Scale-Up Synthesis and Identification of GLYX-13, a NMDAR Glycine-Site Partial Agonist for the Treatment of Major Depressive Disorder


Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Dipeptide Fragment 3
2.2. Synthesis of Dipeptide Segment 5
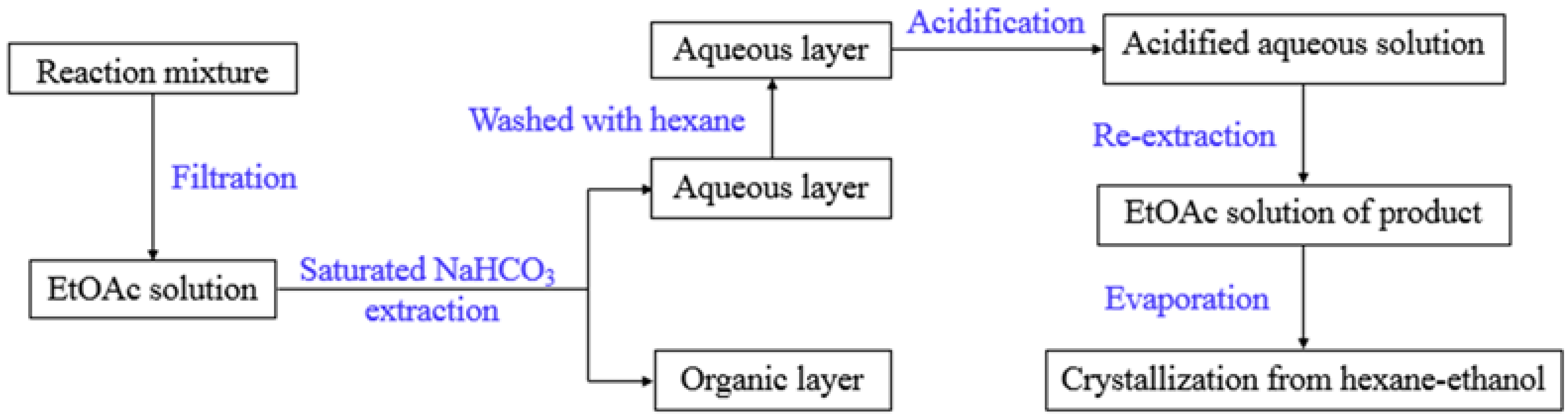
2.3. “2 + 2” Fragment Coupling Reaction and the Preparation of GLYX-13
2.4. X-ray Single Crystal Diffraction and Powder Diffraction of 7
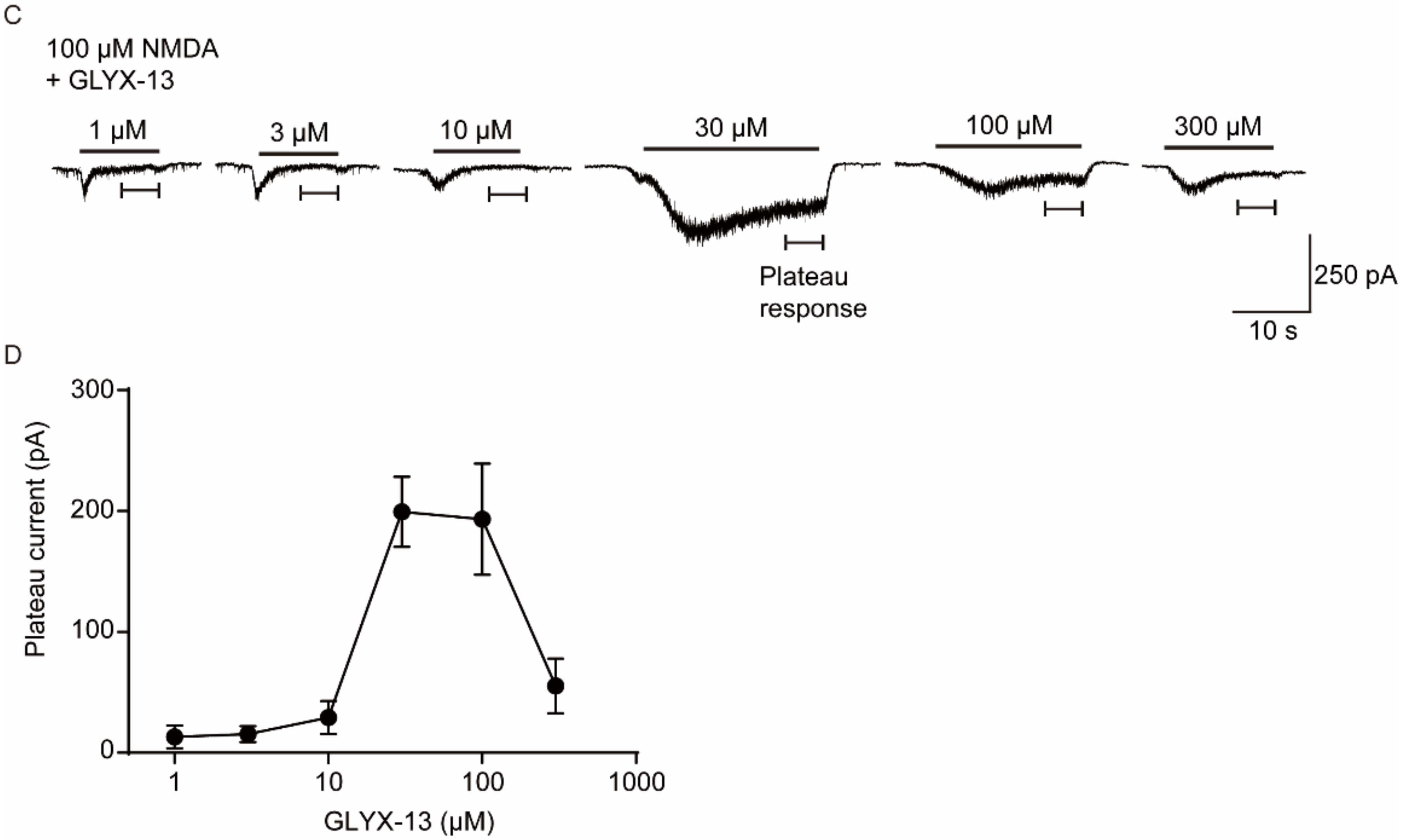
2.5. Verification the Activity of GLYX-13 on Cortical Neurons
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Thr-NH2·HCl (1b)
3.2.2. Fmoc-Pro-ThrNH2 (2b)
3.2.3. Pro-ThrNH2 (3)
3.2.4. Fmoc-Thr(tBu)-Pro-OH (5)
3.2.5. Fmoc-Thr-Pro-Pro-ThrNH2 (7)
3.2.6. Thr-Pro-Pro-ThrNH2 (GLYX-13)
3.3. Cortical Neuronal Cultures
3.4. Whole-Cell Voltage-Clamp Recordings
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Jing, L.; Toledo-Salas, J.C.; Xu, L. Rapid-onset antidepressant efficacy of glutamatergic system modulators: The neural plasticity hypothesis of depression. Neurosci. Bull. 2015, 31, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Iasevoli, F.; Buonaguro, E.F.; De Berardis, D.; Fornaro, M.; Fiengo, A.L.; Martinotti, G.; Orsolini, L.; Valchera, A.; Di Giannantonio, M.; et al. Treating the synapse in major psychiatric disorders: The role of postsynaptic density network in dopamine-glutamate interplay and psychopharmacologic drugs molecular actions. Int. J. Mol. Sci. 2017, 18, 135. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. Nmda receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Campanella, D.; Gambi, F.; La Rovere, R.; Carano, A.; Conti, C.M.; Sivestrini, C.; Serroni, N.; Piersanti, D.; Di Giuseppe, B.; et al. The role of c-reactive protein in mood disorders. Int. J. Immunopathol. Pharmacol. 2006, 19, 721–725. [Google Scholar] [CrossRef]
- Marini, S.; Vellante, F.; Matarazzo, I.; De Berardis, D.; Serroni, N.; Gianfelice, D.; Olivieri, L.; Di Renzo, F.; Di Marco, A.; Fornaro, M.; et al. Inflammatory markers and suicidal attempts in depressed patients: A review. Int. J. Immunopathol. Pharmacol. 2016, 29, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Phillips, C.; Fahimi, A.; McNerney, M.W.; Salehi, A. Mechanisms of action and clinical efficacy of NMDA receptor modulators in mood disorders. Neurosci. Biobehav. Rev. 2017, 80, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Hayley, S.; Litteljohn, D. Neuroplasticity and the next wave of antidepressant strategies. Front. Cell. Neurosci. 