1. Introduction
Recent reports have described transition-metal-free reactions, such as the coupling of haloarenes with arenes [
1,
2,
3,
4], that are promoted by a combination of KO
tBu and an organic additive. The reactions proceed by a radical mechanism, with a single electron transfer (SET) step initiating the formation of the radicals. It has previously been reported that KO
tBu is capable of reductively activating a range of substrates via single electron transfer [
5,
6], and this led some authors to propose that
tert-butoxide anion is responsible for the initiation of these coupling reactions, either alone or in complexation with an additive, such as 1,10-phenanthroline, in the ground state [
2,
3,
7,
8,
9,
10,
11]. However, recent publications have shown that the KO
tBu base reacts with the organic additive to form an electron donor in situ. SET from this electron donor then initiates the radical chain mechanism, not SET from
tert-butoxide anion [
12,
13,
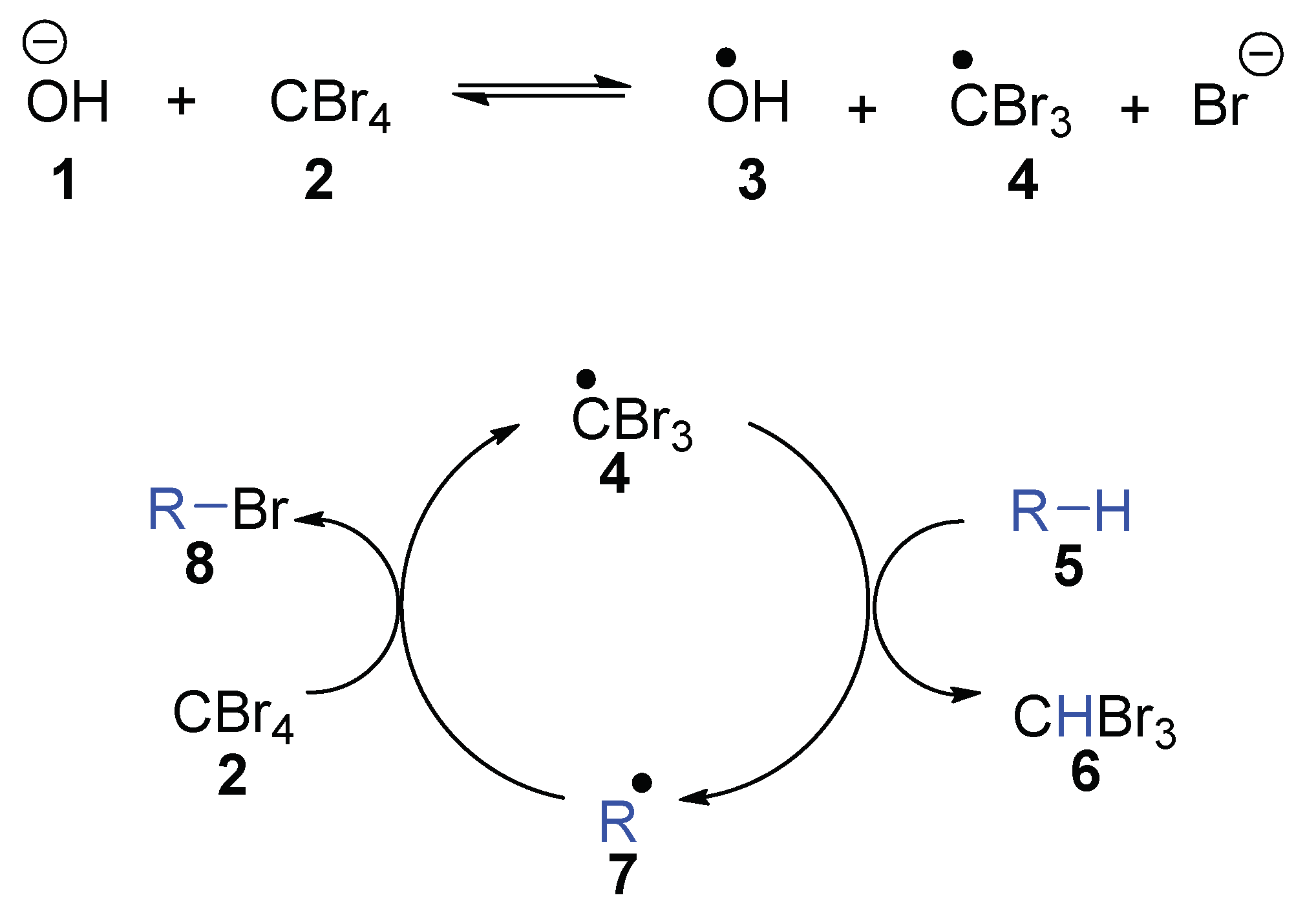
14]. The investigations begged the question: “under what circumstances would KO
tBu behave as a single electron donor?” To answer that question, we were drawn to the research of Schreiner and Fokin et al., who reduced CBr
4 using aqueous NaOH and a phase-transfer catalyst in a two-phase system that led to the bromination of adamantane [
15,
16]. They demonstrated that reaction of hydroxide anion with CBr
4 led to the tribromomethyl radical
4, and concluded that this reflected direct electron transfer from hydroxide to CBr
4 (
Scheme 1). This tribromomethyl radical
4 undergoes a chain reaction, where it abstracts a hydrogen atom from adamantane
5 to form the alkyl radical
7 and bromoform
6. The adamantyl radical
7 abstracts a bromine atom from CBr
4, forming adamantyl bromide
8 and regenerating the tribromomethyl radical
4 [
15]. The reactions of the tribromomethyl radical show diagnostic high selectivities for abstracting tertiary CH hydrogen atoms (to ultimately form 1-bromoadamantane) over secondary (CH
2) counterparts (which would ultimately form 2-bromoadamantane).
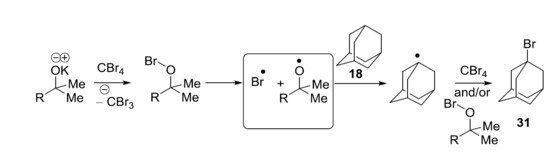
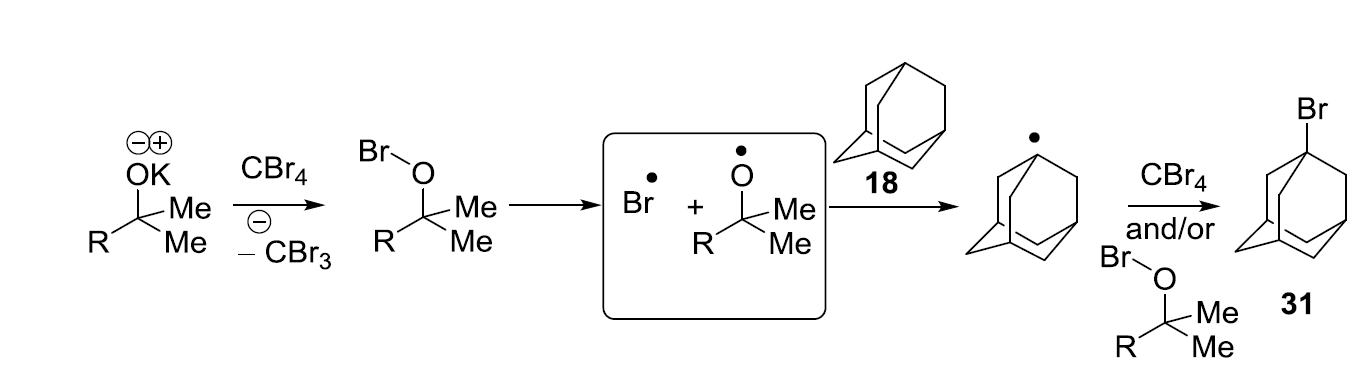
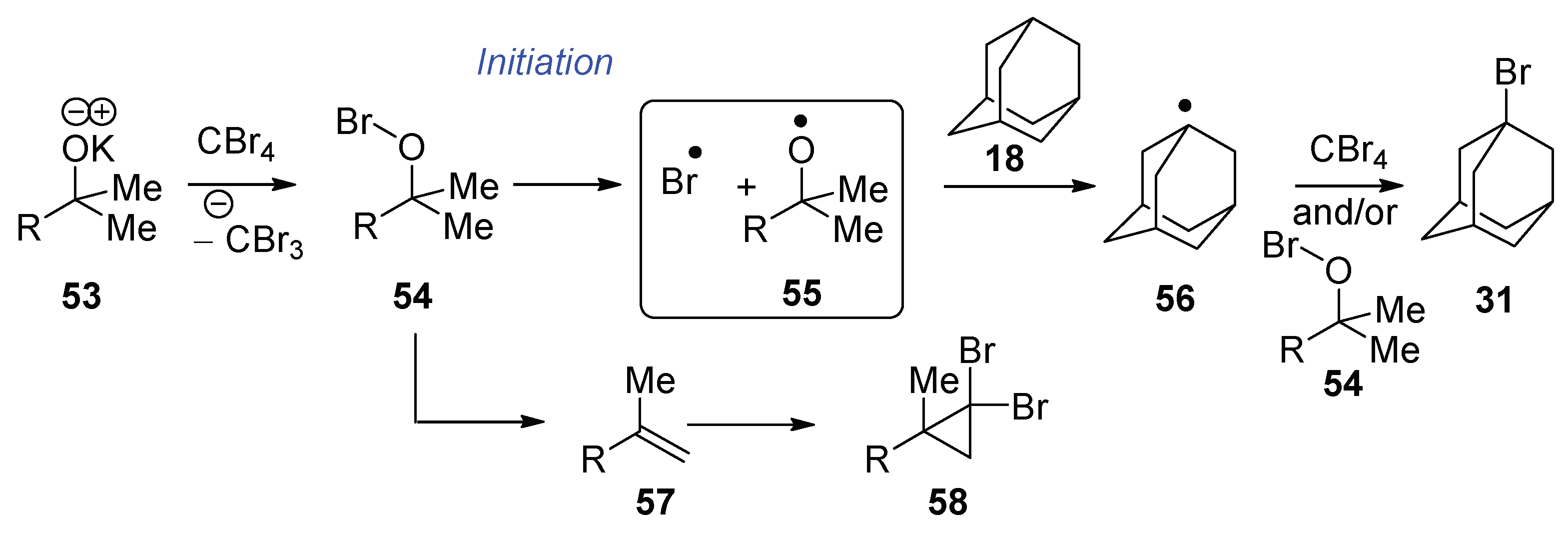
Recently, we showed that KO
tBu reacts with CBr
4 and adamantane (40 °C, 96 h) to afford bromoadamantane—in a reaction that appears to mirror the work of Schreiner and Fokin et al.—with NaOH, although phase-transfer conditions were not used [
