Direct (Hetero)Arylation for the Synthesis of Molecular Materials: Coupling Thieno[3,4-c]pyrrole-4,6-dione with Perylene Diimide to Yield Novel Non-Fullerene Acceptors for Organic Solar Cells


Abstract
:
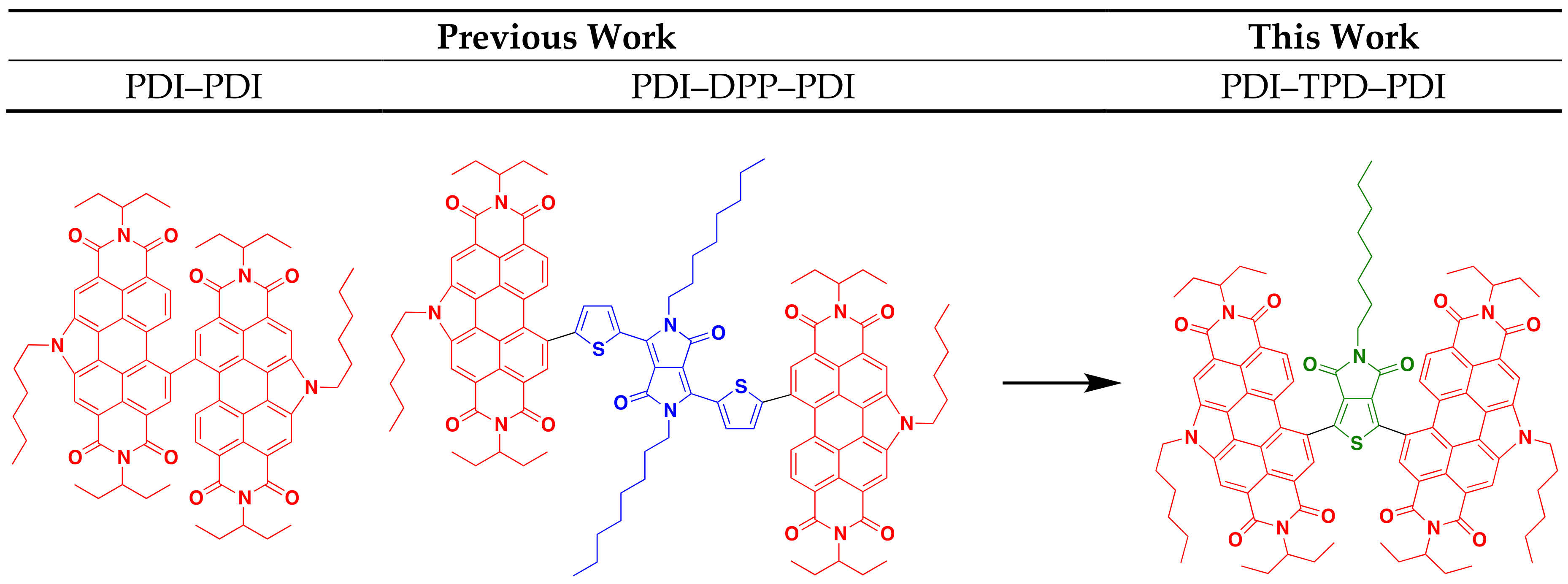
1. Introduction
2. Results and Discussion
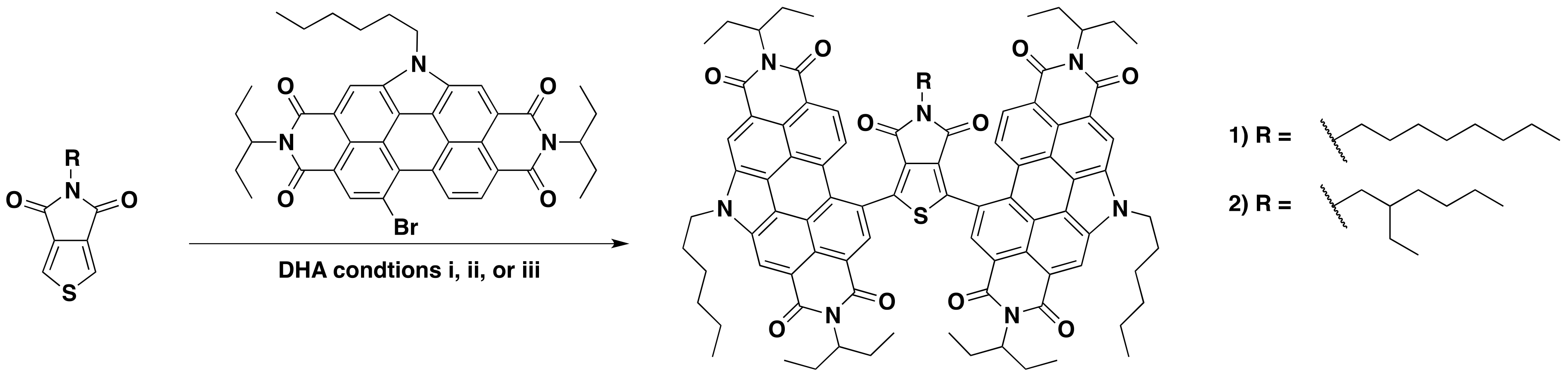
2.1. Synthesis and Characterization
2.2. Characterization
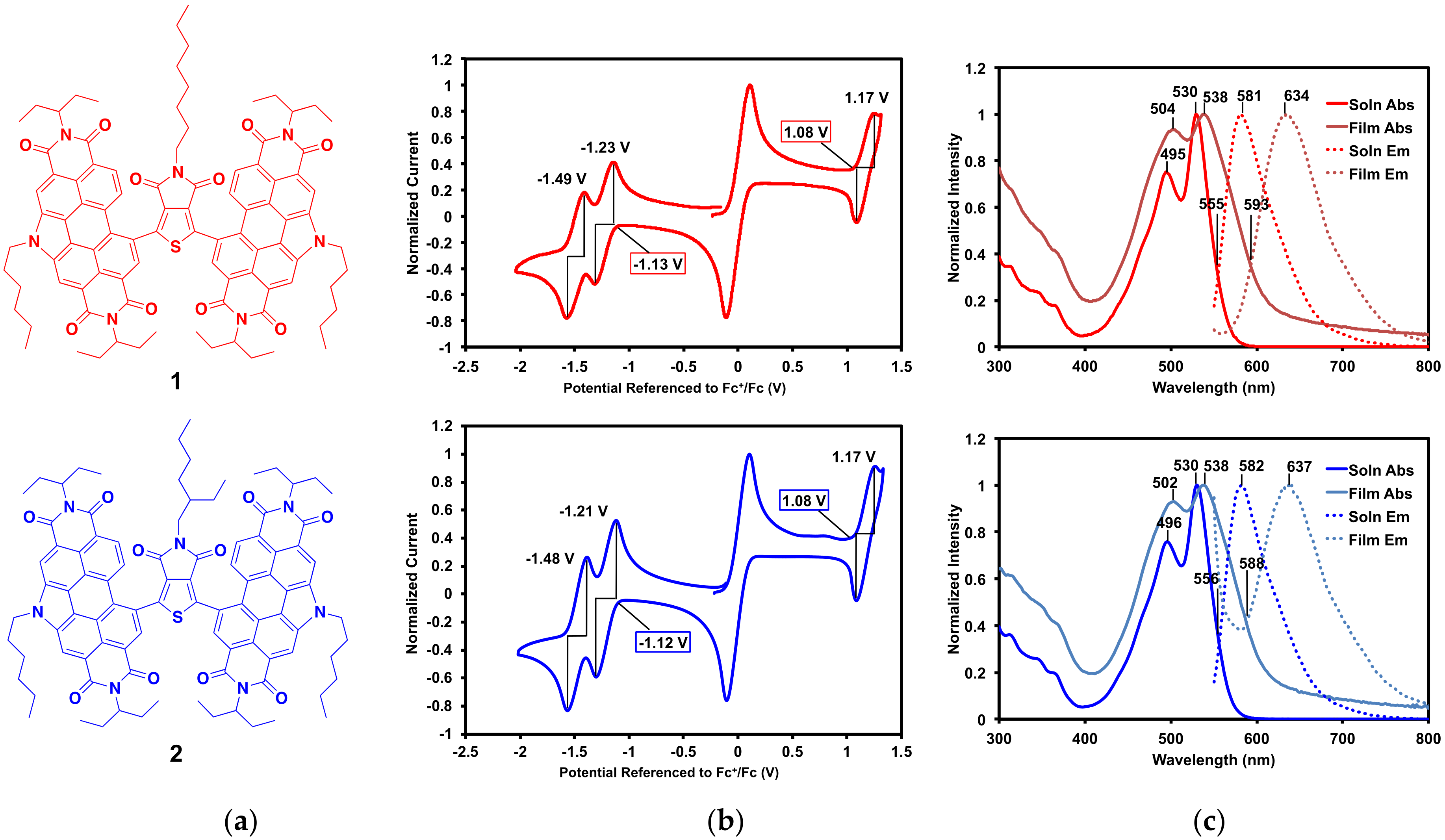
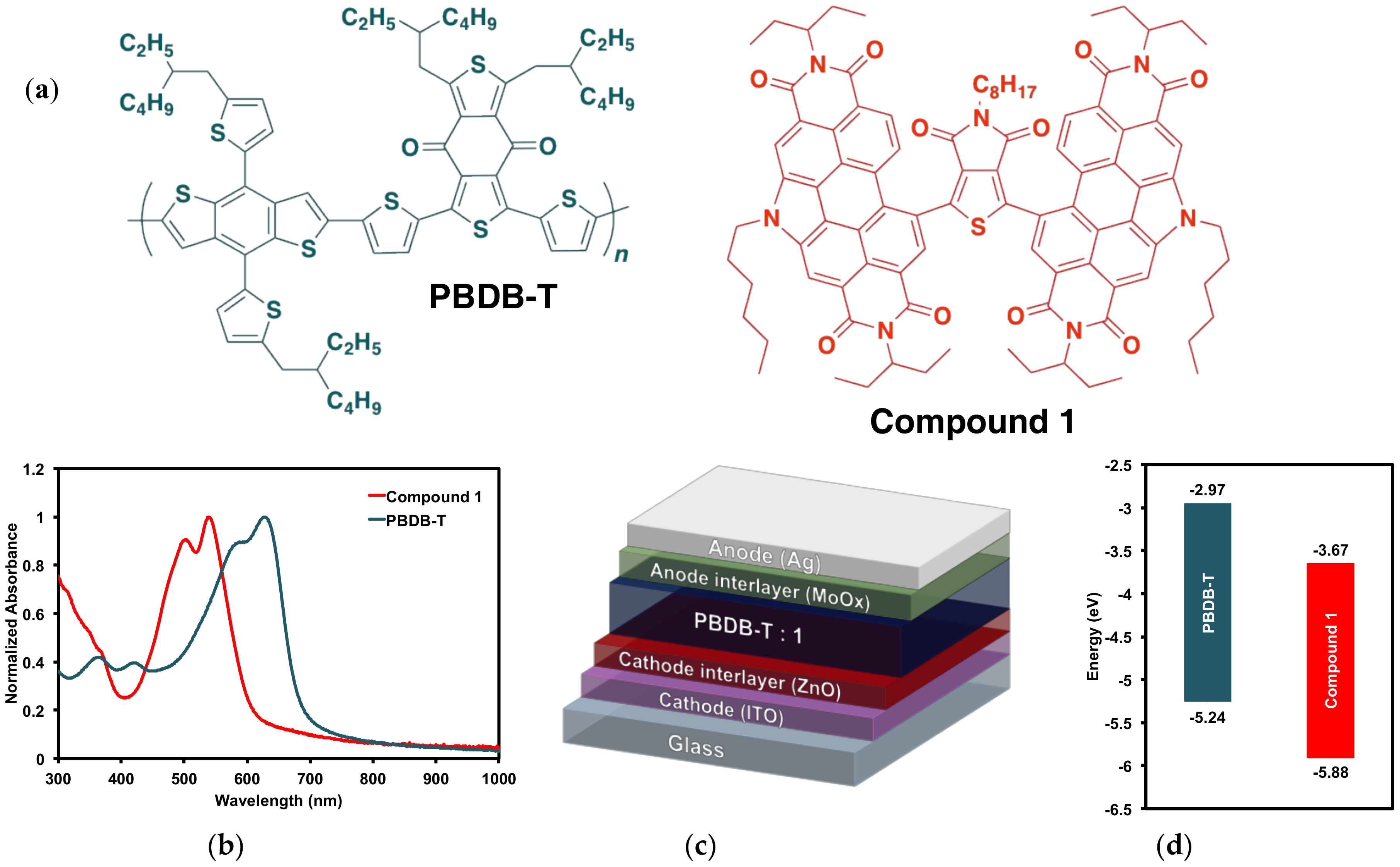
2.2.1. Optical and Electrochemical Properties
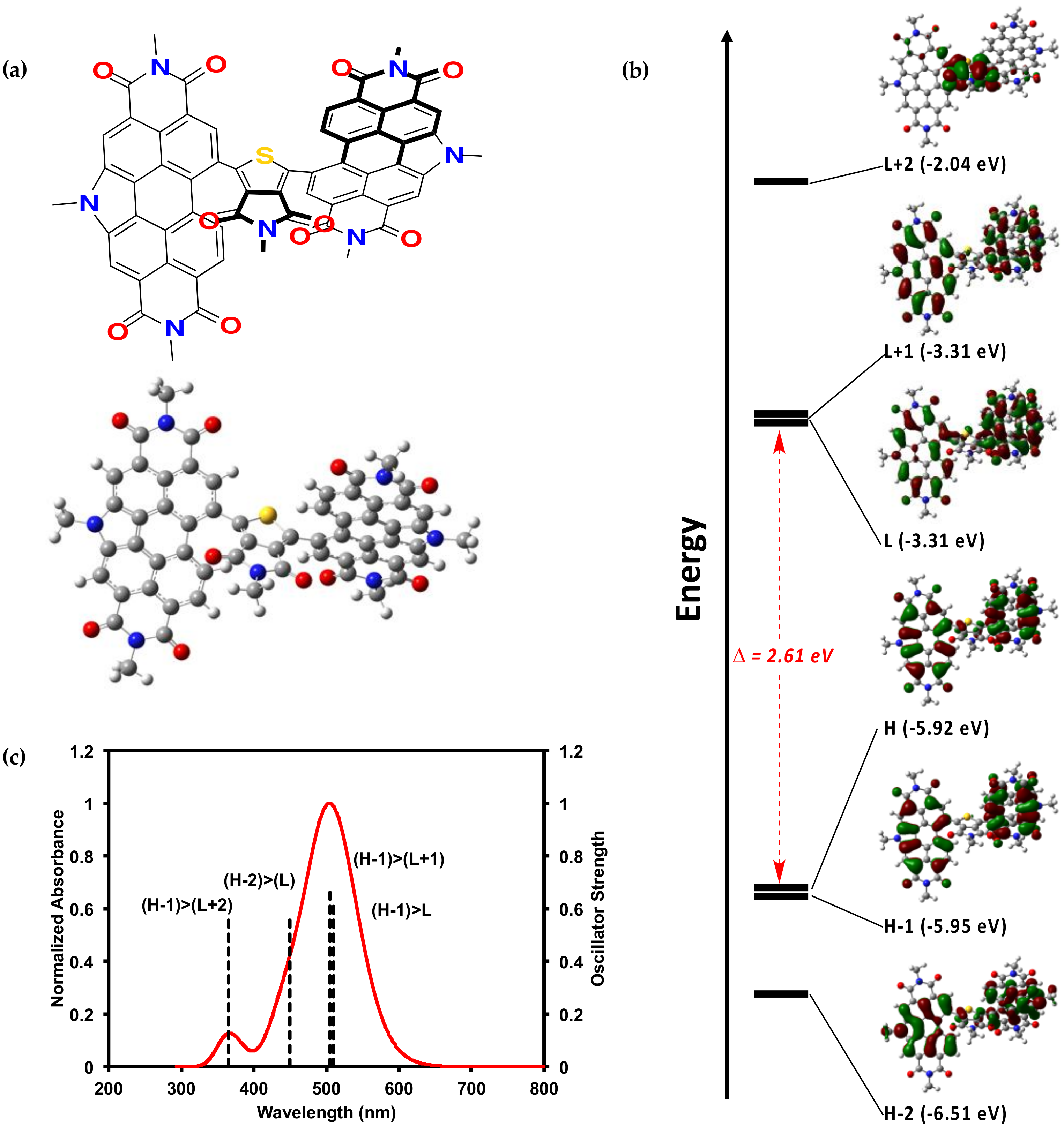
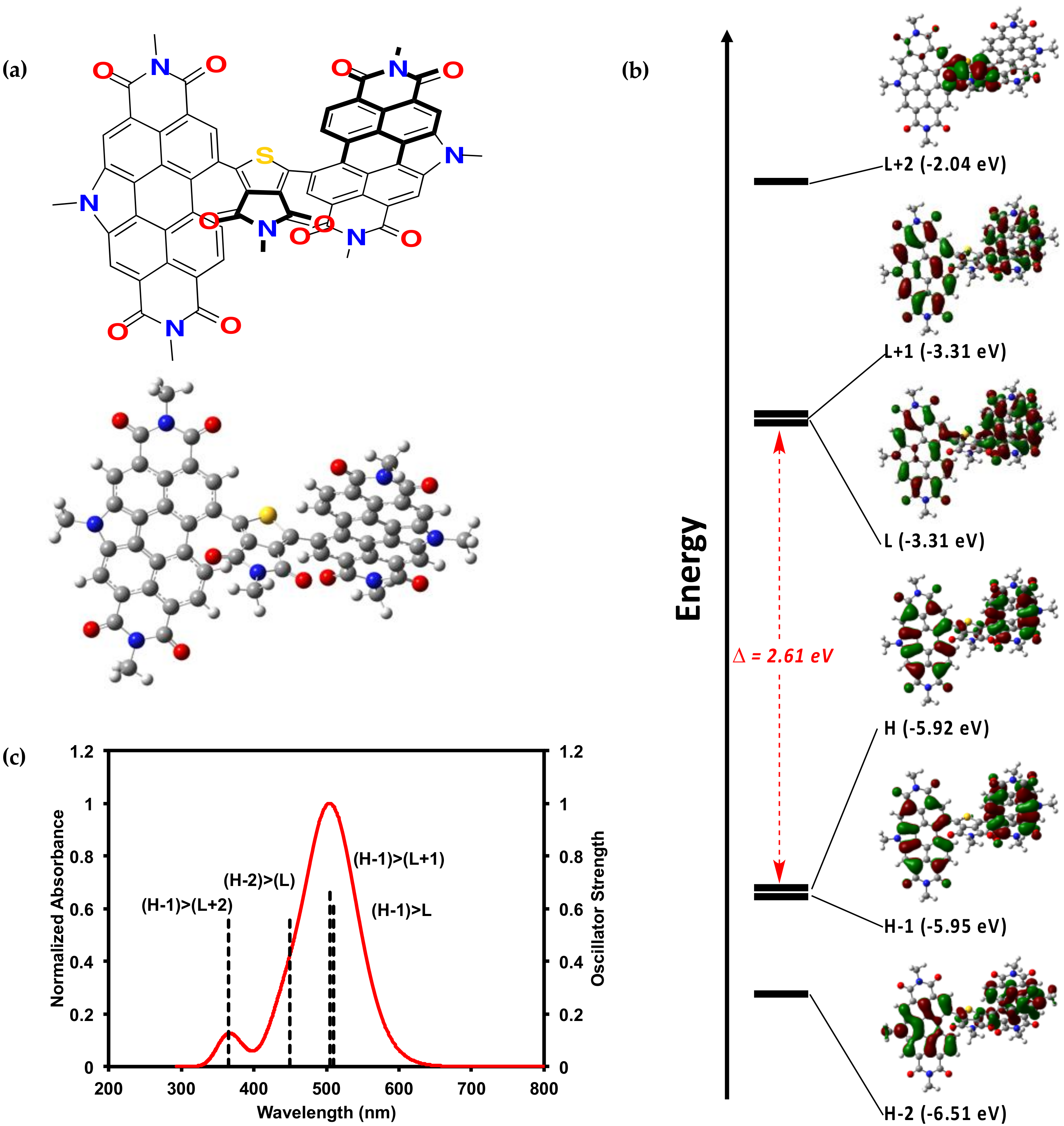
2.2.2. Theoretical Calculations
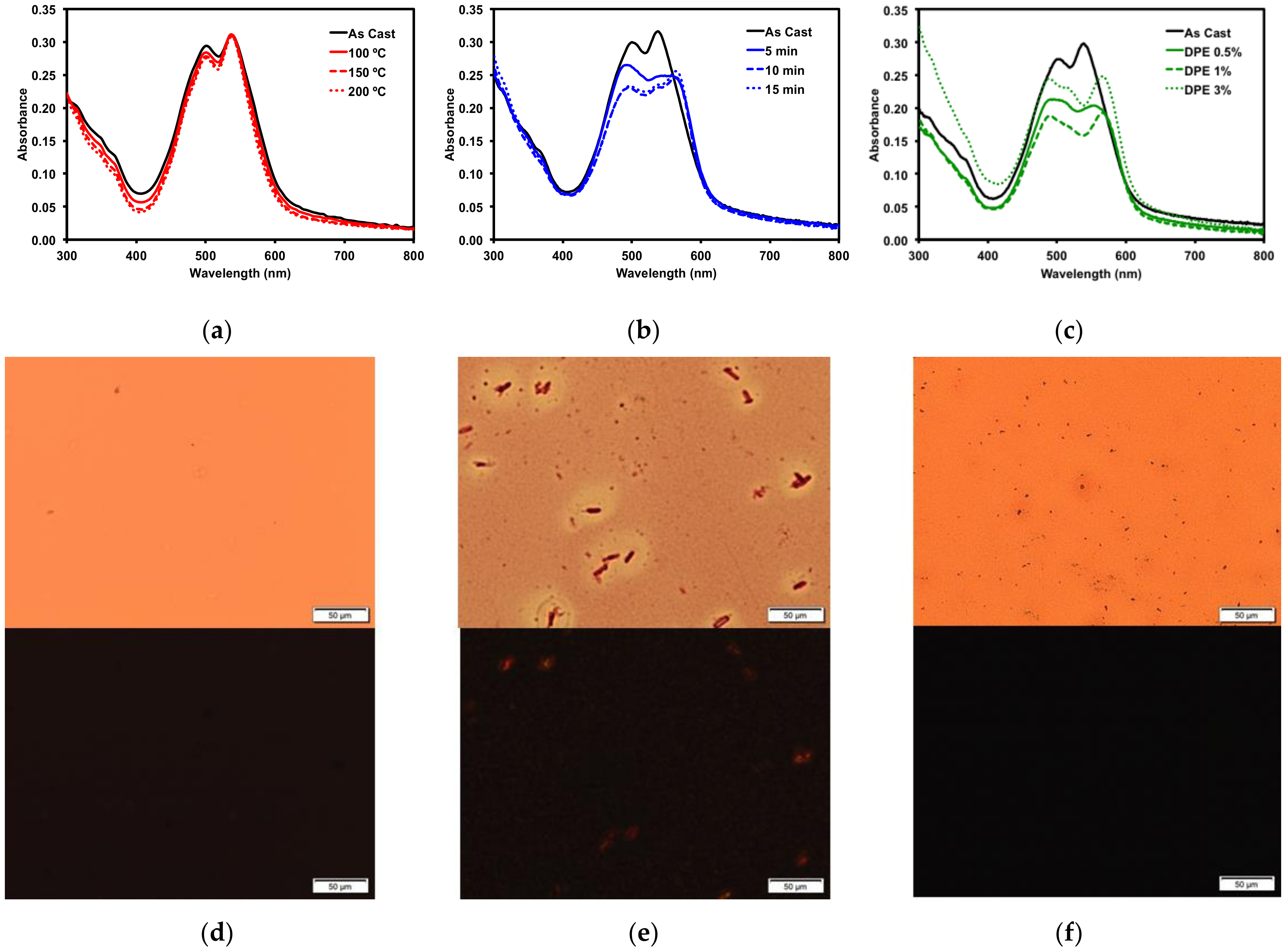
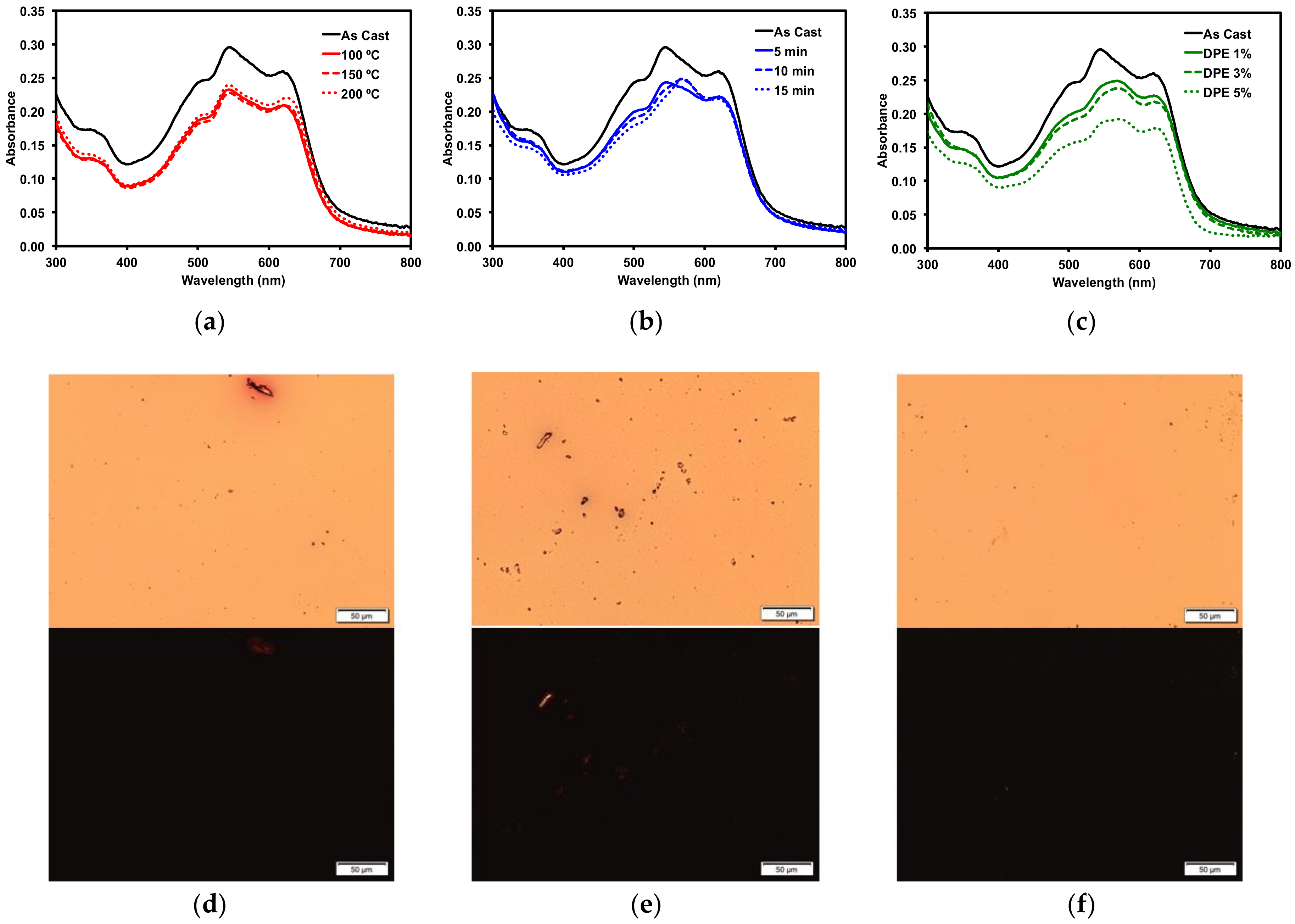
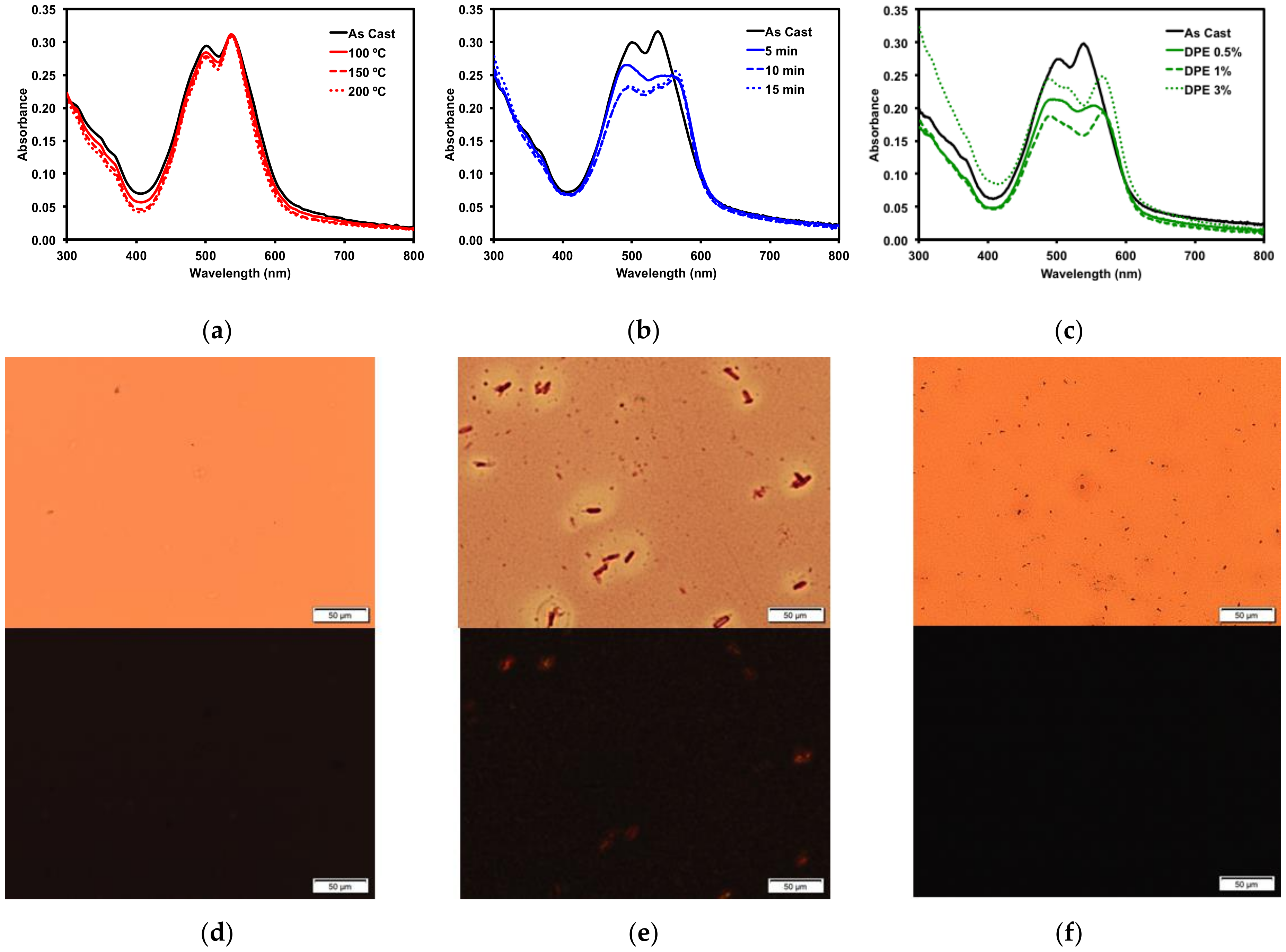
2.2.3. Thin Film Post-Deposition Treatment
2.3. Organic Solar Cells
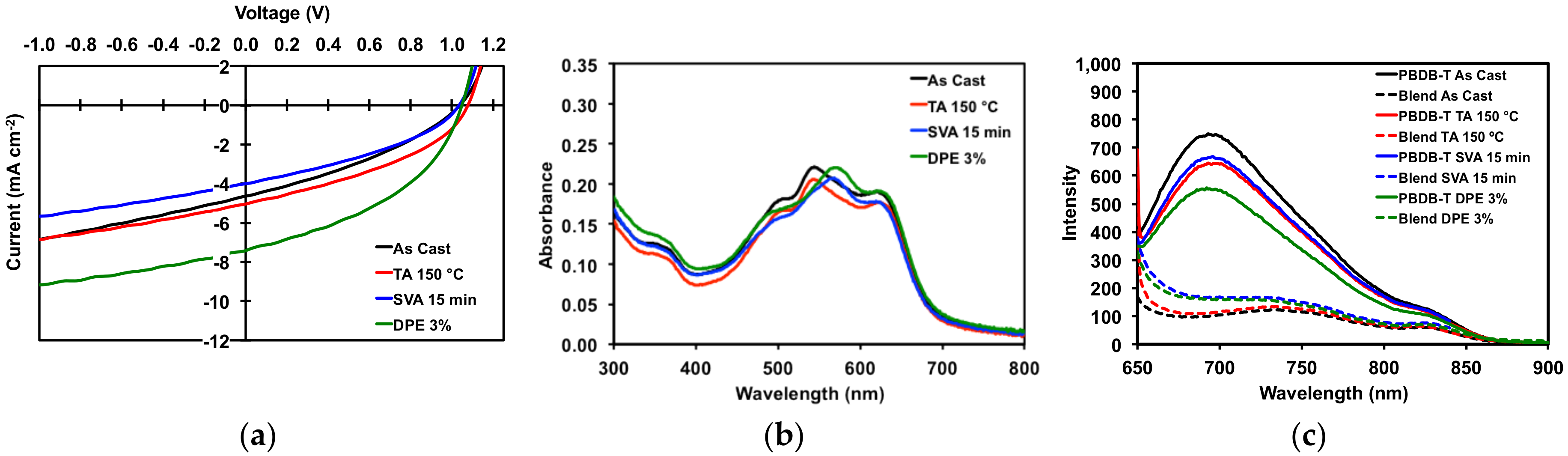
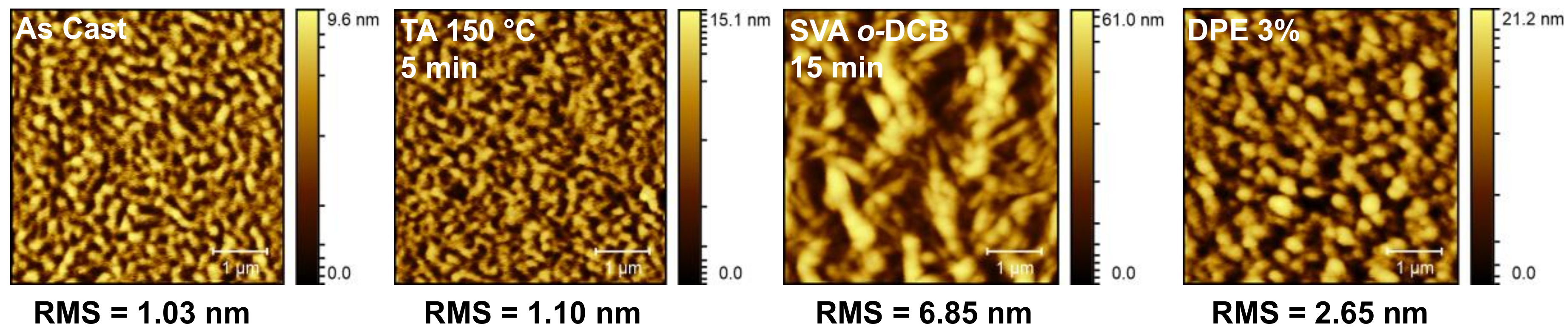
2.3.1. Donor/Acceptor Blend Thin Film Post-Deposition Treatment
2.3.2. Organic Solar Cell Devices
3. Materials and Methods
3.1. Materials
3.2. Characterization
3.3. Synthesis and Device Fabrication
3.3.1. Synthesis of (PDI)2octTPD (1)
3.3.2. Synthesis of (PDI)2ehTPD (2)
3.3.3. Device Fabrication
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl-Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef] [PubMed]
- Mercier, L.G.; Leclerc, M. Direct (Hetero)Arylation: A New Tool for Polymer Chemists. Acc. Chem. Res. 2013, 46, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Schipper, D.J.; Fagnou, K. Direct Arylation as a Synthetic Tool for the Synthesis of Thiophene-Based Organic Electronic Materials. Chem. Mater. 2011, 23, 1594–1600. [Google Scholar] [CrossRef]
- Pouliot, J.R.; Grenier, F.; Blaskovits, J.T.; Beaupré, S.; Leclerc, M. Direct (Hetero)arylation Polymerization: Simplicity for Conjugated Polymer Synthesis. Chem. Rev. 2016, 116, 14225–14274. [Google Scholar] [CrossRef] [PubMed]
- Bohra, H.; Wang, M. Direct C–H arylation: A “Greener” approach towards facile synthesis of organic semiconducting molecules and polymers. J. Mater. Chem. A 2017, 5, 11550–11571. [Google Scholar] [CrossRef]
- Lafrance, M.; Fagnou, K. Palladium-Catalyzed Benzene Arylation: Incorporation of Catalytic Pivalic Acid as a Proton Shuttle and a Key Element in Catalyst Design. J. Am. Chem. Soc. 2006, 128, 16496–16497. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Suzuki, A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc. Chem. Commun. 1979, 866–867. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, J.K. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar] [CrossRef]
