Treatment of Multidrug-Resistant Leukemia Cells by Novel Artemisinin-, Egonol-, and Thymoquinone-Derived Hybrid Compounds

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Culture
2.3. Resazurin Reduction Assay
2.4. Molecular Docking
2.5. Flow Cytometry
2.6. Single Cell Gel Electrophoresis (Alkaline Comet Assay)
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Daddy, N.B.; Kalisya, L.M.; Bagire, P.G.; Watt, R.L.; Towler, M.J.; Weathers, P.J. Artemisia annua dried leaf tablets treated malaria resistant to ACT and i.v. artesunate: Case reports. Phytomedicine 2017, 32, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Woerdenbag, H.J.; Moskal, T.A.; Pras, N.; Malingre, T.M.; el-Feraly, F.S.; Kampinga, H.H.; Konings, A.W. Cytotoxicity of artemisinin-related endoperoxides to Ehrlich ascites tumor cells. J. Nat. Prod. 1993, 56, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Singh, N.P. Selective cancer cell cytotoxicity from exposure to dihydroartemisinin and holotransferrin. Cancer Lett. 1995, 91, 41–46. [Google Scholar] [CrossRef]
- Efferth, T.; Rücker, G.; Falkenberg, M.; Manns, D.; Olbrich, A.; Fabry, U.; Osieka, R. Detection of apoptosis in KG-1a leukemic cells treated with investigational drugs. Arzneimittelforschung 1996, 46, 196–200. [Google Scholar] [PubMed]
- Efferth, T. Artemisinin—Second career as anticancer drug? World J. Tradit. Chin. Med. 2015, 1, 2–25. [Google Scholar] [CrossRef]
- Konstat-Korzenny, E.; Ascencio-Aragón, J.; Niezen-Lugo, S.; Vázquez-López, R. Artemisinin and Its Synthetic Derivatives as a Possible Therapy for Cancer. Med. Sci. 2018, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. Cancer combination therapies with artemisinin-type drugs. Biochem. Pharmacol. 2017, 139, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.C.; Lai, H.; Li, J.R.; Ren, R.L.; McDougall, J.A.; Singh, N.P.; Chou, C.K. Oral administration of dihydroartemisinin and ferrous sulfate retarded implanted fibrosarcoma growth in the rat. Cancer Lett. 1995, 98, 83–87. [Google Scholar] [CrossRef]
- Dell’Eva, R.; Pfeffer, U.; Vene, R.; Anfosso, L.; Forlani, A.; Albini, A.; Efferth, T. Inhibition of angiogenesis in vivo and growth of Kaposi’s sarcoma xenograft tumors by the anti-malarial artesunate. Biochem. Pharmacol. 2004, 68, 2359–2366. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.G.; Dieckmann, D.; Efferth, T.; Schultz, E.S.; Funk, J.O.; Baur, A.; Schuler, G. Artesunate in the treatment of metastatic uveal melanoma—First experiences. Oncol. Rep. 2005, 14, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Yu, S.Q.; Miao, L.Y.; Huang, X.Y.; Zhang, X.P.; Zhu, Y.P.; Xia, X.H.; Li, D.Q. Artesunate combined with vinorelbine plus cisplatin in treatment of advanced non-small cell lung cancer: A randomized controlled trial. Zhong Xi Yi Jie He Xue Bao 2008, 6, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.H.; Adoubi, I.; Kouassi, J.C.; DE, C.; Jansen, N.; Tschulakow, A.; Efferth, T. First study of oral Artenimol-R in advanced cervical cancer: Clinical benefit, tolerability and tumor markers. Anticancer Res. 2011, 31, 4417–4422. [Google Scholar] [PubMed]
- Rutteman, G.R.; Erich, S.A.; Mol, J.A.; Spee, B.; Grinwis, G.C.; Fleckenstein, L.; London, C.A.; Efferth, T. Safety and efficacy field study of artesunate for dogs with non-resectable tumours. Anticancer Res. 2013, 33, 1819–1827. [Google Scholar] [PubMed]
- Breuer, E.; Efferth, T. Treatment of iron-loaded veterinary sarcoma by Artemisia annua. Nat. Prod. Bioprospect. 2014, 4, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. Cancer combination therapy of the sesquiterpenoid artesunate and the selective EGFR-tyrosine kinase inhibitor erlotinib. Phytomedicine 2017, 37, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Ganapathi, S.; Ster, I.C.; Saeed, M.E.; Cowan, M.; Finlayson, C.; Kovacsevics, H.; Jansen, H.; Kremsner, P.G.; Efferth, T.; et al. A randomised, double blind, placebo-controlled pilot study of oral artesunate therapy for colorectal cancer. EBioMedicine 2015, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Osburg, J.; Storch-Hagenlocher, B.; Sertel, S.; Elsässer, M.; Remppis, B.A.; Edler, L.; Munzinger, J.; et al. Prospective open uncontrolled phase I study to define a well-tolerated dose of oral artesunate as add-on therapy in patients with metastatic breast cancer (ARTIC M33/2). Breast Cancer Res. Treat. 2017, 164, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Romero, M.R.; Wolf, D.G.; Stamminger, T.; Marin, J.J.; Marschall, M. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 2008, 47, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.E.; Krishna, S.; Greten, H.J.; Kremsner, P.G.; Efferth, T. Antischistosomal activity of artemisinin derivatives in vivo and in patients. Pharmacol. Res. 2016, 110, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Cen, Y.; Song, Y.; Li, P.; Qin, R.; Liu, C.; Zhao, Y.; Zheng, J.; Zhou, H. Artesunate attenuated progression of atherosclerosis lesion formation alone or combined with rosuvastatin through inhibition of pro-inflammatory cytokines and pro-inflammatory chemokines. Phytomedicine 2016, 23, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.R.M.; Bilia, A.R.; Osman, A.G.; Elsohly, M.; Wink, M.; Bauer, R.; Khan, I.; Bergonzi, M.C.; Marin, J.J.G. Expanding the therapeutic spectrum of artemisinin: Activity against infectious diseases beyond malaria and novel pharmaceutical developments. World J. Tradit. Chin. Med. 2016, 2, 1–23. [Google Scholar] [CrossRef]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honore, C.; Courtney, M.; Huber, K.V.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins target GABAA receptor signaling and impair α cell identity. Cell 2017, 168, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Sauerbrey, A.; Olbrich, A.; Gebhart, E.; Rauch, P.; Weber, H.O.; Hengstler, J.G.; Halatsch, M.E.; Volm, M.; Tew, K.D.; et al. Molecular modes of action of artesunate in tumor cell lines. Mol. Pharmacol. 2003, 64, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, I.R.; Olliaro, P. Safety of artemisinin and its derivatives. A review of published and unpublished clinical trials. Med. Trop. 1998, 58 (Suppl. 3), 50–53. [Google Scholar]
- Tu, Y. Artemisinin-A Gift from Traditional Chinese Medicine to the World (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 10210–10226. [Google Scholar] [CrossRef] [PubMed]
- Berman, P.A.; Adams, P.A. Artemisinin enhances heme-catalysed oxidation of lipid membranes. Free Radic. Biol. Med. 1997, 22, 1283–1288. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Yang, Y.Z.; Lima, V.; Kuypers, F.; Kamchonwongpaisan, S.; Yuthavong, Y. Iron-dependent free radical generation from the antimalarial agent artemisinin (qinghaosu). Antimicrob. Agents Chemother. 1993, 37, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Asawamahasakda, W.; Ittarat, I.; Pu, Y.M.; Ziffer, H.; Meshnick, S.R. Reaction of antimalarial endoperoxides with specific parasite proteins. Antimicrob. Agents Chemother. 1994, 38, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Eckstein-Ludwig, U.; Webb, R.J.; Van Goethem, I.D.; East, J.M.; Lee, A.G.; Kimura, M.; O’Neill, P.M.; Bray, P.G.; Ward, S.A.; Krishna, S. Artemisinins target the SERCA of Plasmodium falciparum. Nature 2003, 424, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Shterman, N.; Kupfer, B.; Moroz, C. Comparison of transferrin receptors, iron content and isoferritin profile in normal and malignant human breast cell lines. Pathobiology 1991, 59, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Judd, W.; Poodry, C.A.; Strominger, J.L. Novel surface antigen expressed on dividing cells but absent from nondividing cells. J. Exp. Med. 1980, 152, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.; Delia, D.; Schneider, C.; Newman, R.; Kemshead, J.; Greaves, M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Gatter, K.C.; Brown, G.; Trowbridge, I.S.; Woolston, R.E.; Mason, D.Y. Transferrin receptors in human tissues: Their distribution and possible clinical relevance. J. Clin. Pathol. 