17]. The oxidation potential of KO
tBu in DMF (0.10 V vs. SCE) [
8] is not too different from the reduction potential of CBr
4 in DMF (−0.31 V vs. SCE) [
18]. It was therefore proposed that KO
tBu might reduce CBr
4 through an electron transfer mechanism, but the door was left open to further investigation. The aim of this paper is to explore the chemistry of KO
tBu and related tertiary alkoxides in this context.
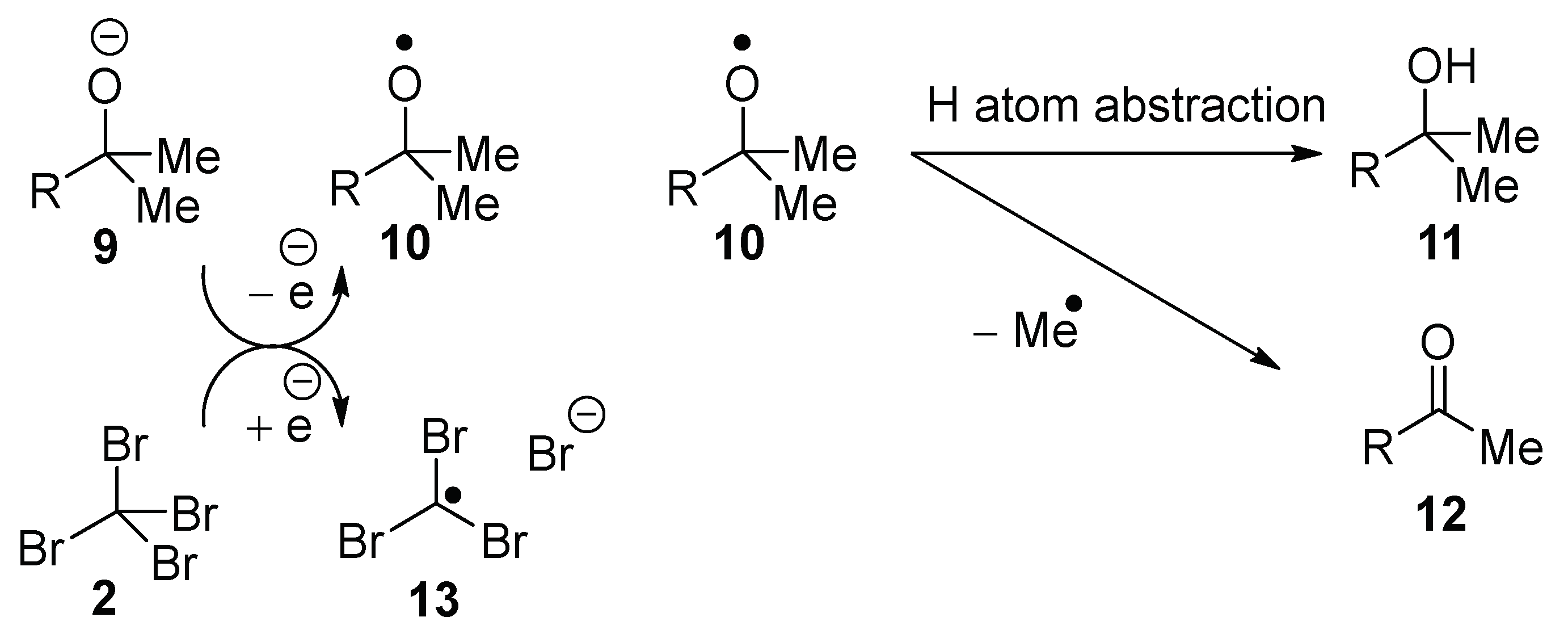
2. Results
The analogy of the proposed reaction of KO
tBu with CBr
4 or CCl
4 (
Scheme 2) to the Schreiner–Fokin reaction of NaOH with this halide is striking (
Scheme 1). Upon donation of an electron, the alkoxide anion
9 would form the corresponding alkoxyl radical
10, which can undergo either hydrogen atom transfer (HAT) to form
11, or β-scission to form the derived ketone
12 (for appropriate R). Either the alkoxyl radical
10 or the methyl radical can abstract a hydrogen atom from adamantane; the adamantyl radical would then propagate the reaction as in
Scheme 1. In order to detect the product from the β-scission of the alkoxyl radical
10, and bearing in mind the volatility of acetone from β-scission of
tert-butoxyl radicals, we synthesized potassium 2-phenylpropan-2-olate
14 and used it as a base to explore a range of reaction conditions (
Table 1).
Surprisingly, and in contrast to the case for KO
tBu, exposure of
14 to CBr
4 in dichloromethane solvent at 40 °C led to no bromination of adamantane (
Table 1, entry 1). The products (2,2-dibromo-1-methylcyclopropyl)benzene
16, methylstyrene
17 and (2,2-dichloro-1-methylcyclo-propyl)benzene
19 were observed, in addition to 2-phenylpropanol
15 and unreacted adamantane
18 (
Table 1, entry 1). To avoid the complexity of having compounds bearing different halogens in the reaction mixture, the reaction was repeated in dichloromethane using CCl
4 as the reagent, instead of CBr
4 (
Table 1, entry 2). Although we had no evidence of light-sensitivity, we conducted this and all future experiments in foil-covered flasks [
19]. The reaction yielded (2,2-dichloro-1-methylcyclopropyl)-benzene
19 as the major product. Note that a blank reaction in dichloromethane (absence of CX
4) resulted in the formation of product, bis((2-phenylpropan-2-yl)oxy)methane (
20, 52%) (
Table 1, entry 3) [
20].
