Steroidal Constituents from Roots and Rhizomes of Smilacina japonica

Abstract
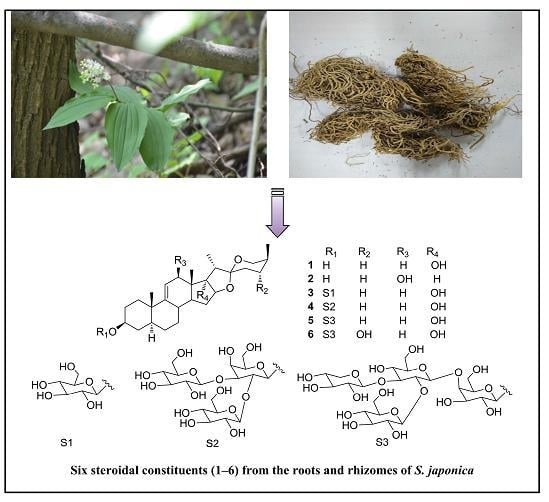
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Plant Material
3.3. Extraction and Isolation
3.4. (25S)-5α-Spirostan-9(11)-en-3β,17α-diol (Compound 1)
3.5. (25S)-5α-Spirostan-9(11)-en-3β,12β-diol (Compound 2)
3.6. (25S)-5α-Spirostan-9(11)-en-3β,17α-diol 3-O-β-d-glucopyranoside (Compound 3)
3.7. (25S)-5α-Spirostan-9(11)-en-3β,17α-diol3-O-β-d-glucopyranosyl-(1→2)-[β-d-glucopyranosyl-(1→3)]-β-d-galactopyranoside (Compound 4)
3.8. Acid Hydrolysis of Compounds 3–4 and Determination of Absolute Configuration of Sugars
3.9. Cytotoxicity Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Song, X.M.; Liu, H.J. Research and Application of “Qi-Medicines” in Taibai Mountains; People’s Medical Publishing House: Beijing, China, 2011. [Google Scholar]
- Jie, W.Y.; Zeng, C.J.; Yao, Z.; Zhang, J.; Zhang, Y.; Zhang, F. Diterpene alkaloids from the roots and processed products of Aconitum pendulum. Chin. Tradit. Herbal Drugs 2010, 41, 347–351. [Google Scholar]
- Yang, S.L.; Liu, X.K.; Wu, H.; Wang, H.B.; Qing, C. Steroidal saponins and cytoxicity of the wild edible vegetable-Smilacina atropurpurea. Steroids 2009, 74, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, H.Z.; Zhang, Y.J.; Jacob, M.R.; Khan, S.I.; Li, X.C.; Yang, C.R. Atropurosides A–G, new steroidal saponins from Smilacina atropurpurea. Steroids 2006, 71, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, Y.F.; Chen, L.; Huang, X.; Yan, S.L.; Chai, X.; Gao, X.M. Steroidal Saponins from the Rhizomes of Smilacina henryi. Helv. Chim. Acta 2013, 96, 478–487. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Niu, X.F.; Xin, W.; Qi, L. Steroidal saponins from Smilacina japonica. Fitoterapia 2012, 83, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.J.; Hong, B.; Yang, L.M.; Han, Z.M. Chemical constituents and their anti-tumor activities of Smilacina japonica. Chin. Tradit. Pat. Med. 2016, 38, 332–335. [Google Scholar]
- Zhao, S.J.; Yang, L.M.; Han, Z.M.; Han, M. Furosteroidal saponin from Smilacina japonica. China J. Chin. Mater. Med. 2011, 36, 3453–3456. [Google Scholar]
- Li, Y.; Wang, X.; He, H.; Zhang, D.; Jiang, Y.; Yang, X.; Wang, F.; Tang, Z.; Song, X.; Yue, Z. Steroidal Saponins from the Roots and Rhizomes of Tupistra chinensis. Molecules 2015, 20, 13659–13669. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, Y.; Zhang, D.; Jiang, Y.; Wang, W.; Song, B.; Tang, Z.; Cui, J.; Yue, Z. Two new spirostanol saponins from the the roots and rhizomes of Tupistra chinensis. Phytochem. Lett. 2015, 13, 6–10. [Google Scholar] [CrossRef]