1983, 36, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Benakis, A.; Romero, M.R.; Tomicic, M.; Rauh, R.; Steinbach, D.; Hafer, R.; Stamminger, T.; Oesch, F.; Kaina, B.; et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radic. Biol. Med. 2004, 37, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Kelter, G.; Steinbach, D.; Konkimalla, V.B.; Tahara, T.; Taketani, S.; Fiebig, H.H.; Efferth, T. Role of transferrin receptor and the ABC transporters ABCB6 and ABCB7 for resistance and differentiation of tumor cells towards artesunate. PLoS ONE 2007, 2, e798. [Google Scholar] [CrossRef] [PubMed]
- Ooko, E.; Saeed, M.E.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, T.E. Cecil Essentials of Medicine; Saunders: Philadelphia, PA, USA, 1997. [Google Scholar]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Enhancement of vinblastine-induced cytotoxicity by lysolecithin and phosphatidylinositol. Cancer Lett. 1981, 13, 133–137. [Google Scholar] [CrossRef]
- Ford, J.M.; Yang, J.M.; Hait, W.N. P-glycoprotein-mediated multidrug resistance: Experimental and clinical strategies for its reversal. Cancer Treat. Res. 1996, 87, 3–38. [Google Scholar] [PubMed]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar] [CrossRef] [PubMed]
- Barthomeuf, C.; Bourguet-Kondracki, M.L.; Kornprobst, J.M. Marine metabolites overcoming or circumventing multidrug resistance mediated by ATP-dependent transporters: A new hope for patient with tumors resistant to conventional chemotherapy. Anticancer Agents Med. Chem. 2008, 8, 886–903. [Google Scholar] [CrossRef] [PubMed]
- Molnar, J.; Engi, H.; Hohmann, J.; Molnar, P.; Deli, J.; Wesolowska, O.; Michalak, K.; Wang, Q. Reversal of multidrug resitance by natural substances from plants. Curr. Top. Med. Chem. 2010, 10, 1757–1768. [Google Scholar] [PubMed]
- Eichhorn, T.; Efferth, T. P-glycoprotein and its inhibition in tumors by phytochemicals derived from Chinese herbs. J. Ethnopharmacol. 