2013, 7, 218. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Fogaca, M.V.; Deyama, S.; Li, X.Y.; Fukumoto, K.; Duman, R.S. BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Moskal, J.R.; Burch, R.; Burgdorf, J.S.; Kroes, R.A.; Stanton, P.K.; Disterhoft, J.F.; Leander, J.D. GLYX-13, an NMDA receptor glycine site functional partial agonist enhances cognition and produces antidepressant effects without the psychotomimetic side effects of NMDA receptor antagonists. Expert Opin. Investig. Drugs 2014, 23, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.C.; Pierantoni, G.M.; Gerlini, R.; Iorio, R.; Olabinjo, Y.; Giovane, A.; Di Domenico, M.; Sogos, C. GLYX-13, a new drug acting on glutamatergic pathways in children and animal models of autism spectrum disorders. BioMed. Res. Int. 2014, 2014, 234–295. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Henter, I.D.; Zarate, C.A. New targets for rapid antidepressant action. Prog. Neurobiol. 2017, 152, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, J.; Zhang, X.L.; Weiss, C.; Gross, A.; Boikess, S.R.; Kroes, R.A.; Khan, M.A.; Burch, R.M.; Rex, C.S.; Disterhoft, J.F.; et al. The long-lasting antidepressant effects of rapastinel (GLYX-13) are associated with a metaplasticity process in the medial prefrontal cortex and hippocampus. Neuroscience 2015, 308, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.L.; Liu, J.X.; Liu, X.; Zhang, L.M.; Ran, Y.H.; Zheng, Y.Y.; Tang, Y.; Li, Y.F.; Xiong, J. Anxiolytic effects of GLYX-13 in animal models of posttraumatic stress disorder-like behavior. J. Psychopharmacol. 2016, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Moskal, J.R.; Burgdorf, J.; Kroes, R.A.; Brudzynski, S.M.; Panksepp, J. A novel NMDA receptor glycine-site partial agonist, GLYX-13, has therapeutic potential for the treatment of autism. Neurosci. Biobehav. Rev. 2011, 35, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Weiguo, S.; Bohua, Z.; Shiyan, F.; Yishan, Y. Process for Preparation of H-Thr-Pro-Pro-Thr-Nh2 Peptide by Liquid-Phase Synthesis Method. CN 104109189A, 22 October 2014. [Google Scholar]
- McDermott, T.S.; Bhagavatula, L.; Borchardt, T.B.; Engstrom, K.M.; Gandarilla, J. Scalable synthesis of dipeptidyl peptidase-4 inhibitor ABT-279. Org. Process Res. Dev. 2009, 13, 1145–1155. [Google Scholar] [CrossRef]
