
Targeting GLI Transcription Factors in Cancer


Abstract
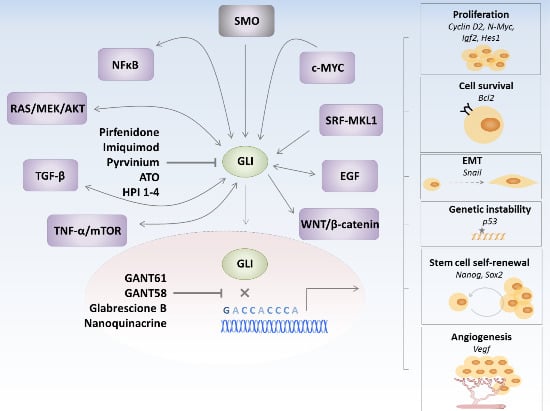
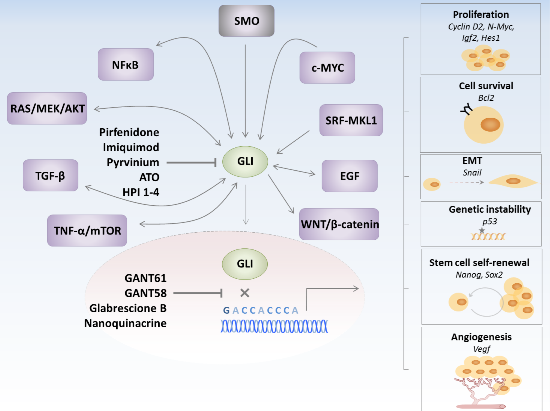
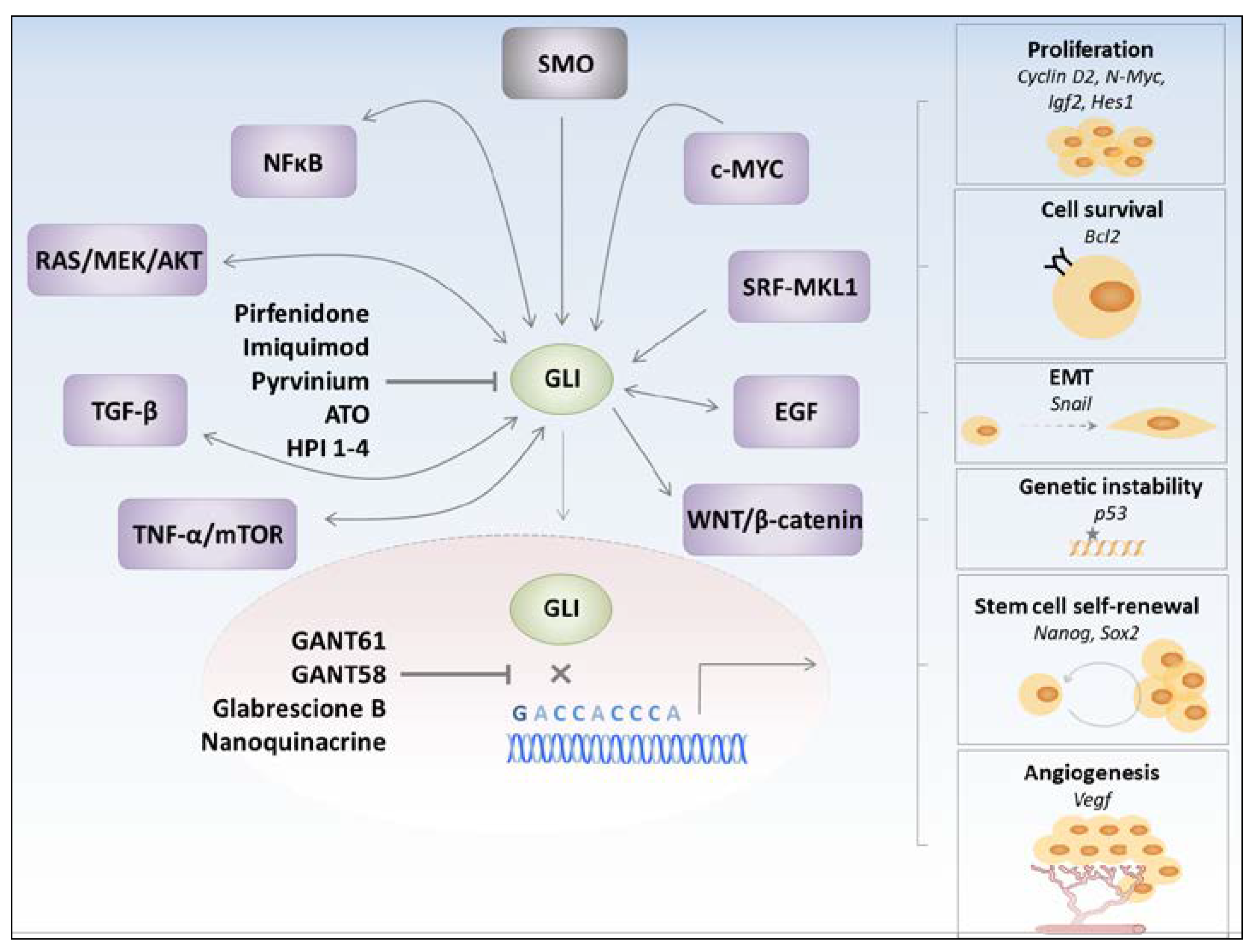
:
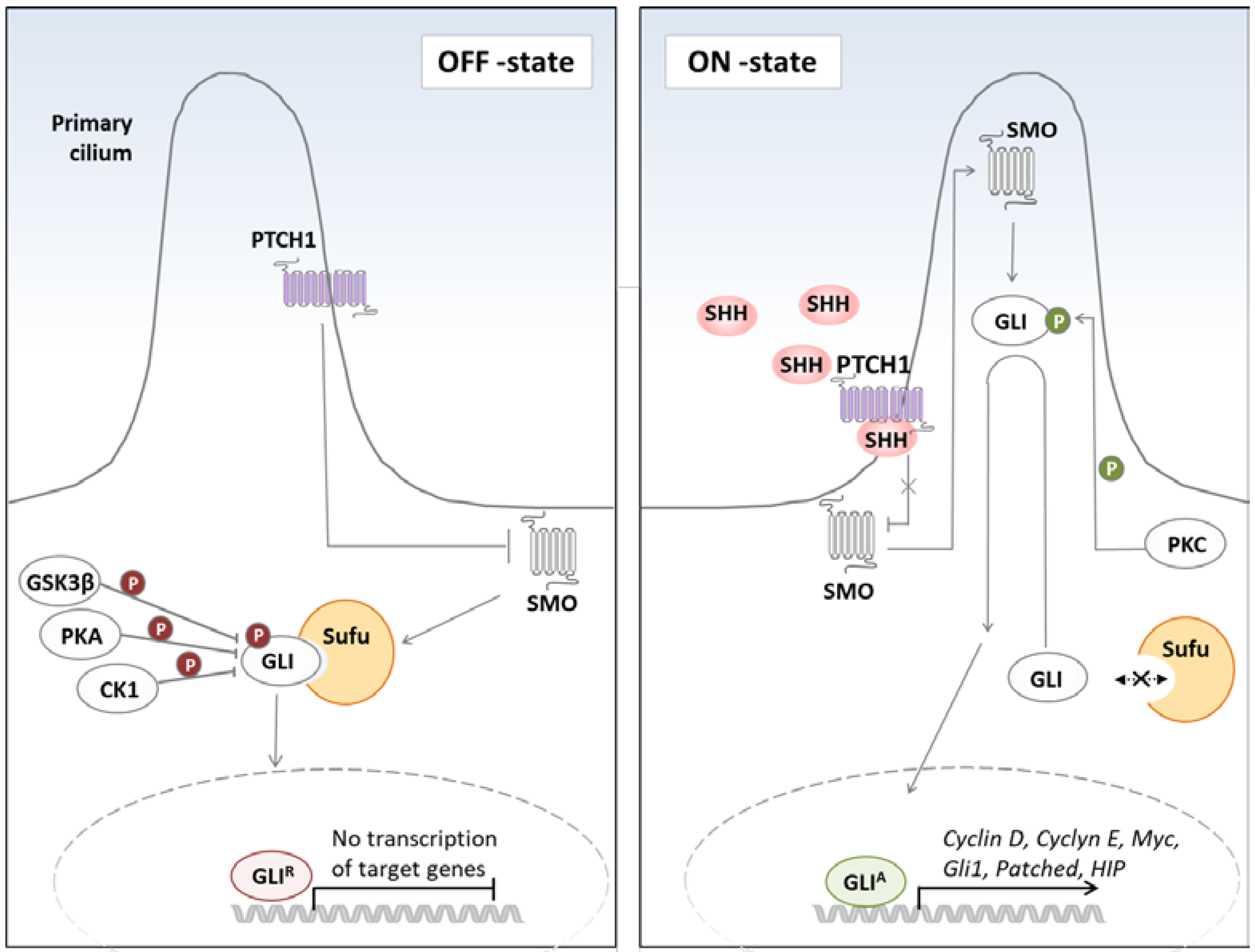
1. Hedgehog Signaling in Cancer
2. Mechanism of Hh Signaling Crosstalk with Other Pro-Tumorigenic Pathways
2.1. Non-Canonical Activation of the Hh Pathway
2.2. GLIs-Dependent Regulation of Pro-Tumorigenic Pathways
3. GLI Transcription Factors Inhibitors
4. Hh Pathway in Cancer Stem Cells
5. Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Wilson, C.W.; Li, Y.J.; Law, K.K.; Lu, C.S.; Gacayan, R.; Zhang, X.; Hui, C.C.; Chuang, P.T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Chuang, P.T. Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 2010, 137, 2079–2094. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.F.; Beachy, P.A. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, C.; Wang, B. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev. Biol. 2009, 326, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Amanai, K.; Wang, G.; Tang, J.; Wang, B.; Jiang, J. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 2002, 416, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, L.; Zhang, Q.; Tong, C.; Wang, B.; Hou, F.; Amanai, K.; Jiang, J. Phosphorylation by double-time/CKIepsilon and CKIα targets cubitus interruptus for Slimb/β-TRCP-mediated proteolytic processing. Dev. Cell 2005, 9, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Maye, P.; Kogerman, P.; Tejedor, F.J.; Toftgard, R.; Xie, W.; Wu, G.; Wu, D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 2002, 277, 35156–35161. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Liu, Y.; Fan, J.; Epperly, G.; Gao, T.; Jiang, J.; Jia, J. Hedgehog-regulated atypical PKC promotes phosphorylation and activation of Smoothened and Cubitus interruptus in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, E4842–E4850. [Google Scholar] [CrossRef] [PubMed]
- Heiden, K.B.; Williamson, A.J.; Doscas, M.E.; Ye, J.; Wang, Y.; Liu, D.; Xing, M.; Prinz, R.A.; Xu, X. The sonic hedgehog signaling pathway maintains the cancer stem cell self-renewal of anaplastic thyroid cancer by inducing snail expression. J. Clin. Endocrinol. Metab. 2014, 99, E2178–E2187. [Google Scholar] [CrossRef] [PubMed]
- Kawahira, H.; Scheel, D.W.; Smith, S.B.; German, M.S.; Hebrok, M. Hedgehog signaling regulates expansion of pancreatic epithelial cells. Dev. Biol. 2005, 280, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Kruger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology 2013, 58, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Lui, V.C.; Sham, M.H.; Pachnis, V.; Tam, P.K. Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. J. Cell Biol. 2004, 166, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.L.; Luo, M.Q.; Peng, W.X.; Li, Q.X.; Feng, Z.Y.; Li, Z.X.; Wang, M.X.; Feng, X.X.; Liu, F.; Huang, J.L. Sonic hedgehog signalling pathway regulates apoptosis through Smo protein in human umbilical vein endothelial cells. Rheumatology 2015, 54, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Feng, J.; Seidel, K.; Shi, S.; Klein, O.; Sharpe, P.; Chai, Y. Secretion of shh by a neurovascular bundle niche supports mesenchymal stem cell homeostasis in the adult mouse incisor. Cell Stem Cell 2014, 14, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Kusano, K.F.; Pola, R.; Murayama, T.; Curry, C.; Kawamoto, A.; Iwakura, A.; Shintani, S.; Ii, M.; Asai, J.; Tkebuchava, T.; et al. Sonic hedgehog myocardial gene therapy: Tissue repair through transient reconstitution of embryonic signaling. Nat. Med. 2005, 11, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Aszterbaum, M.; Rothman, A.; Johnson, R.L.; Fisher, M.; Xie, J.; Bonifas, J.M.; Zhang, X.; Scott, M.P.; Epstein, E.H., Jr. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J. Investig. Dermatol. 1998, 110, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Gessler, M.; Grzeschik, K.H. GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature 1991, 352, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Bonifas, J.M.; Pennypacker, S.; Chuang, P.T.; McMahon, A.P.; Williams, M.; Rosenthal, A.; De Sauvage, F.J.; Epstein, E.H., Jr. Activation of expression of hedgehog target genes in basal cell carcinomas. J. Investig. Dermatol. 2001, 116, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Ehtesham, M.; Sarangi, A.; Valadez, J.G.; Chanthaphaychith, S.; Becher, M.W.; Abel, T.W.; Thompson, R.C.; Cooper, M.K. Ligand-dependent activation of the hedgehog pathway in Glioma progenitor cells. Oncogene 2007, 26, 5752–5761. [Google Scholar] [CrossRef] [PubMed]
- Giroux Leprieur, E.; Vieira, T.; Antoine, M.; Rozensztajn, N.; Rabbe, N.; Ruppert, A.M.; Lavole, A.; Cadranel, J.; Wislez, M. Sonic Hedgehog Pathway Activation Is Associated With Resistance to Platinum-Based Chemotherapy in Advanced Non-Small-Cell Lung Carcinoma. Clin. Lung Cancer 2016, 17, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.S.; Uddin, M.; Rahman, M.Z.; Nayeem, M.J.; Alam, S.S.; Khatun, Z.; Wahiduzzaman, M.; Sultana, A.; Rahman, M.L.; Ali, M.Y.; et al. Overexpression of sonic hedgehog in the triple negative breast cancer: Clinicopathological characteristics of high burden breast cancer patients from Bangladesh. Sci. Rep. 2016, 6, 18830. [Google Scholar] [CrossRef] [PubMed]
- Lum, L.; Beachy, P.A. The Hedgehog response network: Sensors, switches, and routers. Science 2004, 304, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Trnski, D.; Sabol, M.; Gojevic, A.; Martinic, M.; Ozretic, P.; Musani, V.; Ramic, S.; Levanat, S. GSK3β and Gli3 play a role in activation of Hedgehog-Gli pathway in human colon cancer—Targeting GSK3β downregulates the signaling pathway and reduces cell proliferation. Biochim. Biophys. Acta 2015, 1852, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Gipp, J.; Bushman, W. The Sonic Hedgehog pathway stimulates prostate tumor growth by paracrine signaling and recapitulates embryonic gene expression in tumor myofibroblasts. Oncogene 2009, 28, 4480–4490. [Google Scholar] [CrossRef] [PubMed]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; van Sluis, P.; Troost, D.; Meeteren, N.S.; Caron, H.N.; Cloos, J.; et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Nitzki, F.; Becker, M.; Frommhold, A.; Schulz-Schaeffer, W.; Hahn, H. Patched knockout mouse models of Basal cell carcinoma. J. Skin Cancer 2012, 2012, 907543. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Chen, B.Y.; Wu, C.Y.; Tsao, Z.J.; Chen, Y.Y.; Chang, C.P.; Yang, C.R.; Lin, D.P. Hedgehog overexpression leads to the formation of prostate cancer stem cells with metastatic property irrespective of androgen receptor expression in the mouse model. J. Biomed. Sci. 2011, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Ai, K.; Du, Y.; Chen, G. Sonic hedgehog expression correlates with distant metastasis in pancreatic adenocarcinoma. Pancreas 2011, 40, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Pignot, G.; Vieillefond, A.; Vacher, S.; Zerbib, M.; Debre, B.; Lidereau, R.; Amsellem-Ouazana, D.; Bieche, I. Hedgehog pathway activation in human transitional cell carcinoma of the bladder. Br. J. Cancer 2012, 106, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, J.; He, J.; Zheng, Y.; Li, H.; Lu, Y.; Qian, J.; Lin, P.; Weber, D.M.; Yang, J.; et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood 2014, 124, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.A.; Kang, M.H.; Lee, H.J.; Kim, B.H.; Park, J.K.; Kim, H.K.; Kim, J.S.; Oh, S.C. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011, 71, 7061–7070. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, L.H.; Wen, Y.Y.; Song, M.; Li, B.L.; Chen, X.L.; Xu, M.; An, S.X.; Zhao, J.; Lu, Y.Y.; et al. Expression and regulation mechanisms of Sonic Hedgehog in breast cancer. Cancer Sci. 2010, 101, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Ertao, Z.; Jianhui, C.; Chuangqi, C.; Changjiang, Q.; Sile, C.; Yulong, H.; Hui, W.; Shirong, C. Autocrine Sonic hedgehog signaling promotes gastric cancer proliferation through induction of phospholipase Cgamma1 and the ERK1/2 pathway. J. Exp. Clin. Cancer Res. 2016, 35, 63. [Google Scholar] [CrossRef] [PubMed]
- Maitah, M.Y.; Ali, S.; Ahmad, A.; Gadgeel, S.; Sarkar, F.H. Up-regulation of sonic hedgehog contributes to TGF-β1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS ONE 2011, 6, e16068. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Ma, Q.; Xu, Q.; Liu, H.; Duan, W.; Lei, J.; Ma, J.; Wang, X.; Lv, S.; et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin. Cancer Res. 2014, 20, 4326–4338. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Gipp, J.; Bushman, W. Exploration of Shh and BMP paracrine signaling in a prostate cancer xenograft. Differentiation 2010, 79, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, O.; Hennen, E.; Koch, I.; Lindner, M.; Eickelberg, O. Gli1 mediates lung cancer cell proliferation and Sonic Hedgehog-dependent mesenchymal cell activation. PLoS ONE 2013, 8, e63226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, K.; Omura, N.; Hong, S.M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Zhang, Y.; Holtz, A.M.; Kane, K.T.; Song, J.Y.; Allen, B.L.; di Magliano, M.P. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 2014, 9, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Mao, J.; Zhang, Q.; Li, L. Overexpression of Hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncol. Lett. 2011, 2, 995–1001. [Google Scholar] [PubMed]
- Huang, S.; Yang, L.; An, Y.; Ma, X.; Zhang, C.; Xie, G.; Chen, Z.Y.; Xie, J.; Zhang, H. Expression of Hedgehog signaling molecules in lung cancer. Acta Histochem. 2011, 113, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Bieber, M.; Zhang, F.; Zhang, M.; He, B.; Jablons, D.; Teng, N.N. Wnt and hedgehog gene pathway expression in serous ovarian cancer. Int. J. Gynecol. Cancer 2011, 21, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.S.; Shen, Y.; Li, X.; Zhou, C.Z.; Wen, Y.G.; Jin, Y.B.; Li, J.K. Significance and prognostic value of Gli-1 and Snail/E-cadherin expression in progressive gastric cancer. Tumour Biol. 2014, 35, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Kai, M.; Odate, S.; Iwasaki, H.; Morifuji, Y.; Ogino, T.; Morisaki, T.; Nakashima, Y.; Katano, M. Hypoxia activates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci. 2011, 102, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Zhu, H.; Cao, X.; Aldrich, A.; Ali-Osman, F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009, 69, 6790–6798. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Duman-Scheel, M.; Weng, L.; Xin, S.; Du, W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 2002, 417, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Laurendeau, I.; Ferrer, M.; Garrido, D.; D’Haene, N.; Ciavarelli, P.; Basso, A.; Vidaud, M.; Bieche, I.; Salmon, I.; Szijan, I. Gene expression profiling of the hedgehog signaling pathway in human meningiomas. Mol. Med. 2010, 16, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Kita, Y.; Frank, D.J.; Majewski, R.R.; Konicek, B.A.; Nobrega, M.A.; Jacob, H.; Walterhouse, D.; Iannaccone, P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J. Biol. Chem. 2002, 277, 5548–5555. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.A.; Kang, M.H.; Kim, J.S.; Oh, S.C. Sonic hedgehog signaling promotes motility and invasiveness of gastric cancer cells through TGF-β-mediated activation of the ALK5-Smad 3 pathway. Carcinogenesis 2008, 29, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Pierrat, M.J.; Marsaud, V.; Mauviel, A.; Javelaud, D. Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor GLI2 and transforming growth factor-β. J. Biol. Chem. 2012, 287, 17996–18004. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2017, 8, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Zuo, M.; Rashid, A.; Churi, C.; Vauthey, J.N.; Chang, P.; Li, Y.; Hung, M.C.; Li, D.; Javle, M. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. Br. J. Cancer 2015, 112, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, G.; Li, Q.; Wang, Z.; Hu, W.; Li, P.; Li, S.; Wu, H.; Kong, X.; Gao, J.; et al. Hedgehog Signaling Non-Canonical Activated by Pro-Inflammatory Cytokines in Pancreatic Ductal Adenocarcinoma. J. Cancer 2016, 7, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.A.A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgard, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gallant, M.; Lamm, M.L.; Iannaccone, S.; Vieux, K.F.; Proytcheva, M.; Hyjek, E.; Iannaccone, P.; Walterhouse, D. Noncanonical regulation of the Hedgehog mediator GLI1 by C-MYC in Burkitt lymphoma. Mol. Cancer Res. 2013, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Edson, M.A.; Nalam, R.L.; Clementi, C.; Franco, H.L.; Demayo, F.J.; Lyons, K.M.; Pangas, S.A.; Matzuk, M.M. Granulosa cell-expressed BMPR1A and BMPR1B have unique functions in regulating fertility but act redundantly to suppress ovarian tumor development. Mol. Endocrinol. 2010, 24, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; He, M.; Sheng, T.; Zhang, X.; Sinha, M.; Luxon, B.; Zhao, X.; Xie, J. Requirement of TGFβ signaling for SMO-mediated carcinogenesis. J. Biol. Chem. 2010, 285, 36570–36576. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.K.; Qu, C.; Kunkalla, K.; Liu, Y.; Vega, F. Transcriptional regulation of serine/threonine protein kinase (AKT) genes by glioma-associated oncogene homolog 1. J. Biol. Chem. 2013, 288, 15390–15401. [Google Scholar] [CrossRef] [PubMed]
- Rajurkar, M.; Dang, K.; Fernandez-Barrena, M.G.; Liu, X.; Fernandez-Zapico, M.E.; Lewis, B.C.; Mao, J. IKBKE Is Required during KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2017, 77, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [PubMed]
- Louro, I.D.; Bailey, E.C.; Li, X.; South, L.S.; McKie-Bell, P.R.; Yoder, B.K.; Huang, C.C.; Johnson, M.R.; Hill, A.E.; Johnson, R.L.; et al. Comparative gene expression profile analysis of GLI and c-MYC in an epithelial model of malignant transformation. Cancer Res. 2002, 62, 5867–5873. [Google Scholar] [PubMed]
- Stemmer, V.; de Craene, B.; Berx, G.; Behrens, J. Snail promotes Wnt target gene expression and interacts with β-catenin. Oncogene 2008, 27, 5075–5080. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Kasai, K.; Ikeda, H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene 2011, 30, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by β-catenin. Oncogene 2007, 26, 4489–4498. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. WNT antagonist, SFRP1, is Hedgehog signaling target. Int. J. Mol. Med. 2006, 17, 171–175. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Sheng, T.; Stelter, A.A.; Li, C.; Zhang, X.; Sinha, M.; Luxon, B.A.; Xie, J. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J. Biol. Chem. 2006, 281, 35598–35602. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Ormestad, M.; Astorga, J.; Landgren, H.; Wang, T.; Johansson, B.R.; Miura, N.; Carlsson, P. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development 2006, 133, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Lewis, K.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; Marmur, E.; Rudin, C.M.; et al. Pivotal ERIVANCE basal cell carcinoma (BCC) study: 12-month update of efficacy and safety of vismodegib in advanced BCC. J. Am. Acad. Dermatol. 2015, 72, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Demirci, H.; Worden, F.; Nelson, C.C.; Elner, V.M.; Kahana, A. Efficacy of Vismodegib (Erivedge) for Basal Cell Carcinoma Involving the Orbit and Periocular Area. Ophthalmic Plast. Reconstr. Surg. 2015, 31, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Papastefanou, V.P.; Rene, C. Secondary Resistance to Vismodegib After Initial Successful Treatment of Extensive Recurrent Periocular Basal Cell Carcinoma with Orbital Invasion. Ophthalmic Plast. Reconstr. Surg. 2017, 33, S68–S70. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Latuske, E.M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jucker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, M.; Dyberg, C.; Shimokawa, T.; Milosevic, J.; Baryawno, N.; Fuskevag, O.M.; Larsson, R.; Kogner, P.; Zaphiropoulos, P.G.; Johnsen, J.I. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int. J. Cancer 2013, 132, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Chen, Z. Differentiation and apoptosis induction therapy in acute promyelocytic leukaemia. Lancet Oncol. 2000, 1, 101–106. [Google Scholar] [CrossRef]
- Nasr, R.; Guillemin, M.C.; Ferhi, O.; Soilihi, H.; Peres, L.; Berthier, C.; Rousselot, P.; Robledo-Sarmiento, M.; Lallemand-Breitenbach, V.; Gourmel, B.; et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 2008, 14, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Cavigelli, M.; Li, W.W.; Lin, A.; Su, B.; Yoshioka, K.; Karin, M. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 1996, 15, 6269–6279. [Google Scholar] [PubMed]
- Hayashi, T.; Hideshima, T.; Akiyama, M.; Richardson, P.; Schlossman, R.L.; Chauhan, D.; Munshi, N.C.; Waxman, S.; Anderson, K.C. Arsenic trioxide inhibits growth of human multiple myeloma cells in the bone marrow microenvironment. Mol. Cancer Ther. 2002, 1, 851–860. [Google Scholar] [PubMed]
- Kapahi, P.; Takahashi, T.; Natoli, G.; Adams, S.R.; Chen, Y.; Tsien, R.Y.; Karin, M. Inhibition of NF-κB activation by arsenite through reaction with a critical cysteine in the activation loop of IκB kinase. J. Biol. Chem. 2000, 275, 36062–36066. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.K.; Davison, K.; Colombo, M.; Colosimo, A.L.; Diaz, Z.; Padovani, A.M.; Guo, Q.; Scrivens, P.J.; Gao, W.; Mader, S.; et al. Antimony trioxide-induced apoptosis is dependent on SEK1/JNK signaling. Toxicol. Lett. 2006, 160, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Lopez-de la Mora, D.A.; Sanchez-Roque, C.; Montoya-Buelna, M.; Sanchez-Enriquez, S.; Lucano-Landeros, S.; Macias-Barragan, J.; Armendariz-Borunda, J. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int. J. Med. Sci. 2015, 12, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Yoshida, S.; Fujiwara, T.; Wada, H.; Nakajima, T.; Suzuki, H.; Yoshino, I. Effect of Perioperative Pirfenidone Treatment in Lung Cancer Patients With Idiopathic Pulmonary Fibrosis. Ann. Thorac. Surg. 2016, 102, 1905–1910. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Saito, T.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Inomata, M.; Nei, T.; Saito, Y.; Gemma, A.; Azuma, A. Reduced incidence of lung cancer in patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir. Investig. 