2012, 141, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting MDR in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updates 2012, 15, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Zeino, M.; Saeed, M.E.; Kadioglu, O.; Efferth, T. The ability of molecular docking to unravel the controversy and challenges related to P-glycoprotein–A well-known, yet poorly understood drug transporter. Investig. New Drugs 2014, 32, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, S.A.; Efferth, T. Cytotoxicity of the indole alkaloid reserpine from Rauwolfia serpentina against drug-resistant tumor cells. Phytomedicine 2015, 22, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ding, Y.; Zhang, T.; An, H. Sinapine reverses multi-drug resistance in MCF-7/dox cancer cells by downregulating FGFR4/FRS2α-ERK1/2 pathway-mediated NF-κB activation. Phytomedicine 2016, 23, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.A.; Ahmed, O.B.; Spengler, G.; Molnár, J.; Lage, H.; Ferreira, M.J. Jatrophane diterpenes and cancer multidrug resistance—ABCB1 efflux modulation and selective cell death induction. Phytomedicine 2016, 23, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.N.; Sheu, M.J.; Hsieh, Y.W.; Wang, R.Y.; Chiang, Y.C.; Hung, C.C. β-carotene reverses multidrug resistant cancer cells by selectively modulating human P-glycoprotein function. Phytomedicine 2016, 23, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, Y.; Sun, M.; Liu, M.; Wang, X. Comprehensive assessment of Cucurbitacin E related hepatotoxicity and drug-drug interactions involving CYP3A and P-glycoprotein. Phytomedicine 2017, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Umsumarng, S.; Pitchakarn, P.; Yodkeeree, S.; Punfa, W.; Mapoung, S.; Ramli, R.A.; Pyne, S.G.; Limtrakul, P. Modulation of P-glycoprotein by Stemona alkaloids in human multidrug resistance leukemic cells and structural relationships. Phytomedicine 2017, 34, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.J.; Ferreira, M.J.; dos Santos, D.J. Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Ed. 2003, 42, 3996–4028. [Google Scholar] [CrossRef] [PubMed]
- Tsogoeva, S.B. Recent progress in the development of synthetic hybrids of natural or unnatural bioactive compounds for medicinal chemistry. Mini-Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, T.; Capci Karagoz, A.; Reiter, C.; Tsogoeva, S.B. Artemisinin-derived dimers: Potent antimalarial and anticancer agents. J. Med. Chem. 2016, 59, 7360–7388. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Olbrich, A.; Bauer, R. mRNA expression profiles for the response of human tumor cell lines to the antimalarial drugs artesunate, arteether, and artemether. Biochem. Pharmacol. 2002, 64, 617–623. [Google Scholar] [CrossRef]
- Li, P.C.; Lam, E.; Roos, W.P.; Zdzienicka, M.Z.; Kaina, B.; Efferth, T. Artesunate derived from traditional Chinese medicine induces DNA damage and repair. Cancer Res. 2008, 68, 4347–4351. [Google Scholar] [CrossRef] [PubMed]