- Chulin, A.N.; Rodionov, I.L.; Baidakova, L.K.; Rodionova, L.N.; Balashova, T.A.; Ivanov, V.T. Preparation and reactivity of aminoacyl pyroglutamates. Facile synthesis of 10-membered-ring cyclic dipeptides derived from 1,4-diaminobutyric and glutamic acids. J. Pept. Sci. 2005, 11, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.L.; He, B.F.; Xu, J.X.; Wu, B.; Ouyang, P. Efficient chemo-enzymatic synthesis of endomorphin-1 using organic solvent stable proteases to green the synthesis of the peptide. Green Chem. 2011, 13, 1680–1685. [Google Scholar] [CrossRef]
- Kudryavtsev, K.V.; Shulga, D.A.; Chupakhin, V.I.; Sinauridze, E.I.; Ataullakhanov, F.I.; Vatsadze, S.Z. Synthesis of novel bridged dinitrogen heterocycles and their evaluation as potential fragments for the design of biologically active compounds. Tetrahedron 2014, 70, 7854–7864. [Google Scholar] [CrossRef]
- Arnett, E.M.; Miller, J.G.; Day, A.R. Effect of structure on reactivity.1 iii. Aminolysis of esters with primary amines. J. Am. Chem. Soc. 1950, 72, 5635–5638. [Google Scholar] [CrossRef]
- Gordon, M.; Miller, J.G.; Day, A.R. Effect of structure on reactivity. Ii. Influence of solvents on ammonolysis of esters. J. Am. Chem. Soc. 1949, 71, 1245–1250. [Google Scholar] [CrossRef]
- Gordan, M.; Miller, J.G.; Day, A.R. Effect of structure on reactivity. I. Ammonolysis of esters with special reference to the electron release effects of alkyl and aryl groups. J. Am. Chem. Soc. 1948, 70, 1946–1953. [Google Scholar] [CrossRef]
- Marco, R.D.; Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A new non-natural arginine-like amino acid derivative with a sulfamoyl group in the side-chain. Amino Acids 2010, 38, 691–700. [Google Scholar] [CrossRef]
- Flegel, M.; Flegelova, Z.; Malon, P.; Klenerova, V.; Hynie, S. Fmoc deprotection by tert-butylamine and its comparison in solution and solid phase synthesis. Collect. Symp. Ser. 2011, 13, 41–44. [Google Scholar]
- Leggio, A.; Liguori, A.; Napoli, A.; Siciliano, C.; Sindona, G. New strategies for an efficient removal of the 9-fluorenylmethoxycarbonyl (Fmoc) protecting group in the peptide synthesis. Eur. J. Org. Chem. 2000, 573–575. [Google Scholar] [CrossRef]
- Maegawa, T.; Fujiwara, Y.; Ikawa, T.; Hisashi, H.; Monguchi, Y.; Sajiki, H. Novel deprotection method of Fmoc group under neutral hydrogenation conditions. Amino Acids 2009, 36, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Stefan, H.; Roger, M.; Rainer, R.; Simeunovic, M. Thermal cleavage of the Fmoc protection group. Chimia 2010, 64, 200–202. [Google Scholar]