2018, 56, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Boateng, K.; Noyes, D.; Antonia, S.J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, I.; Tritschler, F.; Opitz, C.A.; Frank, B.; Weller, M.; Wick, W. Pirfenidone inhibits TGF-β expression in malignant glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.J.; Huang, Z.; Jiang, T.P.; Shen, Y.P.; Zhao, A.S.; Zhou, S.; Zhang, S. Pirfenidone Inhibits Proliferation and Promotes Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the Wnt/β-Catenin Signaling Pathway. Med. Sci. Monit. 2017, 23, 6107–6113. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fei, D.L.; Flaveny, C.A.; Dahmane, N.; Baubet, V.; Wang, Z.; Bai, F.; Pei, X.H.; Rodriguez-Blanco, J.; Hang, B.; et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014, 74, 4811–4821. [Google Scholar] [CrossRef] [PubMed]
- Wolff, F.; Loipetzberger, A.; Gruber, W.; Esterbauer, H.; Aberger, F.; Frischauf, A.M. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene 2013, 32, 5574–5581. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Satapathy, S.R.; Das, D.; Siddharth, S.; Tripathi, N.; Bharatam, P.V.; Kundu, C. Nanoquinacrine induced apoptosis in cervical cancer stem cells through the inhibition of hedgehog-GLI1 cascade: Role of GLI-1. Sci. Rep. 2016, 6, 20600. [Google Scholar] [CrossRef] [PubMed]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.; Oro, A.; et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Babovic-Vuksanovic, D.; Ballman, K.; Michels, V.; McGrann, P.; Lindor, N.; King, B.; Camp, J.; Micic, V.; Babovic, N.; Carrero, X.; et al. Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology 2006, 67, 1860–1862. [Google Scholar] [CrossRef] [PubMed]
- Salazar, L.G.; Lu, H.; Reichow, J.L.; Childs, J.S.; Coveler, A.L.; Higgins, D.M.; Waisman, J.; Allison, K.H.; Dang, Y.; Disis, M.L. Topical Imiquimod Plus Nab-paclitaxel for Breast Cancer Cutaneous Metastases: A Phase 2 Clinical Trial. JAMA Oncol. 2017, 3, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Kozhaya, L.; Martiniuk, F.; Meng, T.C.; Chiriboga, L.; Liebes, L.; Hochman, T.; Shuman, N.; Axelrod, D.; Speyer, J.; et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 2012, 18, 6748–6757. [Google Scholar] [CrossRef] [PubMed]
- Pachman, D.R.; Barton, D.L.; Clayton, A.C.; McGovern, R.M.; Jefferies, J.A.; Novotny, P.J.; Sloan, J.A.; Loprinzi, C.L.; Gostout, B.S. Randomized clinical trial of imiquimod: An adjunct to treating cervical dysplasia. Am. J. Obstet. Gynecol. 2012, 206, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Polterauer, S.; Natter, C.; Rahhal, J.; Hefler, L.; Tempfer, C.B.; Heinze, G.; Stary, G.; Reinthaller, A.; Speiser, P. Treatment of cervical intraepithelial neoplasia with topical imiquimod: A randomized controlled trial. Obstet. Gynecol. 2012, 120, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, V.; Esni, F.; Garay, M.V.; Feldmann, G.; Habbe, N.; Jensen, J.N.; Dor, Y.; Stoffers, D.; Jensen, J.; Leach, S.D.; et al. Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology 2008, 135, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Pola, R.; Ling, L.E.; Aprahamian, T.R.; Barban, E.; Bosch-Marce, M.; Curry, C.; Corbley, M.; Kearney, M.; Isner, J.M.; Losordo, D.W. Postnatal recapitulation of embryonic hedgehog pathway in response to skeletal muscle ischemia. Circulation 2003, 108, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Renault, M.A.; Roncalli, J.; Tongers, J.; Misener, S.; Thorne, T.; Jujo, K.; Ito, A.; Clarke, T.; Fung, C.; Millay, M.; et al. The Hedgehog transcription factor Gli3 modulates angiogenesis. Circ. Res. 2009, 105, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Chechneva, O.V.; Deng, W. Empowering sonic hedgehog to rescue brain cells after ischemic stroke. Neural Regen Res. 2015, 10, 360–362. [Google Scholar] [PubMed]