- Facchetti, A.; Vaccaro, L. Marrocchi Assunta Semiconducting Polymers Prepared by Direct Arylation Polycondensation. Angew. Chem. Int. Ed. 2012, 51, 3520–3523. [Google Scholar] [CrossRef] [PubMed]
- Marrocchi, A.; Facchetti, A.; Lanari, D.; Petrucci, C.; Vaccaro, L. Current methodologies for a sustainable approach to π-conjugated organic semiconductors. Energy Environ. Sci. 2016, 9, 763–786. [Google Scholar] [CrossRef]
- Lombeck, F.; Marx, F.; Strassel, K.; Kunz, S.; Lienert, C.; Komber, H.; Friend, R.; Sommer, M. To branch or not to branch: C–H selectivity of thiophene-based donor–acceptor–donor monomers in direct arylation polycondensation exemplified by PCDTBT. Polym. Chem. 2017, 8, 4738–4745. [Google Scholar] [CrossRef]
- Chávez, P.; Ngov, C.; de Frémont, P.; Lévêque, P.; Leclerc, N. Synthesis by Direct Arylation of Thiazole–Derivatives: Regioisomer Configurations–Optical Properties Relationship Investigation. J. Org. Chem. 2014, 79, 10179–10188. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, A.; Perea, A.; Bélanger, D.; Leclerc, M. Poly(5-alkyl-thieno[3,4-c]pyrrole-4,6-dione): A study of π-conjugated redox polymers as anode materials in lithium-ion batteries. J. Mater. Chem. A 2017, 5, 18088–18094. [Google Scholar] [CrossRef]
- Garcias-Morales, C.; Romero-Borja, D.; Maldonado, J.L.; Roa, A.E.; Rodríguez, M.; García-Merinos, J.P.; Ariza-Castolo, A. Small Molecules Derived from Thieno[3,4-c]pyrrole-4,6-dione (TPD) and Their Use in Solution Processed Organic Solar Cells. Molecules 2017, 22, 1607. [Google Scholar] [CrossRef] [PubMed]
- McAfee, S.M.; Payne, A.J.; Dayneko, S.V.; Kini, G.P.; Song, C.E.; Lee, J.C.; Welch, G.C. A non-fullerene acceptor with a diagnostic morphological handle for streamlined screening of donor materials in organic solar cells. J. Mater. Chem. A 2017, 5, 16907–16913. [Google Scholar] [CrossRef]
- McAfee, S.M.; Dayneko, S.V.; Josse, P.; Blanchard, P.; Cabanetos, C.; Welch, G.C. Simply Complex: The Efficient Synthesis of an Intricate Molecular Acceptor for High-Performance Air-Processed and Air-Tested Fullerene-Free Organic Solar Cells. Chem. Mater. 2017, 29, 1309–1314. [Google Scholar] [CrossRef]
- Uy, R.L.; Price, S.C.; You, W. Structure-Property Optimizations in Donor Polymers via Electronics, Substituents, and Side Chains Toward High Efficiency Solar Cells. Macromol. Rapid Commun. 2012, 33, 1162–1177. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Najari, A.; Berrouard, P.; Beaupré, S.; Réda Aïch, B.; Tao, Y.; Leclerc, M.A. Thieno[3,4-c]pyrrole-4,6-dione-Based Copolymer for Efficient Solar Cells. J. Am. Chem. Soc. 2010, 132, 5330–5331. [Google Scholar] [CrossRef] [PubMed]