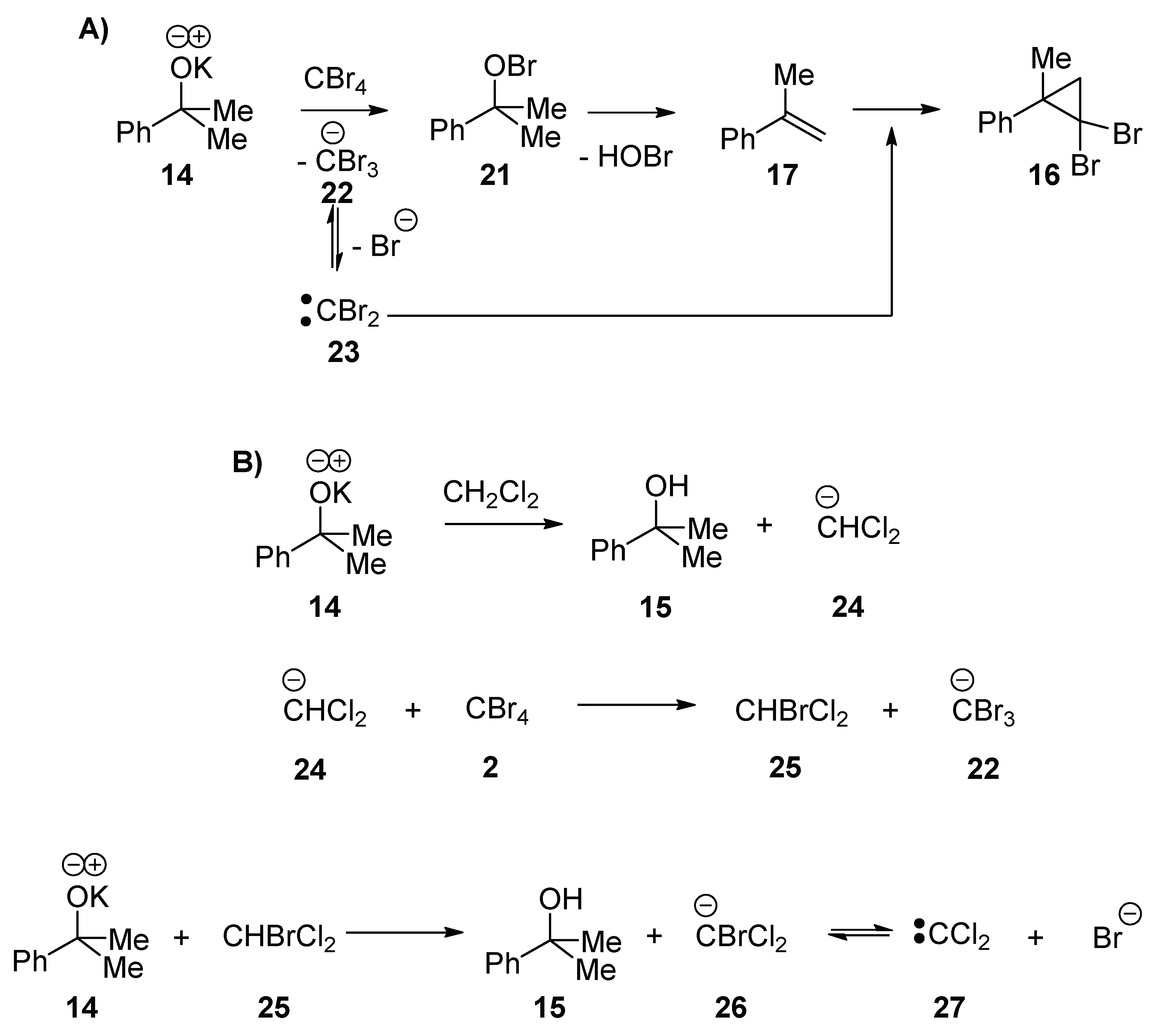
We propose that both (2,2-dibromo-1-methylcyclopropyl)-benzene
16 and (2,2-dichloro-1-methylcyclopropyl)benzene
19 are formed from methylstyrene
17. The formation of methylstyrene is therefore greatly encouraged in the presence of CBr
4, consistent with the conversion of the alkoxide to a leaving group (i.e., a hypobromite) (
Scheme 3A). It is known that hypobromites are formed from reaction of KO
tBu with halogens, X
2 [
21] (and that hypohalites are precursors to alkene halogenation by radical mechanisms [
21,
22]), so this reaction shows that they also form from reaction with tetrahalomethanes. The potassium 2-phenylpropan-2-olate
14 nucleophilically attacks a molecule of CBr
4 to form the hypobromite
21 and eliminate a CBr
3 anion
22. The hypobromite
21 then undergoes an elimination to form methylstyrene
17. The formation of (2,2-dibromo-1-methylcyclopropyl)benzene
16 occurs by decomposition of the tribromomethyl anion to CBr
2 23, which attacks methylstyrene
17 (
Scheme 3A).
Analogous to the formation of (2,2-dibromo-1-methylcyclo-propyl)benzene
16, the isolation of (2,2-dichloro-1-methylcyclo-propyl)benzene
19 means that carbenes (in this case, CCl
2 27) are formed under the reaction conditions (
Scheme 3B). The carbene
27 forms when the potassium 2-phenylpropan-2-olate
14 deprotonates the solvent, CH
2Cl
2, and the resulting CHCl
2 anion
24 undergoes halogen exchange with CBr
4 to form bromodichloromethane
25. A second deprotonation would lead to the bromodichloromethyl anion
26, and decomposition of this anion would lead to the dichlorocarbene
27.
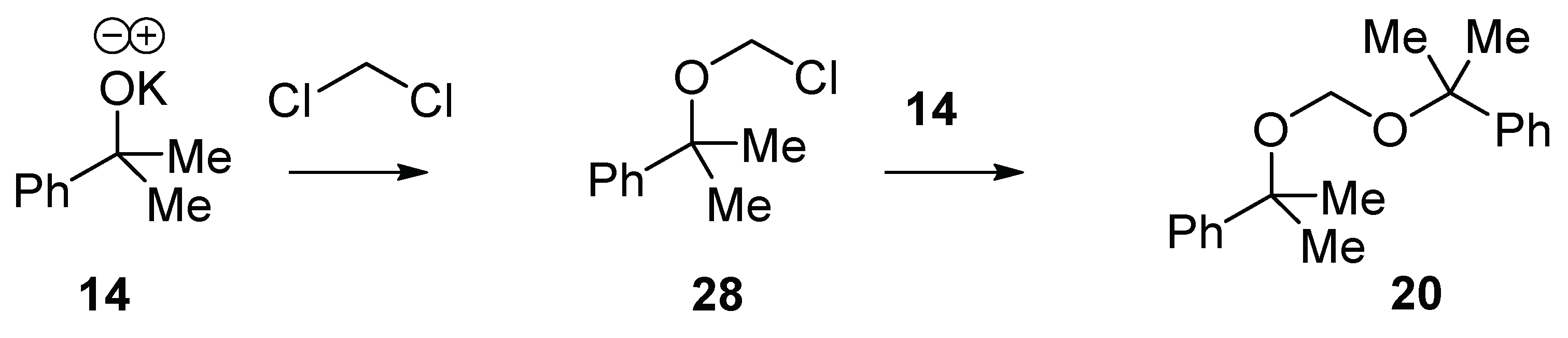
In the blank reaction (without CBr
4) (
Table 1, entry 3), bis((2-phenylpropan-2-yl)oxy)methane
20 is formed by the nucleophilic displacement of chloride from dichloromethane by two molecules of potassium 2-phenylpropan-2-olate
14 (
Scheme 4) [
23,
24]. The fact that bis((2-phenylpropan-2-yl)oxy)methane
20 is only observed in the absence of CBr
4 suggests that the reaction of potassium 2-phenylpropan-2-olate
14 with dichloromethane is slower than the reaction with CBr
4.
Reflecting on the fact that potassium 2-phenylpropan-2-olate
14 did not give rise to halogenation of adamantane (
Table 1), while halogenation was observed when KO
tBu was used as the alkoxide (the ratio of adamantane
18:1-bromoadamantane
31:2-bromoadamantane
51 was determined to be 39:3.3:1 [
17]), this suggests that either the two alkoxide bases do not react with CBr
4 through analogous mechanisms, or that the halogenation reaction was intercepted and inhibited when
14 was used. Interception could occur if methylstyrene
17—formed in the reaction from potassium 2-phenylpropan-2-olate
14—shuts down the radical chain pathway. To probe this possibility, the reaction of KO
tBu with CBr
4 and adamantane
18 was repeated in the presence of methylstyrene
17 (1 equiv.,
Scheme 5).
