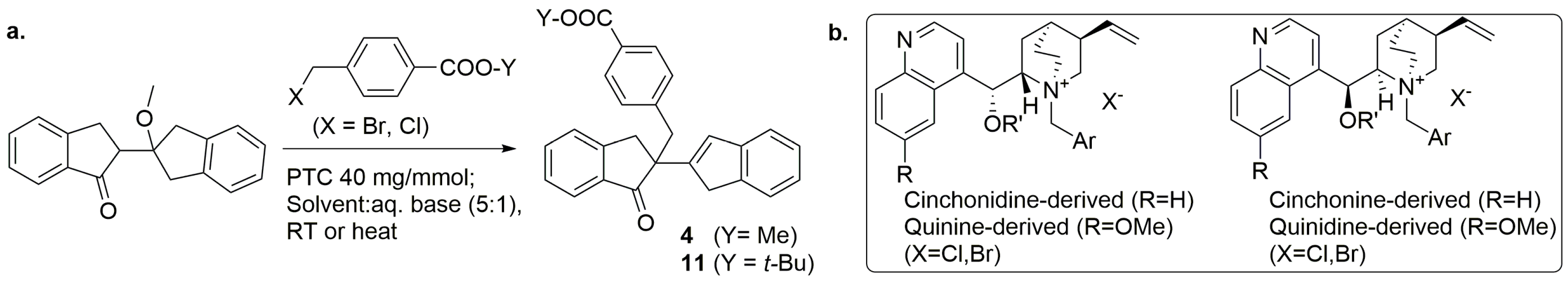
2.2. PTC-Promoted Alkylation of Ketone 3
Based on the concern of the potential (Me)-ester moiety instability in methyl(4-bromomethyl)benzoate (MBMB) under PTC conditions, a more robust analogous, named
t-butyl (
tBu)-(4-bromomethyl)benzoate (TBBMB), was chosen for initial testing (
Scheme 4a). While a variety of parent structures for chiral PTCs are described in the literature [
21,
22,
23,
24], in practice those derived from cinchona alkaloids (
Scheme 4b) are often preferred for reactions that could potentially be carried out on kilo scales. The cinchona alkaloids represent inexpensive sources of chiral information and are widely available, while the PTC derivatives themselves are generally straightforward to prepare, usually without the need for purification [
25,
26,
27]. There are a wide variety of others available, perhaps most notably chiral crown ethers [
28,
29] or Maruoka’s biaryl catalysts [
30,
31], however, many of these would be costly to source or prepare. In our hands, a small set of PTCs derived from cinchona alkaloids was available and it was selected for the first screen, using 25% aqueous (aq.) NaOH/toluene (1:5,
v/
v) as for the
tBu-ester
11 reaction described in
Scheme 4.
The details of the PTCs preparation are described in the
Supplementary Materials. Most of the screening reactions were quite rapid (
Table 1). However, enantioselectivity was lower than envisaged. Interestingly, Entry 1D was much slower than the others, nevertheless, it gave slightly higher enantioselectivity. The reaction rate with this particular catalyst relative to others in this small set cannot be readily correlated to selectivity, as the slower reaction in this case could simply be due to increased water solubility of the catalyst/base ion pair due to the nitro group. Subsequent investigation with the
N-(2-NO
2-Bn)-quinidinium bromide catalyst under a variety of conditions failed to identify any significant improvements (Entries 2A–K). Indeed, Entry 2G in dichloromethane (DCM) gave the unwanted (
S)-enantiomer (with poor selectivity), while Entries 2F and 2H gave complex HPLC profiles without any of the desired product.
Based on the most efficient reaction time observed in
Table 1, the aq. NaOH/toluene solvent/base system with
t-butyl ester alkylating agent was used to screen a further dozen catalysts covering a wider range of benzylic substituents (e.g., 2-CN-Bn and anthracenyl) and O-allyl derivatives. The reaction conditions were kept consistent. Unfortunately, the effects observed for the cinchona alkaloid-based catalysts could not be fully rationalised (Entries 3G to 3R in
Table 2).
