Molybdenum-Catalyzed Enantioselective Sulfoxidation Controlled by a Nonclassical Hydrogen Bond between Coordinated Chiral Imidazolium-Based Dicarboxylate and Peroxido Ligands



Abstract
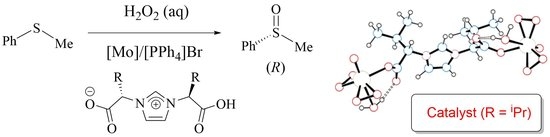
:
1. Introduction
2. Results and Discussion
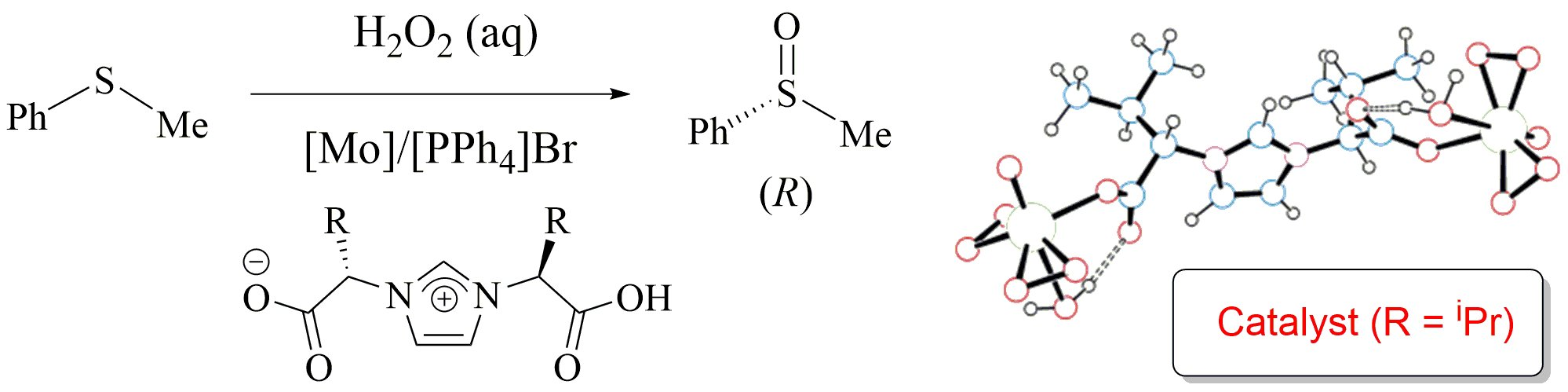
2.1. Enantioselective Oxidation of Different Sulfides with Aqueous Hydrogen Peroxide Catalyzed by the System [Mo(O)(O2)2(H2O)n]/HLR/[PPh4]Br
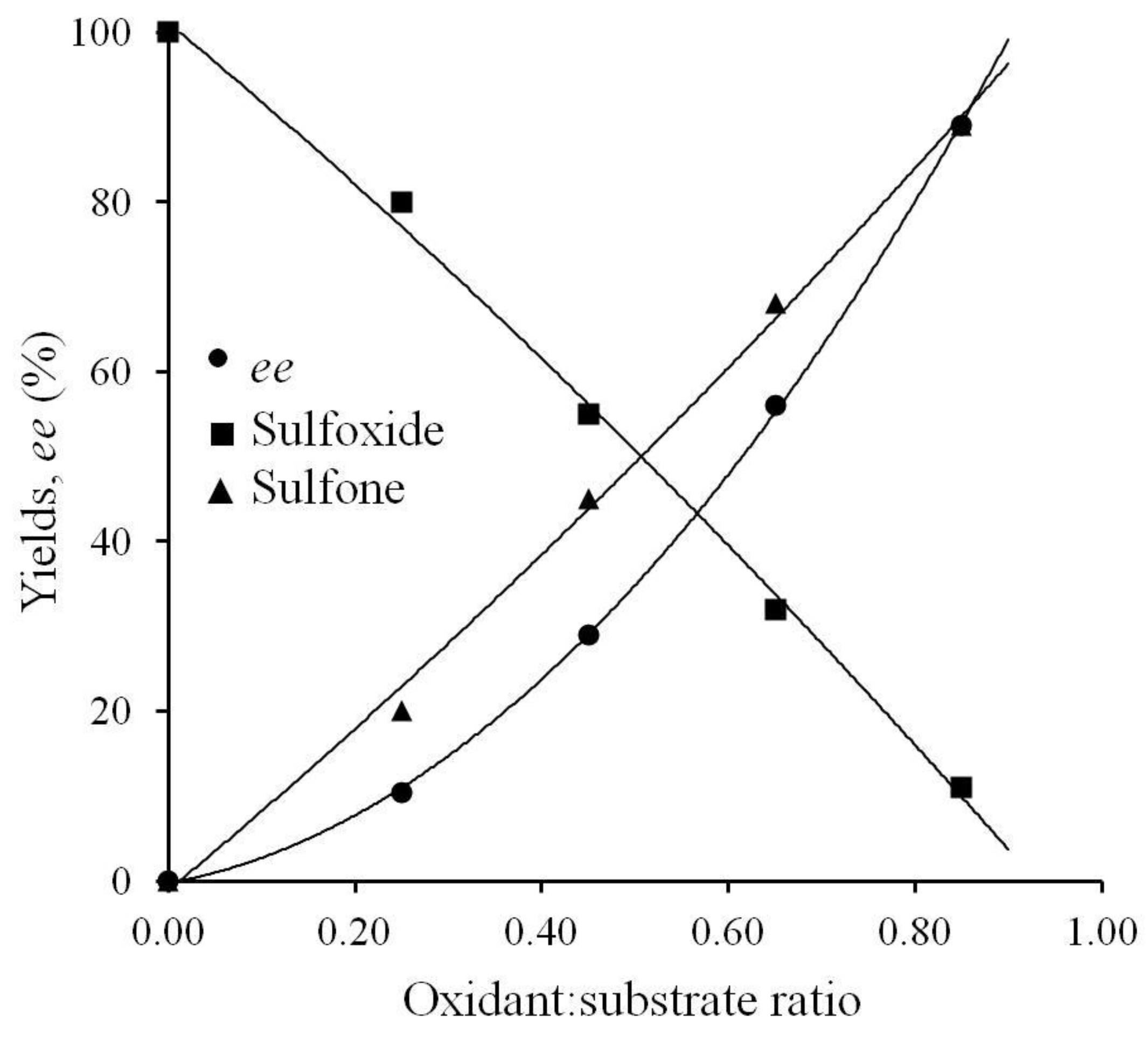
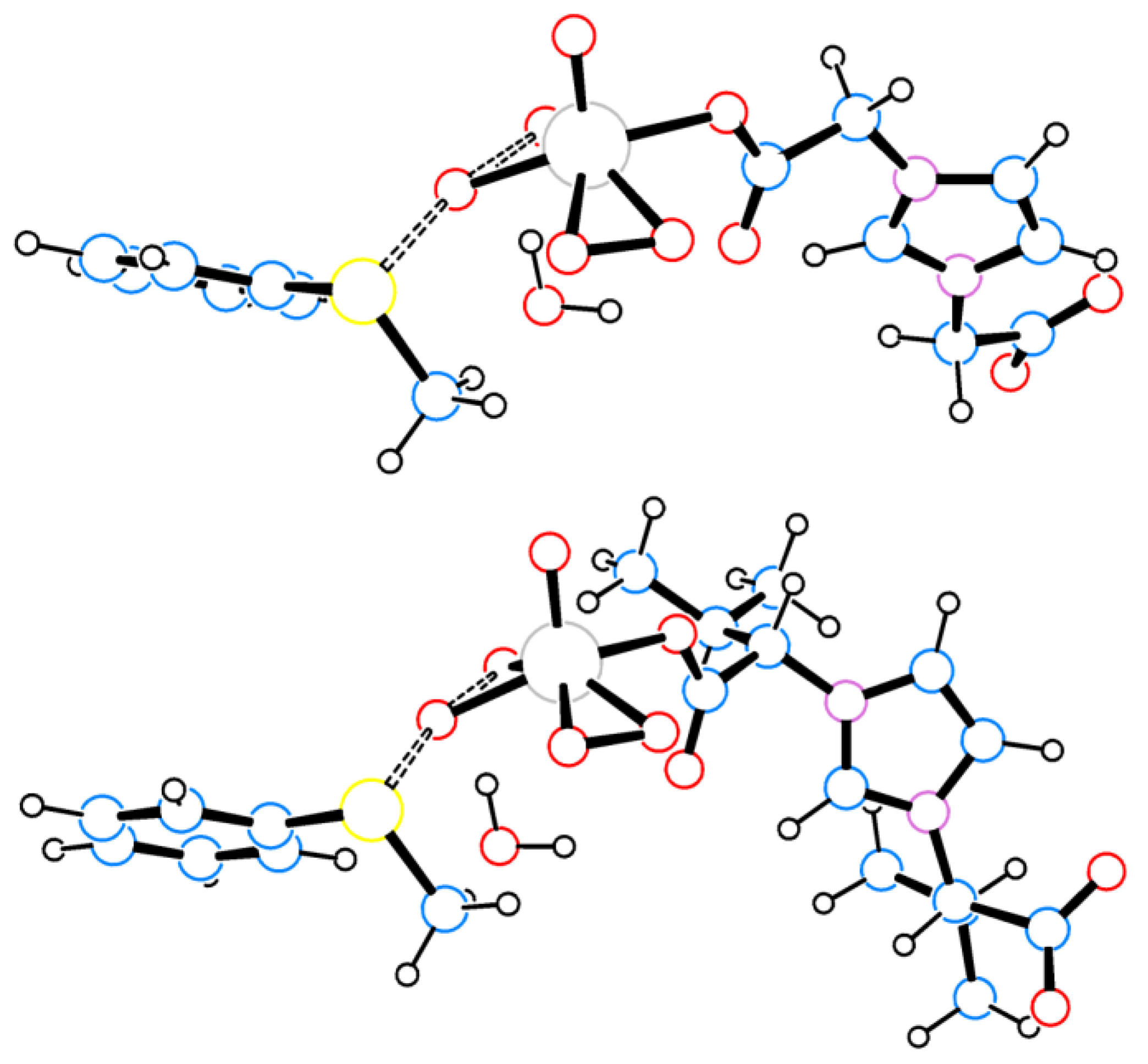
2.2. Nature of the Molybdenum Catalyst and Origin of the Enantioselectivity
3. Materials and Methods
3.1. General
3.2. Synthesis of Chiral Imidazolium-Based Zwitterionic Dicarboxylic Acids HLR
3.3. Preparation and Titration of [Mo(O)(O2)2(H2O)n] Solution
3.4. Synthesis of Complex Na{[Mo(O)(O2)2(H2O)]2(μ-LiPr)}
3.5. General Procedure for Enantioselective Mo-Catalyzed Oxidation of Sulfides in the Presence of HLR
3.6. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patai, S.; Rappoport, Z. (Eds.) Syntheses of Sulphones, Sulphoxides and Cyclic Sulphides; John Wiley & Sons, Ltd.: Chichester, UK, 1995; ISBN 9780470666357. [Google Scholar]
- Wojaczyńska, E.; Wojaczyński, J.; Wojaczyńska, E. Enantioselective Synthesis of Sulfoxides: 2000–2009. Chem. Rev. 2010, 110, 4303–4356. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, G.E.; Ford, A.; Maguire, A.R. Asymmetric oxidation of sulfides. J. Sulfur Chem. 2012, 34, 301–341. [Google Scholar] [CrossRef] [Green Version]
- Kagan, H.B. Asymmetric Synthesis of Chiral Sulfoxides. In Organosulfur Chemistry in Asymmetric Synthesis; Toru, T., Bolm, C., Eds.; Wiley: Weinheim, Germany, 2008; pp. 1–30. [Google Scholar]
- Bentley, R. Role of sulfur chirality in the chemical processes of biology. Chem. Soc. Rev. 2005, 34, 609–624. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, G.E.; Kelly, P.; Lawrence, S.E.; Maguire, A.R. Synthesis of enantioenriched sulfoxides. Arkivoc 2011, 2011, 1–110. [Google Scholar] [CrossRef]
- Fernández, I.; Khiar, N. Recent developments in the synthesis and utilization of chiral sulfoxides. Chem. Rev. 2003, 103, 3651–3705. [Google Scholar] [CrossRef] [PubMed]