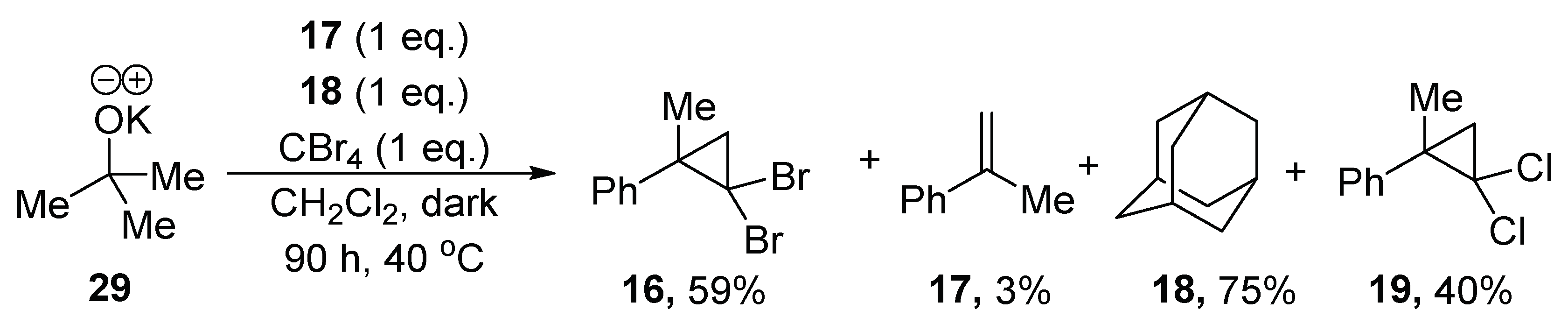
The addition of methylstyrene
17 completely inhibited the bromination of adamantane, which suggests that it can shut down the radical mechanism, perhaps by acting as a preferential source of hydrogen atoms, relative to adamantane
18, for the radical intermediates in the reaction. Computationally, the competition between methylstyrene and adamantane as a source of H atoms was modeled using CBr
3 radicals, which showed that hydrogen atom abstraction by the CBr
3 radical from methylstyrene
17 (ΔG
ǂ = 10.7 kcal/mol and ΔG
rxn = −7.7 kcal/mol) is thermodynamically and kinetically more favorable than from adamantane
18 (ΔG
ǂ = 13.9 kcal/mol and ΔG
rxn = 1.1 kcal/mol) (see
Supplementary Materials).
All yields were calculated using 1,3,5-trimethoxybenzene as the internal standard (10 mol %) in the 1H-NMR. The yield of 18 was calculated based on recovery of adamantane 18, the yield of 16 and 19 was calculated based on CBr4. (Note that alkoxide (4 eq.) is capable of forming methylstyrene 17.)
With the knowledge that methylstyrene 17 is capable of preventing bromination of adamantane, why does bromination succeed when KOtBu 29 + CBr4 alone are used? The two hypobromites, tert-butyl hypobromite and 21, may undergo elimination at different rates; additionally, 2-methylpropene (b.p. −6.9 °C) exists as a gas at the reaction temperature. Such a volatile product would likely be found principally in the headspace above the reaction, rather than in solution, and therefore halogenation of adamantane 18 could occur.
As mentioned earlier, Wirth et al. [
19] used
tert-butyl hypobromite to achieve the bromination of alkanes (while Walling [
20] used
tert-butyl hypochlorite to achieve chlorination) and proposed that the mechanism was initiated via homolysis of the O–Br bond of
tert-butyl hypobromite. Either the bromine radical or the
tert-butoxyl radical could abstract the H atom from adamantane. Propagation could then occur when the adamantyl radical abstracts a Br atom from CBr
4 [
13] or from
tert-butyl hypobromite [
23].
To gain further information on mechanism, an alternative alkoxide, potassium triphenylmethanolate
30, was prepared and subjected to the reaction conditions with CBr
4 and adamantane
18 (
Table 2).
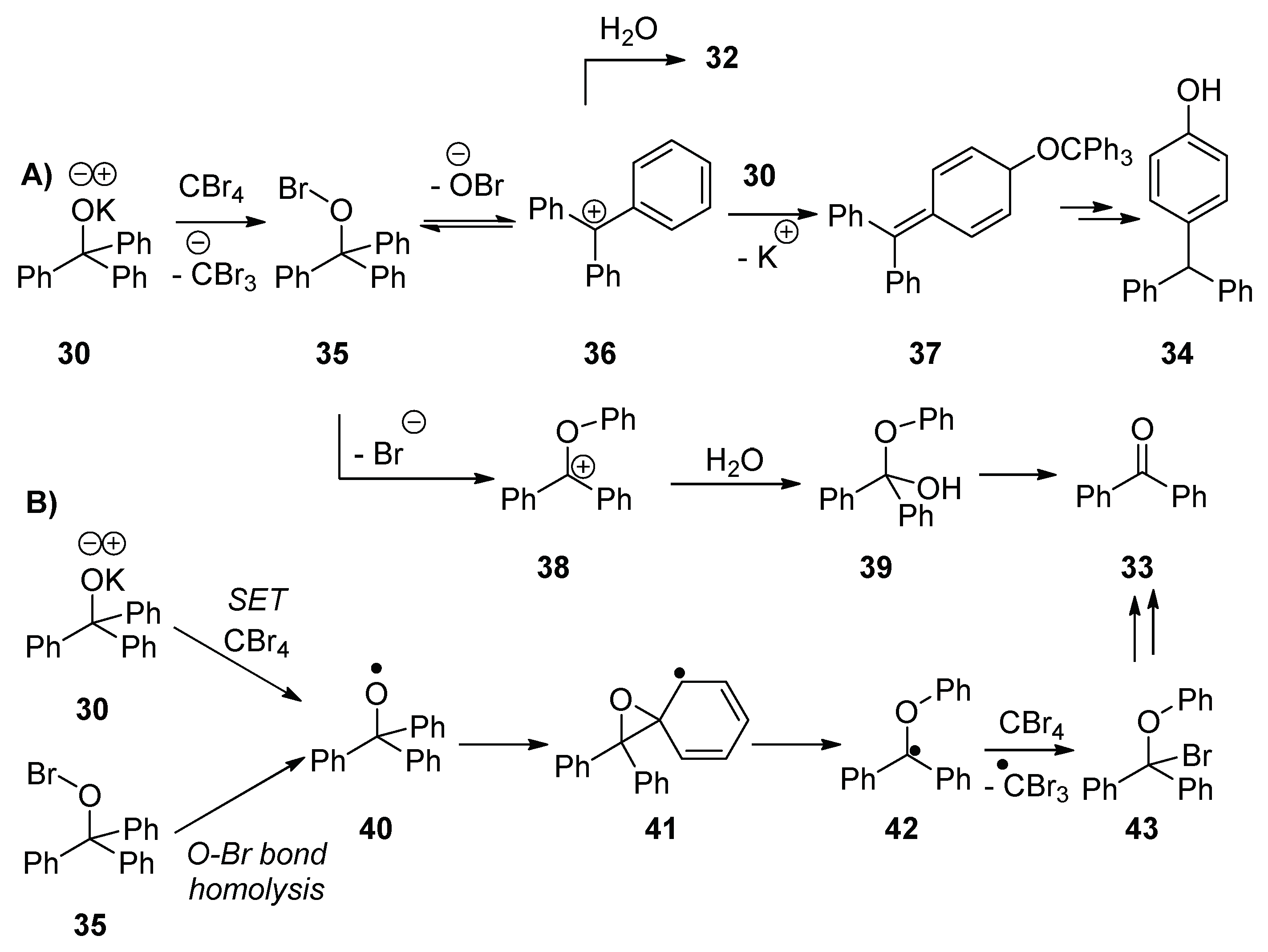
Reaction of potassium triphenylmethanolate
30 with adamantane in the presence of CBr
4 afforded products 1-bromoadamantane
31 (7%) and triphenylmethanol
32 (88%), as well as two additional products, benzophenone
33 (5%) and 4-benzhydrylphenol
34 (12%) (
Table 2, entry 1). When CBr
4 was not present in the reaction mixture, triphenylmethanol
32 (81%) was isolated following workup, together with a trace (1%) of benzophenone
33 (
Table 2, entry 2). Further analysis of the starting material showed trace amounts of
33 present in the commercially supplied triphenylmethanol
32. Background formation of benzophenone
33, in trace amounts from heterolytic fragmentation of similar tertiary alkoxides with expulsion of a phenyl anion, is also precedented [
25,
26].