For instance, comparison of Entries 3Q and 3R showed that the O-allyl substituent had a strong impact upon the e.e., yet the results of a similar comparison between Entries 1B and 3P showed no significant effect. Elucidating the effect of the substituent on the quinoline ring was similarly difficult, e.g., comparing Entries 1A and 1B (~no difference) or Entries 3I with 3L (large difference). However, there seemed to be a definite trend in terms of the steric bulk of the -CH2Ar group at this stage. In particular, Entries 3K and 3R, where the very largest groups were used, showed promising enantioselectivity.
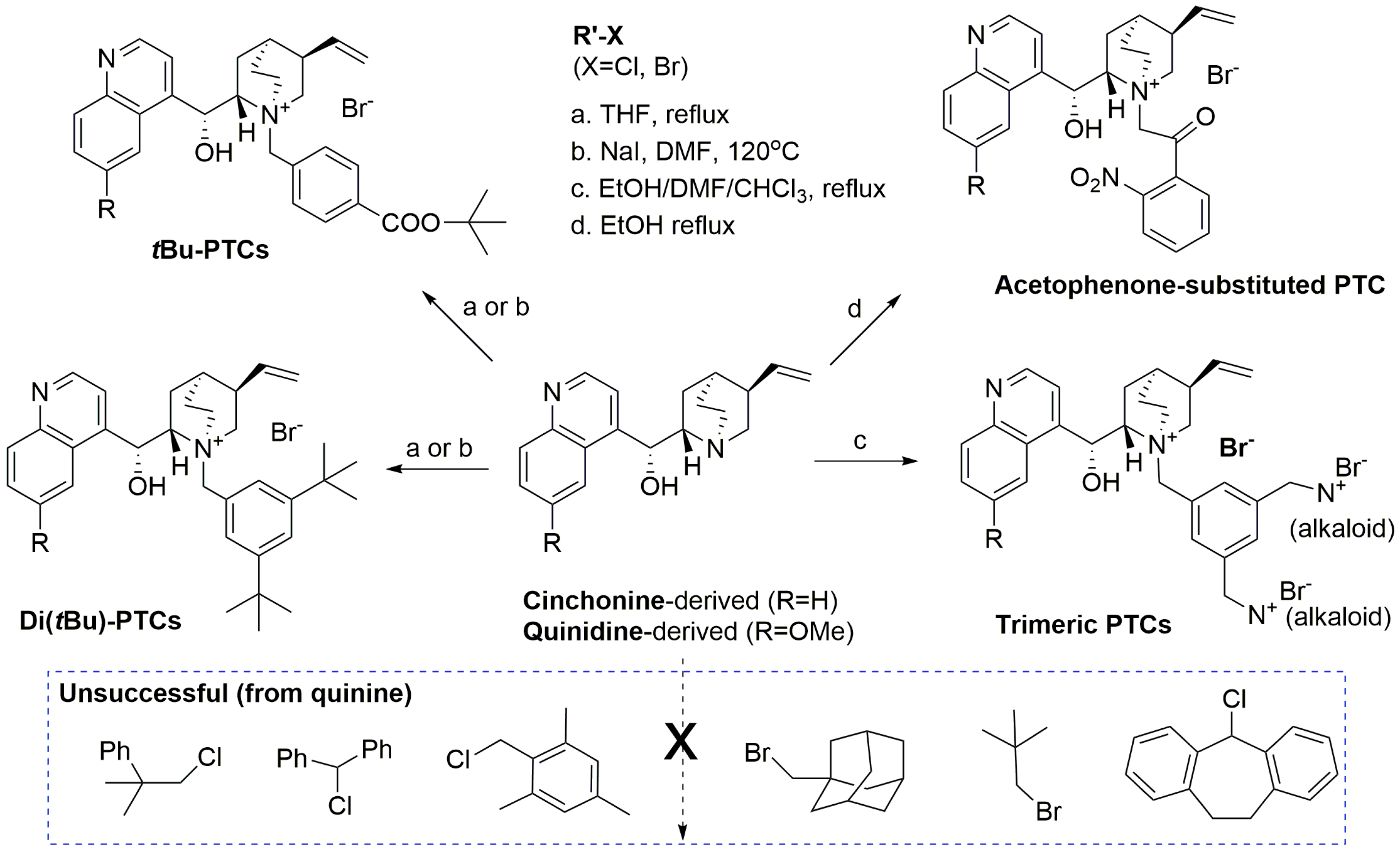
As a result, preparation of super-bulky PTCs was planned for further investigation (
Scheme 5). The work commenced with quinine, followed by cinchonidine. Disappointingly, many of the alkylating agents bearing α-branched or neopentylic groups were very unreactive under both the standard conditions employed [refluxing tetrahydrofuran (THF)) and under more forcing conditions[catalytic sodium iodide, dimethylformamide (DMF), 120 °C, over 48 h]. A few successful reactions were conducted to afford a small set of bulkier PTCs including two literature PTCs, a trimeric PTC made using 1,3,5-tris-(bromomethyl)benzene [
32] and acetophenone-substituted PTCs made using 2-bromo-2′-nitroacetophenone [
33]. These latter PTCs may form a stabilised nitrogen ylide as the active base, given the acidity of the protons of the acetophenone substituent. Whether this would have any effect, however, was unclear.