- Otocka, S.; Kwiatkowska, M.; Madalińska, L.; Kiełbasiński, P. Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef] [PubMed]
- Srour, H.; Le Maux, P.; Chevance, S.; Simonneaux, G. Metal-catalyzed asymmetric sulfoxidation, epoxidation and hydroxylation by hydrogen peroxide. Coord. Chem. Rev. 2013, 257, 3030–3050. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Li, J.; Chen, B.; Li, G.; Lv, Y.; Wang, L.; Gao, S. Asymmetric oxidation catalysis by a porphyrin-inspired manganese complex: Highly enantioselective sulfoxidation with a wide substrate scope. Org. Lett. 2013, 15, 5658–5661. [Google Scholar] [CrossRef] [PubMed]
- Srour, H.; Jalkh, J.; Le Maux, P.; Chevance, S.; Kobeissi, M.; Simonneaux, G. Asymmetric oxidation of sulfides by hydrogen peroxide catalyzed by chiral manganese porphyrins in water/methanol solution. J. Mol. Catal. A Chem. 2013, 370, 75–79. [Google Scholar] [CrossRef]
- Legros, J.; Bolm, C. Iron-Catalyzed Asymmetric Sulfide Oxidation with Aqueous Hydrogen Peroxide. Angew. Chem. Int. Ed. 2003, 42, 5487–5489. [Google Scholar] [CrossRef] [PubMed]
- Legros, J.; Bolm, C. Highly enantioselective iron-catalyzed sulfide oxidation with aqueous hydrogen peroxide under simple reaction conditions. Angew. Chem. Int. Ed. 2004, 43, 4225–4228. [Google Scholar] [CrossRef] [PubMed]
- Legros, J.; Bolm, C. Investigations on the iron-catalyzed asymmetric sulfide oxidation. Chem. Eur. J. 2005, 11, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Katsuki, T. Fe(salan)-Catalyzed Oxidation of Sulfides with Hydrogen Peroxide in Water. J. Am. Chem. Soc. 2007, 129, 8940–8941. [Google Scholar] [CrossRef] [PubMed]
- Buckley, B.R.; Neary, S.P. Organocatalysed Asymmetric Oxidation Reactions. In Enantioselective Organocatalyzed Reactions I; Mahrwald, R., Ed.; Springer: Berlin, Germany, 2011; pp. 1–41. [Google Scholar]
- Basak, A.; Barlan, A.U.; Yamamoto, H. Catalytic enantioselective oxidation of sulfides and disulfides by a chiral complex of bis-hydroxamic acid and molybdenum. Tetrahedron Asymmetry 2006, 17, 508–511. [Google Scholar] [CrossRef]
- Pedrosa, M.R.; Escribano, J.; Aguado, R.; Sanz, R.; Díez, V.; Arnáiz, F.J. Addition compounds of MoO2Cl2 with chiral sulfoxides. First molecular structures of dioxomolybdenum complexes bearing chiral non-racemic sulfoxide as ligand. Inorg. Chim. Acta 2010, 363, 3158–3164. [Google Scholar] [CrossRef]