The important difference between the reaction in the presence of CBr
4 and in its absence (
Table 2, entry 1 and entry 2, respectively) is the formation of 4-benzhydrylphenol
34, which can arise through the hypobromite
35 (
Scheme 6). Potassium triphenylmethanolate
30 reacts with CBr
4 to generate the hypobromite
35. This hypobromite can react by three pathways (1) the OBr anion leaves to form the stabilized cation
36; (2) the O–Br bond fragments ionically with simultaneous migration of a phenyl moiety to form cation
38; or (3) the O–Br bond undergoes homolysis to form alkoxyl radical
40 and a bromine radical, for which there is literature precedent [
23]. If pathway (1) is followed, the carbocation
36 is attacked by another molecule of potassium triphenylmethanolate
30 and, due to steric effects, the alkoxide attacks at the
para position of one of the benzene rings. In doing so, the species
37 is formed, which, following tautomerism and hydrolytic workup, leads to
34. Alternatively,
36 affords triphenylmethanol
32 on workup. Pathways (2) and (3) ultimately lead to the formation of benzophenone
33; pathway (2) involves the formation of intermediate
38, which reacts with water on workup to form
39 and, ultimately, benzophenone
33. Pathway (3) involves formation of the alkoxyl radical
40, which could form via O–Br homolysis of
35 or, alternatively, by SET from potassium triphenylmethanolate
30 to a molecule of CBr
4. This alkoxyl radical might undergo β-scission to form benzophenone
33. However, alternatively, the radical
40 could undergo a neophyl-like rearrangement to form radical
41 (
Scheme 6) [
27]. The product
43 forms when the radical
42 abstracts a bromine atom from either CBr
4 or the hypobromite
35, and
43, in turn, upon workup, undergoes a hydrolytic conversion to benzophenone
33. Previous fragmentations of related alkoxyl radicals have been studied [
28,
29,
30,
31,
32]. The enhanced formation of benzophenone
33 in the presence of CBr
4 could occur through either β-scission of the alkoxyl radical
40 (
Scheme 6B), or the ionic mechanism (
Scheme 6A).
The study above provides evidence for significant formation of hypohalites from alkoxides 14 and 30. However, it might be possible for the OtBu anion in KOtBu to behave differently. The absence of electron-withdrawing aryl groups would render it more electron-rich, and, therefore, it might be a better candidate than the other alkoxides to undergo electron transfer.
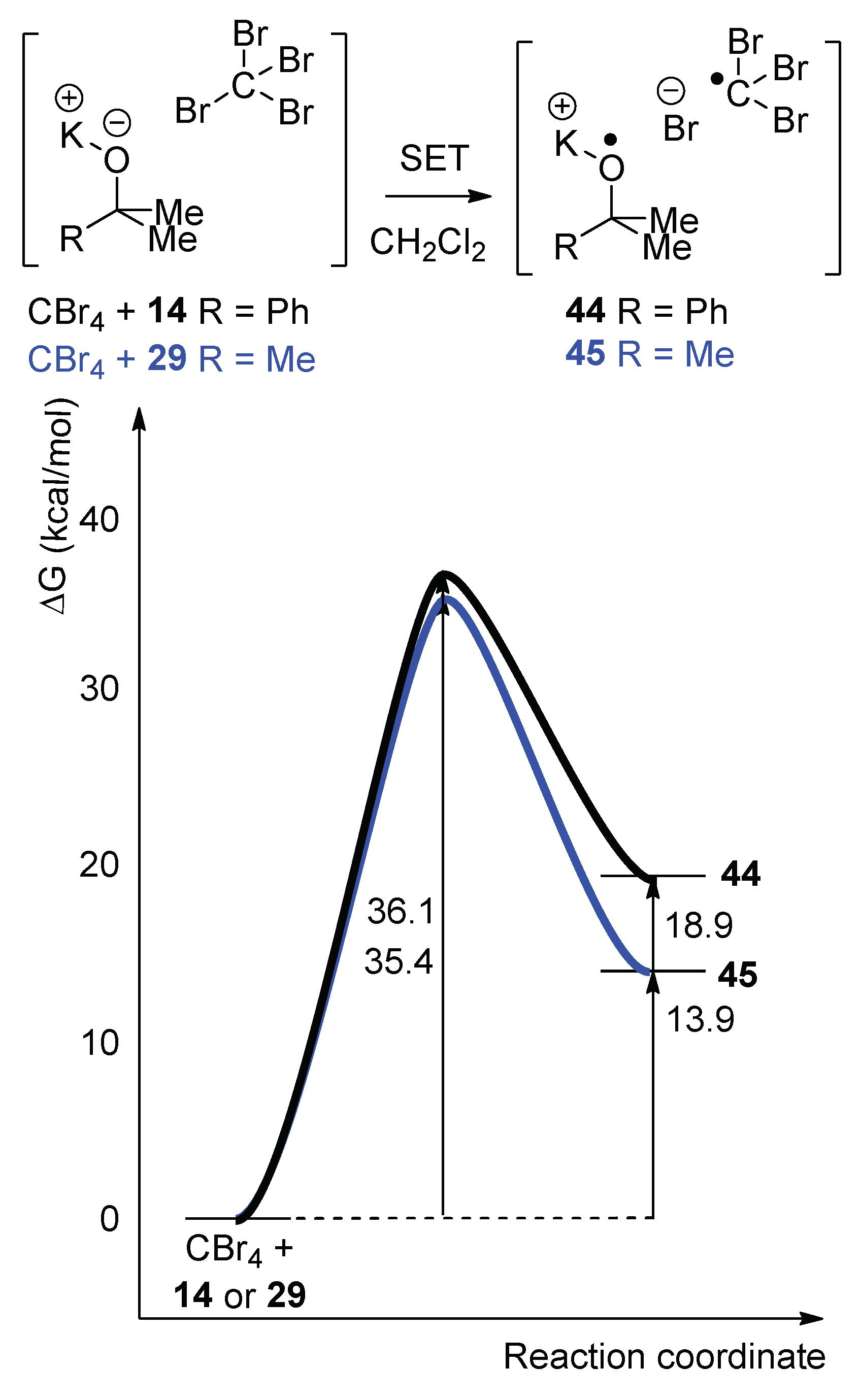
To probe the ability of alkoxides to donate a single electron, computational modeling was implemented to calculate the energy profile for the SET from both KO
tBu and potassium 2-phenylpropan-2-olate
14 to CBr
4 in dichloromethane (
Figure 1) [
33]. Our previous calculations had been based on the classical Nelsen four-point method [
34], but, since that time, we published a more accurate complexation method [
33] for predicting the activation free energy and the relative free energy of reactions. (The calculations were conducted using the M06-2X functional [
35,
36] with the 6-311++G(d,p) basis set [
37,
38,
39,
40,
41] on all atoms, except for the bromine. Bromine was modeled with the MWB28 relativistic pseudo-potential and associated basis set [
42]. All calculations were carried out using the C-PCM implicit solvent model [
43,
44] with the dielectric constant for dichloromethane (ε = 8.93) or carbon tetrachloride (ε = 2.228) as appropriate. All calculations were performed in Gaussian09 [
45]).
The energy barriers for SET from either KO
tBu or potassium 2-phenylpropan-2-olate
14, to a molecule of CBr
4 were calculated to be ΔG
ǂ = +35.4 kcal/mol and +36.1 kcal/mol, respectively (while the corresponding ΔG
rxn values were +13.9 and +18.9 kcal/mol) (
Figure 1). When the energy barrier was calculated for the reactions with CCl
4, a similar energy profile was obtained for the SET from either of the two alkoxides to a molecule of CCl
4 (see
Supplementary Materials). The energy barriers calculated were ΔG
ǂ = 42.5 kcal/mol and 44.5 kcal/mol for SET to CCl
4 from KO
tBu and potassium 2-phenylpropan-2-olate
14, respectively. These barriers for reactions of both CBr4 and CCl
4 are not accessible for a reaction performed at 40 °C, even as an initiation step. These computationally derived energy profiles for the SET step indicate that the initiation for this halogenation of adamantane
18 is not via SET from the alkoxide; the likely alternative radical pathway arises through homolysis of the hypohalite intermediate [
19]. (Importantly, our computation predicts a barrierless, but endergonic, profile for homolysis of the O–Br bond in
tBuO–Br with ΔG
rxn = +27.6 kcal/mol [
34]. This is a highly endergonic reaction, but as it is simply an initiation step, few
tBuO–Br molecules are required to undergo homolysis).
3. Experimental Section
3.1. Computational Methods
The calculations were run using the M06-2X functional [
35,
36] with the 6-311++G(d,p) basis set [
37,
38,
39,
40,
41] on all atoms except bromine. Bromine was modeled with the MWB28 relativistic pseudo-potential and associated basis set [
42]. All calculations were carried out using the C-PCM implicit solvent model [
43,
44] as implemented in Gaussian09 [
45].
3.2. General Experimental Information
All reagents were bought from commercial suppliers and used without further purification unless stated otherwise. All the reactions were carried out under argon atmosphere. Diethyl ether, tetrahydrofuran, dichloromethane and hexane were dried with a Pure-Solv 400 solvent purification system, marketed by Innovative Technology Inc. (Herndon, VA, USA). Organic extracts were, in general, dried over anhydrous sodium sulfate (Na2SO4). A Büchi rotary evaporator was used to concentrate the reaction mixtures. Thin layer chromatography (TLC) was performed using aluminium-backed sheets of silica gel and visualized under a UV lamp (254 nm). The plates were developed using phosphomolybdic acid or KMnO4 solution. Column chromatography was performed to purify compounds by using silica gel 60 (200–400 mesh).