In parallel, since the catalysts were typically formed from the alkaloid and a benzylic halide derivative, several novel catalysts were made from the reaction mixture of TBBMB alkylating agent and the parent alkaloid. All new catalysts were tested (Entries 4 in
Table 2), however, none of these catalysts performed better than the existing ones, although Entries 4U and 4Z were comparable. Overall, few conclusions can be drawn from these results. Interestingly, the catalyst formed in situ using cinchonidine proved moderately selective (Entry 4Z), despite the absence of a bulky substituent in the 2- or 3-position of the aromatic ring. While not quite as selective as others, the ease of use (adding only a cheap alkaloid to the reaction mixture) was certainly attractive.
Chiral PTC-promoted alkylations using the analogous (Me)-ester alkylating agent, such as MBMB, were subsequently investigated. The results in Entries 2D and 2K using weaker carbonate bases suggested (Me)-ester alkylating agent might be compatible with these milder PTC conditions. A small set of reactions using
N-(2-NO
2-Bn)-quinidinium bromide (K
2CO
3 as base) were explored. In parallel, the benzylic chloride electrophile was also tested. MBMB was sufficiently stable under these reaction conditions to afford the desired (Me)-ester
4. However, as expected, the reaction was slow even at 60 °C and after heating for 3 h it reached only ~35% (area) conversion. The measured e.e. after extended heating (~14 h, 60 °C) was 18% of major enantiomer with
R configuration (compared with 30% for the
t-Bu analogue under the same conditions). Meanwhile, under the same conditions, the reaction with analogous methyl(4-chloromethyl)benzoate was too slow to be explored further, giving only a trace of product. Prolonged heating resulted in the generation of significant impurities. Two reactions, 5K & 5R in
Table 2, were set up on 5 mmol scale using the same catalysts as in Entries 3K and 3R, but with (Me)-ester alkylating agent instead. Despite both reactions being slow, they eventually reached >95% conversion of ketone
3 after ~2 days, with Entry 5R completing first. The e.e. measured was 38% after purification, with the later-eluting (
S)-enantiomer being the major, as confirmed by chiral HPLC analysis against (
S)-
4 reference standard. A portion of (
rac)-
4 was carried through the diastereoselective reduction using TiBA with desired (
S)-diastereoisomer (87%) being predominant. Subsequent hydrolysis afforded a mixture consisting primarily of desired diastereoisomer [(
S,
S)-
7 at 6.2 min & (
R,
R)-
7 at 5.9 min] as confirmed by chiral HPLC against the reference standards. This therefore demonstrated that the choice of PTC should be cinchonidine or quinine derived.
Thus far, the catalysts with a free -OH group or substituted with an allyl group have been described. However, it was quite possible that the -OH group of unsubstituted PTCs could be functionalised during the reaction by the reactive alkylating agent used. This could lead to either a more selective or a less selective catalyst, or perhaps both could operate in tandem. At least one literature study has demonstrated facile
O-alkylation of a cinchona alkaloid-derived PTC under similar conditions [
34]. Despite this, most authors continue to describe the catalyst as the -OH form. Indeed a recent paper [
35] included a mechanistic explanation based upon H-bonding with this group, although this explanation seemed to go against the general consensus that it was the cationic N
+ that interacts with the enolate [
36].