- Tana, G.; Kitada, S.; Fujita, S.; Okada, Y.; Kim, S.; Chiba, K. A practical solution-phase synthesis of an antagonistic peptide of TNF-α based on hydrophobic tag strategy. Chem. Commun. 2010, 46, 8219–8221. [Google Scholar] [CrossRef] [PubMed]
- Sheppeck, J.E. A convenient and scaleable procedure removing the Fmoc group in solution. Tetrahedron Lett. 2000, 41, 5329–5333. [Google Scholar] [CrossRef]
- Suhas, R.; Gowda, D.C. Design and synthesis of tryptophan containing peptides as potential analgesic and anti-inflammatory agents. J. Pept. Sci. 2012, 18, 535–540. [Google Scholar] [CrossRef]
- Yao, J.; Liu, H.; Zhou, T.; Chen, H.; Miao, Z.; Dong, G.; Wang, S.; Sheng, C.; Zhang, W. Total synthesis and structure–activity relationships of caspofungin-like macrocyclic antifungal lipopeptides. Tetrahedron 2012, 68, 3074–3085. [Google Scholar] [CrossRef]
- Yao, J.; Liu, H.; Zhou, T.; Chen, H.; Miao, Z.; Sheng, C.; Zhang, W. Total synthesis and structure-activity relationships of new echinocandin-like antifungal cyclolipohexapeptides. Eur. J. Med. Chem. 2012, 50, 196–208. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, Z.X.; Yan, T.; Han, B.; Zhang, N.; Song, W.; Liu, Z.L.; Zhao, J.L.; Liu, J.L. Kilogram-scale synthesis of osteogenic growth peptide (10–14) using a fragment coupling approach. Org. Process Res. Dev. 2015, 19, 1257–1262. [Google Scholar] [CrossRef]
- Nizar, H.; Bo, Q.; Heewon, L.; Lorenz, J.; Varsolona, R.; Kapadia, S.; Sarvestani, M.; Feng, X.W.; Busacca, C.A.; Hebrault, D.; et al. Pat application in the expedited development of a three-step, one-stage synthesis of the dipeptide intermediate of hcv protease inhibitor faldaprevir. Org. Process Res. Dev. 2015, 19, 132–138. [Google Scholar]
- Wakasugi, K.; Akira, L.; Misaki, T.; Nishii, Y.; Tanabe, Y. Simple, mild, and practical esterification, thioesterification, and amide formation utilizingp-toluenesulfonyl chloride andn-methylimidazole. Adv. Synth. Catal. 2003, 345, 1209–1214. [Google Scholar] [CrossRef]
- Rayle, H.L.; Fellmeth, L. Development of a process for triazine-promoted amidation of carboxylic acids. Org. Process Res. Dev. 1999, 3, 172–176. [Google Scholar] [CrossRef]
- Myers, A.G.; Gleason, J.L.; Yoon, T.; Daniel, W.K. Highly practical methodology for the synthesis of d- and l-r-amino acids, N-protected r-amino acids, and N-methyl-r-amino acids. J. Am. Chem. Soc. 1997, 119, 656–673. [Google Scholar] [CrossRef]