The electron transfer reactions were carried out within a glove box (Innovative Technology Inc.) under nitrogen atmosphere, and performed in an oven-dried or flame-dried apparatus using anhydrous solvents, which were degassed under reduced pressure, then purged with argon and dried over activated molecular sieves (3 Å), prior to being sealed and transferred to the glovebox. All solvent or samples placed into the glovebox were transferred through the port, which was evacuated and purged with nitrogen 10 times before entry. When the reaction mixtures were prepared, the reaction vessel was removed from the glove box and the rest of the reaction was performed in a fumehood.
Proton (1H) NMR spectra were recorded at 400.13, 400.03 and 500.16 MHz on Bruker AV3, AV400 and AV500 spectrometers, respectively. Carbon (13C) NMR spectra were recorded using broadband decoupled mode at 100.61, 100.59 and 125.75 MHz on Bruker AV3, AV400 and AV500 spectrometers, respectively. Spectra were recorded in either deuterated chloroform (CDCl3) or deuterated dimethyl sulfoxide (d6-DMSO), depending on the solubility of the compounds. The chemical shifts are reported in parts per million (ppm), calibrated on the residual non-deuterated solvent signal, and the coupling constants, J, are reported in Hertz (Hz). The peak multiplicities are denoted using the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; sx, sextet; m, multiplet; br s, broad singlet; dd, doublet of doublets; dt, doublet of triplets; td, triplet of doublets.
Infrared spectra were recorded on an ATR-IR spectrometer. Melting points were determined on a Gallenkamp melting point apparatus. The mass spectra were recorded by either gas-phase chromatography (GCMS) or liquid-phase chromatography (LCMS), using various ionization techniques, as stated for each compound: atmospheric pressure chemical ionization (APCI), electron ionization (EI), electrospray ionization (ESI). GCMS data were recorded using an Agilent Technologies 7890A GC system coupled to a 5975C inert XL EI/CI MSD detector. Separation was performed using the DB5MS-UI column (30 m × 0.25 mm × 0.25 μm) at a temperature of 320 °C, using helium as the carrier gas. LCMS data were recorded using an Agilent 6130 Dual source mass spectrometer with Agilent 1200, Agilent Poroshell 120ECC 4.6 mm × 75 mm × 2.7 um column.
High-resolution mass spectrometry (HRMS) was performed at the University of Wales, Swansea, in the EPSRC National Mass Spectrometry Centre. Accurate mass was obtained using atmospheric pressure chemical ionization (APCI), chemical ionization (CI), electron ionization (EI), electrospray ionization (ESI) or nanospray ionization (NSI) with a LTQ Orbitrap XL mass spectrometer.
3.3. Synthesis of Alkoxide, Potassium 2-Phenylpropan-2-Olate 14
Potassium hydride (586 mg, 15 mmol, 1.0 eq.) was added to a flame-dried three-necked flask, equipped with a vacuum tap. Under an argon atmosphere, at −78 °C, a solution of 2-phenylpropanol 15 (2.04 g, 15 mmol) in anhydrous diethyl ether (20 mL), as added and the reaction mixture, was stirred at −78 °C for 1 h, then at RT overnight. The solvent was removed under vacuum and the crude material was dried for 1 h to obtain potassium 2-phenylpropan-2-olate 14 (2.46 g, 14.1 mmol, 93%) as an off-white solid m.p. 128–132 °C; (Found: (GCMS-EI) C9H11O (M-K) 135.08); νmax (film)/cm−1 3503, 2972, 1663, 1444, 1433, 1236, 1161, 1067, 1029, 955, 881, 861, 764; 1H-NMR (400 MHz, d6-DMSO) δ 1.41 (6 H, s, 2 × CH3), 7.15–7.19 (1 H, m, ArH), 7.26–7.30 (2 H, m, ArH), 7.45–7.47 (2 H, m, ArH); 13C-NMR (100 MHz, d6-DMSO) δ 31.9 (2 × CH3), 70.5 (C), 124.4 (2 × CH), 125.8 (CH), 127.7 (2 × CH), 150.5 (C). The product was put under an argon atmosphere and transported into the glovebox immediately.
3.4. Blank Reaction (No KOtBu) of CBr4 with Adamantane 18
Adamantane
18 (68 mg, 0.5 mmol), CBr
4 (166 mg, 0.5 mmol, 1.0 eq.) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube and the reaction mixture was stirred at 40 °C for 90 h in the dark. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na
2SO
4, filtered and concentrated in vacuo.
1H-NMR (400 MHz, CDCl
3) δ 1.76–1.75 (12 H, m, C
H2), 1.88 (4 H, br s, C
H);
13C-NMR (100 MHz, CDCl
3) δ 28.5 (6 × CH
2), 37.9 (4 × CH). (The yield of adamantane
18 [
46] (93%) was determined by adding 1,3,5-trimethoxybenzene to the crude mixture as an internal standard for
1H-NMR). These signals are consistent with the literature values and reference samples.
3.5. Reactions of Potassium 2-Phenylpropan-2-Olate 14 with Adamantane 18 and CBr4/CCl4
3.5.1. Table 1, Entry 1
Potassium 2-phenylpropan-2-olate 14 (349 mg, 2 mmol, 4.0 eq.), adamantane 18 (68 mg, 0.5 mmol), CBr4 (166 mg, 0.5 mmol, 1.0 eq.) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube, and the reaction mixture was stirred at 40 °C for 90 h. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na2SO4, filtered and concentrated in vacuo. The yield of 2-phenylpropanol 15 (66%), (2,2-dibromo-1-methylcyclopropyl)-benzene 16 (33%), methylstyrene 17 (18%), adamantane 18 (91%) and (2,2-dichloro-1-methylcyclopropyl)benzene 19 (17%) were determined by adding 1,3,5-trimethoxybenzene to the crude mixture as an internal standard for 1H-NMR. The products were identified by the following characteristic signals; 1H-NMR (400 MHz, CDCl3) δ 1.60 (6 H, s) for 2-phenylpropanol 15; δ 1.72 (3 H, s), 1.78 (1 H, d, J = 7.6 Hz), 2.17 (1 H, d, J = 7.6 Hz) for (2,2-dibromo-1-methylcyclopropyl)benzene 16; δ 2.16 (3 H, s), 5.09 (1 H, s), 5.37 (1 H, s) for methylstyrene 17; δ 1.75–1.77 (12 H, m), 1.88 (4 H, br s) for adamantane 18; δ 1.68 (3 H, s), 1.96 (1 H, d, J = 7.2 Hz) for (2,2-dichloro-1-methylcyclopropyl)benzene 19; 13C-NMR (100 MHz, CDCl3) δ 31.9, 72.7, 124.5, 126.8, 128.4 for 2-phenylpropanol 15; δ 27.9, 33.9, 128.5, 128.6 for (2,2-dibromo-1-methylcyclopropyl)-benzene 16; δ 22.0, 112.5 for methylstyrene 17; δ 28.5, 37.9 for adamantane 18; δ 25.7, 36.6 for (2,2-dichloro-1-methylcyclo-propyl)benzene 19. These signals are consistent with the literature values and reference samples. The compounds 16 and 19 were inseparable, so pure samples of 16 and 19 were prepared for comparison- see below.
3.5.2. Preparation of 16
KO
tBu
29 (224 mg, 2 mmol, 4.0 eq.), HCBr
3 (0.04 mL, 0.5 mmol, 1.0 eq.) and methylstyrene
17 (0.07 mL, 0.5 mmol) were added to an oven-dried pressure tube. Dichloromethane (3.13 mL) was added and the reaction mixture was stirred at 40 °C for 90 h. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na
2SO
4, filtered and concentrated in vacuo. The crude material was purified by column chromatography (100% hexane) to give (2,2-dibromo-1-methylcyclopropyl)benzene
16 [
18] (82.4 mg, 57%) as a colorless oil [Found: (GCMS-CI) C
10H
11Br
2+ (M + H)
+ 288.7]; ν
max (film)/cm
−1 1496, 1445, 1426, 1060, 1019, 763, 691;
1H-NMR (400 MHz, CDCl
3) δ 1.72 (3 H, s, CH
3), 1.78 (1 H, d,
J = 7.6 Hz, CH
2), 2.17 (1 H, d,
J = 7.6 Hz, CH
2), 7.29–7.38 (5 H, m, ArH);
13C{
1H}-NMR (100 MHz, CDCl
3) δ 27.9 (CH
3), 33.8 (CH
2), 35.9 (C), 36.9 (C), 127.4 (CH), 128.5 (2 × CH), 128.6 (2 × CH), 142.5 (C); m/z (CI) 292.6 [(M + H)
+,
81Br
81Br, 61%), 290.6 [(M + H)
+,
79Br
81Br, 100), 288.7 [(M + H)
+,
79Br
79Br, 70)].
