 130.02 (CH); 128.14 (CH); 128.10 (CH); 128.00 (CH); 127.68 (CH); 127.65 (CH); 127.52 (CH); 127.27 (CH); 126.27 (CH); 125.97 (CH); 45.32 (CH2) ppm. HRMS (ESI) calcd. for C34H26N [M + H]+: 448.2065, found 448.2057.
4,8-Di([1,1′-biphenyl]-4-yl)-6,7-dihydro-5H-dibenzo[c,e]azepine (15c): After extractive work-up large amount of product crystallized upon addition of diethylether of a concentrated solution in DCM; from the mother liquor more product was obtained by chromatography; total yield: 85%; m.p.: 250–255 °C. 1H-NMR (CDCl3) δ: 7.63–7.70 (m, 8H); 7.59–7.63 (m, 4H); 7.57 (dd, J = 7.6, 1.5 Hz, 2H); 7.51 (t, J = 7.4 Hz, 2H); 7.44–7.49 (m, 6H); 7.34–7.40 (m, 2H); 3.75 (br.m, 4H) ppm. 13C-NMR (CDCl3) δ: 142.62 (C); 141.63 (C); 140.79 (C); 140.25 (C); 140.04 (C); 134.28 (C); 129.92 (CH); 129.75 (CH); 128.81 (CH); 127.46 (CH); 127.35 (CH); 127.29 (CH); 127.10 (CH); 126.93 (CH); 45.29 (CH2) ppm. HRMS (ESI) calcd. for C38H30N [M + H]+: 500.2378, found: 500.2368.