- Berdelle, N.; Nikolova, T.; Quiros, S.; Efferth, T.; Kaina, B. Artesunate induces oxidative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage response in cancer cells. Mol. Cancer Ther. 2011, 10, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.; Reinboldt, V.; Hehlgans, S.; Efferth, T.; Rodel, C.; Rodel, F. A radiosensitizing effect of artesunate in glioblastoma cells is associated with a diminished expression of the inhibitor of apoptosis protein survivin. Radiother. Oncol. 2012, 103, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.; Fröhlich, T.; Gruber, L.; Hutterer, C.; Marschall, M.; Voigtländer, C.; Friedrich, O.; Kappes, B.; Efferth, T.; Tsogoeva, S.B. Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorg. Med. Chem. 2015, 23, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Efferth, T.; Steinbach, D.; Hamels, J.; de Longueville, F.; Bertholet, V.; Remacle, J. Microarray-based detection of multidrug resistance in human tumor cells by expression profiling of ATP-binding cassette transporter genes. Cancer Res. 2004, 64, 8987–8993. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Konkimalla, V.B.; Wang, Y.F.; Sauerbrey, A.; Meinhardt, S.; Zintl, F.; Mattern, J.; Volm, M. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 2008, 14, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Cao, J.; Kosyakova, N.; Mrasek, K.; Liehr, T.; Efferth, T. Genomic and transcriptomic profiling of resistant CEM/ADR-5000 and sensitive CCRF-CEM leukaemia cells for unravelling the full complexity of multi-factorial multidrug resistance. Sci. Rep. 2016, 6, 36754. [Google Scholar] [CrossRef] [PubMed]
- Kuete, V.; Mbaveng, A.T.; Nono, E.C.; Simo, C.C.; Zeino, M.; Nkengfack, A.E.; Efferth, T. Cytotoxicity of seven naturally occurring phenolic compounds towards multi-factorial drug-resistant cancer cells. Phytomedicine 2016, 23, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Kuete, V.; Mbaveng, A.T.; Sandjo, L.P.; Zeino, M.; Efferth, T. Cytotoxicity and mode of action of a naturally occurring naphthoquinone, 2-acetyl-7-methoxynaphtho[2,3-b]furan-4,9-quinone towards multi-factorial drug-resistant cancer cells. Phytomedicine 2017, 33, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Tajima, Y.; Nakagawa, H.; Tamura, A.; Kadioglu, O.; Satake, K.; Mitani, Y.; Murase, H.; Regasini, L.O.; Bolzani Vda, S.; Ishikawa, T.; et al. Nitensidine A, a guanidine alkaloid from Pterogyne nitens, is a novel substrate for human ABC transporter ABCB1. Phytomedicine 2014, 21, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Chan, A.; Cong Ling Qiu, A.; Wong, V.K.W.; Colligs, V.; Wecklein, S.; Freund-Henni Rached, H.; Efferth, T.; Hsiao, W.W. Artemisinin derivatives target topoisomerase 1 and cause DNA damage in silico and in vitro. Front. Pharmacol. 