- Sakuraba, H.; Maekawa, H. Enantioselective oxidation of sulfides catalyzed by chiral MoV and CuII complexes of catechol-appendedβ-cyclodextrin derivatives in water. J. Incl. Phenom. 2006, 54, 41–45. [Google Scholar] [CrossRef]
- Amini, M.; Haghdoost, M.M.; Bagherzadeh, M. Oxido-peroxido molybdenum(VI) complexes in catalytic and stoichiometric oxidations. Coord. Chem. Rev. 2013, 257, 1093–1121. [Google Scholar] [CrossRef]
- Barlan, A.U.; Zhang, W.; Yamamoto, H. Development and application of versatile bis-hydroxamic acids for catalytic asymmetric oxidation. Tetrahedron 2007, 63, 6075–6087. [Google Scholar] [CrossRef] [PubMed]
- Bellemin-Laponnaz, S.; Coleman, K.S.; Osborn, J.A. Co-ordination of the chiral N,O-ligand 2-[(1S,2S,5R)(−)-menthol]-pyridine to molybdenum(VI) and vanadium(IV) oxo complexes. Polyhedron 1999, 18, 2533–2536. [Google Scholar] [CrossRef]
- Bonchio, M.; Carofiglio, T.; Difuria, F.; Fornasier, R. Supramolecular catalysis: Enantioselective oxidation of thioanisole in water by hydrogen peroxide catalyzed by Mo(VI) in the presence of β-cyclodextrin-based ligands. J. Org. Chem. 1995, 60, 5986–5988. [Google Scholar] [CrossRef]
- Chakravarthy, R.D.; Suresh, K.; Ramkumar, V.; Chand, D.K. New chiral molybdenum complex catalyzed sulfide oxidation with hydrogen peroxide. Inorg. Chim. Acta 2011, 376, 57–63. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Wang, L.; Wang, Y.; Wang, X.; Sun, L. Asymmetric oxidation of sulfides with H2O2 catalyzed by titanium complexes of Schiff bases bearing a dicumenyl salicylidenyl unit. Appl. Organomet. Chem. 2011, 25, 325–330. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Talsi, E.P. Titanium-salan-catalyzed asymmetric oxidation of sulfides and kinetic resolution of sulfoxides with H2O2 as the oxidant. Eur. J. Org. Chem. 2008, 3369–3376. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Talsi, E.P. Asymmetric oxidation of sulfides with H2O2 catalyzed by titanium complexes with aminoalcohol derived Schiff bases. J. Mol. Catal. A Chem. 2007, 264, 280–287. [Google Scholar] [CrossRef]