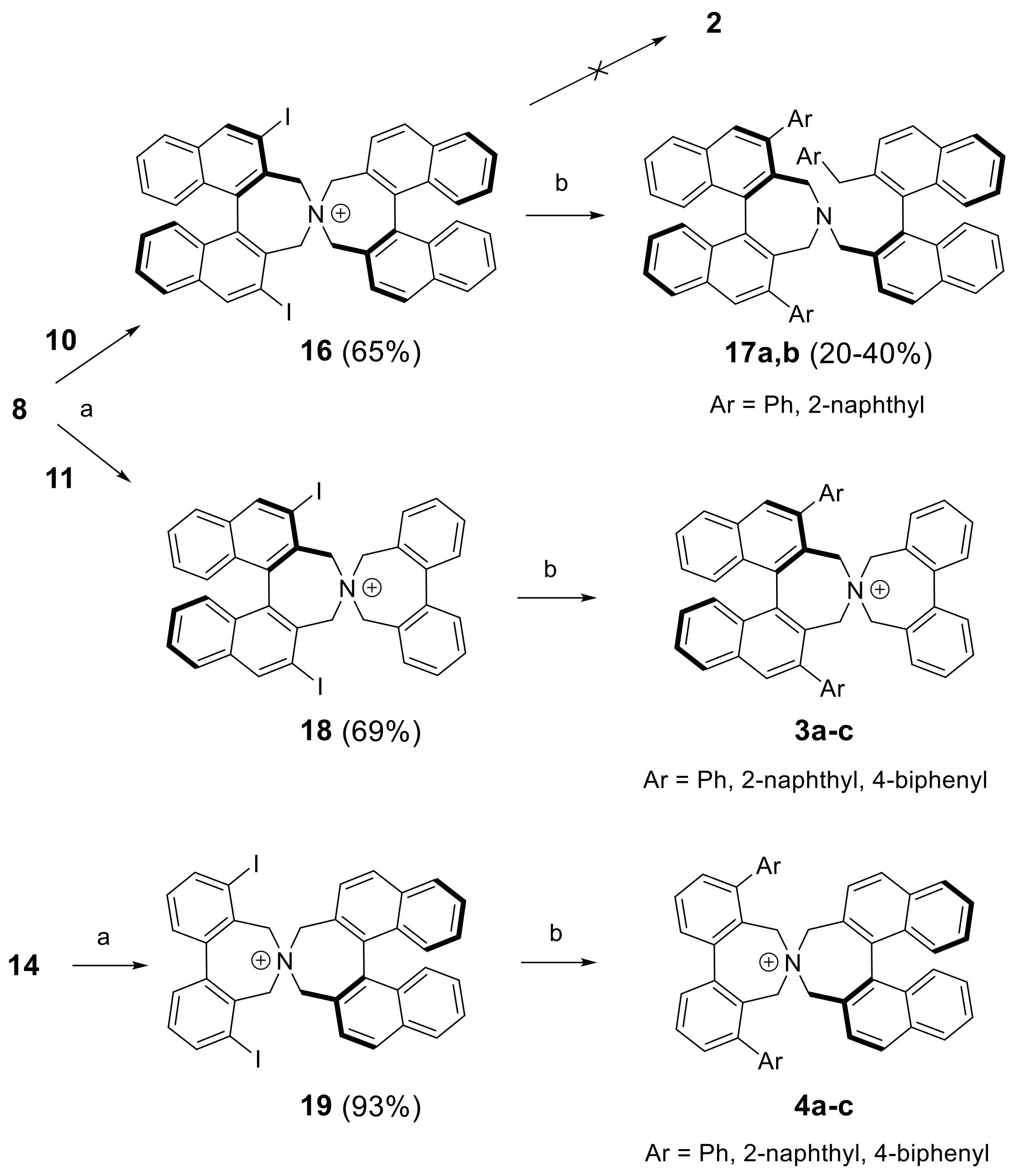
Synthesis of diiodo bisazepinium compounds 16, 18, and 19 (Typical procedure): To diiodoazepine (S)-8 or 14 (0.5 mmol) and dibromide (S)-10 or 11 (0.5 mmol), respectively in acetonitrile (5 mL) was added dry K2CO3 (414 mg, 3 mmol) and the suspension was degassed and stirred at 80 °C under argon overnight. After cooling to room temperature DCM (50 mL) and water (20 mL) was added. The aqueous phase was separated and extracted with DCM (3 × 10 mL). The combined organic layers were evaporated and the crude products purified by MPLC in MeOH (0→10%)/DCM.
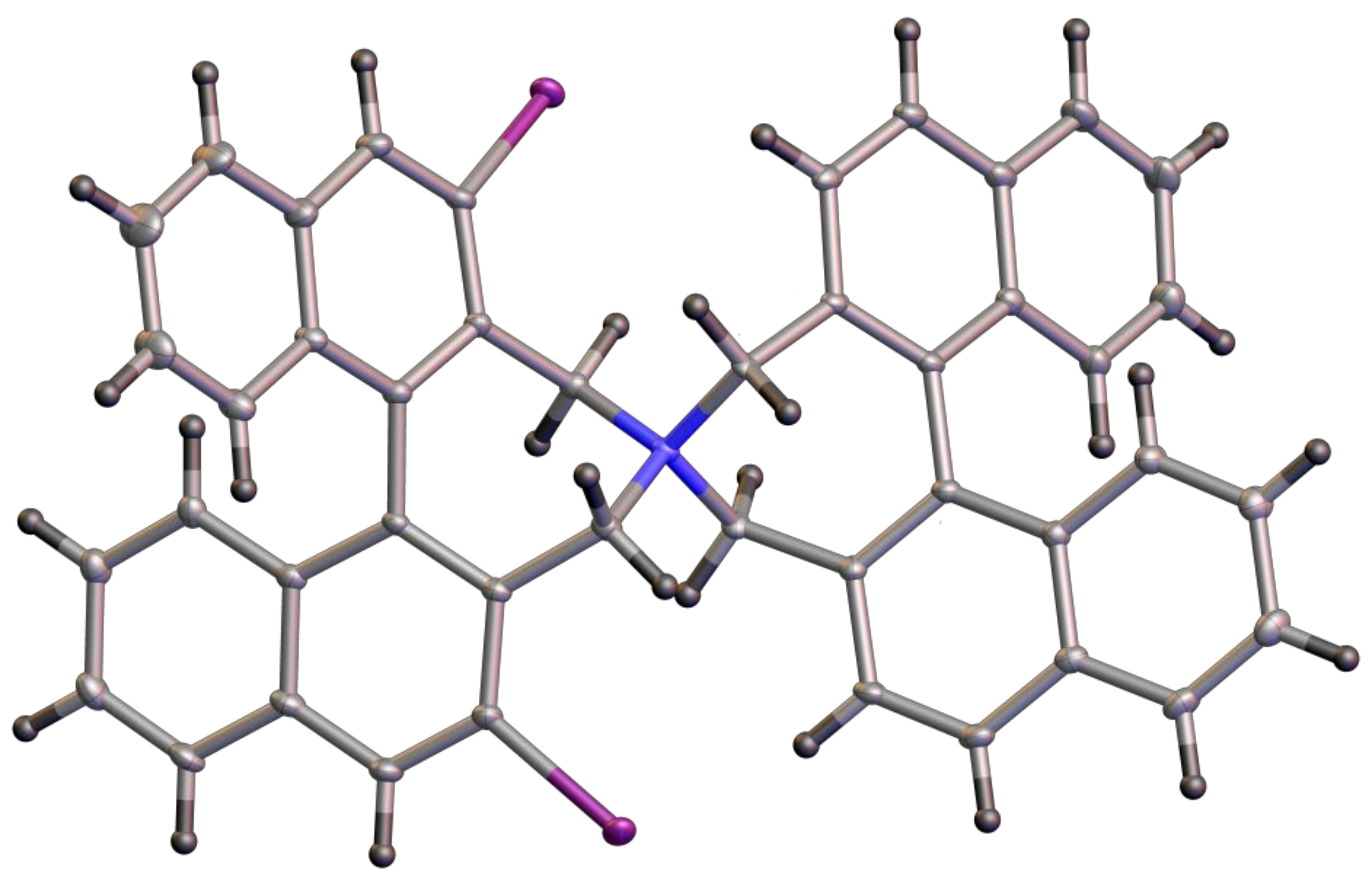
