Development of Thiophene Compounds as Potent Chemotherapies for the Treatment of Cutaneous Leishmaniasis Caused by Leishmania major

 ,
, 

Abstract
:1. Introduction
2. Results
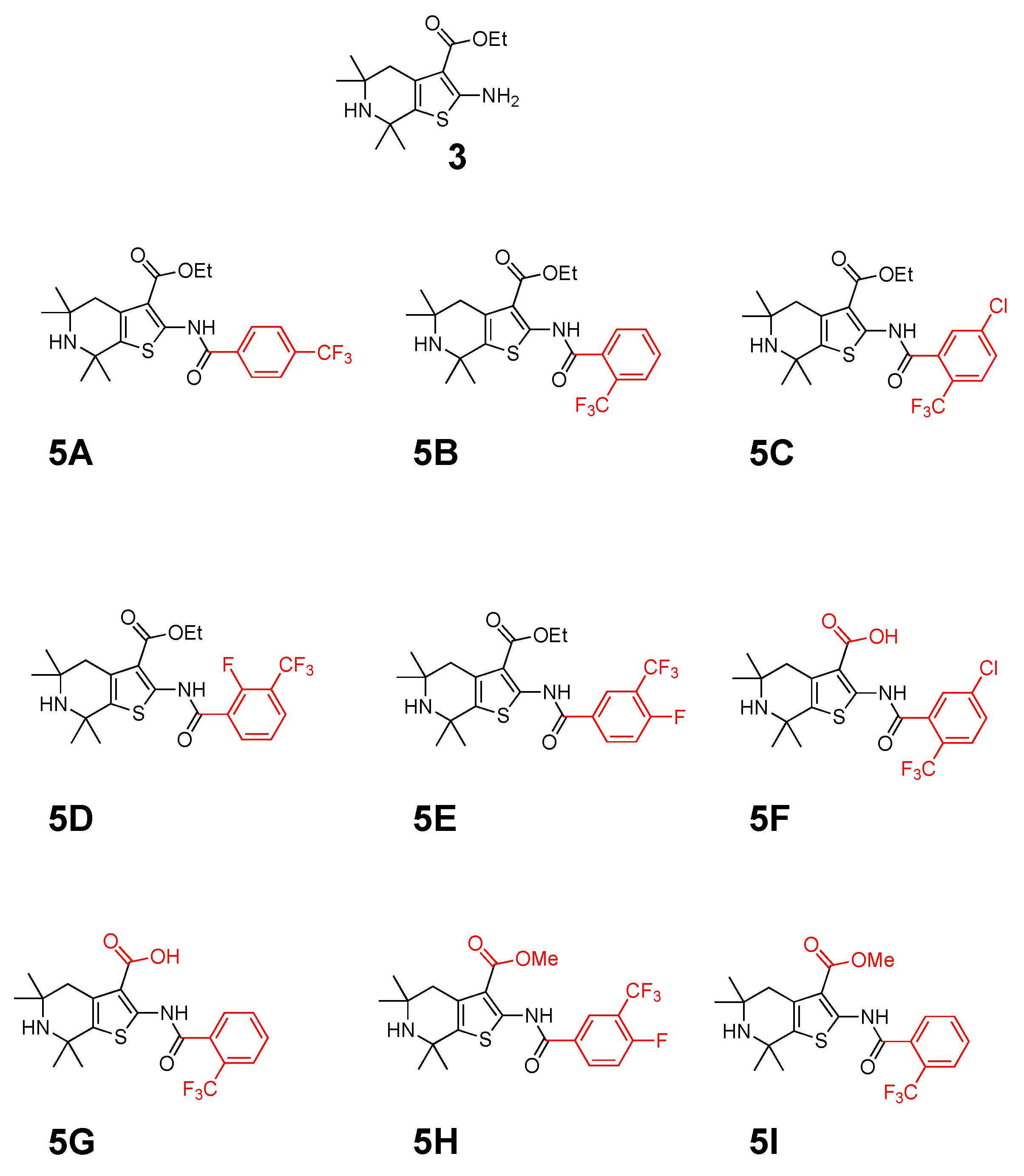
2.1. Synthetic Chemistry
2.2. Efficacy and Cytotoxicity of Parent Compound 5A
2.3. In Vitro Anti-Leishmanial Activity of Thiophene Derivatives and Their Cytotoxicity
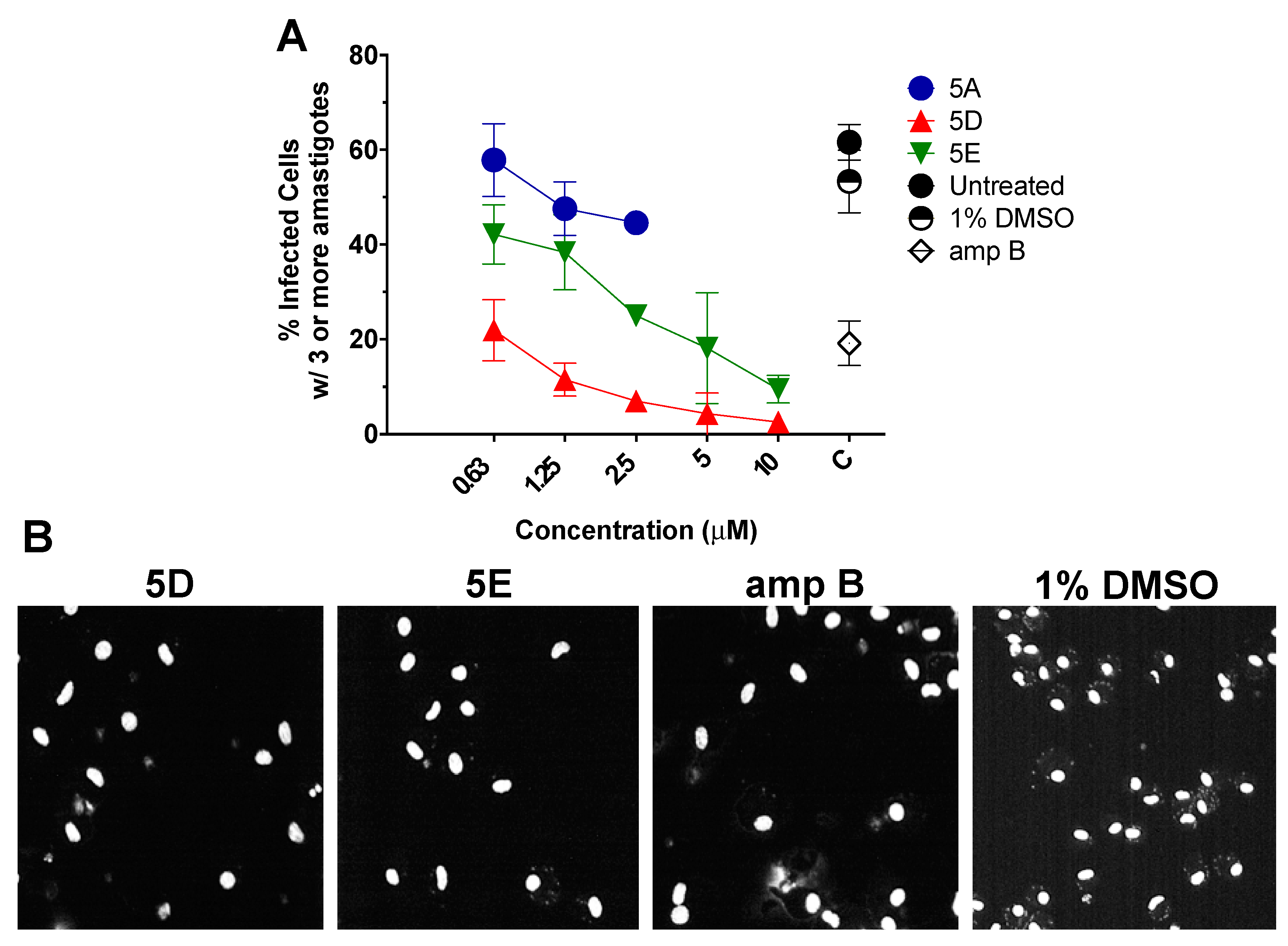
2.4. In Vitro Efficacy of Thiophene 5D Against Intracellular Amastigotes
2.5. Molecule 5D Induces ROS in L. major
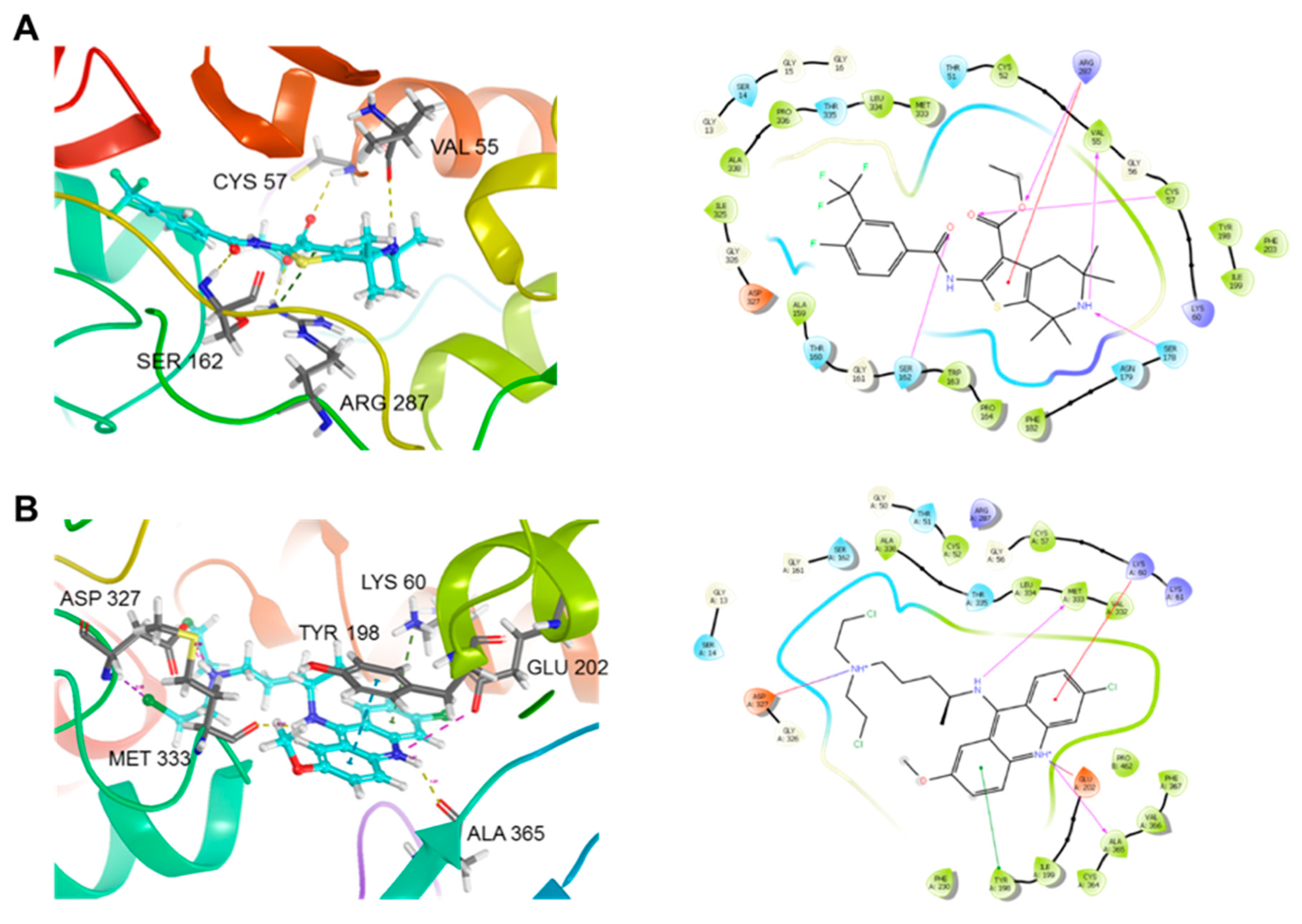
2.6. Docking of 5D on TryR from Leishmania
3. Discussion
4. Materials and Methods
4.1. General
4.2. Chemical Synthesis
4.2.1. General Synthetic Procedure 1
Synthesis of the Ethyl 5,5,7,7-tetramethyl-2-(4-(trifluoromethyl) benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5A)
4.2.2. General Synthetic Procedure 2
Synthesis of 5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno [2,3-c]pyridine-3-carboxylic acid (5G)
Ethyl-5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5B)
Ethyl-5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5C)
Ethyl-2-(2-fluoro-3-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]-pyridine-3-carboxylate (5D)
Ethyl-2-(4-fluoro-3-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]-pyridine-3-carboxylate (5E)
Methyl-2-(4-fluoro-3-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]-pyridine3-carboxylate (5H)
Methyl-5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5I)
2-(5-chloro-2-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylic acid (5F)
5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylic acid (5G)
4.3. Cell Maintenance
4.4. Luciferase Assay—Viability of Leishmania Major promastigotes
4.5. Assessment of Thiophene Compound Mammalian Cell Cytotoxicity
4.6. High-Content Imaging Assay—Proliferation Experiments
4.7. Measurement of Reactive Oxygen Species Levels
4.8. Docking Studies—Pre-Docking Preparation