2017, 8, 711. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Handley, M.D.; Gottesman, M.M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009, 30, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updates 2012, 15, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Ambudkar, S.V.; Xia, D. Structure of a multidrug transporter. Nat. Biotechnol. 2009, 27, 546–547. [Google Scholar] [CrossRef] [PubMed]
- Karwatsky, J.; Lincoln, M.C.; Georges, E. A mechanism for P-glycoprotein-mediated apoptosis as revealed by verapamil hypersensitivity. Biochemistry 2003, 42, 12163–12173. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, H.J.; Pinedo, H.M.; Kuiper, C.M.; Kaptein, L.C.; Schuurhuis, G.J.; Lankelma, J. Induction by verapamil of a rapid increase in ATP consumption in multidrug-resistant tumor cells. FASEB J. 1988, 2, 2278–2282. [Google Scholar] [CrossRef] [PubMed]
- Kimmig, A.; Gekeler, V.; Neumann, M.; Frese, G.; Handgretinger, R.; Kardos, G.; Diddens, H.; Niethammer, D. Susceptibility of multidrug-resistant human leukemia cell lines to human interleukin 2-activated killer cells. Cancer Res. 1990, 50, 6793–6799. [Google Scholar] [PubMed]
- Efferth, T.; Giaisi, M.; Merling, A.; Krammer, P.H.; Li-Weber, M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS ONE 2007, 2, e693. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Barton, V.E.; Panchana, M.; Charoensutthivarakul, S.; Biagini, G.A.; Ward, S.A.; O’Neill, P.M. A Click Chemistry-Based Proteomic Approach Reveals that 1,2,4-Trioxolane and Artemisinin Antimalarials Share a Common Protein Alkylation Profile. Angew. Chem. Int. Ed. Engl. 2016, 55, 6401–6405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, W.; Xiao, Y. Profiling of multiple targets of artemisinin activated by hemin in cancer cell proteome. ACS Chem. Biol. 2016, 11, 882–888. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
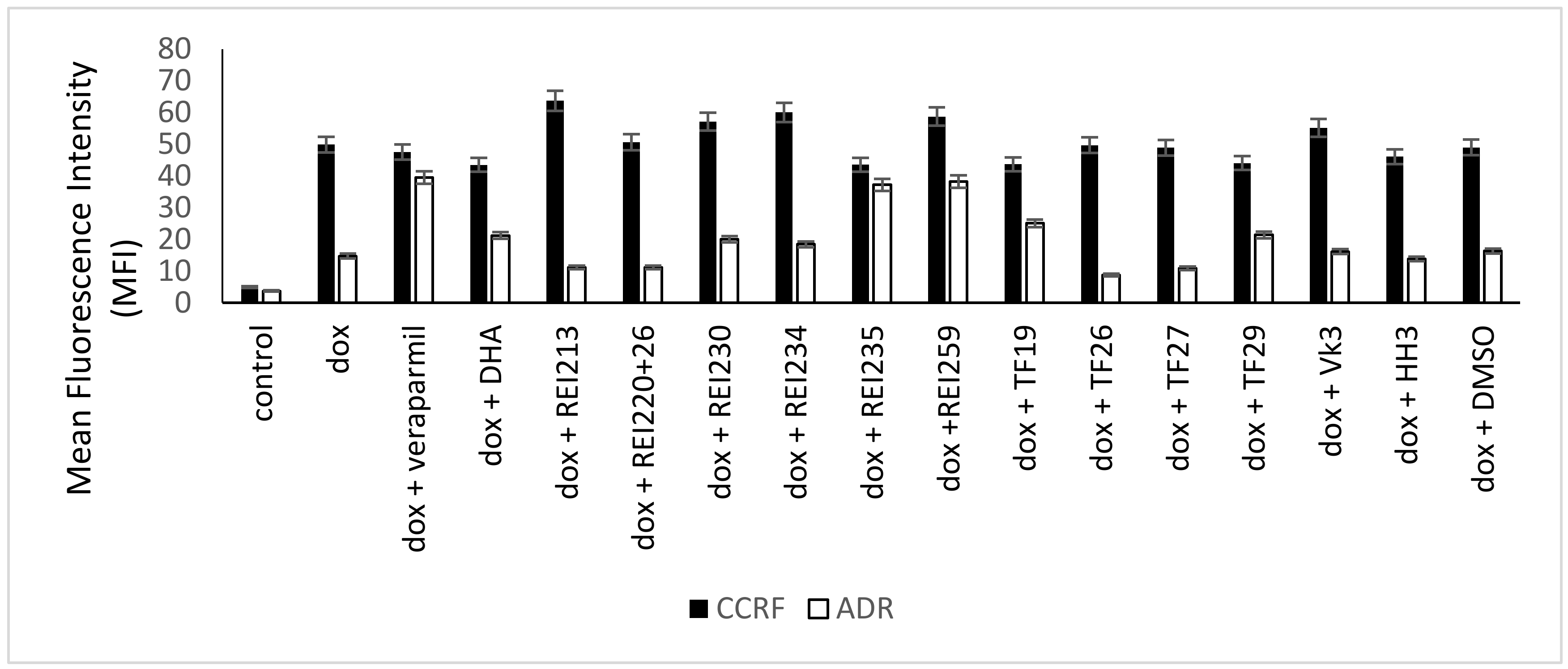{kind=link}
{kind=link}
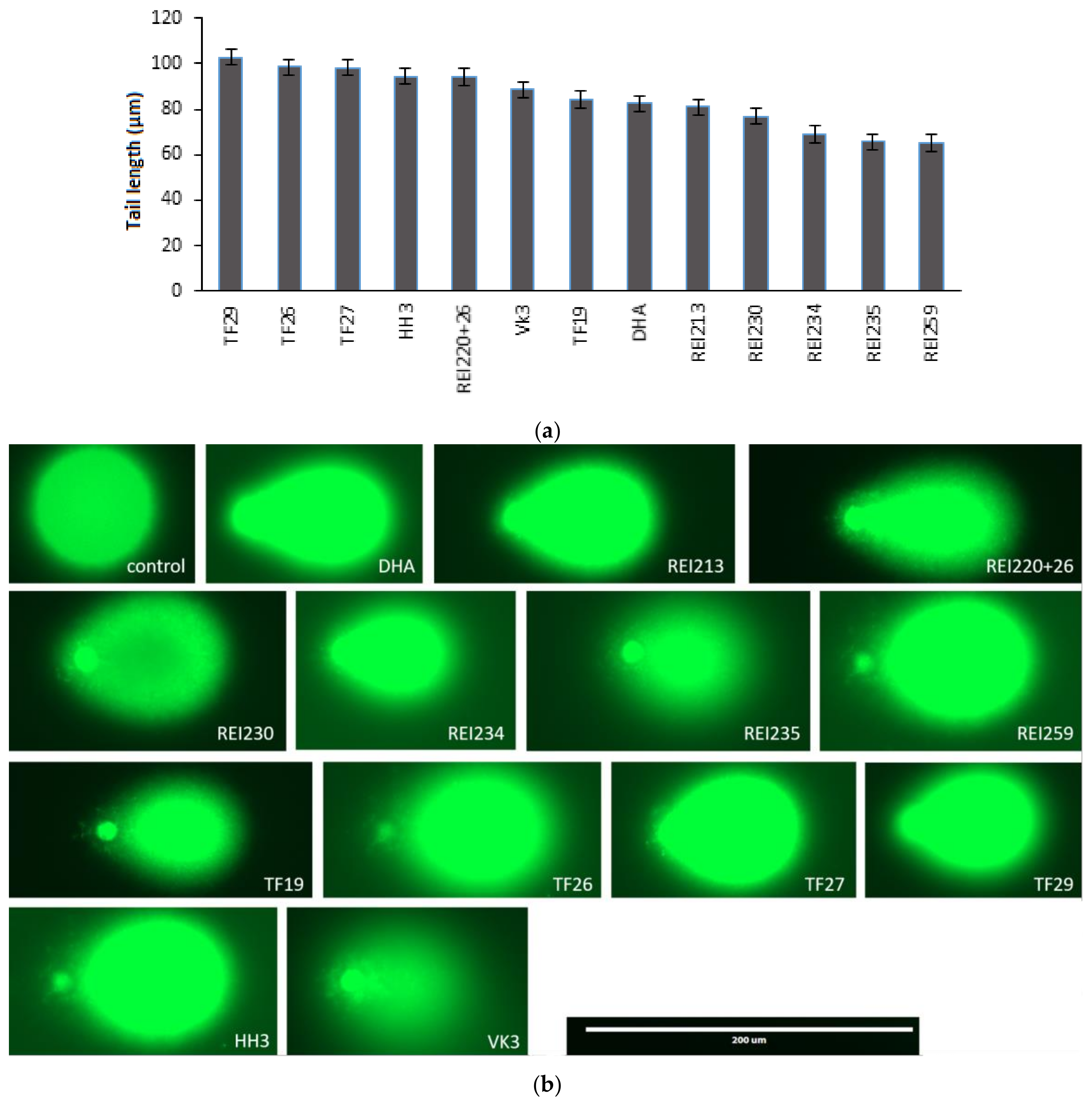{kind=link}
{kind=link}
| Compound | IC50 (µM) ± SD | Degree of Resistance | |
|---|---|---|---|
| CCRF-CEM | CEM/ADR5000 | ||
| Doxorubicin | 0.0033 ± 0.00065 | 1.613 ± 0.166 | 488.79 |
| Artesunic acid | 0.069 ± 0.03 | 0.189 ± 0.003 | 2.739 |
| DHA | 0.085 ± 0.003 | 0.265 ± 0.008 | 3.118 |
