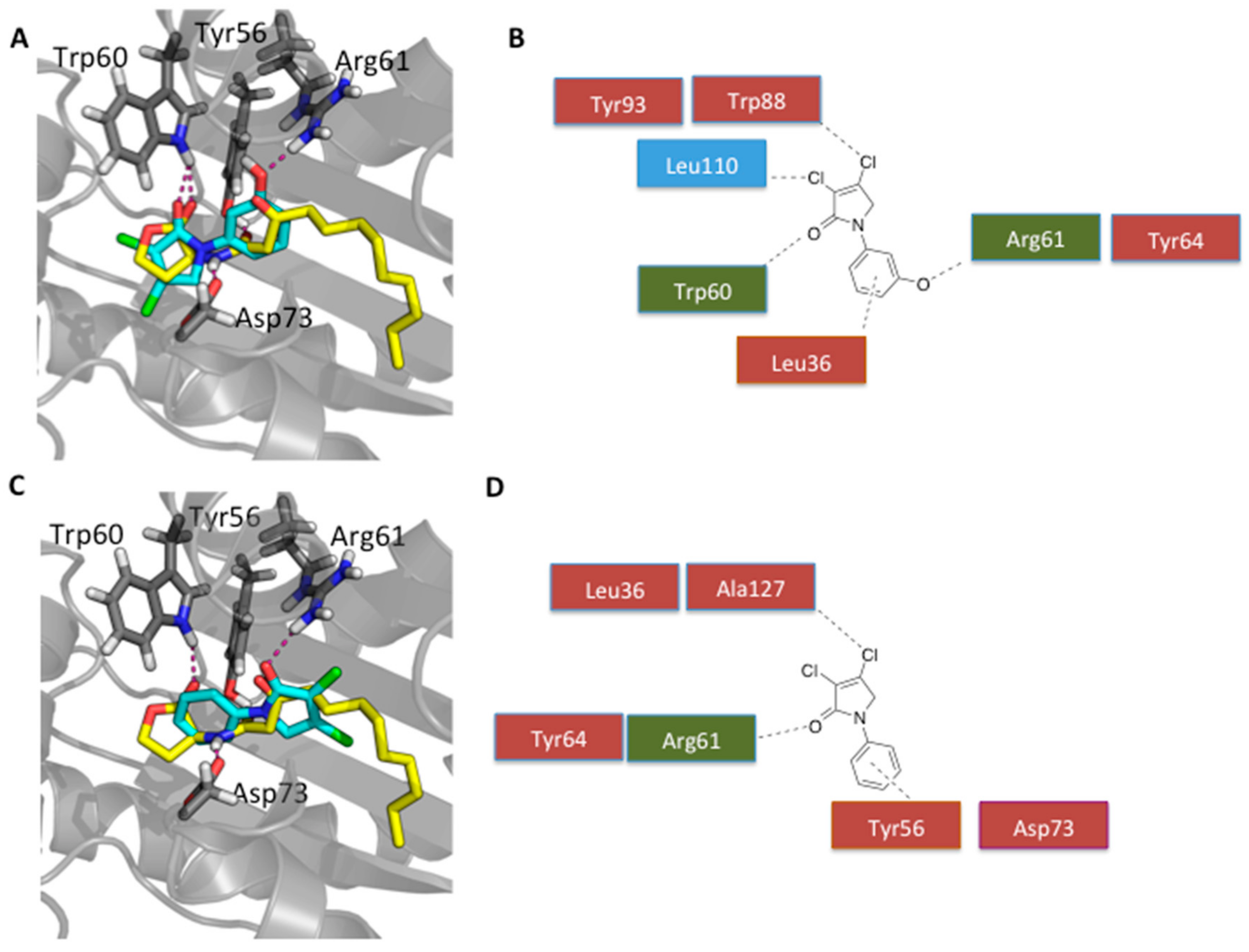
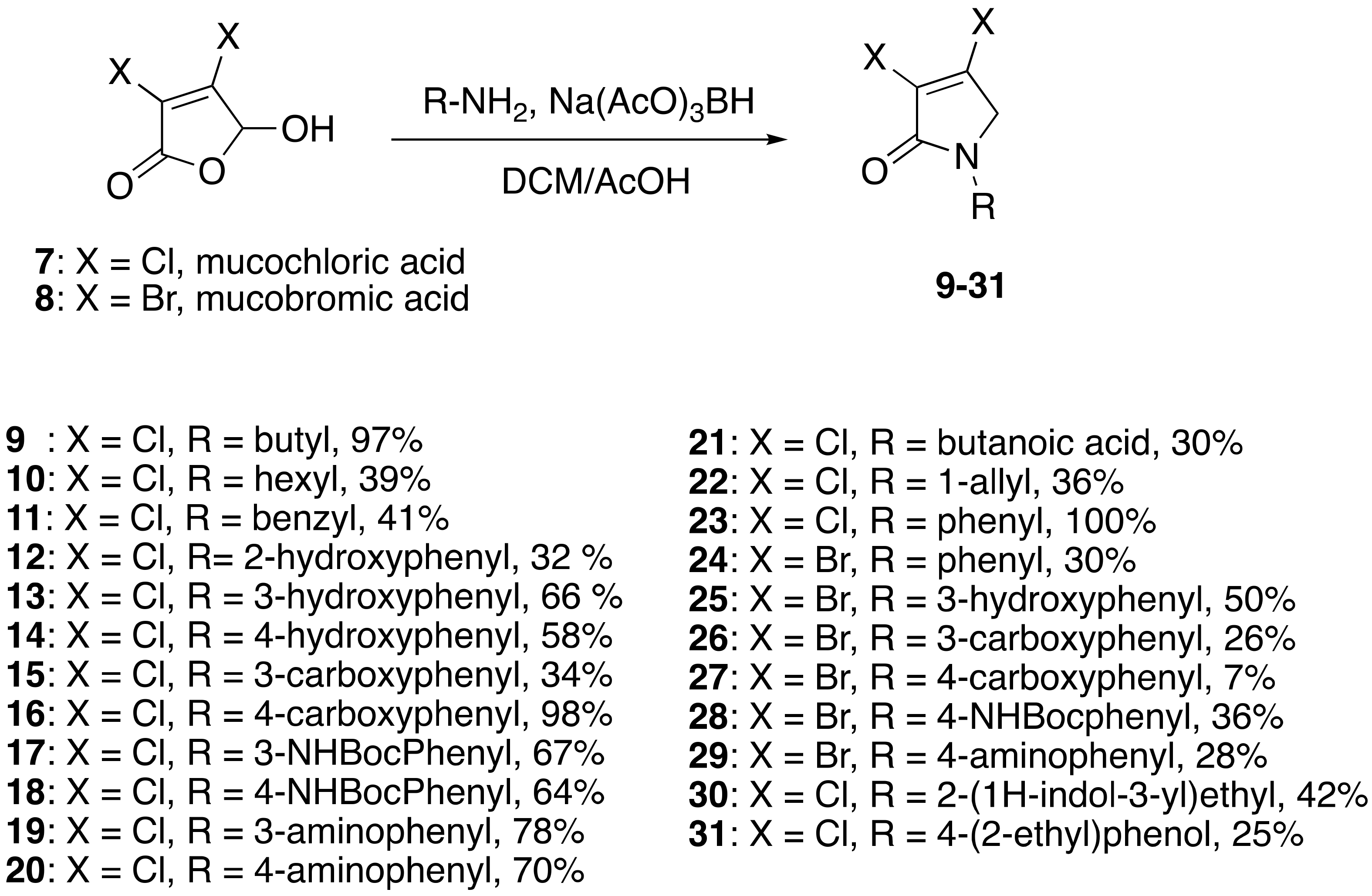
4.2. General Procedure
Procedure A: Reductive amination
Mucohalic acid (1 eq) was added to a solution of 5:3 v/v dichloromethane/glacial acetic acid. Then, an amine (1 eq) was added, and the mixture was stirred for 10 min. To that mixture, sodium triacetoxyborohydride (3 eq) solution in 5:3 v/v dichloromethane and glacial acetic acid was added. The mixture was left to stir at room temperature for 24 h unless otherwise stated.
Procedure B: Amide coupling 1
Acid 16 (1 eq) was treated with thionyl chloride (3 mL) and refluxed for 3 h at 70 °C. The mixture was then cooled and left to stir overnight at room temperature. Thionyl chloride was removed under high vacuum, and the resultant acid chloride was used without further purification. The acid chloride was dissolved in 10 mL of dry tetrahydrofuran, and triethylamine (1 eq) and amine (1 eq) were then added. The reaction mixture was left to stir at room temperature for 24 h.
Procedure C: Amide coupling 2
The amine was dissolved in 10 mL of dry tetrahydrofuran, and then, triethylamine (1 eq) and acid chloride (1.0 eq) were added. The reaction mixture was left to stir at room temperature for 24 h.