4.9. Binding Site Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Aoun, K.; Bouratbine, A. Cutaneous leishmaniasis in North Africa: A review. Parasite 2014, 21, 14. [Google Scholar] [CrossRef] [PubMed]
- Cappai, R.; Morris, L.; Aebischer, T.; Bacic, A.; Curtis, J.M.; Kelleher, M.; McLeod, K.S.; Moody, S.F.; Osborn, A.H.; Handman, E. Ricin-resistant mutants of Leishmania major which express modified lipophosphoglycan remain infective for mice. Parasitology 1994, 108, 397–405. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis-Fact Sheet N’375. 2014. Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 9 May 2016).
- McGwire, B.S.; Satoskar, A.R. Leishmaniasis: Clinical syndromes and treatment. QJM 2014, 107, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, K.A.; Dias, C.N.; Néris, P.L.; Rocha Jda, C.; Scotti, M.T.; Scotti, L.; Mascarenhas, S.R.; Veras, R.C.; de Medeiros, I.A.; Keesen Tde, S.; et al. 2-Amino-thiophene derivatives present antileishmanial activity mediated by apoptosis and immunomodulation in vitro. Eur. J. Med. Chem. 2015, 106, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Coombs, G.H. Leishmaniasis--current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003, 19, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.C.; Morais-Teixeira, E.; Reis, P.G.; Silva-Barcellos, N.M.; Salaun, P.; Campos, P.P.; Dias Correa-Junior, J.; Rabello, A.; Demicheli, C.; Frezard, F. Hepatotoxicity of pentavalent antimonial drug: Possible role of residual Sb(III) and protective effect of ascorbic acid. Antimicrob. Agents Chemother. 2014, 58, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborin, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.G.; Vacchina, P.; Robles-Murguia, M.; Wadsworth, M.; McDowell, M.A.; Morales, M.A. Fitness and Phenotypic Characterization of Miltefosine-Resistant Leishmania major. PLoS Negl. Trop. Dis. 2015, 9, e0003948. [Google Scholar] [CrossRef] [PubMed]