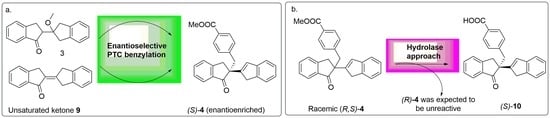
2.3. PTC-Promoted Alkylation of Unsaturated Ketone 9
Chiral analysis of the achiral PTC reaction of ketone
3 with TBBMB proved complex due to the presence of impurities eluting close to the peaks of interest (this was not a problem when analysing the analogous reactions using MBMB, whose products eluted later on the chiral column). It was thought the formation of the unsaturated ketone
9 under basic conditions could account for one of the side-products observed. As a result, it was decided to expand the investigation to the ketone
9 and PTC alkylation of such ketone. Unsaturated ketone
9 was made in moderate yield 73% according to the published method [
37], using ketone
3 in a mixture of methanol (MeOH)/DCM with catalytic trifluoromethanesulfonic acid under reflux conditions. The synthesis of ketone
9 had a further, perhaps more important driver. Previous reactions gave only suggestions as to whether the enolate that reacts with the alkylating agent in PTC reactions was derived from ketone
3, or if unsaturated ketone
9 was formed in situ and was then deprotonated (
Scheme 6).
In fact, either or both routes were possible and it would be challenging to distinguish between these unless intermediates could be isolated. Indeed, the degree to which either route operates could also depend heavily upon catalyst structure, which could be one reason why it was difficult to identify clear trends in selectivity as a function of catalyst structure. Since unsaturated ketone
9 would form an extended and near-planar, highly conjugated enolate while ketone
3 would not, substrate-catalyst interactions could be quite different for each of the two pathways. Alkylation of ketone
9 was tested using TBBMB. The HPLC profiles showed similar results to those of ketone
3, but with slightly fewer impurities (
Table 3). Most importantly, a clear improvement was shown with at least 3 out of 4 catalysts when compared to the reactions of ketone
3 under the same conditions.
In each case, HPLC analysis was performed after 3 h and 21 h showing very little reaction progress after the initial 3 h. The level of both starting materials in Entries 6K, 6R and 6Z were <10% (by area), but the level in Entry 6D was higher (15–20% by area); dissolution of unsaturated ketone 10 also took noticeably longer (>2 h) in this reaction. This suggested that the reaction stalled before completion, possibly due to catalyst decomposition, derivatisation or a change in solubility profile. As with other reactions, precipitation of solids was observed, but these were not further identified.
In the original control reaction with ketone
3 alone under achiral phase transfer conditions, much of the starting material remained following overnight reaction, while the achiral PTC alkylation of ketone
3 was rapid [
10]. This demonstrated that the conversion from ketone
3 to unsaturated ketone
9 under the reaction conditions was not facile, or that the elimination was readily reversible. The results suggested that alkylation takes place via
3-enolate or through both pathways. In contrast, only a single pathway is possible with unsaturated ketone
9. These results suggested that higher selectivity might be possible with this new substrate, while the absence of any ambiguity regarding the reaction pathway could make optimisation more straightforward. The substrate may also be more reactive (it should have a lower pKa than ketone
3 due to the extended conjugation, which could be important if deprotonation is rate-limiting), giving a wider scope for variation of base and temperature for future screening. In addition, it is likely that slight modifications to the synthetic method used to prepare ketone
3 could be made to afford this unsaturated ketone
9. Given the lower solubility of
9 compared to
3, product isolation might even be made easier, or isolated yields higher.