- Song, X.; Zhang, D.; He, H.; Li, Y.; Yang, X.; Deng, C.; Tang, Z.; Cui, J.; Yue, Z. Steroidal glycosides from Reineckia carnea. Fitoterapia 2015, 105, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yue, Z.; Xie, P.; Zhang, L.; Li, Z.; Song, B.; Tang, Z.; Song, X. C19-Norditerpenoid Alkaloids from Aconitum szechenyianum and Their Effects on LPS-Activated NO Production. Molecules 2016, 21, 1175. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Song, X.; Wang, X.; Mei, Q.; Li, Z.; Cui, J.; Tang, Z.; Yue, Z. Two new compounds from the roots and rhizomes of Trillium tschonoskii. Phytochem. Lett. 2014, 10, 113–117. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, W.; Li, Y.; Li, Z.; Jiang, Y.; Tang, Z.; Song, X.; Yue, Z. Two new pregnane glycosides from Reineckia carnea. Phytochem. Lett. 2016, 15, 142–146. [Google Scholar] [CrossRef]
- Yue, Z.; Qin, H.; Li, Y.; Sun, Y.; Wang, Z.; Yang, T.; Liu, L.; Wang, M.; Feng, F.; Mei, Q. Chemical constituents of the root of Jasminum giraldii. Molecules 2013, 18, 4766–4775. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.K. Dependence of 1H NMR chemical shifts of geminal protons of glycosyloxy methylene (H2-26) on the orientation of the 27-methyl group of furostane-type steroidal saponins. Magn. Reson. Chem. MRC 2004, 42, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Feng, S.; Wang, Q.; Cao, Y.; Sun, M.; Zhang, C. Four new furostanol saponins from the rhizomes and roots of Smilax scobinicaulis and their cytotoxicity. Molecules 2014, 19, 20975–20987. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Wang, G.H.; Luo, Q.; Wang, N.L.; Yao, X.S. Two new steroidal saponins from Allium macrostemon bunge and their cytotoxity on different cancer cell lines. Molecules 2009, 14, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Luo, J.; Huang, X.; Kong, L. Four new steroidal glycosides from Solanum torvum and their cytotoxic activities. Steroids 2009, 74, 95. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–6 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | |||||
|---|---|---|---|---|---|---|---|---|
| No. | δH | δC | δH | δC | δH | δC | δH | δC |
| 1 | 1.44, 1H, ca. | 35.3 | 1.46, 1H, ca. | 36.1 | 1.26, 1H, ca. | 35.3 | 1.25, 1H, ca. | 35.3 |
| 1.75, 1H, ca. | 1.66, 1H, ca. | 1.61, 1H, ca. | 1.61, 1H, ca. | |||||
| 2 | 1.73, 1H, ca. | 31.7 | 1.67, 1H, ca. | 32.9 | 1.73, 1H, ca. | 29.5 | 1.73, 1H, ca. | 29.5 |
| 2.06, 1H, ca. | 2.19, 1H, ca. | 2.06, 1H, ca. | 2.06, 1H, ca. | |||||
| 3 | 3.84, 1H, ca. | 69.6 | 3.86, 1H, ca. | 70.4 | 3.97, 1H, ca. | 76.4 | 3.94, 1H, ca. | 76.7 |