- Iniguez, E.A.; Perez, A.; Maldonado, R.A.; Skouta, R. Novel arylalkylamine compounds exhibits potent selective antiparasitic activity against Leishmania major. Bioorg. Med. Chem. Lett. 2015, 25, 5315–5320. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.M.R.A. Preparation of Tetramethyltetrahydrothienopyridine Derivatives for Use as Parasiticides. U.S. Patent Appl. Publ 20160362420 A1, 15 December 2016. [Google Scholar]
- Keri, R.S.; Chand, K.; Budagumpi, S.; Balappa Somappa, S.; Patil, S.A.; Nagaraja, B.M. An overview of benzo[b]thiophene-based medicinal chemistry. Eur. J. Med. Chem. 2017, 138, 1002–1033. [Google Scholar] [CrossRef] [PubMed]
- Sensfuss, U.; Habicher, W.D. 2-aminothiophenes from triacetonamine: A convenient way to novel sterically hindered piperidine derivatives. Heteroat. Chem. 1998, 9, 529–536. [Google Scholar] [CrossRef]
- Thalhofer, C.J.; Graff, J.W; Love-Homan, L.; Hickerson, S.M.; Craft, N.; Beverley, S.M.; Wilson, M.E. In vivo imaging of transgenic Leishmania parasites in a live host. J. Vis. Exp. 2010, 1980. [Google Scholar] [CrossRef]
- Capul, A.A.; Barron, T.; Dobson, D.E.; Turco, S.J.; Beverley, S.M. Two functionally divergent UDP-Gal nucleotide sugar transporters participate in phosphoglycan synthesis in Leishmania major. J. Biol. Chem. 2007, 282, 14006–14017. [Google Scholar] [CrossRef] [PubMed]
- Lara, D.; Feng, Y.; Bader, J.; Savage, P.B.; Maldonado, R.A. Anti-trypanosomatid activity of ceragenins. J. Parasitol. 2010, 96, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.N.; Myburgh, E.; Gao, M.-Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Suite 2018-1 Induced Fit Docking Protocol. Available online: https://www.schrodinger.com/induced-fit (accessed on 10 February 2018).
