
Cytotoxicity-Guided Isolation of Two New Phenolic Derivatives from Dryopteris fragrans (L.) Schott



1
College of Life Science, Northeast Agricultural University, Harbin 150030, China
2
State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(7), 1652; https://doi.org/10.3390/molecules23071652
Submission received: 29 May 2018
/
Revised: 21 June 2018
/
Accepted: 1 July 2018
/
Published: 6 July 2018
(This article belongs to the Special Issue Natural Products and Drug Discovery)
Abstract
:Dryopteris fragrans is a valuable medicinal plant resource with extensive biological activities including anti-cancer, anti-oxidation, and anti-inflammation activities. This work aims to study further the cytotoxic constituents from Dryopteris fragrans. In this work, two new phenolic derivatives known as dryofragone (1) and dryofracoumarin B (2) with six known compounds (3–8) were isolated from the petroleum ether fraction of the methanol extract of the aerial parts of Dryopteris fragrans (L.) Schott by two round cytotoxicity-guided tracking with the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay and cell counting kit-8 (CCK-8) assay. Their structures were elucidated by the extensive spectroscopic analysis (1H-NMR, 13C-NMR, and two dimensions NMR), chemical derivatization, and comparison with data reported in the literature. All the isolates were evaluated for their cytotoxicity against nine cancer cell lines as well as their in vitro immunomodulatory activity. The results showed that compounds have a modest cytotoxicity toward human HeLa cell line with IC50 value below 30 μM and compounds 4 and 5 may modulate immunity to affect the growth of tumor cells.
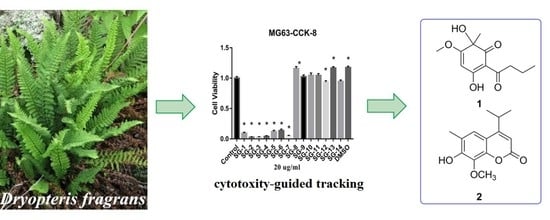
1. Introduction
Dryopteris fragrans (L.) Schott (Figure 1) belonging to the genus Dryopteris is a perennial herb with aroma widely distributed throughout the world and is mostly distributed in the alpine and volcanic regions of Northeast China [1,2]. D. fragrans has been used as folk medicine for treating arthritis and skin diseases such as psoriasis, dermatophytosis, and more [3]. Previous phytochemical investigations on this plant have led to the identification of terpenoids [4], phloroglucinols [5], glucosides [6], and other phenolic derivatives such as coumarin [3]. The earlier biological studies have shown that D. fragrans was a valuable medicinal plant resource with extensive biological activities including anti-cancer, anti-oxidation, insect repellent, anti-microbial, and anti-inflammation activities [3,4,5,6,7].
Of its various biological effects, the mechanism of anti-cancer effects has been studied most. Dryofragin, which is a derivative of phloroglucinol, was found to activate the endogenous pathway of apoptosis by affecting the changes of ROS in mitochondria and inducing changes in mitochondria in breast cancer cell MCF-7 and to cause tumor cell apoptosis by the apoptosis-related protein Bcl-2, Bax, Caspase-9, Caspase-3, and PARP [8]. It has also been reported to be an inhibitor of migration and invasion of the human osteosarcoma cell line U2OS through the PI3K/Akt and MAPK energy pathway involving MMP-2/9 and TIMP-1/2 proteins [9]. Aspidin PB, which is another phloroglucinol derivative from D. fragrans, has been recorded as a tumor cell-inhibiting agent for its impact on cyclin p53/p21 and mitochondrial changes in human osteosarcoma cells Saos-2, U2OS, and HOS [10]. In addition, there have been many other reports on compounds from D. fragrans with cytotoxicity [11,12,13]. To further study cytotoxic constituents from D. fragrans, a cytotoxicity-guided isolation of the extract of D. fragrans was designed. The isolation of two new phenolic derivatives and six known compounds by cytotoxicity-guided tracking as well as their cytotoxicity and immunomodulatory activity detection is described in this paper.