(S,S)-2,6-Diiodo-3,3′,5,5′-tetrahydro-4,4′-spirobi[dinaphtho[2,1-c:1′,2′-e]azepin]-4-ium bromide (16): Yield: 65%; m.p.: 216–219 °C (dec.); = +261 (c: 0.46, DCM). 1H-NMR (DMSO-d6) δ: 9.08 (s, 2H); 8.52 (d, J = 8.5 Hz, 2H); 8.31 (d, J = 8.6 Hz, 2H); 8.18 (d, J = 8.2 Hz, 2H); 8.15 (d, J = 8.3 Hz, 2H); 7.65 (ddd, J = 6.8, 2.9, 1.0 Hz, 2H); 7.63 (ddd, J = 6.8, 3.0, 1.2 Hz, 2H); 7.36 (m, 4H); 7.13 (br.d, J = 8.7 Hz, 2H); 6.80 (br.d, J = 8.6 Hz, 2H); 4.79 (d, J = 14.8 Hz, 2H); 4.43 (d, J = 14.2 Hz, 2H); 4.35 (d, J = 14.2 Hz, 2H); 4.33 (d, J = 14.8 Hz, 2H) ppm. 13C-NMR (DMSO-d6) δ: 141.06 (CH); 137.28 (C); 135.98 (C); 134.93 (C); 134.08 (C); 131.93 (CH); 130.99 (C); 130.93 (C); 128.86 (CH);128.44 (CH); 128.30 (CH); 127.76 (CH); 127.35 (C); 127.33 (CH); 127.29 (CH); 127.17 (CH); 126.80 (CH); 126.68 (CH); 125.69 (C); 97.09 (C); 64.94 (CH2); 62.29 (CH2) ppm. HRMS (ESI) calcd. for C44H30I2N [M − Br]+: 826.0462, found: 826.0480.