- Venalainen, J.I.; Wallen, E.A.; Poso, A.; Garcia-Horsman, J.A.; Mannisto, P.T. Synthesis and characterization of the novel fluorescent prolyl oligopeptidase inhibitor 4-fluoresceinthiocarbamoyl-6-aminocaproyl-l-prolyl-2(s)-(hydroxyacetyl)pyrrolidine. J. Med. Chem. 2005, 48, 7093–7095. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Oda, M.; Sato, M.; Yoshida, N. A facile method for the preparation of t-butyloxycarbonyl amino acid p-nitroanilides. Int. J. Pept. Protein Res. 1990, 36, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.J.; David, J.M. Lithium-initiated imide formation. A simple method for N-acylation of 2-oxazolidinones and bornane-2,10-sultam. J. Org. Chem. 1995, 60, 2271–2273. [Google Scholar] [CrossRef]
- Ryakhovsky, V.V.; Ivanov, A.S. Study of intramolecular aminolysis in peptides containing N-alkylamino acids at position 2. Tetrahedron 2012, 68, 7070–7076. [Google Scholar] [CrossRef]
- Shevchenko, V.P.; Nagaev, I.Y.; Alfeeva, L.Y.; Andreeva, L.A.; Shevchenko, K.V.; Myasoedov, N.F. Synthesis of Tyr-Pro-Phe-Val-Glu-l-[3,4-3H]Pro-Ile, Tyr-d-Ala-Phe-Gly-Tyr-l-[3,4-3H]Pro-Ser-NH2, and Tyr-d-Ala-Phe-Gly-Tyr-d-[3,4-3H]Pro-Ser-NH2, labeled analogs of human β-casomorphin and dermorphin. Radiochemistry 2004, 46, 63–71. [Google Scholar] [CrossRef]
- McMurray, J.S.; Mandal, P.K. Pd-c-induced catalytic transfer hydrogenation with triethylsilane. J. Org. Chem. 2007, 72, 6599–6601. [Google Scholar]
- Martinez, J.; Tolle, J.C.; Miklos, B. Side reactions in peptide synthesis. 12. Hydrogenolysis of the 9-fluorenylmethyloxycarbonyl group. J. Org. Chem. 1979, 44, 3596–3598. [Google Scholar] [CrossRef]
- Somlai, C.; Lovas, S.; Forgo, P.; Murphy, R.F.; Penke, B. Dehydration of threonineine esters during tosylation. Synth. Commun. 2001, 31, 3633–3640. [Google Scholar] [CrossRef]
- Mirza-Aghayan, M.; Boukherroub, R.; Bolourtchian, M. A mild and efficient palladium–triethylsilane system for reduction of olefins and carbon–carbon double bond isomerization. Appl. Organomet. Chem. 2006, 20, 214–219. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Limburg, D.C.; Wilkinson, D.E.; Mark, J.V.; Hamilton, G.S. A mild deprotection procedure for tert-butyl esters and tert-butyl ethers using ZnBr2 in methylene chloride. Tetrahedron Lett. 2000, 41, 2847–2849. [Google Scholar] [CrossRef]