2. Results and Discussion
2.1. Determination of Isolated Compounds
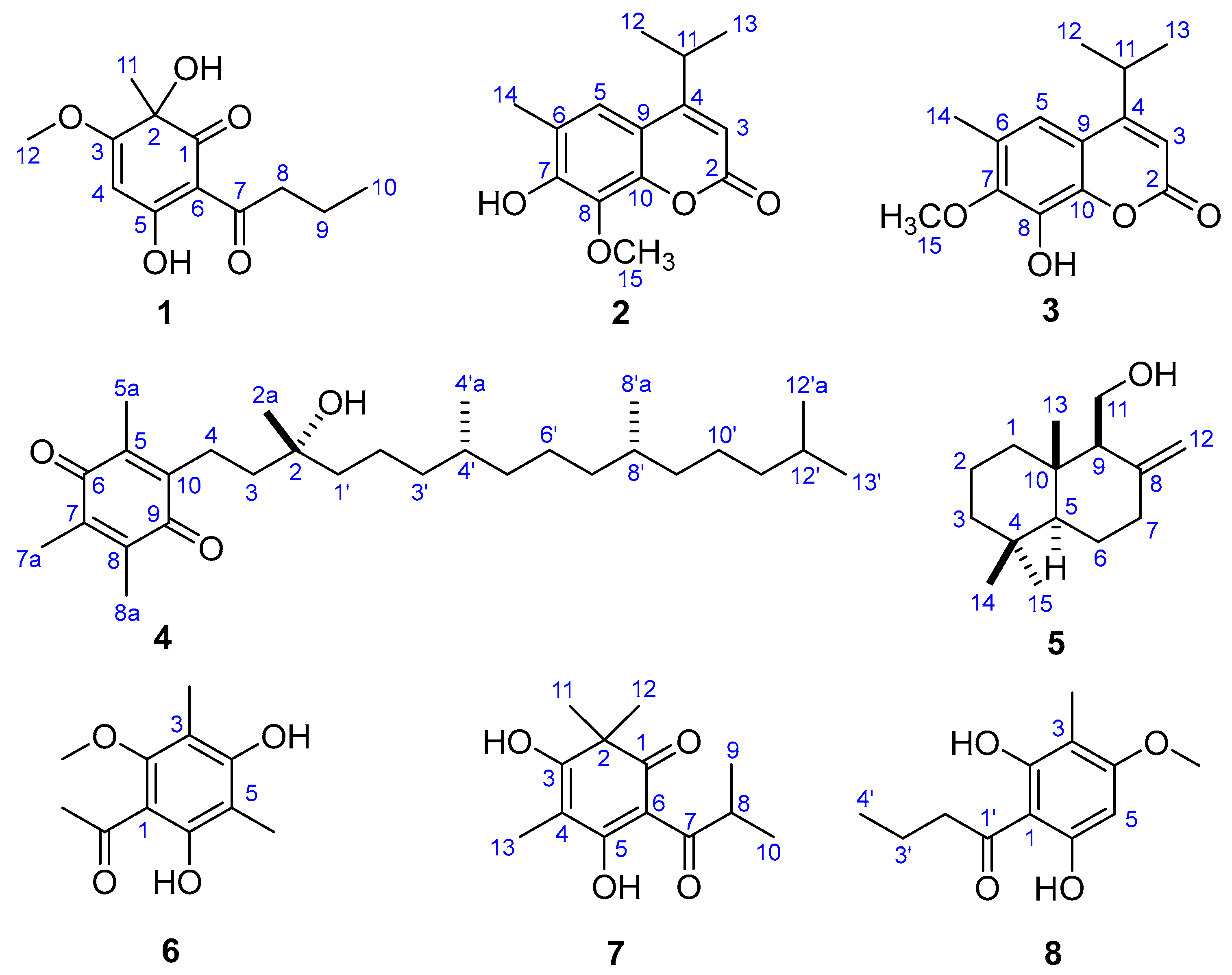
After two round cytotoxicity screening by MTT [14] and CCK-8 [15] assay, Fractions SG1‒SG7 from the petroleum ether-soluble part with prominent cytotoxic activities were selected as the bioactive sites (Figures S1 and S2). Two new phenolic derivatives known as dryofragone (1) and dryofracoumarin B (2) (Figure 2) along with six known compounds (3–8) (Figure 2), were isolated from the above seven bioactive fractions by using extensive chromatographic methods like silica gel, MCI gel, Sephadex LH-20, and HPLC. The known compounds were identified as dryofracoumarin A (3) [3], vitamin E quinone (4) [16], albicanol (5) [5], 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone (6) [17], norflavesone (7) [18], and aspidinol (8) [19] by comparing their 1H- and 13C-NMR data with that reported in the literature.