(1) 1-butyl-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (9)
![Molecules 23 01106 i001]()
The title compound was prepared according to Procedure A from mucochloric acid (400 mg, 2.3 mmol), butyl amine (170 mg, 270 mL, 2.3 mmol) and sodium triacetoxyborohydride (1.46 g, 6.9 mmol). The reaction mixture was left to react for 24 h. The crude mixture was extracted with dichloromethane (30 mL) and washed with water (15 mL) followed by brine (15 mL), dried over sodium sulphate, and the solvent was evaporated. A residue of oil was obtained, which was purified by flash column chromatography using a gradient eluent of 25–50% ethyl acetate in hexane. A yellow oil was obtained (390 mg; 79%). 1H NMR (CDCl3, 400MHz): δ 0.95 (t, J = 7.3, 14.6 Hz, 3H, CH3), δ 1.36 (sext, J = 7.2, 15.0 Hz, 2H, CH2), 1.58 (quint, J = 7.2, 15.0 Hz, 2H, CH2), δ 3.50 (t, J = 7.4, 15.0, 2H, CH2), δ 4.04 (s, 2H, CH2). 13C NMR (CDCl3, 101): 13.7 (CH3), 19.9 (CH2), 30.4 (CH2), 42.8 (CH2), 53.4 (C5-CH2), 125.9 (C), 139.0 (C), 164.2 (C); IR (ATR): υmax 2957.6, 2870.7, 1700.9, 1397.7, 1341.6, 1269.6, 1188.5, 1130.0, 843.9; UV (MeOH) λmax 230.0 nm (ε 7488 cm−1M−1)HRMS (ESI) m/z calcd. for C8H11Cl2N1O1Na1 230.0110 [M + Na]+, found 230.0108.
(2) 3,4-dichloro-1-hexyl-1,5-dihydro-2H-pyrrol-2-one (10)
![Molecules 23 01106 i002]()
The title compound was prepared according to Procedure A from mucochloric acid (400 mg, 2.3 mmol), hexyl amine (300 mg, 270 mL, 2.3 mmol) and sodium triacetoxyborohydride (1.46 g, 6.9 mmol). The reaction mixture was left to react for 24 h. The crude mixture was extracted with dichloromethane (30 mL) and washed with water (15 mL) followed by brine (15 mL), dried over sodium sulphate, and the solvent was evaporated and a residue oil obtained, which was purified by flash column chromatography using a gradient eluent of 25–50% ethyl acetate in hexane. A yellow oil was obtained (220 mg; 39%); 1H NMR (CDCl3, 400 MHz): δ 0.90 (t, J = 6.9, 13.5 Hz, 3H, CH3), δ 1.24 (m, 6H, CH2), δ 1.60 (m, 2H, CH2), δ 3.49 (t, J = 7.4, 14.6 Hz, CH2), δ 4.04 (s, 2H, C5-CH2). 13C NMR (CDCl3, 101): 14.0 (CH3), 22.5 (CH2), 26.3 (CH2), 28.3 (CH2), 31.4 (CH2), 43.1 (CH2), 53.4 (C5-CH2), 125.7 (C), 139.2 (C), 164.2 (C=O); IR (ATR): υmax 2927.2, 2857.2, 1702.3, 1438.2, 1397.4 1367.5, 1131.0, 1171.0, 953.7; UV (MeOH) λmax 235.0 nm (ε 4248 cm−1M−1); HRMS (ESI) m/z calcd. for C10H16Cl2N1O1 236.0603 [M + H]+, found 236.0601.
(3) 1-benzyl-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (11)
The title compound was made according to Procedure A following the reported method [
14]. m.p. 102.1 °C;
1H NMR (CDCl
3, 400 MHz):
δ 3.92 (s, 2H, CH
2), 4.67 (s, 2H, CH
2), 7.26–7.39 (m, 5H, ArH);
13C NMR (CDCl
3, 101): δ 47.0 (CH
2), 52.8 (CH
2), 125.5 (C), 128.2 (ArCH), 129.0 (ArCH), 135.8, 139.9 (C), 164.28 (C=O); IR (ATR): υ
max 2917.9, 2850.9, 1693.6, 1437.5, 1266.9, 966.1, 846.2, 739.1.
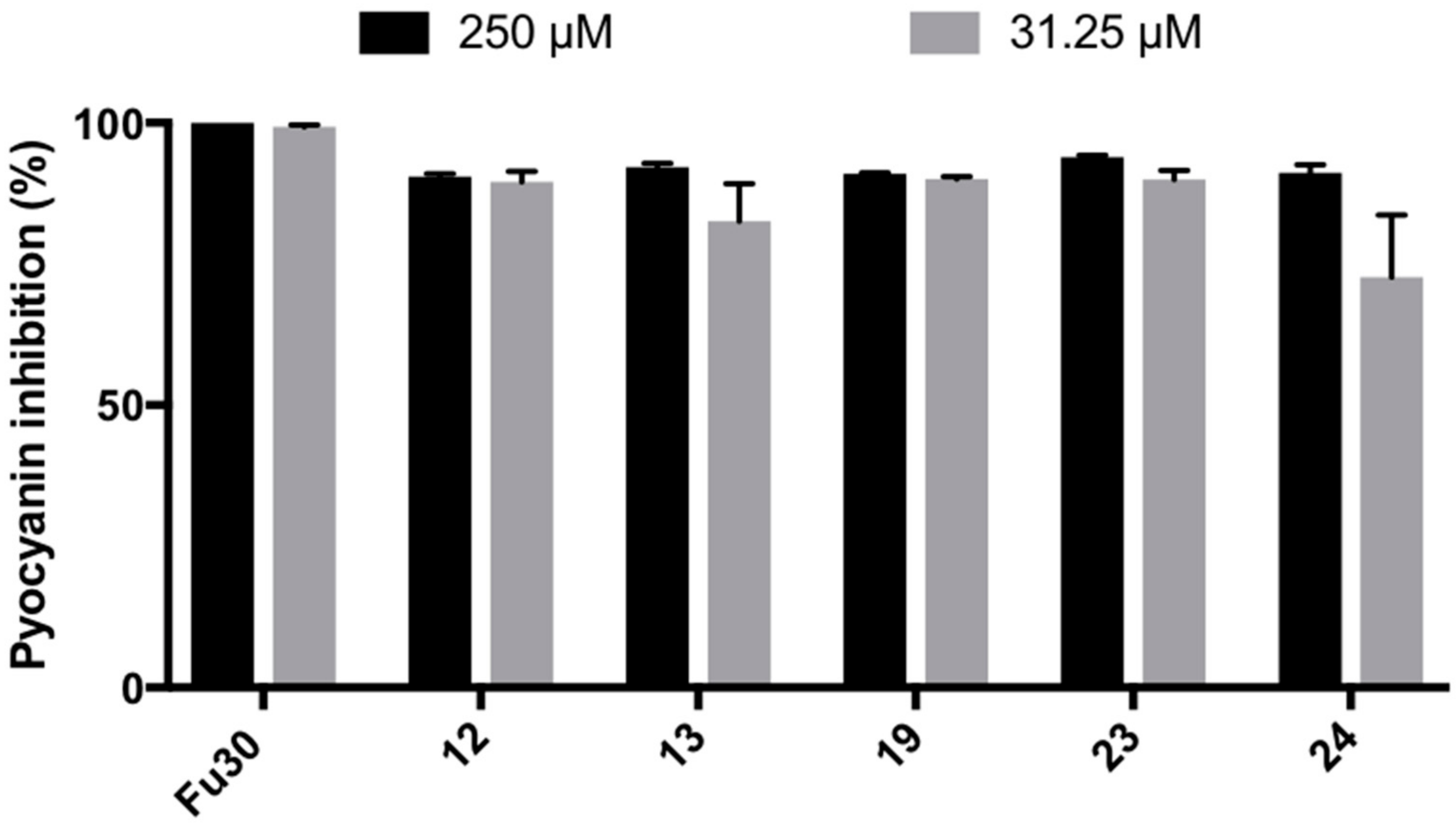
(4) 3,4-dichloro-1-(2-hydroxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (12)
The title compound was prepared according to Procedure A from mucochloric acid (600 mg, 3.55 mmol), ortho-aminophenol (388 mg, 3.55 mmol) and sodium triacetoxyborohydride (2.26 g, 10.65 mmol). The reaction mixture was left to react for 48 h. The precipitated solid was filtered by vacuum filtration, and a white solid was obtained (280 mg; 32%). m.p. 171.6 °C; 1H NMR (DMSO-d6, 300 MHz): δ 4.61 (s, 2H, CH2), δ 6.85–6.97 (m, 2H, ArH), δ 7.17–7.28 (m, 2H, ArH), 9.86 (s, OH); 13C NMR (DMSO, 75.5 MHz): 55.4 (CH2), 117.2 (ArCH), 119.6 (ArCH), 124.3 (C), 124.4 (ArCH), 129.3 (ArCH), 129.6 (ArCH), 142.0 (ArC), 153.5 (C), 163.1 (C=O); IR (ATR): υmax 3105.4, 1666.0, 1591.4, 1509.6, 1445.4, 1275.3, 1198.2, 930.5, 839.1; UV (MeOH): λmax 280.0 nm (ε 2928 cm−1M−1); HRMS (ESI) m/z calcd. for C10H7Cl2N1O2Na1265.9746 [M + Na]+, found 265.9744.