- Clark, A.J.; Tiwary, P.; Borrelli, K.; Feng, S.; Miller, E.B.; Abel, R.; Friesner, R.A.; Berne, B.J. Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J. Chem. Theory Comput. 2016, 12, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.C.; Kelly, J.M.; Chapman, C.J.; Fairlamb, A.H.; Miles, M.A. The structure, organization, and expression of the Leishmania donovani gene encoding trypanothione reductase. Mol. Biochem. Parasitol. 1994, 64, 293–301. [Google Scholar] [CrossRef]
- Tovar, J.; Wilkinson, S.; Mottram, J.C.; Fairlamb, A.H. Evidence that trypanothione reductase is an essential enzyme in Leishmania by targeted replacement of the tryA gene locus. Mol. Microbiol. 1998, 29, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.O. Trypanothione reductase: A viable chemotherapeutic target for antitrypanosomal and antileishmanial drug design. Drug Target Insights 2007, 2, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Feasey, N.; Wansbrough-Jones, M.; Mabey, D.C.; Solomon, A.W. Neglected tropical diseases. Br. Med. Bull. 2010, 93, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Haidle, A.M.; Zabierek, A.A.; Childers, K.K.; Rosenstein, C.; Mathur, A.; Altman, M.D.; Chan, G.; Xu, L.; Bachman, E.; Mo, J.R.; et al. Thiophene carboxamide inhibitors of JAK2 as potential treatments for myleoproliferative neoplasms. Bioorg. Med. Chem. Lett. 2014, 24, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, H.; Yang, S.; Chreifi, G.; Martasek, P.; Roman, L.J.; Meyskens, F.L.; Poulos, T.L.; Silverman, R.B. Potent and selective double-headed thiophene-2-carboximidamide inhibitors of neuronal nitric oxide synthase for the treatment of melanoma. J. Med. Chem. 2014, 57, 686–700. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, J.F.; da Silva, A.L.; Vendramini-Costa, D.B.; da Cruz Amorim, C.A.; Campos, J.F.; Ribeiro, A.G.; de Moura, R.O.; Neves, J.L.; Ruiz, A.L.T.G.; de Carvalho, J.E.; et al. Synthesis of thiophene-thiosemicarbazone derivatives and evaluation of their in vitro and in vivo antitumor activities. Eur. J. Med. Chem. 2015, 104, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Marchais-Oberwinkler, S.; Xu, K.; Wetzel, M.; Perspicace, E.; Negri, M.; Meyer, A.; Odermatt, A.; Moller, G.; Adamski, J.; Hartmann, R.W. Structural