- Ochoa, B.; Syn, W.K.; Delgado, I.; Karaca, G.F.; Jung, Y.; Wang, J.; Zubiaga, A.M.; Fresnedo, O.; Omenetti, A.; Zdanowicz, M.; et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010, 51, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lim, A.; Zhao, C.; Sahoo, D.; Pan, Y.; Spiekerkoetter, E.; Liao, J.C.; Beachy, P.A. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell 2014, 26, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed]
- Gerling, M.; Buller, N.V.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergstrom, A.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.; et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Rothenberg, M.E.; Seeley, E.S.; Zimdahl, B.; Kawano, S.; Lu, W.J.; Shin, K.; Sakata-Kato, T.; Chen, J.K.; Diehn, M.; et al. Control of inflammation by stromal Hedgehog pathway activation restrains colitis. Proc. Natl. Acad. Sci. USA 2016, 113, E7545–E7553. [Google Scholar] [CrossRef] [PubMed]
- Hitzenberger, M.; Schuster, D.; Hofer, T.S. The Binding Mode of the Sonic Hedgehog Inhibitor Robotnikinin, a Combined Docking and QM/MM MD Study. Front. Chem. 2017, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Petrova, E.; Rios-Esteves, J.; Ouerfelli, O.; Glickman, J.F.; Resh, M.D. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat. Chem. Biol. 2013, 9, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Maun, H.R.; Wen, X.; Lingel, A.; de Sauvage, F.J.; Lazarus, R.A.; Scales, S.J.; Hymowitz, S.G. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J. Biol. Chem. 2010, 285, 26570–26580. [Google Scholar] [CrossRef] [PubMed]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449—A potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Song, R.; Xie, J. Sonidegib: Mechanism of action, pharmacology, and clinical utility for advanced basal cell carcinomas. OncoTargets Ther. 2017, 10, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Oguro, Y.; Tanaka, T.; Shiokawa, Z.; Tanaka, Y.; Shibata, S.; Sato, Y.; Yamakawa, H.; Hattori, H.; Yamamoto, Y.; et al. Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: Modification of the core skeleton for improved solubility. Bioorg. Med. Chem. 2012, 20, 5507–5517. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetsky, M.; Zhang, X.M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F.Y.; Jones, S.; Shulok, J.; et al. Small-molecule modulators of Hedgehog signaling: Identification and characterization of Smoothened agonists and antagonists. J. Biol. 2002, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Chen, M.H.; Chuang, P.T. Smoothened adopts multiple active and inactive conformations capable of trafficking to the primary cilium. PLoS ONE 2009, 4, e5182. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, D.; Bahra, M.; Boas-Knoop, S.; Lippert, S.; Bradtmoller, M.; Guse, K.; Seehofer, D.; Bova, R.; Sauer, I.M.; Neuhaus, P.; et al. Hedgehog pathway as a potential treatment target in human cholangiocarcinoma. J. Hepato Biliary Pancreat. Sci. 2014, 21, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Xin, L.; Liang, A.; Fu, Y. Cancer stem cell hypothesis: A brief summary and two proposals. Cytotechnology 2013, 65, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Rauenzahn, S.; Maitra, A. In vitro models of pancreatic cancer for translational oncology research. Expert Opin. Drug Discov. 