- Piliego, C.; Holcombe, T.W.; Douglas, J.D.; Woo, C.H.; Beaujuge, P.M.; Fréchet, J.M.J. Synthetic Control of Structural Order in N-Alkylthieno[3,4-c]pyrrole-4,6-dione-Based Polymers for Efficient Solar Cells. J. Am. Chem. Soc. 2010, 132, 7595–7597. [Google Scholar] [CrossRef] [PubMed]
- Najari, A.; Beaupré, S.; Berrouard, P.; Zou, Y.; Pouliot, J.R.; Lepage-Pérusse, C.; Leclerc, M. Synthesis and Characterization of New Thieno[3,4-c]pyrrole-4,6-dione Derivatives for Photovoltaic Applications. Adv. Funct. Mater. 2011, 21, 718–728. [Google Scholar] [CrossRef]
- Chu, T.Y.; Lu, J.; Beaupré, S.; Zhang, Y.; Pouliot, J.R.; Wakim, S.; Zhou, J.; Leclerc, M.; Li, Z.; Ding, J.; et al. Bulk Heterojunction Solar Cells Using Thieno[3,4-c]pyrrole-4,6-dione and Dithieno[3,2-b:2′,3′-d]silole Copolymer with a Power Conversion Efficiency of 7.3%. J. Am. Chem. Soc. 2011, 133, 4250–4253. [Google Scholar] [CrossRef] [PubMed]
- Su, M.S.; Kuo, C.Y.; Yuan, M.C.; Jeng, U.S.; Su, C.J.; Wei, K.H. Improving Device Efficiency of Polymer/Fullerene Bulk Heterojunction Solar Cells Through Enhanced Crystallinity and Reduced Grain Boundaries Induced by Solvent Additives. Adv. Mater. 2011, 23, 3315–3319. [Google Scholar] [CrossRef] [PubMed]
- Amb, C.M.; Chen, S.; Graham, K.R.; Subbiah, J.; Small, C.E.; So, F.; Reynolds, J.R. Dithienogermole as a Fused Electron Donor in Bulk Heterojunction Solar Cells. J. Am. Chem. Soc. 2011, 133, 10062–10065. [Google Scholar] [CrossRef] [PubMed]
- Small, C.E.; Chen, S.; Subbiah, J.; Amb, C.M.; Tsang, S.W.; Lai, T.H.; Reynolds, J.R.; So, F. High-efficiency inverted dithienogermole–thienopyrrolodione-based polymer solar cells. Nat. Photonics 2012, 6, 115–120. [Google Scholar] [CrossRef]
- Lu, L.P.; Kabra, D.; Johnson, K.; Friend, R.H. Charge-Carrier Balance and Color Purity in Polyfluorene Polymer Blends for Blue Light-Emitting Diodes. Adv. Funct. Mater. 2012, 22, 144–150. [Google Scholar] [CrossRef]
- Aïch, B.R.; Lu, J.; Beaupré, S.; Leclerc, M.; Tao, Y. Control of the active layer nanomorphology by using co-additives towards high-performance bulk heterojunction solar cells. Org. Electron. 2012, 13, 1736–1741. [Google Scholar] [CrossRef]