3.5.3. Preparation of 19
KO
tBu
29 (224 mg, 2 mmol, 4.0 eq.), HCCl
3 (0.04 mL, 0.5 mmol, 1.0 eq.) and methylstyrene
17 (0.07 mL, 0.5 mmol) were added to an oven-dried pressure tube. Dichloromethane (3.13 mL) was added and the reaction mixture was stirred at 40 °C for 90 h. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na
2SO
4, filtered and concentrated in vacuo. The crude material was purified by column chromatography (100% hexane) to give (2,2-dichloro-1-methylcyclopropyl)benzene
19 [
18] (63.7 mg, 63%) as a colorless oil [Found: (HRMS-EI) 200.0157. C
10H
10Cl
2 (M)
•+ requires 200.0160]; ν
max (film)/cm
−1 1497, 1446, 1425, 1075, 1033, 1026, 936, 868, 772, 754, 697, 595;
1H-NMR (400 MHz, CDCl
3) δ 1.60 (1 H, d,
J = 7.2 Hz, CH
2), 1.68 (3 H, s, CH
3), 1.96 (1 H, d,
J = 7.2 Hz, CH
2), 7.27–7.38 (5 H, m, ArH);
13C-NMR (100 MHz, CDCl
3) δ 25.7 (CH
3), 32.0 (CH
2), 36.6 (C), 66.0 (C), 127.4 (CH), 128.6 (2 × CH), 128.7 (2 × CH), 141.4 (C); m/z (CI) 203.9 ((M)
•+,
37Cl
37Cl, 12%), 201.9 ((M)
•+,
35Cl
37Cl, 70), 199.9 ((M)
•+,
35Cl
35Cl, 100).
3.5.4. Table 1, Entry 2
Potassium 2-phenylpropan-2-olate 14 (349 mg, 2 mmol, 4.0 eq.), adamantane 18 (68 mg, 0.5 mmol), CCl4 (0.05 mL, 0.5 mmol, 1.0 eq.) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube and the reaction mixture was stirred at 40 °C for 90 h in the dark. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na2SO4, filtered and concentrated in vacuo. The yield of 2-phenylpropanol 15 (67%), methylstyrene 17 (3%), adamantane 18 (76%), (2,2-dichloro-1-methylcyclopropyl)benzene 19 (50%) and bis((2-phenylpropan-2-yl)oxy)-methane 20 (3%) were determined by adding 1,3,5-trimethoxybenzene to the crude mixture as an internal standard for 1H-NMR. The products were identified by the following characteristic signals; 1H-NMR (400 MHz, CDCl3) δ 1.60 (6 H, s) for 2-phenylpropanol 15; δ 2.17 (3 H, s), 5.10 (1 H, s), 5.38 (1 H, s) for methylstyrene 17; δ 1.76–1.78 (12 H, m), 1.89 (4 H, br s) for adamantane 18; δ 1.68 (3 H, s), 1.97 (1 H, d, J = 7.2 Hz) for (2,2-dichloro-1-methylcyclopropyl)benzene 19; δ 4.51 (2 H, s), 7.20–7.24 (2 H, m) for bis((2-phenylpropan-2-yl)oxy)methane 20; 13C-NMR (100 MHz, CDCl3) δ 31.9, 72.7, 124.5, 126.8, 128.4 for 2-phenylpropanol 15; δ 22.0, 112.6 for methylstyrene 17; δ 28.5, 37.9 for adamantane 18; δ 25.6, 36.6 for (2,2-dichloro-1-methylcyclopropyl)benzene 19. These signals are consistent with the literature values and reference samples.
3.5.5. Table 1, Entry 3
Potassium 2-phenylpropan-2-olate 14 (349 mg, 2 mmol, 4.0 eq.), adamantane 18 (68 mg, 0.5 mmol) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube and the reaction mixture was stirred at 40 °C for 90 h in the dark. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na2SO4, filtered and concentrated in vacuo. The yield of 2-phenylpropanol 15 (39%), methylstyrene 17 (1%), adamantane 18 (84%) and bis((2-phenylpropan-2-yl)oxy)methane 20 (52%) were determined by adding 1,3,5-trimethoxybenzene to the crude mixture as an internal standard for 1H-NMR. The products were identified by the following characteristic signals; 1H-NMR (400 MHz, CDCl3) δ 1.60 (6 H, s) for 2-phenylpropanol 15; δ 2.16 (3 H, s), 5.09 (1 H, s), 5.37 (1 H, s) for methylstyrene 17; δ 1.75–1.77 (12 H, m), 1.88 (4 H, br s) for adamantane 18; δ 4.51 (2 H, s), 7.20–7.24 (2 H, m) for bis((2-phenylpropan-2-yl)oxy)methane 20; 13C-NMR (100 MHz, CDCl3) δ 31.9, 72.7, 124.5, 149.2 for 2-phenyl-2-propanol 15; δ 28.5, 37.9 for adamantane 18; δ 29.5, 77.7, 86.8, 146.9 for bis((2-phenylpropan-2-yl)oxy)methane 20. These signals are consistent with the literature values and reference samples. This crude material was purified by column chromatography (0–5% ethyl acetate in hexane) to give bis((2-phenylpropan-2-yl)oxy)methane 20 (44.7 mg, 26%) as a colorless oil (Found: (HRMS-ESI) 302.2118. C19H28O2N (M + NH4)+ requires 302.2115); νmax (film)/cm−1 2978, 2934, 1493, 1447, 1381, 1364, 1258, 1153, 1072, 1018, 991, 818, 762; 1H-NMR (400 MHz, CDCl3) δ 1.59 (12 H, s, 4 × CH3), 4.50 (2 H, s, CH2), 7.20–7.23 (2 H, m, ArH), 7.27–7.31 (4 H, m, ArH), 7.38–7.40 (4 H, m, ArH); 13C-NMR (100 MHz, CDCl3) δ 29.5 (4 × CH3), 77.7 (2 × C), 86.8 (CH2), 125.9 (4 × CH), 126.9 (2 × CH), 128.2 (4 × CH), 146.9 (2 × C).
3.6. Reaction of KOtBu in Dichloromethane
KO
tBu
29 (224 mg, 2 mmol, 4.0 eq.), CBr
4 (166 mg, 0.5 mmol, 1.0 eq.), adamantane
18 (68 mg, 0.5 mmol) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube and the reaction mixture was stirred at 40 °C for 90 h in the dark. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na
2SO
4, filtered and concentrated in vacuo. The ratio of adamantane
18:1-bromoadamantane
31:2-bromoadamant-ane
51 was determined to be 39:3.3:1 from the
1H-NMR spectrum of the crude mixture. The products were identified by the following characteristic signals;
1H-NMR (400 MHz, CDCl
3) δ 1.74–1.76 (12 H, m, CH
2 × 6), 1.88 (4 H, br s, CH × 4) for adamantane
18; δ 1.73 (6 H, m, CH
2 × 3), 2.10 (3 H, br s, CH × 3), 2.36 (6 H, m, CH
2 × 3) for 1-bromoadamantane
31 [
47]; δ 1.96–2.00 (2 H, m, CH
2), 2.15 (2 H, br s, CH × 2), 2.33 (2 H, br s, CH × 2), 4.68 (1 H, br s, C
H) for 2-bromoadamantane
51 [
48];