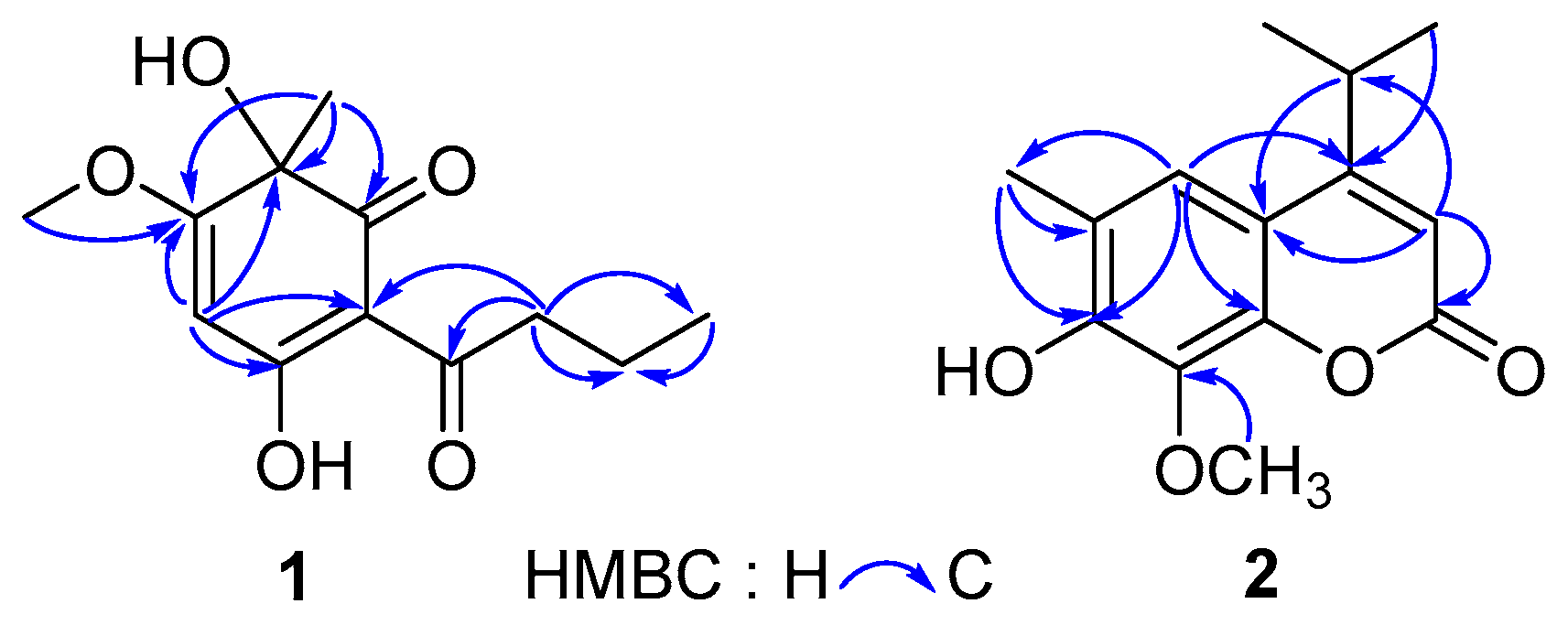
Compound 1 was obtained as yellow powder from CHCl3. The HR-ESI-MS data (m/z 239.0926 [M − H]−, calcd for 239.0925) of 1 showed the molecular formula C12H16O5, which correspond to five degrees of unsaturation. The IR spectrum of 1 displayed hydroxyls (3321 cm−1), carbonyl groups (1714 cm−1), and double bonds (1607 cm−1) absorptions. The red shifted hydroxyl signal (3321 cm−1) also showed that some hydroxyls in 1 were involved in the hydrogen bonding interaction. The 1H-NMR spectrum of 1 (Table 1) showed one 3H-singlet at δH 1.54 for a tertiary methyl group, one 3H-singlet at δH 3.91 for a methoxy group, one 3H-triplet at δH 1.01 for a primary methyl group, and an olefinic proton at δH 5.37. The 13C-NMR spectrum of 1 revealed 12 resonance signals including two ketone carbons at δC 196.3 (conjugated) and 203.7, two pair of olefinic carbons (δC 189.4, 176.0, 104.5 and 94.5) with two oxygenated sites (δC 189.4 and 176.0), an oxygenated tertiary carbon (δC 75.4), a methoxy carbon (δC 57.4), two aliphatic methylene carbon (δC 41.0 and 18.7), and two methyl carbons (δC 30.2 and 14.1). The above evidence indicated that compound 1 presumably possessed an oxygenated phloroglucinol core [20]. This inference was further confirmed by the 2D-NMR spectra (Figure 3). The long-range HMBC couplings—H-4/C-2, C-3, C-5, and C-6 as well as Me-11/C-1, C-2, and C-3 demonstrated the presence of a cyclohexadiene moiety with two oxygen-bearing