- Brunel, J.M.; Diter, P.; Duetsch, M.; Kagan, H.B. Highly enantioselective oxidation of sulfides mediated by a chiral titanium complex. J. Org. Chem. 1995, 60, 8086–8088. [Google Scholar] [CrossRef]
- Choudary, B.M.; Shobha Rani, S.; Narender, N. Asymmetric oxidation of sulfides to sulfoxides by chiral titanium pillared montmorillonite catalyst. Catal. Lett. 1993, 19, 299–307. [Google Scholar] [CrossRef]
- Komatsu, N.; Hashizume, M.; Sugita, T.; Uemura, S. Catalytic asymmetric oxidation of sulfides to sulfoxides with tert-butyl hydroperoxide using binaphthol as a chiral auxiliary. J. Org. Chem. 1993, 58, 4529–4533. [Google Scholar] [CrossRef]
- Cotton, H.; Elebring, T.; Larsson, M.; Li, L.; Sörensen, H.; Von Unge, S. Asymmetric synthesis of esomeprazole. Tetrahedron Asymmetry 2000, 11, 3819–3825. [Google Scholar] [CrossRef]
- Bolm, C.; Bienewald, F. Asymmetric Sulfide Oxidation with Vanadium Catalysts and H2O2. Angew. Chem. Int. Ed. 1995, 34, 2640–2642. [Google Scholar] [CrossRef]
- Liu, G.; Cogan, D.A.; Ellman, J.A. Catalytic Asymmetric Synthesis of tert-Butanesulfinamide. Application to the Asymmetric Synthesis of Amines. J. Am. Chem. Soc. 1997, 119, 9913–9914. [Google Scholar] [CrossRef]
- Karpyshev, N.N.; Yakovleva, O.D.; Talsi, E.P.; Bryliakov, K.P.; Tolstikova, O.V.; Tolstikov, A.G. Effect of portionwise addition of oxidant in asymmetric vanadium-catalyzed sulfide oxidation. J. Mol. Catal. A Chem. 2000, 157, 91–95. [Google Scholar] [CrossRef]
- Blum, S.A.; Bergman, R.G.; Ellman, J.A. Enantioselective oxidation of di-tert-butyl disulfide with a vanadium catalyst: Progress toward mechanism elucidation. J. Org. Chem. 2003, 68, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Bolm, C. Vanadium-catalyzed asymmetric oxidations. Coord. Chem. Rev. 2003, 237, 245–256. [Google Scholar] [CrossRef]
- Zeng, Q.; Wang, H.; Wang, T.; Cai, Y.; Weng, W.; Zhao, Y. Vanadium-catalyzed enantioselective sulfoxidation and concomitant, highly efficient kinetic resolution provide high enantioselectivity and acceptable yields of sulfoxides. Adv. Synth. Catal. 2005, 347, 1933–1936. [Google Scholar] [CrossRef]