(S)-2′,6′-Diiodo-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]azepine-6,4′-dinaphtho[2,1-c:1′,2′-e]azepin]-6-ium bromide (18): Yield: 69%; m.p.: 255–259 °C (dec.); = +74 (c: 0.81, DCM) 1H-NMR (CDCl3) δ: 8.69 (s, 2H); 8.04 (d, J = 8.0 Hz, 2H); 7.92 (d, J = 8.2 Hz, 2H); 7.68–7.73 (m, 4H); 7.62 (t, J = 7.5 Hz, 2H); 7.57 (m, 2H); 7.36 (t, J = 8.0 Hz, 2H); 7.24 (d, J = 8.7 Hz, 2H); 5.12 (d, J = 13.5 Hz, 2H); 5.02 (d, J = 13.5 Hz, 2H); 4.88 (d, J = 12.6 Hz, 2H); 4.38 (d, J = 12.6 Hz, 2H) ppm. 13C-NMR (CDCl3) δ: 141.33 (CH); 141.08 (C); 138.55 (C); 135.73 (C); 132.02 (CH); 131.61 (C); 131.55 (CH); 129.50 (CH); 128.69 (CH); 128.66 (CH); 128.01 (CH); 127.69 (CH); 127.60 (CH); 127.60 (C); 127.15 (C); 96.80 (C); 66.78 (CH2); 63.26 (CH2) ppm. HRMS (ESI) calcd. for C36H26I2N [M − Br]+: 726.0149, found: 726.0152.
(S)-4,8-Diiodo-3′,5,5′,7-tetrahydrospiro[dibenzo[c,e]azepine-6,4′-dinaphtho[2,1-c:1′,2′-e]azepin]-6-ium bromide (19): Yield: 93%; m.p.: 279–281 °C (dec.); = +86 (c: 0.62, DCM). 1H-NMR (CDCl3) δ: 8.27 (d, J = 8.4 Hz, 2H); 8.15 (d, J = 8.4 Hz, 2H); 8.06 (dm, J = 8.4 Hz, 2H); 8.01 (dd, J = 8.1, 1.2 Hz, 2H); 7.63 (ddd, J = 8.1, 6.8, 1.1 Hz, 2H); 7.61 (dd, J = 7.7, 1.2 Hz, 2H); 7.54 (dm, J = 8.6 Hz, 2H); 7.41 (ddd, J = 8.5, 6.8, 1.3 Hz, 2H); 7.36 (t, J = 7.9 Hz, 2H); 5.26 (d, J = 12.7 Hz, 2H); 5.16 (d, J = 13.8 Hz, 2H); 4.70 (d, J = 13.6 Hz, 2H); 4.46 (d, J = 12.7 Hz, 2H) ppm. 13C-NMR (CDCl3) δ: 143.36 (C); 140.73 (CH); 136.92 (C); 134.51 (C); 132.78 (CH); 131.12 (C); 130.39 (CH); 130.24 (CH); 130.01 (C); 128.85 (CH); 127.78 (CH); 127.68 (CH); 127.53 (CH); 127.09 (CH); 126.94 (C); 102.69 (C); 66.43 (CH2); 64.32 (CH2) ppm. HRMS (ESI) calcd. for C36H26I2N [M − Br]+: 726.0149, found: 726.0138.