- Jo, J.; Pron, A.; Berrouard, P.; Leong, W.L.; Yuen, J.D.; Moon, J.S.; Leclerc, M.; Heeger, A.J. A New Terthiophene-Thienopyrrolodione Copolymer-Based Bulk Heterojunction Solar Cell with High Open-Circuit Voltage. Adv. Energy Mater. 2012, 2, 1397–1403. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, N.; Lou, S.J.; Hennek, J.W.; Ponce Ortiz, R.; Butler, M.R.; Boudreault, P.L.T.; Strzalka, J.; Morin, P.O.; Leclerc, M.; et al. Bithiopheneimide–Dithienosilole/Dithienogermole Copolymers for Efficient Solar Cells: Information from Structure–Property–Device Performance Correlations and Comparison to Thieno[3,4-c]pyrrole-4,6-dione Analogues. J. Am. Chem. Soc. 2012, 134, 18427–18439. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xin, H.; Kim, F.S.; Liyanage, A.D.T.; Jenekhe, S.A.; Watson, M.D. Thieno[3,4-c]pyrrole-4,6-dione-Based Donor−Acceptor Conjugated Polymers for Solar Cells. Macromolecules 2011, 44, 269–277. [Google Scholar] [CrossRef]
- Hendsbee, A.D.; Sun, J.P.; Law, W.K.; Yan, H.; Hill, I.G.; Spasyuk, D.M.; Welch, G.C. Synthesis, Self-Assembly, and Solar Cell Performance of N-Annulated Perylene Diimide Non-Fullerene Acceptors. Chem. Mater. 2016, 28, 7098–7109. [Google Scholar] [CrossRef]
- McAfee, S.M.; Dayneko, S.V.; Hendsbee, A.D.; Josse, P.; Blanchard, P.; Cabanetos, C.; Welch, G.C. Applying direct heteroarylation synthesis to evaluate organic dyes as the core component in PDI-based molecular materials for fullerene-free organic solar cells. J. Mater. Chem. A 2017, 5, 11623–11633. [Google Scholar] [CrossRef]
- Dayneko, S.V.; Hendsbee, A.D.; Welch, G.C. Fullerene-free polymer solar cells processed from non-halogenated solvents in air with PCE of 4.8%. Chem. Commun. 2017, 53, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Hendsbee, A.D.; Dayneko, S.V.; Pells, J.A.; Cann, J.R.; Welch, G.C. N-annulated perylene diimide dimers: the effect of thiophene bridges on physical, electronic, optical, and photovoltaic properties. Sustain. Energy Fuels 2017, 1, 1137–1147. [Google Scholar] [CrossRef]
- Cann, J.; Dayneko, S.; Sun, J.P.; Hendsbee, A.D.; Hill, I.G.; Welch, G.C. N-Annulated perylene diimide dimers: acetylene linkers as a strategy for controlling structural conformation and the impact on physical, electronic, optical and photovoltaic properties. J. Mater. Chem. C 2017, 5, 2074–2083. [Google Scholar] [CrossRef]