| 4 | 1.51, 1H, ca. | 34.4 | 1.53, 1H, ca. | 39.3 | 1.36, 1H, ca. | 34.4 | 1.36, 1H, ca. | 34.4 |
| 1.76, 1H, ca. | 1.84, 1H, ca. | 1.87, 1H, ca. | 1.88, 1H, ca. | |||||
| 5 | 1.25, 1H, ca. | 42.9 | 1.23, 1H, ca. | 43.7 | 1.03, 1H, ca. | 42.6 | 1.06, 1H, ca. | 42.7 |
| 6 | 1.22, 1H, ca. | 28.1 | 1.24, 1H, ca. | 28.9 | 1.16, 1H, ca. | 28.2 | 1.18, 1H, ca. | 28.2 |
| 1.38, 1H, ca. | 1.28, 1H, ca. | 1.25, 1H, ca. | 1.24, 1H, ca. | |||||
| 7 | 0.95, 1H, ca. | 33 | 0.94, 1H, ca. | 33.5 | 0.92, 1H, ca. | 33 | 0.93, 1H, ca. | 33 |
| 1.75, 1H, ca. | 1.81, 1H, ca. | 1.73, 1H, ca. | 1.73, 1H, ca. | |||||
| 8 | 2.24, 1H, ca. | 36.1 | 2.18, 1H, ca. | 36.3 | 2.09, 1H, ca. | 36.2 | 2.08, 1H, ca. | 36.2 |
| 9 | − | 145.5 | − | 148.5 | − | 145.5 | − | 145.5 |
| 10 | − | 37.5 | − | 38.2 | − | 37.6 | − | 37.6 |
| 11 | 5.51, 1H, d, J = 5.3 Hz | 116.2 | 5.59, 1H, brs | 123.9 | 5.45, 1H, d, J = 5.3 Hz | 116.4 | 5.45, 1H, d, J = 5.3 Hz | 116.4 |
| 12 | 1.78, 1H, ca. | 32.8 | 4.33, 1H, brs | 78.6 | 1.77, 1H, ca. | 33 | 1.78, 1H, ca. | 33 |
| 3.07, 1H, d, J = 17.4 Hz | 3.06, 1H, d, J = 17.4 Hz | 3.06, 1H, d, J = 17.4 Hz | ||||||
| 13 | − | 42.9 | − | 45.2 | − | 43.1 | − | 43.1 |
| 14 | 2.13, 1H, ca. | 50.5 | 1.48, 1H, ca. | 53.2 | 2.13, 1H, ca. | 50.6 | 2.13, 1H, ca. | 50.6 |
| 15 | 1.51, 1H, ca. | 31.9 | 1.52, 1H, ca. | 32.4 | 1.53, 1H, ca. | 32.1 | 1.52, 1H, ca. | 32.1 |
| 2.32, 1H, ca. | 2.07, 1H, ca. | 2.32, 1H, ca. | 2.31, 1H, ca. | |||||
| 16 | 4.45, 1H, d, J = 7.2 Hz | 90.1 | 4.62, 1H, ca. | 81.5 | 4.48, 1H, d, J = 7.2 Hz | 90.3 | 4.45, 1H, d, J = 7.2 Hz | 90.3 |
| 17 | − | 88.9 | 2.34, 1H, t, J = 6.0 Hz | 61.9 | − | 89.1 | − | 89.1 |
| 18 | 0.92, 3H, s | 16.4 | 0.1.09, 3H, s | 10.9 | 0.92, 3H, s | 16.6 | 0.92, 3H, s | 16.6 |
| 19 | 0.98, 3H, s | 17.4 | 0.99, 3H, s | 18.0 | 0.68, 3H, s | 17.4 | 0.84, 3H, s | 17.4 |
| 20 | 2.23, 1H, ca. | 44.8 | 2.14, 1H, ca. | 43.8 | 2.21, 1H, ca. | 45.1 | 2.22, 1H, ca. | 45.1 |
| 21 | 1.21, 3H, d, J = 7.1 Hz | 8.2 | 1.42, 3H, d, J = 6.9 Hz | 14.0 | 1.23, 3H, d, J = 7.1 Hz | 8.5 | 1.22, 3H, d, J = 7.1 Hz | 8.5 |
| 22 | − | 110.5 | − | 110.1 | − | 109.8 | − | 109.8 |
| 23 | 1.45, 1H, ca. | 25.6 | 1.47, 1H, ca. | 26.4 | 1.46, 1H, ca. | 25.8 | 1.46, 1H, ca. | 25.8 |
| 1.92, 1H, ca. | 1.92, 1H, ca. | 1.92, 1H, ca. | 1.91, 1H, ca. | |||||
| 24 | 1.32, 1H, ca. | 24.8 | 1.34, 1H, ca. | 26.2 | 1.33, 1H, ca. | 25.1 | 1.32, 1H, ca. | 25.1 |
| 2.06, 1H, ca. | 2.04, 1H, ca. | 2.05, 1H, ca. | 2.03, 1H, ca. | |||||
| 25 | 1.55, 1H, ca. | 26.5 | 1.59, 1H, ca. | 27.6 | 1.55, 1H, ca. | 26.3 | 1.54, 1H, ca. | 26.7 |
| 26 | 3.24, 1H, d, J = 11.0 Hz | 64.2 | 3.33, 1H, d, J = 11.0 Hz | 65.2 | 3.24, 1H, d, J = 11.0 Hz | 64.4 | 3.24, 1H, d, J = 11.0 Hz | 64.4 |