carbon at C-2 and C-5. The HMBC correlation from a methoxy at δH 3.91 (Me-12) to a quaternary olefinic carbon at δC 176.0 (C-3) revealed that a methoxy was located at C-3. Furthermore, a butyryl was linked to C-6, which was supported by the HMBC correlations from protons at C-8 (δH 2.99 and 2.92) to carbons at δC 104.5 (C-6), 203.7 (C-7), 18.7 (C-9), and 14.1 (C-10), respectively. The CD experiment towards compound 1 was performed. However, the CD spectrum (Figure S9) of 1 showed no characteristic cotton effect. Compound 1 was considered to be a pair of enantiomers.
Therefore, the structure of 1 was concluded to be a new acylphloroglucinol, 6-isobutyryl-2,5-dihydroxy-2-methyl-3-methoxy-cyclohexa-3,5-dien-1-one, and was named dryofragone.
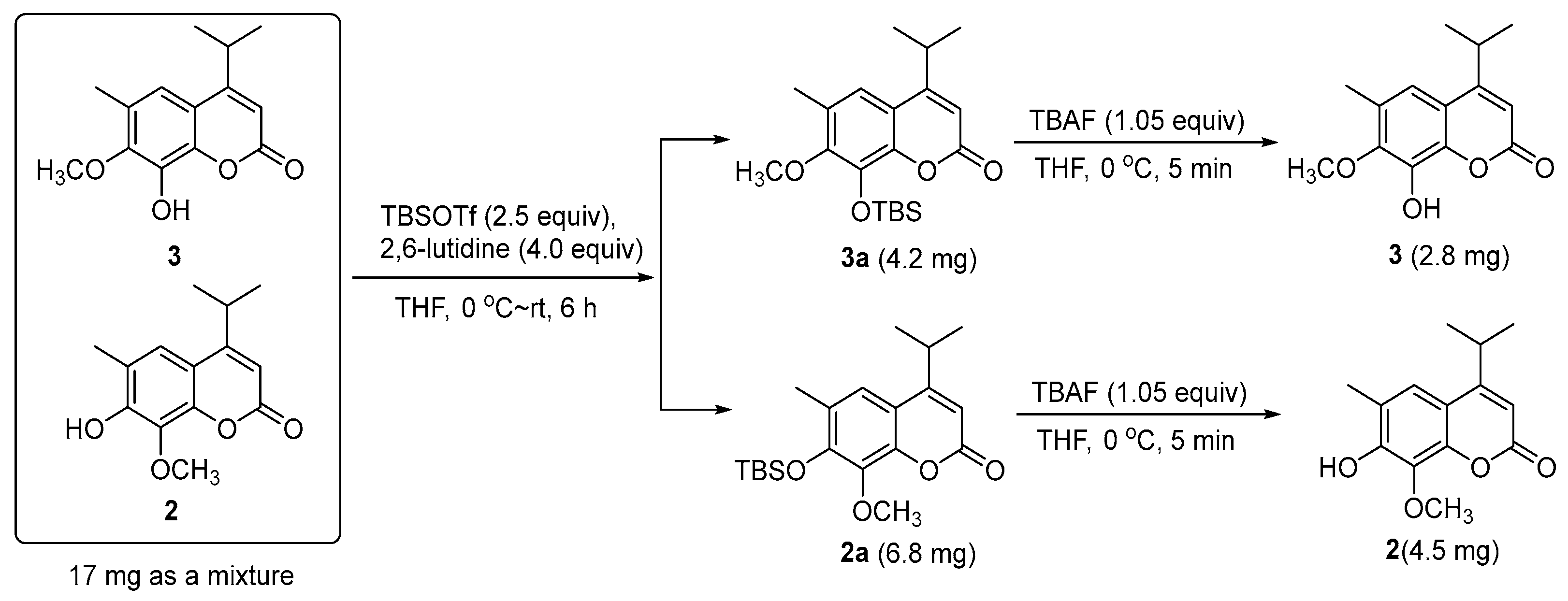
Compound 2 was obtained as a mixture with compound 3 initially. The 13C-NMR spectrum of the mixture revealed 28 resonance signals (Figure S12) in which half were consistent with the data reported for a coumarin and dryofracoumarin A (3) [3]. However, the ESI-MS data (m/z 249[M + H]+, 271[M + Na]+, 287[M + K]+) of the mixture showed only one molecular weight (248 Da), which aligned with that of 3. Consequently, the other half of carbon resonance signals in the 13C-NMR for the mixture, which were highly similar with that of 3, were supposed to be of an isomer of 3 featuring exchanged positions of hydroxyl and methoxy groups in the coumarin core. Based on the large space size of