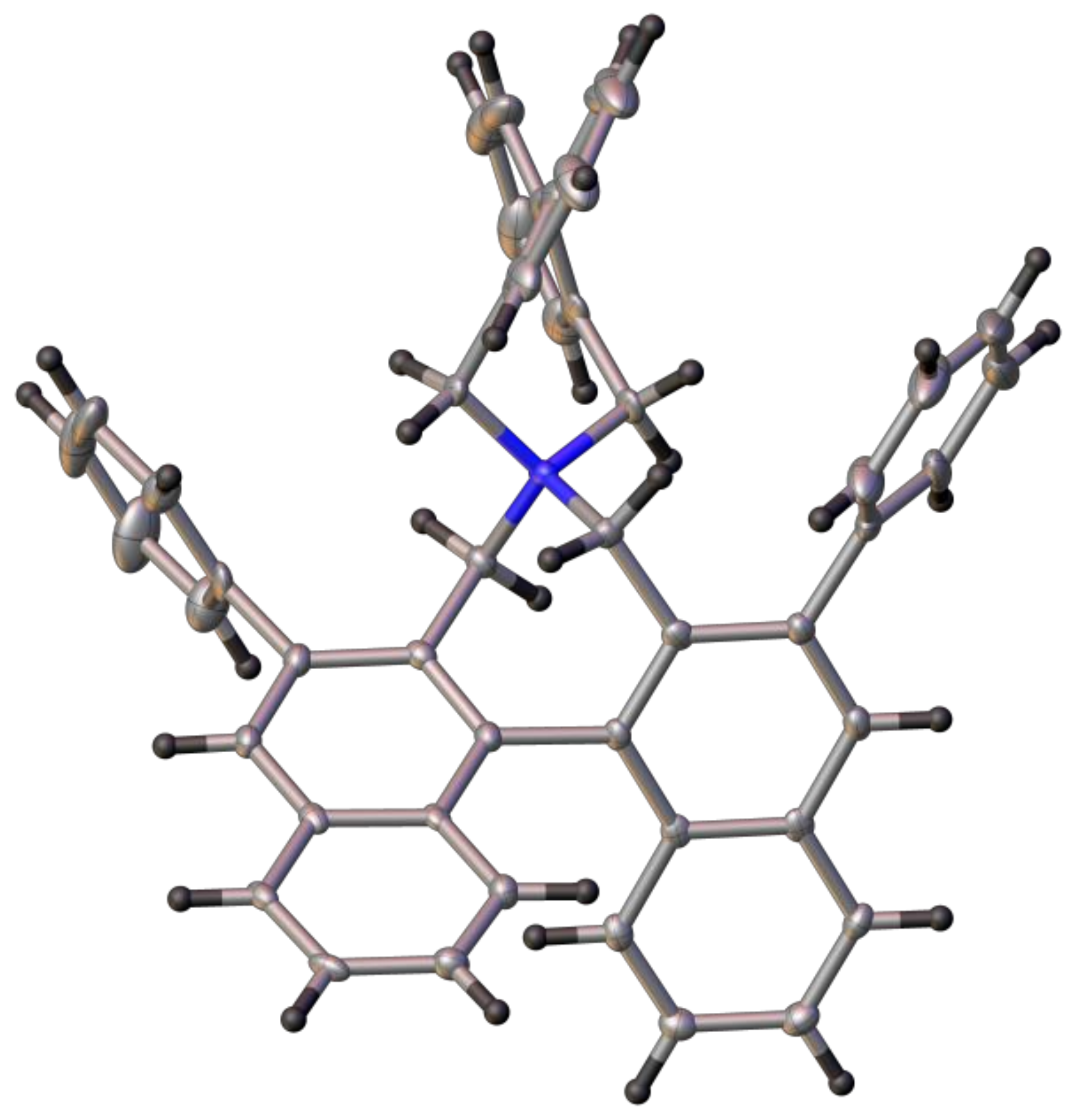
(R)-4-(((S)-2′-Benzyl-[1,1′-binaphthalen]-2-yl)methyl)-2,6-diphenyl-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepine (17a): Yield: 20–40%. 1H-NMR (CDCl3) δ: 8.03 (d, J = 8.0 Hz, 1H); 7.96 (d, J = 8.0 Hz, 2H); 7.89 (d, J = 8.5 Hz, 1H); 7.80 (d, J = 8.3 Hz, 1H); 7.77 (s, 2H); 5.58 (s, 2H); 7.54 (m, 3H); 7.50 (d, J = 8.7 Hz, 2H); 7.33 (m, 3H); 7.23 (m, 2H); 6.97–7.18 (m, 10H); 7.95 (d, J = 8.5 Hz, 1H); 6.86 (d, J = 8.7 Hz, 1H); 6.81 (t, J = 7.3 Hz, 1H); 6.73 (d, J = 8.5 Hz, 1H); 6.63 (t, J = 7.6 Hz, 2H); 6.29 (d, J = 7.6 Hz, 2H); 3.77 (d, J = 12.8 Hz, 2H); 3.16 (d, J = 16.0 Hz, 1H); 2.99 (d, J = 14.9 Hz, 1H); 2.84–2.94 (br.m, 4H) ppm. 13C-NMR (CDCl3) δ: 140.90 (C); 140.45 (C); 139.65 (C); 136.22 (C); 136.04 (C); 135.00 (C); 134.12 (C); 133.85 (C); 133.22 (C); 132.52 (C); 132.43 (C); 132.42 (C); 132.32 (C); 132.06 (C); 130.71 (C); 129.70 (CH); 129.10 (CH); 128.55 (CH); 128.29 (CH); 127.99 (CH); 127.91 (CH); 127.84 (CH); 127.79 (CH); 127.59 (CH); 127.53 (CH); 127.15 (CH); 126.73 (CH); 126.26 (CH); 126.21 (CH); 125.79 (CH); 125.76 (CH); 125.74 (CH); 125.65 (CH); 125.61 (CH); 125.57 (CH); 125.37 (CH); 125.14 (CH); 57.49 (CH2); 51.72 (CH2); 39.31 (CH2) ppm. HRMS (ESI) calcd. for C62H46N [M + H]+: 804.3625, found: 804.3640.