Optimization of 2,5-Thiophene Amides as Highly Potent and Selective 17 beta-Hydroxysteroid Dehydrogenase Type 2 Inhibitors for the Treatment of Osteoporosis. J. Med. Chem. 2013, 56, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Opsenica, I.M.; Verbic, T.Z.; Tot, M.; Sciotti, R.J.; Pybus, B.S.; Djurkovic-Djakovic, O.; Slavic, K.; Solaja, B.A. Investigation into novel thiophene- and furan-based 4-amino-7-chloroquinolines afforded antimalarials that cure mice. Bioorg. Med. Chem. 2015, 23, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.; Dash, R.P.; Thakur, S.K.; Pandya, A.N.; Venkatesh, P.; Vasu, K.K.; Nivsarkar, M. Implication of novel thiazolo-thiophene derivative (MCD-KV-10) for management of asthma. J. Enzym. Inhib. Med. Chem. 2015, 30, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Al-Dhfyan, A.; Al-Dosari, M.S.; El-Gazzar, M.G.; AlSaid, M.S. Antiproliferative activity of novel thiophene and thienopyrimidine derivatives. Drug Res. 2014, 64, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Ashok, P.; Lu, C.L.; Chander, S.; Zheng, Y.T.; Murugesan, S. Design, Synthesis, and Biological Evaluation of 1-(thiophen-2-yl)-9H-pyrido[3,4-b]indole Derivatives as Anti-HIV-1 Agents. Chem. Biol. Drug Des. 2015, 85, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Leishmania survival in the macrophage: Where the ends justify the means. Curr. Opin. Microbiol. 2015, 26, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Butler, W.T. Pharmacology, toxicity, and therapeutic usefulness of amphotericin B. JAMA 1966, 195, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Amer, A.O.; Swanson, M.S. A phagosome of one’s own: A microbial guide to life in the macrophage. Curr. Opin. Microbiol. 2002, 5, 56–61. [Google Scholar] [CrossRef]