tert-butyl dimethyl silicyl group, which can strike the balance of molecular polarity for compounds 2 and 3 and the high yield of the desilication step, a silicon etherification-desilication procedure was designed for the isolation of the mixture (See Section 3.5 and Figure 4). NMR data of compounds 2 and 3 are shown in Table 2. After the chemical derivatization, compound 2 was afforded as a simplex. The IR spectrum of 2 exhibited a signal of hydroxyl with no hydrogen bonds (3548 cm−1), a strong band at 1668 cm−1 for the lactone subunit in coumarin core, and absorptions (1636, 1602, 1572 cm−1) of benzene ring moiety in coumarin. The HR-ESI-MS data (m/z 247.0975 [M − H]−, calcd for 247.0976) indicated a molecular formula C14H16O4 with seven degrees of unsaturation for 2. The HMBC correlations (Figure 3) from Me-12 and -13 (δH 1.30 × 2) to C-4 (δC 163.0) as well as the correlations from H-11(δH 3.25) to C-3 (δC 107.8), C-4 (δC 163.0), and C-9 (δC 112.2), which suggests that an isopropyl was fused to C-4. Another HMBC correlation Me-15/C-8 verified that a methoxyl group was linked to C-8. In addition, the HMBC correlations from an isolated methyl (δH 2.31) to C-5 (δC 120.0), C-6 (δC 121.1), and C-7 (δC 150.0) inferred a methyl at C-6 in 2. The above analyses disclosed our former hypothesis. As a result, the structure of 2 was determined to be 7-hydroxy-6-methyl-8-methoxy-4-isopropyl-2H-chromen-2-one, which was given the trivial name of dryofracoumarin B.