- Bryan, L.; Berliner, M.; Buzon, R.; Stephen, T.C.; Kaneko, T.; Keene, N.; Kissel, W.; Tung, L.; Leeman, K.R. Aqueous phosphoric acid as a mild reagent for deprotection of tert-butyl carbamates, esters, and ethers. J. Org. Chem. 2006, 71, 9045–9050. [Google Scholar]
- Bartoli, G.; Bosco, M.; Carlone, A.; Locatelli, M.; Marcantoni, E.; Melchiorre, P.; Sambri, L. Tert-butyl ethers: Renaissance of an alcohol protecting group. Facile cleavage with cerium(iii) chloride/sodium iodide. Adv. Synth. Catal. 2006, 348, 905–910. [Google Scholar] [CrossRef]
- Moskal, J.R.; Kuo, A.G.; Weiss, C.; Wood, P.L.; O’Connor Hanson, A.; Kelso, S.; Harris, R.B.; Disterhoft, J.F. GLYX-13: A monoclonal antibody-derived peptide that acts as an N-methyl-d-aspartate receptor modulator. Neuropharmacology 2005, 49, 1077–1087. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Method | Base | Reaction Solvent a | Impurity b |
|---|---|---|---|---|
| 1 | EDCI/HOBt | DIPEA | DCM | Obvious |
| 2 | EDCI/DMAP | DIPEA | DCM | Obvious |
| 3 | EDCI/HOSu | DIPEA NaHCO3 | DCM/THF | Obvious |
| 4 | EDCI/HONp | DIPEA NaHCO3 | DCM/THF | Obvious |
| 5 | TsCl | NMM | DCM, THF, ACN | Obvious |
| 6 | TsCl | NMI | DCM, THF, ACN | Obvious |
| 7 | Cyanuric chloride | NMM | ACN/Acetone/DMF | Obvious |
| 8 | Cyanuric chloride | NMM | THF/toluene | Trace |
| 9 | PivCl | TEA | THF/DCM | Trace |
| 10 | PivCl | Imidazole | THF/DCM | Trace |
| Entry | Method | Reaction Solvent | Yield (%) |
|---|---|---|---|
| 1 | Pd/C, H2 | MeOH | - |
| 2 | Pd/C, H2 | EtOH | - |
| 3 | Pd/C, HCOONH4 | MeOH | - |
| 4 | Pd/C, HCOOH | MeOH | - |
| 5 | Pd/C, Et3SiH | MeOH | 65% |
| 6 | Pd/C, Et3SiH | EtOH | 68% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Liu, J.; Fan, M.; Li, Z.; Chen, Y.; Zhang, G.; Huang, Z.; Zhang, L. Scale-Up Synthesis and Identification of GLYX-13, a NMDAR Glycine-Site Partial Agonist for the Treatment of Major Depressive Disorder. Molecules 2018, 23, 996. https://doi.org/10.3390/molecules23050996
Li W, Liu J, Fan M, Li Z, Chen Y, Zhang G, Huang Z, Zhang L. Scale-Up Synthesis and Identification of GLYX-13, a NMDAR Glycine-Site Partial Agonist for the Treatment of Major Depressive Disorder. Molecules. 2018; 23(5):996. https://doi.org/10.3390/molecules23050996
Chicago/Turabian StyleLi, Wenchao, Jingjian Liu, Minghua Fan, Zhongtang Li, Yin Chen, Guisen Zhang, Zhuo Huang, and Liangren Zhang. 2018. "Scale-Up Synthesis and Identification of GLYX-13, a NMDAR Glycine-Site Partial Agonist for the Treatment of Major Depressive Disorder" Molecules 23, no. 5: 996. https://doi.org/10.3390/molecules23050996