- Hinch, M.; Jacques, O.; Drago, C.; Caggiano, L.; Jackson, R.F.W.; Dexter, C.; Anson, M.S.; Macdonald, S.J.F. Effective asymmetric oxidation of enones and alkyl aryl sulfides. J. Mol. Catal. A Chem. 2006, 251, 123–128. [Google Scholar] [CrossRef]
- Adão, P.; Pessoa, J.C.; Henriques, R.T.; Kuznetsov, M.L.; Avecilla, F.; Maurya, M.R.; Kumar, U.; Correia, I. Synthesis, characterization, and application of vanadium-salan complexes in oxygen transfer reactions. Inorg. Chem. 2009, 48, 3542–3561. [Google Scholar] [CrossRef] [PubMed]
- Aydin, A.E. Synthesis of novelβ-amino alcohols and their application in the catalytic asymmetric sulfoxidation of sulfides. Tetrahedron Asymmetry 2013, 24, 444–448. [Google Scholar] [CrossRef]
- Carrasco, C.J.; Montilla, F.; Galindo, A. Molybdenum-catalyzed asymmetric sulfoxidation with hydrogen peroxide and subsequent kinetic resolution, using an imidazolium-based dicarboxylate compound as chiral inductor. Catal. Commun. 2016, 84, 134–136. [Google Scholar] [CrossRef]
- Carrasco, C.J.; Montilla, F.; Alvarez, E.; Mealli, C.; Manca, G.; Galindo, A. Experimental and theoretical insights into the oxodiperoxomolybdenum-catalysed sulphide oxidation using hydrogen peroxide in ionic liquids. Dalton Trans. 2014, 43, 13711–13730. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, C.J.; Montilla, F.; Bobadilla, L.; Ivanova, S.; Odriozola, J.A.; Galindo, A. Oxodiperoxomolybdenum complex immobilized onto ionic liquid modified SBA-15 as an effective catalysis for sulfide oxidation to sulfoxides using hydrogen peroxide. Catal. Today 2014, 255, 102–108. [Google Scholar] [CrossRef]
- Kühl, O.; Palm, G. Imidazolium salts from amino acids—A new route to chiral zwitterionic carbene precursors? Tetrahedron: Asymmetry 2010, 21, 393–397. [Google Scholar] [CrossRef]
- Kühl, O.; Millinghaus, S.; Wehage, P. Functionalised, chiral imidazolium compounds from proteinogenic amino acids. Cent. Eur. J. Chem. 2010, 8, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Keith, J.M.; Larrow, J.F.; Jacobsen, E.N. Practical considerations in kinetic resolution reactions. Adv. Synth. Catal. 2001, 343, 5–26. [Google Scholar] [CrossRef]
- Montilla, F.; Galindo, A. Oxidodiperoxidomolybdenum Complexes: Properties and Their Use as Catalysts in Green Oxidations. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: New York City, NY, USA, 2017; pp. 1–17. [Google Scholar]