- McAfee, S.M.; Topple, J.M.; Payne, A.J.; Sun, J.P.; Hill, I.G.; Welch, G.C. An Electron-Deficient Small Molecule Accessible from Sustainable Synthesis and Building Blocks for Use as a Fullerene Alternative in Organic Photovoltaics. Chem. Phys. Chem. 2015, 16, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, J.R.; Mercier, L.G.; Caron, S.; Leclerc, M. Accessing New DPP-Based Copolymers by Direct Heteroarylation Polymerization. Macromol. Chem. Phys. 2013, 214, 453–457. [Google Scholar] [CrossRef]
- Welsh, T.A.; Laventure, A.; Baumgartner, T.; Welch, G.C. Dithienophosphole-based molecular electron acceptors constructed using direct (hetero)arylation cross-coupling methods. J. Mater. Chem. C 2018, 6, 2148–2154. [Google Scholar] [CrossRef]
- Payne, A.J.; Li, S.V.; Dayneko, S.; Risko, C.C.; Welch, G. An unsymmetrical non-fullerene acceptor: synthesis via direct heteroarylation, self-assembly, and utility as a low energy absorber in organic photovoltaic cells. Chem. Commun. 2017, 53, 10168–10171. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.J.; Welch, G.C. Optimized synthesis of π-extended squaraine dyes relevant to organic electronics by direct (hetero)arylation and Sonogashira coupling reactions. Org. Biomol. Chem. 2017, 15, 3310–3319. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.J.; Hendsbee, A.D.; McAfee, S.M.; Paul, D.K.; Karan, K.; Welch, G.C. Synthesis and structure–property relationships of phthalimide and naphthalimide based organic π-conjugated small molecules. Phys. Chem. Chem. Phys. 2016, 18, 14709–14719. [Google Scholar] [CrossRef] [PubMed]
- McAfee, S.M.; McCahill, J.S.J.; Macaulay, C.M.; Hendsbee, A.D.; Welch, G.C. Utility of a heterogeneous palladium catalyst for the synthesis of a molecular semiconductor via Stille, Suzuki, and direct heteroarylation cross-coupling reactions. RSC Adv. 2015, 5, 26097–26106. [Google Scholar] [CrossRef]
- Lemay, M.; Pandarus, V.; Simard, M.; Marion, O.; Tremblay, L.; Béland, F. SiliaCat® S-Pd and SiliaCat DPP-Pd: Highly Reactive and Reusable Heterogeneous Silica-Based Palladium Catalysts. Top. Catal. 2010, 53, 1059–1062. [Google Scholar] [CrossRef]
- Beaupré, S.; Pron, A.; Drouin, S.H.; Najari, A.; Mercier, L.G.; Robitaille, A.; Leclerc, M. Thieno-, Furo-, and Selenopheno[3,4-c]pyrrole-4,6-dione Copolymers: Effect of the Heteroatom on the Electrooptical Properties. Macromolecules 2012, 45, 6906–6914. [Google Scholar] [CrossRef]
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; Robert McElroy, C.; Sherwood, J. Tools and techniques for solvent selection: green solvent selection guides. Sustain. Chem. Process. 2016, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, X.; Burgers, M.A.; Huang, Y.; Bazan, G.C. Green-Solvent-Processed Molecular Solar Cells. Angew. Chem. Int. Ed. 2014, 53, 14378–14381. [Google Scholar] [CrossRef] [PubMed]