2.2. In Vitro Cytotoxicity and Immunomodulatory Activity Detection
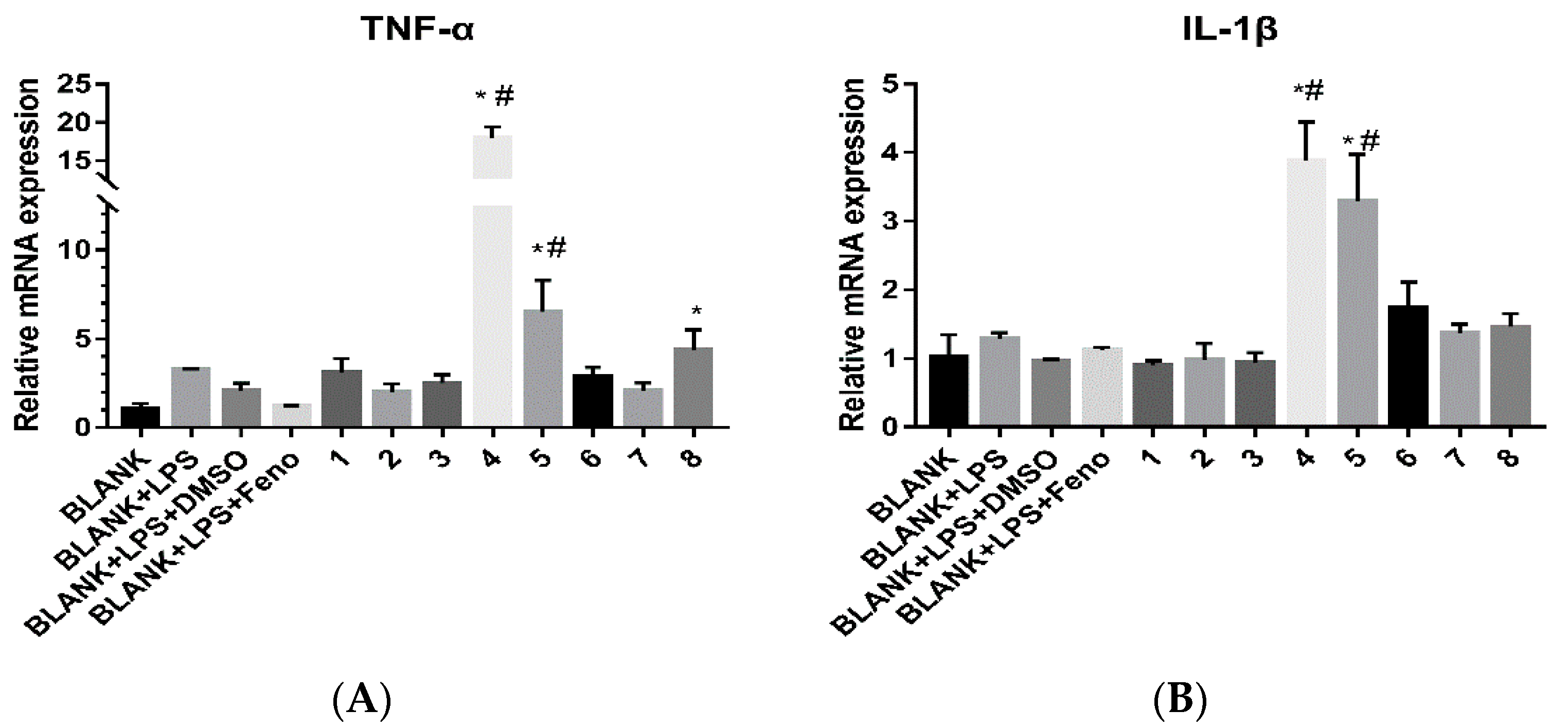
For all the isolates, their cytotoxicities against nine human cancer cell lines known as HepG2, A549, HeLa, U251, HOS, MG63, U2OS, MB231, and SKBR-3 as well as their immuno-regulation activities were evaluated. The cytotoxicities were screened using the CCK-8 assay [15]. The IC50 values of cytotoxicities for the eight compounds are shown in Table 3. For compounds isolated by cytotoxicity-guided tracking, they exhibited moderate activities to the HeLa cell line and weak activities to glioma, liver cancer, and lung cancer cell lines. However, they were not very sensitive to osteosarcoma and breast cancer cell lines when compared to the crude extract. For their immuno-regulation activities, LPS stimulated THP-1 cells were used as the in vitro model for the detection [21]. Fenofibrate (Feno) pre-treatment (20 μM) was used as a positive control [21]. The results for immuno-regulation activities are shown in Figure 5. Only compounds 4 and 5 could enhance the secretion of the factors TNF-α and IL-1β. The results showed that compounds 4 and 5 may activate the LPS signaling pathway, which may modulate immunity to affect the growth of tumor cells.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were recorded in MeOH using a JASCO P-1020 Polarimeter (Jasco Corp., Tokyo, Japan). UV spectra were acquired in MeOH with a Shimadzu UV-2401PC UV-VIS spectrophotometer (Shimadzu Corp., Kyoto, Japan). IR spectra were measured on a Bruker Tensor 27 FTIR Spectrometer with KBr disks (Bruker Corp., Karlsruhe, Germany). 1H-NMR, 13C-NMR, and 2D NMR spectra were recorded in CDCl3 using a Bruker AVANCE III-600 spectrometer or a Bruker DRX-400 spectrometer (Bruker Corp., St. Gallen, Switzerland). TMS was used as the internal standard. ESI-MS spectra were recorded using a Waters Xevo TQ-S Ultra High Pressure Liquid Chromatography Triple Quadrupole Mass Spectrometer (Waters Corp., Manchester, UK). HR-ESI-MS data were obtained using an Agilent G6230 Q-TOF mass instrument (Agilent Corp., Santa Clara, CA, USA). Column chromatography (CC) was performed using a silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), MCI gel CHP 20P (75–150 µm, Mitsubishi Corp., Tokyo, Japan), and Sephadex LH-20 (25–100 mm, Pharmacia Biotech Ltd., Uppsala, Sweden). Thin-layer chromatography (TLC) was performed using pre-coated silica gel GF254 plates (0.25 mm in thickness for analysis and 0.60 mm thickness for preparation, Qingdao Marine Chemical Inc., Qingdao, China) with various solvent systems. Spots were visualized under UV light (254 nm) and colored by iodine and by spraying silica gel plates with 10% H2SO4 in MeOH followed by heating. Preparative HPLC separations were performed on a CXTH system equipped with a UV3000 detector (Beijing Chuangxintongheng Instruments Co. Ltd., Beijing, China), and a Kromasil C18 column (250 mm × 20 mm i.d., 5 mm, EKA Chemicals Corp., Bohus, Sweden) using a flow rate of 8.0 mL/min at a column temperature of 25 °C. Semi-preparative HPLC was conducted on a HITACHI Chromaster system (Hitachi Ltd., Tokyo, Japan) equipped with an Agilent ZORBAXSB-C18 column (150 mm × 9.4 mm i.d., 5 mm, Agilent Corp., Santa Clara, CA, USA) using a flow rate of 3.0 mL/min at a column temperature of 25 °C. The detection was performed with a DAD detector.
3.2. Plant Material
The aerial parts of Dryopteris fragrans (L.) Schott were collected in June 2016 from the Wudalianchi scenic area, Heihe City, Helongjiang Province, China and identified by Prof. Baodong Liu from the Harbin Normal University. A voucher specimen (No. df-20070702-9) was deposited in the Plant Herbarium of Northeast Agricultural University in Harbin, China.
3.3. Determination of Anti-Tumor Fraction of Dryopteris fragrans
After methanol extraction, the crude extract was then partitioned with petroleum ether (rt), dichloromethane (DCM) (rt), EtOAc (rt), and n-BuOH (rt) in sequence. The crude extract was divided into six parts (the whole extracts, petroleum ether layer, DCM layer, EtOAc layer, n-BuOH layer, and water phase). Each fraction was dissolved in DMSO and the final concentration of DMSO in the cell culture medium was no more than 0.1%. The osteosarcoma cell lines HOS and MG63 were used as the first round screening target in MTT [14] for the above six parts of the crude extracts. As shown in Figure S1, petroleum ether fraction of crude extracts had the most obvious cytotoxic effects at the point of 48 h. The petroleum ether fraction was then divided into 14 sub-fractions (Fr SG1‒SG14) by using silica gel column chromatography. The above 14 fractions were then subjected to MTT or CCK-8 assay [15] against HepG2, MB231, and MG63 cell lines, respectively. As shown in Figure S2, Fractions SG1–SG7 from the petroleum ether-soluble part exhibited prominent cytotoxic activities.