| REI213 | 0.568 ± 0.215 | 0.582 ±0.224 | 1.025 |
| REI220+26 | 43.685 ± 4.385 | 17.450 ± 1.010 | 0.399 |
| REI230 | 2.748 ± 0.021 | 2.789 ± 0.018 | 1.015 |
| REI234 | 0.0018 ± 0.0001 | 0.0068 ± 0.0006 | 3.778 |
| REI235 | 0.092 ± 0.006 | 0.199 ± 0.023 | 2.163 |
| REI259 | 0.876 ± 0.192 | 3.852 ± 1.021 | 4.397 |
| TF19 | 6.071 ± 0.247 | 5.663 ± 0.190 | 0.933 |
| TF26 | 0.0027 ± 0.001 | 7.872 ± 0.594 | 2915.556 |
| TF27 | 0.0024 ± 0.0001 | 0.196 ± 0.008 | 81.667 |
| TF29 | 0.0021 ± 0.0003 | 0.485 ± 0.210 | 230.952 |
| HH3 | 7.343 ± 0.911 | 7.071 ± 1.408 | 0.963 |
| VK3 | 0.134 ± 0.140 | 5.210 ± 0.153 | 38.881 |
| Modulator | IC50 (µM) in Combination with Doxorubicin ± SD | Fold Change in IC50 (Degree of Resistance Reversal) |
|---|---|---|
| - | 2.19 ± 0.041 | - |
| Verapamil | 0.69 ± 0.170 | 3.17 |
| REI259 | 0.84 ± 0.020 | 2.61 |
| REI235 | 0.85 ± 0.077 | 2.58 |
| TF19 | 1.43 ± 0.034 | 1.53 |
| Compound | Lowest Binding Energy (kcal/mol) | Mean Binding Energy (Kcal/mol) | Number of Interacting AA | AA Involved in H-Bond | pKi |
|---|---|---|---|---|---|
| REI213 | −12.945 (± 0.015) | −11.83 (± 0.11) | 13 | Gln725, Tyr953 | 9.4 |
| REI20+26 | −11.055 (± 0.105) | −10.31 (± 0.0) | 13 | - | 7.8 |
| REI230 | −12–78 (± 0.01) | 12.34 (± 0.12) | 11 | - | 9.3 |
| REI234 | −12.84 (± 0.035) | 12.185 (± 0.095) | 13 | Tyr953 | 9.5 |
| REI235 | −13.01 (± 0.01) | 12.24 (± 0.0) | 13 | - | 9.5 |
| TF19 | 11.425 (± 0.755) | 11.015 (± 0.345) | 17 | - | 8.9 |
| TF26 | −13.68 (± 0.425) | 13.3 (± 0.81) | 15 | Gln195 | 9.7 |
| TF27 | −14.815 (± 0.205) | −12.75 (± 0.37) | 15 | Gln990 | 11.0 |
| TF29 | −15.545 (± 0.049) | −13.845 (± 0.665) | 10 | Gln990 | 11.75 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruber, L.; Abdelfatah, S.; Fröhlich, T.; Reiter, C.; Klein, V.; Tsogoeva, S.B.; Efferth, T. Treatment of Multidrug-Resistant Leukemia Cells by Novel Artemisinin-, Egonol-, and Thymoquinone-Derived Hybrid Compounds. Molecules 2018, 23, 841. https://doi.org/10.3390/molecules23040841
Gruber L, Abdelfatah S, Fröhlich T, Reiter C, Klein V, Tsogoeva SB, Efferth T. Treatment of Multidrug-Resistant Leukemia Cells by Novel Artemisinin-, Egonol-, and Thymoquinone-Derived Hybrid Compounds. Molecules. 2018; 23(4):841. https://doi.org/10.3390/molecules23040841
Chicago/Turabian StyleGruber, Lisa, Sara Abdelfatah, Tony Fröhlich, Christoph Reiter, Volker Klein, Svetlana B. Tsogoeva, and Thomas Efferth. 2018. "Treatment of Multidrug-Resistant Leukemia Cells by Novel Artemisinin-, Egonol-, and Thymoquinone-Derived Hybrid Compounds" Molecules 23, no. 4: 841. https://doi.org/10.3390/molecules23040841