- Pommerehne, J.; Vestweber, H.; Guss, W.; Mahrt, R.F.; Bässler, H.; Porsch, M.; Daub, J. Efficient two layer leds on a polymer blend basis. Adv. Mater. 1995, 7, 551–554. [Google Scholar] [CrossRef]
- Rutledge, L.R.; McAfee, S.M.; Welch, G.C. Design and Computational Characterization of Non-Fullerene Acceptors for Use in Solution-Processable Solar Cells. J. Phys. Chem. A 2014, 118, 7939–7951. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xiao, C.; Hao, L.; Wang, Z.; Ceymann, H.; Lambert, C.; DiMotta, S.; Negri, F. Localization/Delocalization of Charges in Bay-Linked Perylene Bisimides. Chem. Eur. J. 2012, 18, 6764–6775. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Geng, Y.; Sun, G.Y.; Su, Z.M. Theoretical Design of Perylene Diimide Dimers with Different Linkers and Bridged Positions as Promising Non-Fullerene Acceptors for Organic Photovoltaic Cells. J. Phys. Chem. C 2017, 121, 2125–2134. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Binning, R.C.; Curtiss, L.A. Compact contracted basis sets for third-row atoms: Ga–Kr. J. Comput. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 16; revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView Version 5; Semichem, Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Welch, G.C.; Perez, L.A.; Hoven, C.V.; Zhang, Y.; Dang, X.D.; Sharenko, A.; Toney, M.F.; Kramer, E.J.; Nguyen, T.Q.; Bazan, G.C. A modular molecular framework for utility in small-molecule solution-processed organic photovoltaic devices. J. Mater. Chem. 2011, 21, 12700–12709. [Google Scholar] [CrossRef]
- Wessendorf, C.D.; Schulz, G.L.; Mishra, A.; Kar, P.; Ata, I.; Weidelener, M.; Urdanpilleta, M.; Hanisch, J.; Mena-Osteritz, E.; Lindén, M.; et al. Efficiency Improvement of Solution-Processed Dithienopyrrole-Based A-D-A Oligothiophene Bulk-Heterojunction Solar Cells by Solvent Vapor Annealing. Adv. Energy Mater. 2014, 4, 14. [Google Scholar] [CrossRef]
- Sun, Y.; Welch, G.C.; Leong, W.L.; Takacs, C.J.; Bazan, G.C.; Heeger, A.J. Solution-processed small-molecule solar cells with 6.7% efficiency. Nat. Mater. 2012, 11, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, C.; Zhan, X. Morphology Control in Organic Solar Cells. Adv. Energy Mater. 2018, 1703147. [Google Scholar] [CrossRef]
- Huang, Y.; Kramer, E.J.; Heeger, A.J.; Bazan, G.C. Bulk Heterojunction Solar Cells: Morphology and Performance Relationships. Chem. Rev. 2014, 114, 7006–7043. [Google Scholar] [CrossRef] [PubMed]