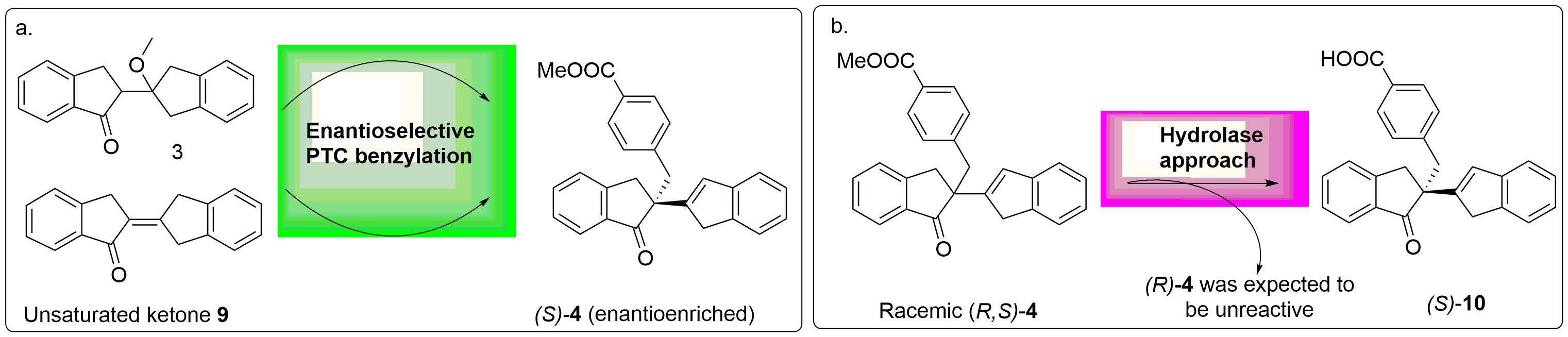
2.4. Hydrolase Screening
Enzyme catalysed kinetic bio-resolution is largely used in organic and medicinal chemistry, especially in the highly controlled enantiomeric synthesis of chiral carboxylic acids [
38,
39,
40,
41,
42]. Due to the chirality of the active site of the enzyme, one enantiomer fits better than its counterpart and therefore converts at a higher rate. As a result, a kinetic resolution of the racemate is achieved [
43,
44]. For high selectivity, the large difference in the reaction rates of the individual enantiomers should be achieved [
45]. However, in many cases, the resolution does not show such differences in rates. In 1982, C. J. Sih [
46] introduced a useful treatment of the kinetics of enzymatic resolutions, describing the dependency of the enantiomeric excess of substrate (eeS) and product (eeP) and the reaction conversion based on a theoretical basis laid by Sharpless [
47] and Fajans [
48]. Enantiomeric ratio (E), a selectivity parameter of a resolution, was introduced, which remains constant throughout the reaction and is only determined by the environment of the system. This method was further developed in the 1990s [
49,
50]. The relationship between the selectivity of a reaction (E value) and the optical purity of both substrate (eeS) and product (eeP) was expressed in the following equation:
The expected optical purity of a substrate can be calculated for a chosen point of conversion and the E value can be determined as a convenient constant value for the “selectivity” of the resolution. It has well been accepted that E-values of <8 is not a useful resolution; ~8–30 is regarded to yield e.e. from moderate to good; >30–100 is regarded to yield e.e. from good to excellent; >100 is regarded to yield excellent e.e. of both enantiomers [
51].
(
Rac)-
4 was screened against a panel of 48 commercial hydrolase enzymes in the presence of two different organic cosolvents, since organic cosolvents strongly influence the activity and/or selectivity of many enzymes for a given substrate. Hydrolase screening using DMSO as cosolvent resulted in four positive hits. AH-06 and AH-24 exhibited only trace levels of conversion and no discernible e.e. was observed in either case. AH-09 and AH-46 however showed some selectivity for the desired transformation (
Table 4).
No positive hits were observed for the screen using 2-MeTHF as cosolvent. In an effort to improve upon the modest enantio-selectivity observed with DMSO, a further extensive solvent screen was carried out on (
rac)-4 with the most promising enzymes: AH-09, AH-24 and AH-46. Eighteen cosolvent systems were investigated in total, including hexane, pentane, toluene, DMSO, DMF, diethyl ether, THF, dioxane, 2-MeTHF, methyl
tert-butyl ether (MtBE), DCM, chloroform, ethyl acetate, acetonitrile, ethanol, Isopropanol (IPA), butanol and a reaction with no organic cosolvent present. Some representative results of these screens after 72 h reaction time are outlined in
Table 5.
Significant improvement was achieved in Entries 18 and 19, where MtBE as cosolvent gave the best result with a selectivity factor of 8.4 (
Figure 1). As AH-46 hydrolyses (
R)-
4 preferentially over its enantiomer (
Table 5), it is possible to isolate (
S)-
4 with an e.e. of 95% in approximately 35% yield by driving the reaction to 68% conversion as shown in
Scheme 2. It was believed that further optimisation screening reaction could be performed on the bio-resolution reaction of (
rac)-
4 using AH-46 with MtBE as cosolvent to further polish the E value, which includes a number of parameters, namely, temperature, concentration, pH, % cosolvent loading, % enzyme loading, % substrate loading, salt additives and the organic cosolvent:aqueous buffer ratio. These investigations are currently ongoing.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}