(5) 3,4-dichloro-1-(3-hydroxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (13)
The title compound was prepared according to Procedure A following the reported method [
14] from mucochloric acid (600 mg, 3.55 mmol),
meta-aminophenol (388 mg, 3.55 mmol) and sodium triacetoxyborohydride (2.26 g, 10.65 mmol). The reaction mixture was left to react for 24 h. As the reaction progresses, a yellow precipitated solid was evident. The solid was filtered by vacuum filtration and a yellow solid was obtained (578 mg; 66%); m.p. 163.6 °C;
1H NMR (DMSO-
d6, 400 MHz):
δ 4.80 (s, 2H, CH
2),
δ 6.59 (d,
J = 8.0 Hz, 1H, ArH), 7.04 (d,
J = 9.3 Hz, 1H, ArH), 7.18 (t,
J = 8.0 Hz, 1H, ArH), 7.28 (s, 1H, ArH), 9.71 (brs, OH);
13C NMR (DMSO, 100 MHz): 54.0 (CH
2), 106.6 (ArCH), 109.8 (ArCH), 112.3 (ArCH), 124.3 (C), 130.2 (ArCH), 139.8 (C), 141.9 (C), 158.4 (C), 162.4 (C=O); UV (MeOH): λ
max 280.0 nm (ε 3854 cm
−1M
−1); HRMS (ESI)
m/
z calcd. for C
10H
7Cl
2N
1O
2Na
1 265.9746 [M + Na]
+, found 265.9745.
(6) 3,4-dichloro-1-(4-hydroxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (14)
The title compound was prepared according to Procedure A from mucochloric acid (600 mg, 3.55 mmol), 4-aminophenol (388 mg, 3.55 mmol) and sodium triacetoxyborohydride (2.26 g, 10.65 mmol). The reaction mixture was left to react for 24 h. As the reaction progressed, a yellow precipitated solid was evident. The solid was filtered by vacuum filtration, and a yellow solid was obtained (500 mg; 58%); m.p. 126.0 °C; 1H NMR (DMSO-d6, 400 MHz): δ 4.77 (s, 2H, CH2), δ 6.81 (d, J = 8.9 Hz, 2H, ArH), δ 7.44 (d, J = 8.9 Hz, 2H, ArH), 9.8 (brs, 1H, OH); 13C NMR (DMSO, 100 MHz): 54.5 (CH2), 115.8 (ArCH), 122.2 (ArCH), 124.3 (C), 130.3 (ArC), 141.2 (ArC), 155.4 (C), 162.1 (C=O), IR (ATR): υmax 3277.8, 1682.9, 1630.2, 1510.6, 1270.8, 1219.3, 1046.9, 934.0, 830.7; UV (MeOH): λmax 280.0 nm (ε 1090.2 cm−1M−1); HRMS (ESI) m/z calcd. for C10H7Cl2N1O2Na1 265.9746 [M + Na]+, found 265.9745.
(7) N-(3-carboxyphenyl)-3,4-dibromo-1,5-dihydro-2H-pyrrol-2-one (DHP phenyl acid-2) (15)
![Molecules 23 01106 i007]()
The title compound was synthesized according to Procedure A from mucochloric acid (1 g, 5.91 mmol), 3-aminobenzoic acid (0.81 g, 5.91 mmol) and sodium triacetoxyborohydride (3.76 g, 17.75 mmol) in 5:3 v/v dichloromethane/glacial acetic acid (12 mL). The reaction mixture was left to stir at room temperature for 18 h, during which time, a yellow precipitate was evident. The mixture was filtered under vacuum, and the filtered solid was purified by flash chromatography. The solid was then recrystallized in methanol after chromatography to yield the pure title product as a white solid (0.54g; 34%); m.p. 214–216 °C; 1H NMR (300 MHz, DMSO-d6) δ 4.93 (s, 2H, CH2), δ 7.55 (t, J = 7.98 Hz, 1H, ArH), δ 7.74 (tt, J = 7.98 and 1.48 Hz, 1H, ArH), δ 7.89–7.93 (m, 1H, ArH), δ 8.34 (t, J = 1.8 Hz, 1H, ArH), δ 13.13 (brs, 1H, COOH); 13C NMR (75.5 MHz, DMSO-d6) δ 54.0 (CH2), 120.0 (ArCH), 123.4 (ArCN), 125.8 (ArC), 129.9 (ArCH), 132.1 (ArCH), 138.9 (ArCH), 142.4 (2 × CCl), 162.7 (C=O), 167.3 (C=O); IR (ATR): υmax 2969, 2824, 2654, 2539, 1699, 1586, 1491, 1433, 1382, 1313, 1272, 1158, 1051, 938, 899, 818, 757, 739, 677 cm−1; UV (THF): λmax 236 nm (ε 20671 cm−1M−1), 288 (32,346); HRMS (ESI) m/z calcd. for C11H7Cl2NO3Na 293.9695 [M + Na]+, found 293.9698.
(8) N-(4-carboxyphenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (16)
![Molecules 23 01106 i008]()
The title compound was synthesized according to Procedure A, by first dissolving mucochloric acid (1 g, 5.91 mmol) in 5:3 v/v dichloromethane/glacial acetic acid (12 mL). To this mixture, a solution of p-aminobenzoic acid (0.81 g, 5.91 mmol) in dichloromethane (8 mL) was added followed by sodium triacetoxyborohydride (3.76 g, 17.75 mmol). The reaction mixture was stirred at room temperature for 18 h, during which time a yellow precipitate was evident. The mixture was filtered under vacuum and washed with dichloromethane and distilled water to yield a yellow solid (0.6 g; 37%). m.p. 229 °C; 1H NMR (300 MHz, DMSO-d6) δ 4.91 (s, 2H, CH2), δ 7.84–7.88 (m, 2H, ArH), δ 7.96–8.01 (m, 2H, ArH), δ 12.8 (brs, 1H, COOH); 13C NMR (75.5 MHz, DMSO-d6) δ 53.9 (CH2), 118.3 (2 × ArCH), 124.2 (ArCN), 126.7 (ArC), 130.9 (2 × ArCH), 142.4 (CCl), 142.8 (CCl), 162.8 (C=O), 167.1 (C=O); IR (ATR): υmax 2939, 2814, 2659, 2537, 1679, 1604, 1516, 1425, 1375, 1279, 1042, 934, 860, 769, 741, 718 cm−1; UV (THF): λmax 236 nm (ε 5338 cm−1M−1), 289 (2736); HRMS (ESI) m/z calcd. for C11H7Cl2NO3Na 293.9695 [M + Na]+, found 293.9696.
(9) N-(3-tert-butylphenylcarbamate)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (17)
![Molecules 23 01106 i009]()
The title compound was prepared from mucochloric acid (1.0 g, 5.91 mmol), N-Boc-m-phenylenediamine (1.2 g, 5.91 mmol) and sodium triacetoxyborohydride (3.7 g, 17.75 mmol) in 5:3 v/v dichloromethane/glacial acetic acid (12 mL). The mixture was stirred at room temperature for 3.5 h. The reaction mixture was washed with water and brine and then extracted into ethyl acetate. The organic layer was dried over sodium sulphate and evaporated in vacuo. The solid was recrystallized in DCM and hexane to yield the title compound as a yellow solid (1.36 g, 67.17%). m.p. 131–132 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.48 (s, 9H, 3 × CH3), δ 4.80 (s, 2H, CH2), δ 7.29 (m, 3H, ArH), 7.97 (s, 1H, ArH), δ 9.45 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 28.6 (3 × CH3), δ 54.1 (CH2), δ 79.6.0 (C-O), 113.2 (ArC), 115.1 (ArC), 124.2 ©, 129.6 (ArCH), 139.0 (ArCH), 140.7 (ArC), 153.2 (C=O), 162.4 (C=O); IR (ATR): υmax 3305, 2977, 1695, 1604, 1498, 1421, 1388, 1285, 1227, 1151, 945, 876, 683, cm−1; HRMS (ESI) m/z calcd. for C15H16Cl2N2O3Na 365.0430 [M + Na]+, found 381.0428.
(10) N-(4′-tert-butylphenylcarbamate)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (18)
![Molecules 23 01106 i010]()
The title compound was prepared according to Procedure A from mucochloric acid (0.9 g, 5.36 mmol), N-Boc-p-phenylenediamine (1.1 g, 5.36 mmol) and sodium triacetoxyborohydride (3.38 g, 15.98 mmol) in 5:3 v/v dichloromethane/glacial acetic acid (12 mL). The mixture was stirred at room temperature for 3 h. The reaction mixture was washed with water and brine and then extracted into ethyl acetate. The organic layer was dried over sodium sulphate and evaporated in vacuo to yield the title compound as a dark red solid (1.16 g, 64%). m.p. 130–131 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.48 (s, 9H, 3 × CH3), δ 4.8 (s, 2H, CH2), δ 7.46 (dd, J = 41.9 and 9 Hz, 4H, ArH), δ 9.38 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 28.5 (3 × CH3), δ 54.1 (CH2), δ 82.0 (C-O), δ 118.9 (2 × ArCH), 120.3 (ArC), 124.2 (ArC), 133.1 (2 × ArCH), 136.9 (CCl), 141.5 (CCl), 153.2 (C=O), 162.2 (C=O); IR (ATR): υmax 3345, 2923, 2321, 1698, 1589, 1519, 1385, 1312, 1228, 1151, 1025, 932, 831, 758 cm−1; UV (ACN): λmax 245 nm (ε 13,990 cm−1M−1); HRMS (ESI) m/z calcd. for C15H16Cl2N2O3Na 365.0430 [M + Na]+, found 381.0426.