- Taylor, R.; Kennard, O. Crystallographic evidence for the existence of CH…O, CH…N and CH…Cl hydrogen bonds. J. Am. Chem. Soc. 1982, 104, 5063–5070. [Google Scholar] [CrossRef]
- Steiner, T. C–H···O hydrogen bonding in crystals. Crystallogr. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Steiner, T.; Desiraju, G.R. Distinction between the weak hydrogen bond and the van der Waals interaction. Chem. Commun. 1998, 891–892. [Google Scholar] [CrossRef]
- Steiner, T. Effect of acceptor strength on C–H···O hydrogen bond lengths as revealed by and quantified from crystallographic data. J. Chem. Soc. Chem. Commun. 1994, 2341–2342. [Google Scholar] [CrossRef]
- Steiner, T. Weak hydrogen bonding. Part 1. Neutron diffraction data of amino acid Cα–H suggest lengthening of the covalent C–H bond in C–H···O interactions. J. Chem. Soc. Perkin Trans. 1995, 2, 1315–1319. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Role of C-H…O hydrogen bonds in the coordination of water molecules. Analysis of neutron diffraction data. J. Am. Chem. Soc. 1993, 115, 4540–4547. [Google Scholar] [CrossRef]
- Herbert, M.; Montilla, F.; Galindo, A. Olefin epoxidation in solventless conditions and apolar media catalysed by specialised oxodiperoxomolybdenum complexes. J. Mol. Catal. A Chem. 2011, 338, 111–120. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Plenum: New York, NY, USA, 1976; p. 1. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09 (Revision B.01); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | HLR | Sulfide | Conversion (%) b | Selectivity to Sulfoxide (%) b | Selectivity to Sulfone (%) b | Sulfoxide Yield (%) | Sulfoxide ee (%) and Configuration c |
|---|---|---|---|---|---|---|---|
| 1 | HLH, 1a | PhMeS | 93 | 95 | 5 | 88 | Racemic |
| 2 | (S,S)-HLMe, 1b | PhMeS | 93 | 95 | 5 | 88 | 2 (R) |
| 3 | (S,S)-HLiPr, 1c | PhMeS | 94 | 95 | 5 | 89 | 40 (R) |
| 4 | (R,R)-HLiPr, 1c’ | PhMeS | 95 | 95 | 5 | 90 | 42 (S) |
| 5 | (S,S)-HLCH2Ph, 1d | PhMeS | 67 | 100 | 0 | 67 | 5 (R) |
| 6 | (S,S)-HLiBu, 1e | PhMeS | 88 | 96 | 4 | 85 | 14 (R) |
| 7 | (S,S)-HLsec-Bu, 1f | PhMeS | 95 | 95 | 5 | 90 | 47 (R) |