- Fonseca-Silva, F.; Inacio, J.D.F.; Canto-Cavalheiro, M.M.; Almeida-Amaral, E.E. Reactive oxygen species production and mitochondrial dysfunction contribute to quercetin induced death in Leishmania amazonensis. PLoS ONE 2011, 6, e14666. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Irigoin, F.; Cibils, L.; Comini, M.A.; Wilkinson, S.R.; Flohe, L.; Radi, R. Insights into the redox biology of Trypanosoma cruzi: Trypanothione metabolism and oxidant detoxification. Free Radic. Biol. Med. 2008, 45, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Krauth-Siegel, R.L. Enzymes of the trypanothione metabolism as targets for antitrypanosomal drug development. Curr. Top. Med. Chem. 2002, 2, 1239–1259. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.R.; Horn, D.; Prathalingam, S.R.; Kelly, J.M. RNA interference identifies two hydroperoxide metabolizing enzymes that are essential to the bloodstream form of the african trypanosome. J. Biol. Chem. 2003, 278, 31640–31646. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Carreon, T.; Iniguez, E.; Anzellotti, A.; Sanchez, A.; Tyan, M.; Sattler, A.; Herrera, L.; Maldonado, R.A.; Sanchez-Delgado, R.A. Searching for new chemotherapies for tropical diseases: Ruthenium-clotrimazole complexes display high in vitro activity against Leishmania major and Trypanosoma cruzi and low toxicity toward normal mammalian cells. J. Med. Chem. 2012, 55, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Capul, A.A.; Hickerson, S.; Barron, T.; Turco, S.J.; Beverley, S.M. Comparisons of mutants lacking the Golgi UDP-galactose or GDP-mannose transporters establish that phosphoglycans are important for promastigote but not amastigote virulence in Leishmania major. Infect. Immun. 2007, 75, 4629–4637. [Google Scholar] [CrossRef] [PubMed]
- Lema, C.; Varela-Ramirez, A.; Aguilera, R.J. Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. Curr. Cell. Biochem. 2011, 1, 1–14. [Google Scholar] [PubMed]