(11) N-(3-aminophenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (19)
The title compound was prepared according to Procedure A using Compound 17. The Boc group was cleaved by treating 17 (1.36 g) with trifluoroacetic acid (6 mL) at room temperature for 1 h followed by evaporation of TFA under high vacuum. The residue was neutralized and washed with saturated solution of sodium bicarbonate, and the solid obtained was filtered under vacuum to yield a red solid (0.75 g, 78%). m.p. 109 °C; 1H NMR (300 MHz, DMSO-d6) δ 4.75 (s, 2H, CH2), 5.21 (brs, 2H, NH2), δ 6.43 (dd, J = 6.42, 7.7 Hz, 1H, ArH), 6.78 (dd, J = 6.4, 7.7 Hz, 1H, ArH), 7.04–7.12 (m, 2H, ArH); 13C NMR (75.5 MHz, DMSO-d6) δ 54.0 (CH2), δ 105.4 (ArCH), 107.3 (ArCH), 111.4 (ArCH), 124.3 (C), 129.8 (ArCH), 139.4 (ArC), 141.6 (ArC), 149.0 (C), 162.2 (C=O); IR (ATR): υmax 3340, 1687, 1466, 1389, 1268, 949, 758, HRMS (ESI) m/z calcd. for C10H8Cl2N2O1 243.0086 [M + 1]+, found 243.0084.
(12) N-(4-aminophenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (20)
The title compound was prepared according to Procedure A from the Boc group, which was cleaved by treating intermediate 18 (0.53 g) with trifluoroacetic acid (5 mL) at room temperature for 1 h followed by evaporation of TFA under high vacuum. The residue was neutralized and washed with saturated solution of sodium bicarbonate, and the solid obtained was filtered under vacuum to yield a red solid (0.25 g, 64%). m.p. 106 °C; 1H NMR (300 MHz, DMSO-d6) δ 4.7 (s, 2H, CH2), δ 5.13 (brs, 1H, NH2), δ 6.55–6.6 (m, 2H, ArH), δ 7.24–7.29 (m, 2H, ArH); 13C NMR (75.5 MHz, DMSO-d6) δ 54.6 (CH2), δ 114.2 (2 × ArCH), 121.5 (ArC), 122.3 (2 × ArCH), 127.5 (ArC), 140.6 (CCl), 146.9 (CCl), 163.0 (C=O); IR (ATR): υmax 3297, 3205, 2921, 1697, 1636, 1515, 1400, 1389, 1300, 1175, 1043, 928, 812 cm−1; UV (ACN): λmax 247 nm (ε 10,817 cm−1M−1), 307 (5783); HRMS (ESI) m/z calcd. for C10H8Cl2N2ONa 264.9906 [M + Na]+, found 264.9909.
(13) 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)butanoic acid (21)
The title compound was synthesized according to Procedure A, by first dissolving mucochloric acid (1.0 g, 5.9 mmol) in 5:3 v/v dichloromethane/glacial acetic acid (12 mL). To this mixture, a solution of aminobutanoic acid (0.61 g, 5.9 mmol) in dichloromethane (8 mL) was added followed by sodium triacetoxyborohydride (3.76 g, 17.75 mmol) to yield a white solid (0.41 g, 30%). m.p. 106 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.91 (m, CH2), δ 2.42 (m, CH2), δ 3.56 (t, J = 7.0 Hz), δ 4.07 (s, C5-CH2); 13C NMR (75.5 MHz, DMSO-d6) δ 23.42 (CH2), 30.82 (CH2), 42.40 (CH2), 53.56 (C5-CH2), 125.7 (C), 139.7 (C), 164.7 (C=O), 176.9 (C=O); IR (ATR): υmax 3936, 2641, 1735, 1339, 1289, 1195, 859, 745 cm−1; HRMS (ESI) m/z calcd. for C8H9Cl2N1O3Na1259.9852 [M + Na]+, found 259.9850.
(14) 1-allyl-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (22)
![Molecules 23 01106 i014]()
The title compound was prepared according to Procedure A from mucochloric acid (600 mg, 3.55 mmol), allylamine (202.7 mg, 3.55 mmol) and sodium triacetoxyborohydride (2.26 g, 10.65 mmol). The reaction mixture was left to react for 3 days. The crude mixture was extracted with dichloromethane (30 mL) and washed with water (15 mL) followed by brine (15 mL), dried over sodium sulphate, and the solvent was evaporated and a residue oil obtained, which was purified by flash column chromatography using a gradient eluent of 5% to 25% ethyl acetate and hexane. A yellow oil was obtained (244 mg, 36%); 1H NMR (CDCl3, 300 MHz): 4.01 (s, 2H, C5-CH2), 4.11 (d, J = 6.1 Hz, 2H, N-CH2), 5.20–5.80 (m, 2H, HC=CH2), 5.74–5.80 (m, 1H, HC=CH2); 13C NMR (CDCl3, 75.5MHz): 45.5 (CH2), 52.9 (C5-CH2), 119.0 (HC=CH2), 125.6 (CCl), 132.0 (HC=CH2), 139.8 (C) 164.0 (C); IR (ATR): υmax 3290.1, 1685.5, 1621.0, 1402.1, 1270.5, 1157.0, 1100.6, 929.8, 841.8; UV (MeOH): λmax 280.0 nm (ε 4928 cm−1M−1); HRMS (ESI) m/z calcd. for C7H7Cl2N1O1Na1 213.9797 [M + Na]+, found 213.9794.
(15) 3,4-dichloro-1-phenyl-1,5-dihydro-2H-pyrrol-2-one (23)
![Molecules 23 01106 i015]()
The title compound was prepared according to Procedure A from mucochloric acid (1g, 5.92 mmol), aniline (551.2 mg, 5.92 mmol) and sodium triacetoxyborohydride (3.76 g, 17.76 mmol). The reaction mixture was left to react for 48 h. The reaction mixture was extracted with dichloromethane (30 mL) and washed with water (15 mL) followed by brine (15 mL), dried over sodium sulphate, and the solvent was evaporated. The obtained semi solid was triturated with methanol, and the precipitated solid was collected by vacuum filtration and dried over silica in the desiccator. A white small needle solid was obtained (680 mg, 99%). m.p. 201.2 °C; 1H NMR (DMSO-d6, 300 MHz): δ 4.86 (s, 2H, CH2), 7.20 (t, J = 9.0 Hz, 1H, ArH), 7.43 (t, J = 9.00 Hz, 2H, ArH), 7.73 (d, J = 9.0 Hz, 2H, ArH); 13C NMR (DMSO, 75.5MHz): 54.0 (CH2), 119.5 (ArCH), 124.3 (C), 125.1 (ArC)), 129.6 (ArCH), 139.0 (C), 142.0 (C), 162.5 (C=O); IR (ATR): υmax 3060.4, 2918.3, 1688.4, 1628.9, 1500.0, 1437.1, 1298.7, 1153.3, 1049.2, 927.4, 755.6; UV (MeOH): λmax 280.0 nm (ε 3854.4 cm−1M−1); HRMS (ESI) m/z calcd. for C10H7Cl2N1O1Na1 249.9797 [M + Na]+, found 249.9794.
(16) 3,4-dibromo-1-phenyl-1,5-dihydro-2H-pyrrol-2-one (24)
The title compound was prepared according to Procedure A from mucobromic acid (1.20 g, 4.65 mmol), aniline (433.3 mg, 4.65 mmol) and sodium triacetoxyborohydride (3.60 g, 13.95 mmol). The reaction mixture was left to stir for 48 h. The precipitated solid was filtered by vacuum filtration, and a white solid was obtained (440 mg; 30%); m.p. 160.7 °C; 1H NMR (DMSO-d6, 300 MHz): δ 4.88 (s, 2H, CH2), 7.18 (t, J = 7.5 Hz, 1H, ArH), 7.43 (t, J = 7.5 Hz, 2H, ArH), 7.72 (d, J = 8.7 Hz, 1H, ArH); 13C NMR (DMSO, 75.5 MHz): 57.3 (CH2), 119.4 (ArCH), 119.8 (ArC), 125.0 (C), 129.5 (ArCH), 136.4 (C), 138.8 (C), 163.3 (C); IR (ATR): υmax 3059.5, 1692.5, 1589.5, 1421.5, 1374.4, 1145.1, 1035.1, 885.4, 753.7; UV (MeOH): λmax 280.0 nm (ε 7829 cm−1M−1); HRMS (ESI) m/z calcd. for C10H7Br2N1O1Na1 337.8787 [M + Na]+, found 337.8789.