| 8 | (S,S)-HLtBu, 1g | PhMeS | 92 | 96 | 4 | 88 | 32 (R) |
| 9 | (S,S)-HLsec-Bu, 1f | (p-Me-C6H4)MeS | 90 | 91 | 9 | 82 | 55 (R) |
| 10 | (S,S)-HLsec-Bu, 1f | (p-Cl-C6H4)MeS | 89 | 96 | 4 | 85 | 44 (R) |
| 11 | (S,S)-HLsec-Bu, 1f | (p-Br-C6H4)MeS | 91 | 87 | 13 | 79 | 51 (R) |
| 12 | (S,S)-HLsec-Bu, 1f | Ph(PhCH2)S | 90 | 64 | 36 | 58 | 53 (R) |
| 13 | (S,S)-HLsec-Bu, 1f | Ph(HOCH2CH2)S | 81 | 36 | 0 | 29 | 43 (S) |
| R (μ–LR)− | Distances, Å | Angles, ° | |||
|---|---|---|---|---|---|
| C–H···O | O–H···O | C–H···O | O–H···O | ||
| H | 2a | - | 1.814, 1.822 | - | 158 |
| Me | 2b | >4 | 1.814, 1.831 | - | 158, 159 |
| iPr | 2c | 2.509, 2.521 | 1.807, 1.817 | 168 | 159, 160 |
| CH2Ph | 2d | 2.352 (C–Harom.), 3.570 | 1.803, 1.815 | 138 (C–Harom.), 111 | 160 |
| iBu | 2e | 2.552, 2.558 | 1.796, 1.811 | 170, 171 | 160 |
| secBu | 2f | 2.621, 2.656 | 1.793, 1.803 | 160 | 160, 161 |
| tBu | 2g | 2.509, 2.521 | 1.807, 1.817 | 166 | 160 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco, C.J.; Montilla, F.; Galindo, A. Molybdenum-Catalyzed Enantioselective Sulfoxidation Controlled by a Nonclassical Hydrogen Bond between Coordinated Chiral Imidazolium-Based Dicarboxylate and Peroxido Ligands. Molecules 2018, 23, 1595. https://doi.org/10.3390/molecules23071595
Carrasco CJ, Montilla F, Galindo A. Molybdenum-Catalyzed Enantioselective Sulfoxidation Controlled by a Nonclassical Hydrogen Bond between Coordinated Chiral Imidazolium-Based Dicarboxylate and Peroxido Ligands. Molecules. 2018; 23(7):1595. https://doi.org/10.3390/molecules23071595
Chicago/Turabian StyleCarrasco, Carlos J., Francisco Montilla, and Agustín Galindo. 2018. "Molybdenum-Catalyzed Enantioselective Sulfoxidation Controlled by a Nonclassical Hydrogen Bond between Coordinated Chiral Imidazolium-Based Dicarboxylate and Peroxido Ligands" Molecules 23, no. 7: 1595. https://doi.org/10.3390/molecules23071595
APA StyleCarrasco, C. J., Montilla, F., & Galindo, A. (2018). Molybdenum-Catalyzed Enantioselective Sulfoxidation Controlled by a Nonclassical Hydrogen Bond between Coordinated Chiral Imidazolium-Based Dicarboxylate and Peroxido Ligands. Molecules, 23(7), 1595. https://doi.org/10.3390/molecules23071595