- Josse, P.; Labrunie, A.; Dalinot, C.; McAfee, S.M.; Dabos-Seignon, S.; Roncali, J.; Welch, G.C.; Blanchard, P.; Cabanetos, C. Effect of side chains on the electronic and photovoltaic properties of diketopyrrolopyrrole-based molecular acceptors. Org. Electron. 2016, 37, 479–484. [Google Scholar] [CrossRef]
- Lan, S.C.; Chang, C.K.; Lu, Y.H.; Lin, S.W.; Jen, A.K.Y.; Wei, K.H. Side chain structure affects the molecular packing and photovoltaic performance of oligothiophene-based solution-processable small molecules. RSC Adv. 2015, 5, 67718–67726. [Google Scholar] [CrossRef]
- Kan, B.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Yang, X.; Zhang, M.; Zhang, H.; Russell, T.P.; et al. Small Molecules Based on Alkyl/Alkylthio-thieno[3,2-b]thiophene-Substituted Benzo[1,2-b:4,5-b′]dithiophene for Solution-Processed Solar Cells with High Performance. Chem. Mater. 2015, 27, 8414–8423. [Google Scholar] [CrossRef]
- Graham, K.R.; Cabanetos, C.; Jahnke, J.P.; Idso, M.N.; El Labban, A.; Ngongang Ndjawa, G.O.; Heumueller, T.; Vandewal, K.; Salleo, A.; Chmelka, B.F.; et al. Importance of the Donor:Fullerene Intermolecular Arrangement for High-Efficiency Organic Photovoltaics. J. Am. Chem. Soc. 2014, 136, 9608–9618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Jiang, K.; Yang, G.; Liu, J.; Li, Z.; Lin, H.; Liu, Y.; Zhao, J.; Zhang, J.; Huang, F.; et al. Terthiophene-Based D–A Polymer with an Asymmetric Arrangement of Alkyl Chains That Enables Efficient Polymer Solar Cells. J. Am. Chem. Soc. 2015, 137, 14149–14157. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Lee, O.P.; Niskala, J.R.; Yiu, A.T.; Tassone, C.J.; Schmidt, K.; Beaujuge, P.M.; Onishi, S.S.; Toney, M.F.; Zettl, A.; et al. Enhanced Solid-State Order and Field-Effect Hole Mobility through Control of Nanoscale Polymer Aggregation. J. Am. Chem. Soc. 2013, 135, 19229–19236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells. Nat. Commun. 2014, 5, 5293. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Choulis, S.A.; Nelson, J.; Bradley, D.D.C.; Cook, S.; Durrant, J.R. Device annealing effect in organic solar cells with blends of regioregular poly(3-hexylthiophene) and soluble fullerene. Appl. Phys. Lett. 2005, 86, 063502. [Google Scholar] [CrossRef]
- Aïch, R.B. Solvent effect and device optimization of diketopyrrolopyrrole and carbazole copolymer based solar cells. Org. Electron. 2010, 11, 1053–1058. [Google Scholar] [CrossRef]