(17) 3,4-dibromo-1-(3-hydroxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (25)
The title compound was prepared according to Procedure A from mucobromic acid (1.20 g, 4.65 mmol), meta-aminophenol (507.8 mg, 4.65 mmol) and sodium triacetoxyborohydride (3.60 g, 13.95 mmol). The reaction mixture was left to stir for 48 h. The precipitated solid was filtered by vacuum filtration, and a light brown solid was obtained (780 mg, 50%); m.p. 164.0 °C; 1H NMR (DMSO-d6, 300 MHz): δ 4.80 (s, 2H, CH2), 6.57 (m, 2H, ArH), 7.05–7.27 (m, 2H, ArH), δ 9.59 (s, 1H, OH); 13C NMR (DMSO, 75.5 MHz): δ 57.4 (CH2), 106.6 (ArCH), 109.8 (ArCH), 112.2 (ArCH), 119.8 (C), 130.2 (ArCH), 136.4 (C), 139.9 (C), 158.3 (C), 163.2 (C=O); IR (ATR): υmax 3257.5, 1664.0, 1597.9, 1456.1, 1392.4, 1255.0, 1213.9, 1039.0, 929.8, 867.1; UV (MeOH): λmax 290.0 nm (ε 5893 cm−1M−1); HRMS (ESI) m/z calcd. for C10H7Br2N1O2Na1 353.8736 [M + Na]+, found 353.8733.
(18) N-(3-carboxyphenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (26)
![Molecules 23 01106 i018]()
The title compound was prepared from mucobromic acid (2.0 g, 7.75 mmol), meta-aminobenzoic acid (1.06 g, 7.75 mmol) and sodium triacetoxyborohydride (4.93 g, 23.26 mmol). The reaction mixture was left to stir for 24 h. The precipitated solid was filtered by vacuum filtration to afford the product as pale yellow solid (0.36 g, 26%). m.p. 186 °C; 1H NMR (300 MHz, DMSO-d6) δ 4.93 (s, 2H, CH2), δ 7.55 (t, J = 8.0 Hz, 1H, ArH), δ 7.73 (tt, J = 8.0 and 1.47 Hz, 1H, ArH), δ 7.89–7.93 (m, 1H, ArH), δ 8.33 (t, J = 1.8 Hz, 1H, ArH), δ 13.09 (brs, 1H, COOH); 13C NMR (75.5 MHz, DMSO-d6) δ 57.3(CH2), 119.9 (ArCH), 123.3 (ArCN), 125.7 (ArC), 129.8 (ArCH), 136.9 (ArCH), 139.0 (ArCH), 146.0 (2 × CBr), 163.5 (C=O), 167.4 (C=O); IR (ATR): υmax 2821, 2551, 2321, 1698, 1584, 1490, 1425, 1380, 1312, 1289, 1227, 1151, 1032, 939, 901, 840, 757, 673 cm−1; UV (THF): λmax 239 nm (ε 15,683cm−1M−1), 291 (6639); HRMS (ESI) m/z calcd. for C11H7Br2NO3Na 381.8685 [M + Na]+, found 381.8685.
(19) N-(4-carboxyphenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (27)
![Molecules 23 01106 i019]()
The title compound was synthesized from mucochloric acid (3 g, 11.63 mmol), p-aminobenzoic acid (1.59 g, 11.63 mmol) and sodium triacetoxyborohydride (7.39 g, 34.9 mmol) in 5:3 v/v dichloromethane/glacial acetic acid (30 mL). The reaction mixture was stirred and heated at 30 °C for 3 h during which time a precipitate was evident. The mixture was filtered under vacuum, and the solid was recrystallized in 1:9 acetone/methanol to get the desired product as a white solid (0.296 g, 7%). m.p. 221 °C; 1H NMR (300 MHz, DMSO-d6) δ 4.9 (s, 2H, CH2), δ 7.83 (dd, J = 37.8 and 8.7 Hz, 4H, ArH), δ 12.8 (brs, 1H, COOH); 13C NMR (75.5 MHz, DMSO-d6) δ 57.2 (CH2), 118.2 (2 × ArCH), 120.0 (ArCN), 126.6 (ArC), 130.9 (2 × ArCH), 137.4 (CBr), 142.5 (CBr), 163.7 (C=O), 167.1 (C=O); IR (ATR): υmax2811, 2659, 2535, 2112, 1679, 1601, 1516, 1423, 1371, 1275, 1188, 1145, 1017, 929, 889, 757, 704 cm−1; UV (THF): λmax 245 nm (ε 14,965cm−1M−1), 288 (15,575); HRMS (ESI) m/z calcd. for C11H7Br2NO3Na 381.8685 [M + Na]+, found 381.8684.
(20) N-(4′-tert-butylphenylcarbamate)-3,4-dibromo-1,5-dihydro-2H-pyrrol-2-one (28)
![Molecules 23 01106 i020]()
The title compound was synthesized by reacting mucobromic acid (1 g, 3.87 mmol), N-Boc-p-phenylenediamine (0.8 g, 3.87 mmol) and sodium triacetoxyborohydride (2.46 g, 11.63 mmol) in 5:3 v/v dichloromethane/glacial acetic acid (10 mL). The reaction mixture was left to stir at room temperature for 18 h. The mixture was washed with water and brine and then extracted into ethyl acetate. The organic layer was dried over sodium sulphate and chromatographed on silica gel to yield the desired product as a yellow solid (0.6 g, 36%). 1H NMR (300 MHz, CDCl3) δ 1.54 (s, 9H, 3 × CH3), δ 4.50 (s, 2H, CH2), δ 6.51 (s, 1H, NH), δ 7.39–7.42 (m, 2H, ArH), δ 7.54–7.59 (m, 2H, ArH). 13C NMR (75.5 MHz, CDCl3) δ 28.3 (3 × CH3), 57.2 (CH2), 80.8 (C-O), 119.2 (2 × ArCH), 119.9 (ArC), 121.5 (ArC), 132.9 (2 × ArCH), 133.3 (CBr), 135.5 (CBr), 152.6 (C=O), 163.1 (C=O); IR (ATR): υmax 3348, 3099, 2973, 1768, 1688, 1605, 1518, 1430, 1364, 1283, 1232, 1146, 1020, 846, 739, 680 cm−1; HRMS (ESI) m/z calcd. for C15H16Br2N2O3Na 452.9420 [M + Na]+, found 452.9422.
(21) N-(4-aminophenyl)-3,4-dibromo-1,5-dihydro-2H-pyrrol-2-one (29)
The title compound was synthesized by following the same method used to synthesize Compound 19 to afford a yellow solid (0.13 g, 28%). m.p. 163 °C; 1H NMR (300 MHz, DMSO-d6) δ 4.70 (s, 2H, CH2), δ 5.16 (brs, 1H, NH2), δ 6.55–6.6 (m, 2H, ArH), 7.25–7.28 (m, 2H, ArH); 13C NMR (75.5 MHz, DMSO-d6) δ 57.0 (CH2), 115.4 (2 × ArCH), 121.6 (2 × ArCH), 127.0 (ArC), 129.4 (ArC), 132.4 (CBr), 144.2 (CBr), 164.2 (C=O); IR (ATR): υmax 3305, 3208, 2920, 2287, 1693, 1611, 1512, 1442, 1383, 1280, 1150, 1023, 900, 830 cm−1; UV (ACN): λmax 243 (ε 8973 cm−1M−1), 306 (3430); HRMS (ESI) m/z calcd. for C10H9Br2N2O 330.9076 [M + H]+, found 330.9075.
(22) 3,4-dichloro-1-(4-hydroxyphenethyl)-1,5-dihydro-2H-pyrrol-2-one (30)
The title compound was prepared according to Procedure A from mucochloric acid (700 mg, 4.14 mmol) and tyramine (568.4 mg, 4.14 mmol) and was left to react for 3 days. The crude mixture was poured into icy water, and the solid was collected and washed twice with ether. A light green solid was obtained (280 mg; 25%). m.p. 157.8 °C; 1H NMR (CDCl3, 300 MHz): δ 2.72 (t, J = 7.2 Hz, 2H, CH2), δ 3.55 (t, J = 7.2 Hz, 2H, CH2), δ 4.2 (s, 2H, C5-CH2), 6.67 (d, J = 8.4 Hz, 2H, ArH), δ 7.00 (d, J = 8.4 Hz, 2H, ArH), 9.22 (s, 1H, OH). 13C NMR (DMSO, 75.5): 33.3 (CH2), 44.6 (CH2), 53.7 (C5-CH2), 115.7 (ArCH), 124.1 ©, 129.0 (C), 130.0 (ArCH), 140.9 (C), 156.3 (C), 163.4 (C=O); IR (ATR): υmax 3237.6, 1670.5, 1513.3, 1451.15, 1279.3, 1219.1, 1169.4, 827.6; UV (MeOH): λmax 277.0 nm (ε 19075.9 cm−1M−1), 320 (2585.1); HRMS (ESI) m/z calcd. for C12H11Cl2N1O2Na1294.0059 [M + Na]+, found 294.0060.