(R)-2,6-Di(naphthalen-2-yl)-4-(((S)-2′-(naphthalen-2-ylmethyl)-[1,1′-binaphthalen]-2-yl)methyl)-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepine (17b): Yield: 20–30%. 1H-NMR (CDCl3) δ: 8.04 (d, J = 8.0 Hz, 1H); 8.01 (d, J = 8.2 Hz, 2H); 7.89 (s, 2H); 7.81 (d, J = 8.6 Hz, 1H); 7.79 (br.d, J = 7.8 Hz, 2H); 7.72 (br.s, 2H); 7.69 (br.d, J = 8.0 Hz, 2H); 7.45–7.61 (m, ~11H); 7.28–7.42 (m, 8H); 7.22 (ddd, J = 8.0, 6.8, 1.1 Hz, 1H); 7.08 (ddd, J = 8.3, 6.8, 1.2 Hz, 1H); ~7.0 (br.m, 1H); 7.00 (ddd, J = 8.3, 6.8, 1.3 Hz, 1H); 6.94 (d, J = 8.6 Hz, 1H); 6.90 (br.d, J = 8.4 Hz, 1H); 6.83 (s, 1H); 6.79 (d, J = 8.4 Hz, 1H); ~6.7 (br.m, 1H); 6.69 (d, J = 8.6 Hz, 1H); 6.36 (dd, J = 8.5, 1.6 Hz, 1H); 3.84 (d, J = 12.9 Hz, 2H); 3.24 (d, J = 15.5 Hz, 1H); ~3.1 (br.m, 2H); 3.01 (d, J = 15.6 Hz, 1H); 3.00 (d, J = 14.6 Hz, 1H); 2.97 (d, J = 14.8 Hz, 1H) ppm. 13C-NMR (CDCl3) δ: 140.39 (C); 138.47 (C); 137.39 (C); 136.29 (C); 136.15 (C); 135.07 (C); 134.25 (C); 133.79 (C); 133.31 (C); 133.13 (C); 132.94 (C); 132.54 (C); 132.53 (C); 132.32 (C); 132.31 (C); 131.71 (C); 130.88 (C); 128.87 (CH); 128.42 (CH); 128.39 (CH); 128.11 (CH); 128.07 (CH); 128.01 (CH); 127.77 (CH); 127.71 (CH); 127.68 (CH); 127.62 (CH); 127.60 (CH); 127.52 (CH); 127.45 (CH); 127.37 (CH); 127.27 (CH); 127.24 (CH); 126.46 (CH); 126.21 (CH); 126.10 (CH); 125.88 (CH); 125.82 (CH); 125.80 (CH); 125.77 (CH); 125.60 (CH); 125.56 (CH); 125.43 (CH); 125.00 (CH); 124.93 (CH); 57.67 (CH2); 51.90 (CH2); 39.59 (CH2) ppm (2C, 1CH not observed). HRMS (ESI) calcd. for C74H52N [M − Br]+: 954.4094, found: 954.4095.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}