| 4.04, 1H, dd, J = 2.4, 11.0 Hz | 4.09, 1H, dd, J = 2.4, 11.0 Hz | 4.03, 1H, dd, J = 2.4, 11.0 Hz | 4.04, 1H, dd, J = 2.4, 11.0 Hz | |||||
| 27 | 1.05, 3H, d, J = 7.1 Hz | 15.4 | 1.08, 3H, d, J = 6.9 Hz | 16.3 | 1.05, 3H, d, J = 7.1 Hz | 15.6 | 1.06, 3H, d, J = 7.1 Hz | 15.6 |
| Sugar | ||||||||
| Gal-1 | 4.98, 1H, d, J = 7.6 Hz | 102.2 | 4.91, 1H, d, J = 7.6 Hz | 101.8 | ||||
| 2 | 4.47, 1H, ca. | 72.1 | 4.62, 1H, ca. | 80.5 | ||||
| 3 | 4.18, 1H, t, J = 6.0 Hz | 76.4 | 4.18, 1H, ca. | 85.5 | ||||
| 4 | 4.61, 1H, d, J = 3.0 Hz | 69.8 | 4.53, 1H, ca. | 72.7 | ||||
| 5 | 4.24, 1H, dd, J = 3.0, 9.4 Hz | 74.9 | 4.01, 1H, ca. | 77.6 | ||||
| 6 | 4.48, 2H, ca. | 62 | 4.26, 1H, ca. | 59.9 | ||||
| 4.80, 1H, ca. | ||||||||
| Glc-1′ | 5.16, 1H, d, J = 7.5 Hz | 104.6 | ||||||
| 2′ | 4.13, 1H, ca. | 75 | ||||||
| 3′ | 3.81, 1H, ca. | 78.3 | ||||||
| 4′ | 3.99, 1H, ca. | 71.2 | ||||||
| 5′ | 4.07, 1H, ca. | 76.2 | ||||||
| 6′ | 4.02, 1H, ca. | 60.9 | ||||||
| 4.65, 1H, ca. | ||||||||
| Glc-1′′ | 5.27, 1H, d, J = 7.5 Hz | 106.4 | ||||||
| 2′′ | 4.06, 1H, ca. | 74.5 | ||||||
| 3′′ | 3.97, 1H, ca. | 77.9 | ||||||
| 4′′ | 4.26, 1H, ca. | 69.6 | ||||||
| 5′′ | 4.12, 1H, ca. | 77 | ||||||
| 6′′ | 4.37, 1H, ca. | 62.6 | ||||||
| 4.59, 1H, ca. | ||||||||
| Compounds | Cell Lines | ||||
|---|---|---|---|---|---|
| SMMC-7721 | Bel-7402 | A549 | H460 | K562 | |
| b 5-Fu | 2.4 ± 1.9 | 4.3 ± 2.1 | 4.0 ± 1.6 | 1.7 ± 2.8 | 1.0 ± 0.9 |
| 1 | 10.4 ± 3.9 | 13.3 ± 4.6 | >100 | 33.7 ± 2.2 | 25.5 ± 3.5 |
| 2 | 11.2 ± 4.4 | 6.9 ± 1.7 | 14.4 ± 2.8 | 26.2 ± 2.7 | 29.3 ± 5.3 |
| 3 | 8.6 ± 3.4 | 11.4 ± 4.1 | >100 | 31.4 ± 1.7 | 24.2 ± 2.9 |
| 4 | 7.2 ± 4.6 | 4.4 ± 1.1 | 12.3 ± 2.5 | 21.8 ± 2.1 | >100 |
| 5 | 7.4 ± 5.8 | 31.9 ± 2.1 | >100 | 34.4 ± 3.2 | 15.8 ± 5.4 |
| 6 | 8.8 ± 5.3 | 30.3 ± 2.4 | >100 | 30.3 ± 3.7 | 12.3 ± 5.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.; Yang, X.; Zhang, D.; Li, Y.; Zhang, L.; Song, B.; Yue, Z.; Song, X.; Tang, H. Steroidal Constituents from Roots and Rhizomes of Smilacina japonica. Molecules 2018, 23, 798. https://doi.org/10.3390/molecules23040798
Cui Y, Yang X, Zhang D, Li Y, Zhang L, Song B, Yue Z, Song X, Tang H. Steroidal Constituents from Roots and Rhizomes of Smilacina japonica. Molecules. 2018; 23(4):798. https://doi.org/10.3390/molecules23040798
Chicago/Turabian StyleCui, Yuwen, Xinjie Yang, Dongdong Zhang, Yuze Li, Li Zhang, Bei Song, Zhenggang Yue, Xiaomei Song, and Haifeng Tang. 2018. "Steroidal Constituents from Roots and Rhizomes of Smilacina japonica" Molecules 23, no. 4: 798. https://doi.org/10.3390/molecules23040798