- Sun, Y.; Seo, J.H.; Takacs, C.J.; Seifter, J.; Heeger, A.J. Inverted Polymer Solar Cells Integrated with a Low-Temperature-Annealed Sol-Gel-Derived ZnO Film as an Electron Transport Layer. Adv. Mater. 2011, 23, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds (PDI)2octTPD and (PDI)2ehTPD are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPD R Group | Conditions | Yield (%) 1 |
|---|---|---|
| octyl | i | 49 |
| octyl | ii | 0 |
| octyl | iii | 46 |
| ethylhexyl | i | 56 |
| ethylhexyl | ii | 0 |
| ethylhexyl | iii | 0 |
| Property | Compound 1 | Compound 2 |
|---|---|---|
| EOx Onset (V) | 1.08 | 1.08 |
| E1/2 Ox (V) | 1.17 | 1.17 |
| ERed Onset (V) | −1.13 | −1.12 |
| E1/2 Red (V) | −1.23, −1.49 | −1.21, −1.48 |
| IP (eV) 1 | 5.88 | 5.88 |
| EA (eV) 1 | 3.67 | 3.68 |
| Eelec (eV) | 2.21 | 2.20 |
| Solution λabs (nm) | 530 | 530 |
| Solution λem (nm) | 581 | 582 |
| Solution Eopt (eV) 2 | 2.24 | 2.23 |
| Solution Stokes Shift (eV) 3 | 0.21 | 0.21 |
| Thin Film λabs (nm) | 538 | 538 |
| Thin Film λem (nm) | 634 | 637 |
| Thin Film Eopt (eV) 2 | 2.09 | 2.11 |
| Thin Film Stokes Shift (eV) 3 | 0.35 | 0.36 |
| Parameters | VOC (V) Avg. (Best) | Jsc (mA/cm2) Avg. (Best) | FF (%) Avg. (Best) | PCE (%) Avg. (Best) |
|---|---|---|---|---|
| As Cast | 1.06 (1.07) | 4.66 (4.84) | 34.66 (35.15) | 1.71 (1.81) |
| TA 150 °C 5 min | 1.07 (1.07) | 4.91 (5.04) | 37.14 (38.59) | 1.96 (2.09) |
| SVA o-DCB 15 min | 1.03 (1.03) | 3.92 (4.00) | 36.33 (36.52) | 1.46 (1.50) |
| DPE 3% | 1.04 (1.05) | 7.02 (7.40) | 42.85 (42.37) | 3.14 (3.28) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welsh, T.A.; Laventure, A.; Welch, G.C. Direct (Hetero)Arylation for the Synthesis of Molecular Materials: Coupling Thieno[3,4-c]pyrrole-4,6-dione with Perylene Diimide to Yield Novel Non-Fullerene Acceptors for Organic Solar Cells. Molecules 2018, 23, 931. https://doi.org/10.3390/molecules23040931
Welsh TA, Laventure A, Welch GC. Direct (Hetero)Arylation for the Synthesis of Molecular Materials: Coupling Thieno[3,4-c]pyrrole-4,6-dione with Perylene Diimide to Yield Novel Non-Fullerene Acceptors for Organic Solar Cells. Molecules. 2018; 23(4):931. https://doi.org/10.3390/molecules23040931
Chicago/Turabian StyleWelsh, Thomas A., Audrey Laventure, and Gregory C. Welch. 2018. "Direct (Hetero)Arylation for the Synthesis of Molecular Materials: Coupling Thieno[3,4-c]pyrrole-4,6-dione with Perylene Diimide to Yield Novel Non-Fullerene Acceptors for Organic Solar Cells" Molecules 23, no. 4: 931. https://doi.org/10.3390/molecules23040931