(23) 1-(2-(1H-indol-3-yl)ethyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (31)
![Molecules 23 01106 i023]()
The title compound was prepared according to Procedure A from mucochloric acid (200 mg, 1.18 mmol) and tryptamine (189.7 mg, 1.18 mmol) and was left to react for 24 h. The crude mixture was extracted with dichloromethane (30 mL) and washed with water (15 mL) followed by brine (15 mL), dried over sodium sulphate, and the solvent was evaporated. The oil residue was then triturated with methanol, and the precipitated solid was filtered to yield a green solid (145 mg; 42%). m.p. 159.3 °C; 1H NMR (DMSO, 400 MHz,): δ 2.96 (t, J = 7.3 Hz, CH2), 3.67 (t, J = 7.3 > N-CH2), 7.07 (s, 1H, ArH), δ 7.16 (t, J = 7.7, 14.8 Hz, 1H, ArH), δ 7.22 (t, J = 7.0, 14.8 Hz, 1H, ArH), δ 7.40 (d, J = 8.1 Hz, 1H), δ 7.61 (d, J = 8.1 Hz, 1H, ArH), 10.84 (1H, NH). 13C NMR (DMSO, 100 MHz): δ 24.2 (CH2), 43.6 (CH2), 54.0 (C5-CH2), 111.3 ©, 111.9 (ArCH), 118.6 (ArCH), 118.8 (ArCH), 121.5 (ArCH), 123.3 (ArCH), 124.2 (C), 127.5 (ArC), 136.7 (ArC), 140.9 (C), 163.5 (C=O); IR (ATR): υmax 3277.5, 3058.0, 2918.1, 1772.8, 1678.8, 1452.1, 1364.8, 1298.4, 1099.1, 1036.7, 974.5, 852.9, 738.8; UV (MeOH): λmax 275.0 nm (ε 3463.2 cm−1M−1), 220.0 nm (15204.8); HRMS (ESI) m/z calcd. for C14H12Cl2N2O1Na1 317.02189 [M + Na]+, found 317.02188.
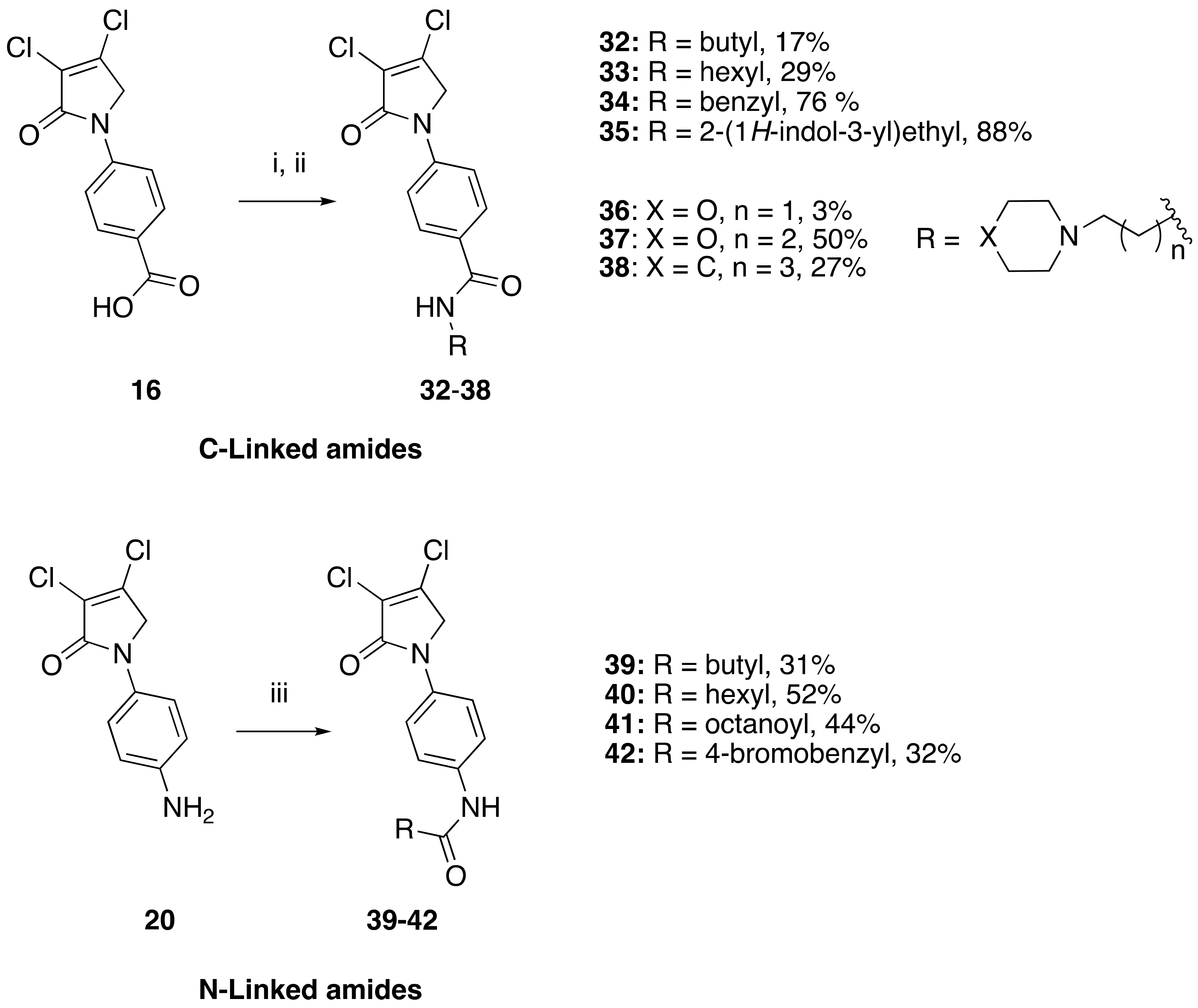
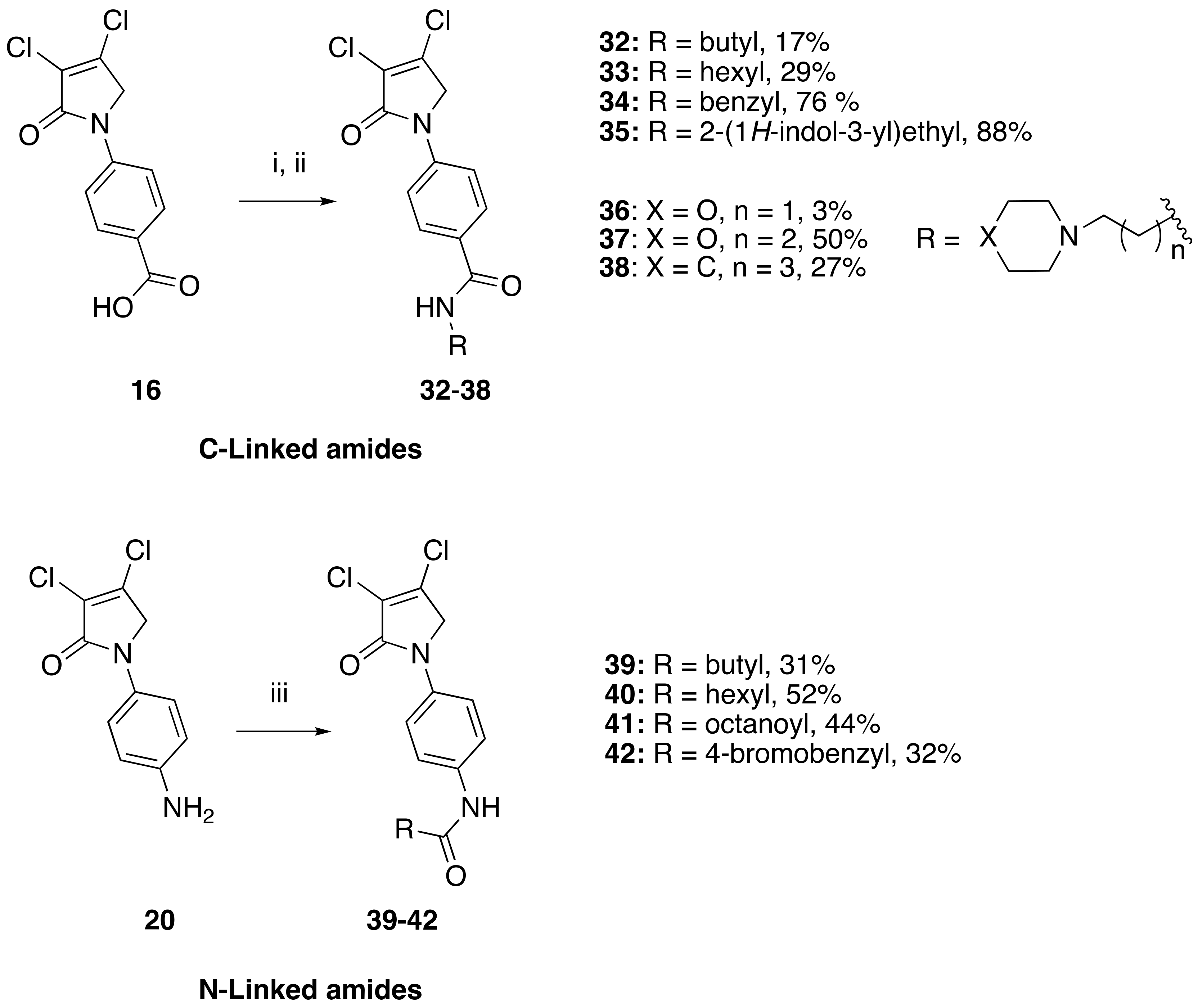
(24) N-butyl-4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzamide (32)
![Molecules 23 01106 i024]()
The title compound was prepared from acid 19 (200 mg, 0.74 mmol) and butylamine (53.8 mg, 0.74 mmol) according to the general Procedure B. The solvent of the reaction mixture was evaporated then triturated with methanol, and the precipitated solid was filtered. A beige solid was obtained (40 mg; 17%). m.p. 168.5 °C; 1H NMR (DMSO-d6, 600 MHz): δ 0.99 (t, J = 7.4 Hz, 3H, CH3), 1.23–1.34 (m, 4H, 2 × CH2), 1.50 (m, 2H, CH2), 4.89 (s, 2H, C5-CH2), 7.80 (d, J = 8.9 Hz, 2H, ArH), 7.89 (d, J = 8.9 Hz, 2H, ArH), 8.41 (s, 1H, NH). 13C NMR (DMSO-d6, 150.9 MHz): δ 14.11 (CH3), 20.11 (CH2), 31.71 (CH2), 53.92 (C5-CH2), 118.26 (ArCH), 124.2 (C), 129.60 (ArCH), 130.8 (C), 141.0 (C), 142.5 (C), 162.7 (C=O), 165.7 (C=O); IR (ATR): υmax 3314.7, 3078.0, 2955.3, 1696.7, 1606.9, 1508.1, 1381.5, 1152.3, 835.7, 763.2; UV (MeOH): λmax 300.0 nm (ε 10,897.6 cm−1M−1); HRMS (ESI) m/z calcd. for C15H16Cl2N2O2Na1 349.0481 [M + Na]+, found 349.0482.