- Iniguez, E.; Sanchez, A.; Vasquez, M.A.; Martinez, A.; Olivas, J.; Sattler, A.; Sanchez-Delgado, R.A.; Maldonado, R.A. Metal-drug synergy: New ruthenium(II) complexes of ketoconazole are highly active against Leishmania major and Trypanosoma cruzi and nontoxic to human or murine normal cells. J. Biol. Inorg. Chem. 2013, 18, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Leishmania major-luc | Mammalian Cells | Mammalian Cells |
|---|---|---|---|
| Promastigotes | IPΦ | LLC-MK2 | |
| EC50 (μM) | CC50 (μM) [S.I.] | CC50 (μM) [S.I.] | |
| 5A | ~0.3410 | ~ 10.40 [30.50] | 17.69 ± 1.12 [52.85] |
| 5B | 5.98 ± 1.72 | N/A | N/A |
| 5C | 4.73 ± 0.69 | N/A | N/A |
| 5D | 0.09 ± 0.02 | 27.89 ± 3.19 [310] | >80 |
| 5E | 0.78 ± 0.11 | 16.59 ± 1.52 [21.27] | 80 ± 4.45 [102.56] |
| 5F | >12.50 | N/A | N/A |
| 5G | >12.50 | N/A | N/A |
| 5H | 3.05 ± 0.47 | N/A | N/A |
| 5I | 5.5 ± 1.80 | N/A | N/A |
| Receptor | Ligand | Structure | Glide SP (XP) | IFD XP (IFD Score) |
|---|---|---|---|---|
| TryR (2JK6) | 5D |  | −4.6 (−5.5) | −10.0 |
| Quinacrine Mustard (control) |  | −6.9 (−6.7) | −9.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, F.; Iniguez, E.; Pena Contreras, G.; Ahmed, H.; Costa, T.E.M.M.; Skouta, R.; Maldonado, R.A. Development of Thiophene Compounds as Potent Chemotherapies for the Treatment of Cutaneous Leishmaniasis Caused by Leishmania major. Molecules 2018, 23, 1626. https://doi.org/10.3390/molecules23071626
Rodriguez F, Iniguez E, Pena Contreras G, Ahmed H, Costa TEMM, Skouta R, Maldonado RA. Development of Thiophene Compounds as Potent Chemotherapies for the Treatment of Cutaneous Leishmaniasis Caused by Leishmania major. Molecules. 2018; 23(7):1626. https://doi.org/10.3390/molecules23071626
Chicago/Turabian StyleRodriguez, Felipe, Eva Iniguez, Guadalupe Pena Contreras, Haidar Ahmed, Thadeu E. M. M. Costa, Rachid Skouta, and Rosa A. Maldonado. 2018. "Development of Thiophene Compounds as Potent Chemotherapies for the Treatment of Cutaneous Leishmaniasis Caused by Leishmania major" Molecules 23, no. 7: 1626. https://doi.org/10.3390/molecules23071626