13C-NMR (100 MHz, CDCl
3) δ 28.5, 37.9 adamantane
18; δ 32.8, 35.7, 49.5 for 1-bromoadamantane
31; 36.6, 39.1 for 2-bromoadamantane
51. These signals are consistent with the literature values and reference samples.
3.7. Reactions of Adamantane 18 with CBr4, Methylstyrene 17 and KOtBu 29 (Scheme 5)
KO
tBu
29 (224 mg, 2 mmol, 4.0 eq.), CBr
4 (166 mg, 0.5 mmol, 1.0 eq.), methylstyrene
17 (0.07 mL, 0.5 mmol, 1.0 eq.), adamantane
18 (68 mg, 0.5 mmol) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube and the reaction mixture was stirred at 40 °C for 90 h in the dark. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na
2SO
4, filtered and concentrated in vacuo. The yield of (2,2-dibromo-1-methylcyclopropyl)benzene
16 (59%), methylstyrene
17 (3%), adamantane
18 (75%) and (2,2-dichloro-1-methylcyclopropyl)benzene
19 (40%) were determined by adding 1,3,5-trimethoxybenzene to the crude mixture as an internal standard for
1H-NMR. The products were identified by the following characteristic signals;
1H-NMR (400 MHz, CDCl
3) δ 1.72 (3 H, s), 1.78 (1 H, d,
J = 7.6 Hz) for (2,2-dibromo-1-methylcyclopropyl)benzene
16; δ 5.09 (1 H, s), 5.37 (1 H, s) for methylstyrene
17; δ 1.75–1.78 (12 H, m), 1.88 (4 H, br s) for adamantane
18; δ 1.60 (1 H, d,
J = 7.2 Hz), 1.69 (3 H, s), 1.97 (1 H, d,
J = 7.2 Hz) for (2,2-dichloro-1-methylcyclopropyl)benzene
19;
13C-NMR (100 MHz, CDCl
3) δ 27.9, 33.9, 36.9, 142.5 for (2,2-dibromo-1-methylcyclopropyl)benzene
16; δ 28.5, 37.9 for adamantane
18; δ 25.7, 32.0, 36.6, 141.4 for (2,2-dichloro-1-methylcyclopropyl)benzene
19. These signals are consistent with the literature values and reference samples [
49,
50].
3.8. Synthesis of Alkoxide, Potassium Triphenylmethanolate 30
Potassium hydride (802 mg, 20 mmol, 1.0 eq.) was added to a flame-dried three-necked flask, equipped with a vacuum tap. Under an argon atmosphere, at –78 °C, a solution of triphenylmethanol 32 (5.21 g, 20 mmol) in anhydrous tetrahydrofuran (25 mL) was added and the reaction mixture was stirred at −78 °C for 1 h, then at RT overnight. The solvent was removed on the house vacuum line and the crude material was dried for 1 h to give potassium triphenylmethanolate 30 (5.07 g, 17 mmol, 85%) as an off-white solid, m.p. 238 °C (dec.); (Found: (GCMS-EI) C19H16O (M)•+ 260.1 (under the MS analysis 30 is protonated to the alcohol)); νmax (film)/cm−1 3057, 3022, 1595, 1487, 1443, 1414, 1329, 1155, 1053, 1009, 891, 756; 1H-NMR (400 MHz, d6-DMSO) δ 6.93–6.98 (3 H, m, ArH), 7.04–7.08 (6 H, m, ArH), 7.34–7.37 (6 H, m, ArH); 13C-NMR (100 MHz, d6-DMSO) δ 84.7 (C), 123.6 (3 × CH), 126.0 (6 × CH), 128.2 (6 × CH), 157.6 (3 × C). The product was put under an argon atmosphere and transported into the glove box immediately.
3.9. Reactions of Potassium Triphenylmethanolate 30 at 40 °C
3.9.1. Table 2, Entry 1
Potassium triphenylmethanolate
30 (597 mg, 2 mmol, 4.0 eq.), CBr
4 (166 mg, 0.5 mmol, 1.0 eq.), adamantane
18 (68 mg, 0.5 mmol) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube and the reaction mixture was stirred at 40 °C for 90 h in the dark. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na
2SO
4, filtered and concentrated in vacuo. The yield of adamantane
18 (49%), 1-bromoadamantane
31 (7%), triphenylmethanol
32 (88%), benzophenone
33 (5%) and 4-benzhydrylphenol
34 (12%) were determined by adding 1,3,5-trimethoxybenzene to the crude mixture as an internal standard for
1H-NMR. The products were identified by the following characteristic signals;
1H-NMR (400 MHz, CDCl
3) δ 1.75–1.78 (12 H, m), 1.88 (4 H, br s) for adamantane
18; δ 1.74 (6 H, m), 2.12 (3 H, br s), 2.38 (6 H, m) for 1-bromoadamantane
31 [
47]; δ 7.27–7.34 (15 H, m) for triphenylmethanol
32; δ 7.49 (4 H, d,
J = 8.0 Hz), 7.60 (2 H, d,
J = 8.0 Hz), 7.82 (4 H, d,
J = 8.0 Hz) for benzophenone
33 [
51]; δ 5.49 (1 H, s), 6.73–6.77 (2 H, m), 6.97–7.00 (2 H, m) for 4-benzhydrylphenol
34 [
52];
13C-NMR (100 MHz, CDCl
3) δ 28.3, 37.7 for adamantane
18; δ 49.3, 35.5, 32.6 for 1-bromoadamantane
31; δ 82.2, 127.4, 128.0, 147.0 for triphenylmethanol
32; δ 56.1, 115.3, 144.3 for 4-benzhydroxylphenol
34. These signals are consistent with the literature values and reference samples. This crude material was purified by column chromatography (0%–10% ethyl acetate in hexane) to give both benzophen-one
33 [
51] (7 mg, 4%) as a yellow oil (Found: (GCMS-EI) C
13H
10O (M)
•+ 182.0);
νmax (film)/cm
−1 3057, 1655, 1597, 1445, 1275, 1175, 939, 918, 808, 762;
1H-NMR (400 MHz, CDCl
3) δ 7.49 (4 H, t,
J = 8.0 Hz, Ar
H), 7.60 (2 H, t,
J = 8.0 Hz, Ar
H), 7.82 (4 H, d,
J = 8.0 Hz, Ar
H);
13C-NMR (100 MHz, CDCl
3) δ 128.2 (4 × CH), 130.2 (4 × CH), 132.6 (2 × CH), 137.5 (2 × C), 196.7 (C) and 4-benzhydrylphenol
34 [
52] (18.9 mg, 7%) as a yellow oil (Found: (GCMS-EI) C
19H
16O (M)
•+ 260.1);
νmax(film)/cm
−1 3366, 2361, 2336, 1595, 1508, 1491, 1449, 1238, 1173, 1103, 1030, 816, 800, 750, 735;
1H-NMR (400 MHz, CDCl
3) δ 4.82 (1 H, br s, O
H), 5.49 (1 H, s, C
H), 6.73–6.77 (2 H, m, Ar
H), 6.97–7.00 (2 H, m, Ar
H), 7.11–7.13 (4 H, m, Ar
H), 7.19–7.32 (6 H, m, Ar
H);
13C-NMR (100 MHz, CDCl
3) δ 56.1 (CH), 115.3 (2 × CH), 126.4 (2 × CH), 128.4 (4 × CH), 129.5 (4 × CH), 130.7 (2 × CH), 136.4 (C), 144.3 (2 × C), 154.1 (C).
Triphenylmethanol 32 1H-NMR (400 MHz, CDCl3) δ 2.79 (1 H, s, OH), 7.26–7.34 (15 H, m, ArH), 7.57 (2 H, d, J = 8.4 Hz, ArH); 13C-NMR (100 MHz, CDCl3) δ 82.2 (C), 127.4 (CH), 128.1 (6 × CH), 147.0 (9 × C). These signals are consistent with a commercial sample used as a reference.
3.9.2. Table 2, Entry 2
Potassium triphenylmethanolate
30 (597 mg, 2 mmol, 4.0 eq.), adamantane
18 (68 mg, 0.5 mmol) and dichloromethane (3.13 mL) were added to an oven-dried pressure tube and the reaction mixture was stirred at 40 °C for 90 h in the dark. The reaction mixture was cooled to RT and quenched with aqueous hydrochloric acid (1 M, 5 mL) and extracted with diethyl ether (4 × 10 mL). The organic phases were combined, dried over Na
2SO
4, filtered and concentrated in vacuo. The yield of adamantane
18 (87%), triphenylmethanol
32 (81%) and benzophenone
33 (1%) were determined by adding 1,3,5-trimethoxybenzene to the crude mixture as an internal standard for
1H-NMR. The products were identified by the following characteristic signals;
1H-NMR (400 MHz, CDCl
3) δ 1.75–1.78 (12 H, m), 1.88 (4 H, br s) for adamantane
18; δ 7.27–7.34 (15 H, m) for triphenylmethanol
32, δ 7.49 (4 H, d,
J = 8.0 Hz, Ar
H), 7.60 (2 H, d,
J = 8.0 Hz, Ar
H), 7.82 (4 H, d,
J = 8.0 Hz, Ar
H) for benzophenone
33;
13C-NMR (100 MHz, CDCl
3) δ 28.3, 37.7 for adamantane
18; δ 82.2, 127.4, 128.0, 147.0 triphenylmethanol
32 [
49]. These signals are consistent with the literature values and reference samples.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