(25) 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)-N-hexylbenzamide (33)
![Molecules 23 01106 i025]()
The title compound was prepared from acid 19 (200 mg, 0.74 mmol mmol) and hexylamine (74.4 mg, 0.74 mmol) according to the general Procedure B. The solvent of the reaction mixture was evaporated then triturated with methanol, and the precipitated solid was filtered. A white solid was obtained (75 mg; 29%); m.p. 159.5 °C; 1H NMR (DMSO-d6, 400 MHz): δ 0.88 (t, J = 6.9 Hz, 3H, CH3), 1.18–1.21 (m, 6H, 3 × CH2), 1.28–1.53 (m, 2H, CH2), 3.06–3.09 (m, 2H, CH2), 4.90 (s, 2H, CH2), 7.81 (d, J = 8.4 Hz, 2H, ArH), 7.90 (d, J = 8.4 Hz, 2H, ArH), 8.43 (s, NH). 13C NMR (DMSO-d6, 100 MHz): δ 22.5 (CH3), 26.7 (CH2), 29.6 (CH2), 31.5 (CH2), 39.7 (CH2), 45.9 (CH2), 54.0 (C5-CH2), 120.0 (ArCH), 124.2 (C), 128.6 (ArCH), 130.8 (C), 140.9 (C), 142.4 (C), 162.7 (C=O, 165.7 (C=O). IR (ATR): υmax 3331.8, 2929.2, 1691.9, 1630.4, 1503.6, 1377.3, 1270.6, 1151.8, 927.3, 846.7; UV (MeOH): λmax 280.0 nm (ε 6522.5 cm−1M−1); HRMS (ESI) m/z calcd. for C17H20Cl2N2O2Na1 377.0794 [M + H]+, found 377.0792.
(26) N-benzyl-4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzamide (34)
![Molecules 23 01106 i026]()
The title compound was prepared from 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzoic acid (19) (200 mg, 0.74 mmol mmol) and benzylamine (117.8 mg, 1.1 mmol) according to the general Procedure B. The precipitated solid was collected by filtration and washed with 1 mL of water, followed by hexane, then triturated from methanol. A white solid was obtained (230 mg; 76%). m.p. 175.3 °C; 1H NMR (DMSO-d6, 400 MHz): δ 3.96 (s, 2 H, CH2), 4.90 (s, 2H, C5-CH2), 7.21–7.46 (m, 6H, ArH and NH), 7.80 (d, J = 8.8 Hz, 2H, ArH), 7.96 (d, J = 8.8, 2H, ArH). 13C NMR (DMSO-d6, 400 MHz): δ 54.0 (C5-CH2), 67.5 (CH2), 118.2 (ArCH), 124.2 (C), 127.7 (ArC), 128.3 (ArC), 128.4 (ArCH), 128.7 (ArCH), 128.9 (ArCH), 130.8 (C), 142.4 (C), 162.8 (C=O), 167.8 (C=O). IR (ATR): υmax 2824.5, 2641.0, 1699.5, 1604, 1510.0, 1366.2, 1305.9, 1152.1, 782.0; UV (MeOH): λmax 285.0 nm (ε 3272.6 cm−1M−1); HRMS (ESI) m/z calcd. for C18H15Cl2N2O2 361.0505 [M + H]+, found 361.0501.
(27) Synthesis of N-(2-(1H-indol-3-yl)ethyl)-4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzamide (35)
![Molecules 23 01106 i027]()
The title compound was prepared from 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzoic acid (19) (300 mg, 1.10 mmol) and tryptamine (176.7 mg, 1.1 mmol) according to the general Procedure B. The precipitated solid was collected by filtration and washed with (1 mL) water, followed by hexane, then the solid was triturated from methanol to yield a pale pink solid (400 mg, 88%); m.p. 149.8 °C; 1H NMR (DMSO-d6, 400 Hz): δ 2.52 (m, 2H, CH2), 3.32 (m, 2H, CH2), 4.88 (S, 2H, C5-CH2), 6.99 (t, J = 7.0 Hz, 1H, ArH), δ 7.08 (t, J = 7.0 Hz, 1H, ArH)), δ 7.22 (s, 1H, NH), δ 7.36 (d, J = 8.0 Hz, 1H, ArH), δ 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.75 (d, J = 8.8 Hz, 2H, ArH), 7.94 (d, J = 8.8 Hz, 2H, ArH), 10.94 (s, CONH). 13C NMR (DMSO-d6, 100 MHz): δ 23.4 (CH2), 46.1 (CH2), 53.8 (CH2), 83.01 (ArC), 109.82 (ArCH), 112.04 (ArCH), 118.53 (ArCH), 119.03 (ArCH), 120.15 (ArCH), 121.7 (ArCH), 123.8 ©, 130.6 (ArCH), 136.7 (ArCH), 141.2 (ArC), 142.37 (ArC), 142.85 (ArC), 153.0 (C), 166.35 (C=O), 167.4 (C=O); IR (ATR): υmax 3223.4, 2902.4, 1700.1, 1603.1, 1370.0, 1248, 1038.6, 843.4, 781.0, UV (MeOH): λmax 280.0 nm (ε 3802.5 cm−1M−1); HRMS (ESI) m/z calcd. for C21H17Cl2N3O2Na1 436.0590 [M + Na]+, found 436.0583.
(28) 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)-N-(2-morpholinoethyl)benzamide (36)
![Molecules 23 01106 i028]()
The title compound was prepared from 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzoic acid (19) (200 mg, 0.74 mmol mmol) and 2-morpholinoethan-1-amine (95.7 mg, 0.74 mmol) according to Procedure B. After the completion of the reaction, the crude mixture was dissolved in ethanol, and few drops of diethyl ether were added dropwise. Then, the black solid impurity was filtered and discarded. The yellow filtrate was evaporated to dryness then recrystallized from ethanol, and a yellow small needle crystal was obtained (7 mg; 3%). m.p. 129.1 °C; 1H NMR (DMSO-d6, 600 MHz): δ 3.05–3.13 (m, 4H, 2 X CH2), 3.55 (d, J = 12 Hz, 2H, CH2), 3.68 (d, J = 6.0 Hz, 2H, CH2), 3.79 (t, J = 12.0 Hz, 2H, CH2), 3.98 (d, J = 12.0, 2H, CH2), 4.91 (s, 2H, C5-CH2), 7.84 (d, J = 8.6, 2H, ArH), 7.98 (d, J = 8.6 Hz, 2H, ArH), 8.87 (s, NH). 13C NMR (DMSO-d6, 100 MHz): δ 34.2 (CH2), 46.0 (CH2), 51.7 (CH2), 54.0 (CH2), 63.6 (CH2), 118.3 (ArCH), 124.2 ©, 129.0 (ArCH), 131.0 (C), 141.4 (C), 142.7 (C), 162.8 (C=O); IR (ATR): υmax 3307.4, 2923.8, 1699.1, 1603.7, 1509.0, 1370.2, 1110.5, 782.0; UV (MeOH): λmax 285.0 nm (ε 6378.6 cm−1M−1); HRMS (ESI) m/z calcd. for C17H20Cl2N3O3 384.0876 [M + H]+, found 384.0879.
(29) 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)-N-(3-morpholinopropyl)benzamide (37)
![Molecules 23 01106 i029]()
The title compound was prepared from 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzoic acid (19) (300 mg, 1.1 mmol mmol) and 3-morpholinopropan-1-amine (160 mg, 1.1 mmol) according to the general Procedure B. The precipitated solid was filtered, washed with ether and hexane to yield a light beige solid (180 mg; 50%). m.p. 129.1 °C; 1H NMR (DMSO-d6, 400 MHz): δ 1.70–1.74 (m, 2H, CH2), 2.34–2.41 (m, 10 H, 5 × CH2), 2.53 (t, J = 5.4, 10.0, 2H, CH2), 4.88 (s, 2H, C5-CH2), 7.72 (d, J = 8.8, 2H, ArH), 7.91 (d, J = 8.8, 2H ArH and NH), 13C NMR (DMSO-d6, 100 MHz): δ 23.91 (CH2), 38.8 (CH2), 53.3 (CH2), 55.97 (CH2), 66.34 (CH2), 117.9 (ArCH), 124.8 ©, 129.9 (ArCH), 134.2 (ArC), 139.9 (C), 141.44 (C), 163.18 (C=O), 173.09 (C=O). IR (ATR): υmax 3372.1, 3078.2, 2934.0, 1695.2, 1531.4, 1370.7, 1110.8, 1042.2, 778.6; UV (MeOH): λmax 285.0 nm (ε 2695.1 cm−1M−1); HRMS (ESI) m/z calcd. for C18H22Cl2N3O3 398.1033 [M + H]+, found 398.1030.