2009, 4, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Gulino, A.; Ferretti, E.; De Smaele, E. Hedgehog signalling in colon cancer and stem cells. EMBO Mol. Med. 2009, 1, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi-Ihara, N.; Okuhashi, Y.; Itoh, M.; Murohashi, I.; Nara, N.; Tohda, S. Promotion of the self-renewal capacity of human leukemia cells by sonic hedgehog protein. Anticancer Res. 2011, 31, 781–784. [Google Scholar] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Xia, Y.P.; Zhou, Y.N.; Dai, R.L.; Yang, X.; Duan, S.J.; Qiao, X.; Mei, Y.W.; Hu, B.; Cui, H. A critical role of Sonic Hedgehog signaling in maintaining the tumorigenicity of neuroblastoma cells. Cancer Sci. 2009, 100, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Mysliwietz, J.; Ellwart, J.; Gamarra, F.; Huber, R.M.; Bergner, A. Effects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populations. Clin. Exp. Med. 2012, 12, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Alfonsi, R.; Ingallina, C.; Quaglio, D.; Ghirga, F.; D’Acquarica, I.; Bernardi, F.; Di Magno, L.; Canettieri, G.; Screpanti, I.; et al. Inhibition of Hedgehog-dependent tumors and cancer stem cells by a newly identified naturally occurring chemotype. Cell Death Dis. 2016, 7, e2376. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Wang, Q.H.; Brown, P.; Peacock, C.; Merchant, A.A.; Brennan, S.; Jones, E.; McGovern, K.; Watkins, D.N.; Sakamoto, K.M.; et al. Self-renewal of acute lymphocytic leukemia cells is limited by the Hedgehog pathway inhibitors cyclopamine and IPI-926. PLoS ONE 2010, 5, e15262. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Enguita-German, M.; Schiapparelli, P.; Rey, J.A.; Castresana, J.S. CD133+ cells from medulloblastoma and PNET cell lines are more resistant to cyclopamine inhibition of the sonic hedgehog signaling pathway than CD133- cells. Tumour Biol. 2010, 31, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Inhibitor Name | Target | Mode of Action | Reference |
|---|---|---|---|
| Robotnikinin | SHH | binds to the N terminal part of SHH | [147] |
| RU-SKI 43 | SHH | inhibits SHH palmitoylation | [148] |
| 5E1 antibody | SHH | binds to the N terminal part of SHH | [149] |
| GDC-0449 (Vismodegib) | SMO | antagonist of SMO | [150] |
| LDE225 (Sonidegib) | SMO | antagonist of SMO | [151] |
| IPI 926 (Saridegib) | SMO | cyclopamine-derived antagonist of SMO | [152] |
| LY2940680 (Taladegib) | SMO | antagonist of SMO | [153] |
| PF-04449913 (Glasdegib) | SMO | antagonist of SMO | [154] |
| Cyclopamine | SMO | antagonist of SMO, blocks conformational change of SMO into the active form | [155] |
| TAK-441 | SMO | antagonist of SMO | [156] |
| CUR61414 | SMO | antagonist of SMO | [157] |
| Jervine | SMO | antagonist of SMO, blocks conformational change of SMO into the active form | [158] |
| BMS-833923 | SMO | antagonist of SMO | [159] |
| GANT61, GANT58 | GLI | blocks binding of GLIs to DNA | [101] |
| Glabrescione B | GLI | blocks binding of GLIs to DNA | [107] |
| Arsenic trioxide | GLI | reduces stability of GLI2 | [115] |
| Pirfenidone | GLI | reduces stability of GLI2 | [64] |
| HPI 1–4 | GLI | influence stability, degradation rate, and trafficking of GLIs to the primary cilium | [125] |
| Pyrvinium | GLI | induces proteosomal degradation of GLIs through CK1–mediated phosphorylation | [127] |
| Imiquimod | GLI | induces proteosomal degradation of GLIs through ADORA/PKA–mediated phosphorylation | [128] |
| Nanoquinacrine | GLI | increased expressions of GSK3β, PTEN and binds to and destabilizes GLI1-DNA complex | [129] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Didiasova, M.; Schaefer, L.; Wygrecka, M. Targeting GLI Transcription Factors in Cancer. Molecules 2018, 23, 1003. https://doi.org/10.3390/molecules23051003
Didiasova M, Schaefer L, Wygrecka M. Targeting GLI Transcription Factors in Cancer. Molecules. 2018; 23(5):1003. https://doi.org/10.3390/molecules23051003
Chicago/Turabian StyleDidiasova, Miroslava, Liliana Schaefer, and Malgorzata Wygrecka. 2018. "Targeting GLI Transcription Factors in Cancer" Molecules 23, no. 5: 1003. https://doi.org/10.3390/molecules23051003
APA StyleDidiasova, M., Schaefer, L., & Wygrecka, M. (2018). Targeting GLI Transcription Factors in Cancer. Molecules, 23(5), 1003. https://doi.org/10.3390/molecules23051003