(30) 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)-N-(3-(piperidin-1-yl)propyl)benzamide (38)
![Molecules 23 01106 i030]()
The title compound was prepared from 4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)benzoic acid (19) (200 mg, 0.74 mmol mmol) and 3-(piperidin-1-yl)propan-1-amine (104.6 mg, 0.74 mmol) according to the general Procedure B. The solid product was collected by filtration, washed with water and hexane to yield a beige solid (64 mg; 27%). m.p. 165.5 °C; 1H NMR DMSO-d6, 600 MHz): δ 1.24–1.38 (m, 8H, 4 × CH2), 1.46–1.48 (m, 4H, 2 × CH2), 1.98–2.00 (m, 2H, CH2), 2.01–2.3 (m, 2H, CH2), 4.90 (s, 2H, C5-CH2), 5.32 (s, NH), 7.79 (d, J = 8.2 Hz, 2H, ArH), 7.95 (d, J = 8.2 Hz, 2H, ArH). 13C NMR (DMSO-d6, 100 MHz): δ 22.3 (CH2), 25.5 (CH2), 26.4 (CH2), 29.1 (CH2), 31.3 (CH2), 39.7 (CH2), 46.4 (CH2), 118.1 (ArCH), 124.8 ©, 128.0 (ArC), 129.5 (ArCH), 130.5 (C), 140.9 (C), 163.2 (C=O), 167.8 (C=O). IR (ATR): υmax 3355.8, 2936.3, 16887.5, 1540.6, 1375.5, 1042.7, 850.0, 775.0; UV (MeOH): λmax 285.0 nm (ε 7022.6 cm−1M−1); HRMS (ESI) m/z calcd. for C19H24Cl2N3O2 396.1240 [M + H]+, found 396.1234.
(31) N-(4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)phenyl)butyramide (39)
![Molecules 23 01106 i031]()
The title compound was prepared from 1-(4-aminophenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (23) (200 mg, 0.82 mmol) and butyryl chloride (87.7 mg, 0.82 mmol) according to the general Procedure C. After completion of the reaction, the crude mixture was evaporated to complete dryness, followed by trituration from methanol, then washed with 1 mL of ether twice; the solid was collected by filtration and dried in high vac. An off-white solid was obtained (89 mg, 31%). m.p. 195.5 °C; 1H NMR (DMSO-d6, 400 MHz): δ 0.91 (t, J = 7.3 Hz, 3H, CH3), 1.58–1.64 (m, 2H, CH2), 2.26 (t, J = 7.3 Hz, 2H), 4.82 (s, 2H, C5-CH2), 7.62 (brs, 4 H, ArH), 9.92 (s, NH). 13C NMR (DMSO-d6, 100 MHz): δ 14.1 (CH2), 19.0 (CH2), 54.1 (CH2), 120.0 (ArCH), 120.1 (ArCH), 124.3 ©, 134.0 (C), 136.6 (C), 141.6 (C), 162.2 (C), 171.5 (C=O); IR (ATR): υmax 3343.2, 2961.6, 1677.3, 1522.0, 1408.4, 1283.0, 820.6; UV (MeOH): λmax 290.0 nm (ε 8377.5 cm−1M−1); HRMS (ESI) m/z calcd. for C14H24Cl2N2O2Na1 335.0325 [M + Na]+, found 335.0323.
(32) N-(4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)phenyl)hexanamide (40)
![Molecules 23 01106 i032]()
The title compound was prepared from 1-(4-aminophenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (23) (200 mg, 0.82 mmol) and hexanoyl chloride (110.7 mg, 0.82 mmol) according to Procedure C. After completion of the reaction, the crude mixture was evaporated to complete dryness, followed by trituration from methanol, then the solid was washed with 1 mL of ether twice. The solid was collected by filtration and dried in high vac. An off-white solid was obtained (164 mg; 52%). m.p. 225.7 °C; 1H NMR (DMSO-d6, 300 MHz): δ 0.93 (t, J = 7.0 Hz, 3H, CH3), 1.38 (d, J = 3.6 Hz, 4H, 2 × CH2), 1.73–1.79 (m, 2H, CH2), 2.38 (t, J = 7.47 Hz, 2H, CH2), 4.49 (s, 2H, C5-CH2), 7.18 (s, NH), 7.59 (brs, 4H, ArH). 13C NMR (CDCl3, 75.5 MHz): 14.3 (CH2), 22.4 (CH2), 25.2 (CH2), 31.4 (CH2), 36.8 (CH2), 54.1 (CH2), 119.9 (ArCH), 120.1 (ArCH), 124.3 (C), 133.8 (C), 136.7 (C), 141.6 (C), 162.2 (C), 171.7 (C); IR (ATR): υmax 3358.6, 2949.4, 1677.3, 1515.9, 1413.1, 1515.9, 1284.3, 1042.8, 853.0; UV (MeOH): λmax 290.0 nm (ε 8394.3 cm−1M−1); HRMS (ESI) m/z calcd. for C16H18Cl2N2O2Na1 363.0638 [M + Na]+, found 363.0636.
(33) N-(4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)phenyl)octanamide (41)
![Molecules 23 01106 i033]()
The title compound was prepared from 1-(4-aminophenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (23) (200 mg, 0.82 mmol) and octanoyl chloride (133.8 mg, 0.82 mmol) according to Procedure C. After completion of the reaction, the crude mixture was evaporated to complete dryness, followed by trituration from methanol and then washed with 1 mL of ether twice. The solid was collected by filtration and dried in high vac. A white solid was obtained (140 mg; 44%). m.p. 194.1 °C; 1H NMR (CDCl3, 300 MHz): 0.91 (t, J = 6.8 Hz, 3H, CH3), 1.31–1.40 (m, 4H, 2 × CH2), 1.58 (brs, 4H, 2 × CH2), 1.75 (m, 2 H, CH2), 2.38 (t, J = 7.7 Hz, 2H, CH2), 4.50 (s, 2H, C5-CH2), 7.24 (s, NH), 7.58 (brs, 4H, ArH). 13C NMR (CDCl3, 75.5 MHz): 14.4 (CH2), 22.5 (CH2), 25.6 (CH2), 28.9 (CH2), 29.1 (CH2), 31.6 (CH2), 36.8 (CH2), 54.1 (CH2), 119.9 (ArCH), 120.1 (ArCH), 124.3 (C), 133.8 (C), 136.7 (C), 141.6 (C), 162.2 (C), 171.7 (C=O); IR (ATR): υmax 3338.9, 2915.7, 1678.1, 1521.3, 1285.3, 1156.1, 931.4, 822.9; UV (MeOH): λmax 290.0 nm (ε 5982.4 cm−1M−1); HRMS (ESI) m/z calcd. for C18H22Cl2N2O2Na1 391.0951 [M + Na]+, found 391.0952.
(34) 2-(4-bromophenyl)-N-(4-(3,4-dichloro-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)phenyl)acetamide (42)
![Molecules 23 01106 i034]()
The title compound was prepared according to method C from amine 1-(4-aminophenyl)-3,4-dichloro-1,5-dihydro-2H-pyrrol-2-one (23) (200 mg, 0.82 mmol) and 2-(4-bromophenyl)acetic acid (110.7 mg, 0.82 mmol). The latter was first converted to the corresponding acid chloride prior to amide coupling by treatment with thionyl chloride (2 h) on reflux, followed by complete dryness in high vac. After completion of the reaction, the crude mixture was evaporated to complete dryness, followed by trituration from methanol, and the obtained solid was then washed twice with 1 mL of ether. The solid was collected by filtration and dried in high vac. A white solid (100 mg; 32%) was obtained; m.p. 159.2 °C; 1H NMR (CDCl3, 300 MHz): 3.64 (s, 2H, CH2), 4.82 (s, 2H, C5-CH2), 7.30 (d, J = 8.2 Hz, 2H, ArH), 7.52 (d, J = 8.2 Hz, 2H, ArH), 7.63 (brs, 4H, ArH), 10.33 (s, NH). 13C NMR (CDCl3, 75.5 MHz): 45.95 (CH2), 54.09 (CH2), 120.06 (ArCH), 120.21 (ArCH), 124.3 (C), 131.6 (ArCH), 131.9 (ArCH), 134.1 (C), 135.85 (C), 136.40 (C), 141.7 (C), 162.3 (C), 169.0 (C); IR (ATR): υmax 3279.8, 2978.7, 1690.5, 1516.5, 1379.5, 1153.7, 927.8, 824.9; UV (MeOH): λmax 285.0 nm (ε 3375.7 cm−1M−1); HRMS (ESI) m/z calcd. for C18H13Br1Cl2N2O2Na1 460.9430 [M + Na]+, found 460.9431.

 ,
, 






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}