3.4. Extraction and Isolation
The air-dried aerial parts of Dryopteris fragrans (L.) Schott powder (2 kg) were extracted with 100% methanol (20 L × 2 d × 3) and ultrasonized (40 Hz) for 4 h at each time. After filtration, the filtrate was concentrated to yield the crude extract. The crude extract was then suspended in water (1.5 L) and partitioned with petroleum ether (3 × 1.5 L), DCM (3 × 1.5 L), EtOAc (3 × 1.5 L), and n-BuOH (3 × 1.5 L) sequentially. Guided by the first round cytotoxicity screening, the petroleum ether fraction (54 g) was chosen for further isolation. The petroleum ether-soluble part was then subjected to silica gel CC and eluted with petroleum ether–EtOAc (1:0–0:1) to create 14 fractions (SG1–SG14). According to the second round cytotoxicity screening, Fractions SG1–SG7 were selected as the isolation targets for the next step. Fractions SG1 and SG2 were not actually involved in the next step because their low polarities made an effective separation on column chromatography difficult.
Fraction SG3 (5.93 g) was submitted to the silica gel CC (petroleum ether–EtOAc 1:0–0:1) and Sephadex LH-20 CC (MeOH–CHCl3 1:1) and followed by preparative TLC (petroleum ether–EtOAc 11:2, Rf = 0.53) to afford compound 4 (14.4 mg). Compound 5 (103.2 mg) was isolated from Fraction SG4 (5.01 g) by undergoing a protocol of repeated silica gel CC (petroleum ether–EtOAc 1:0–10:1), Sephadex LH-20 CC (MeOH–CHCl3 1:1), and preparative TLC (petroleum ether–EtOAc 8:1, Rf = 0.40). Fraction SG6 (3.54 g) was chromatographed on MCI CC (MeOH–H2O 40:60 to 100:0) to yield 16 sub-fractions (Fr M1–M16). Further purification of Fr. M6 by semi-preparative HPLC (MeOH: H2O 53:47) resulted in the isolation of compounds 6 (15.8 mg) and 7 (10.0 mg). Fraction SG7 (2.49 g) was further separated by MCI CC (MeOH–H2O 20:80 to 100:0) to yield 11 fractions (Fr. M21–M211). Compound 1 (4.5 mg) was purified from Fraction M24 using semi-preparative HPLC (MeOH:H2O 58:42). In the same way, compound 8 (2.0 mg) was obtained from Fraction M27. Compounds 2 and 3 were obtained as a mixture (17.0 mg) from Fraction M26 by semi-preparative HPLC (MeOH:H2O 77:23). They were separated by a silicon etherification-desilication procedure (See Section 3.5).
Dryofragone (1): yellow powder (CHCl3). [α –20.3 (c 0.10, MeOH); UV (MeOH) λmax (log ε): 198 (3.17) nm, 241 (3.38) nm, 276 (3.20) nm, 320 (3.14) nm, IR (KBr) νmax IR (KBr) νmax 3321, 2929, 1714, 1607, 1533, 1442, 1231, 1104; 1H-NMR (600 MHz, CDCl3): δH 5.37 (1H, s, H-4), 3.91 (3H, s, Me-12), 2.99 (1H, m, H-8a), 2.92 (1H ,m, H-8b), 1.69 (2H, m, H-9), 1.54 (3H, s, Me-11), 1.01 (3H, t, J = 7.4 Hz, Me-10); 203.7 (C-7), 196.3 (C-1), 189.4 (C-5), 176.0 (C-3), 104.5 (C-6), 94.5 (C-4), 75.4 (C-2), 57.4 (C-12), 41.0 (C-8), 30.2 (C-11), 18.7 (C-9),14.1 (C-10), ESI-MS m/z 239 [M − H]−, and HR-ESI-MS m/z 239.0926 [M − H]− (calcd for C12H15O5, 239.0925).
3.5. Silicon Etherification Involved Isolation of 2 and 3
3.5.1. Silicon Etherification of the Mixture of Compounds 2 and 3
With regard to the solution of the mixture of Compounds 2 and 3 (17 mg, 0.068 mmol) in dry DCM (0.5 mL), 2,6-luditine (30 μL, 0.27 mmol, 4.0 equiv) was added at 0 °C, which is followed by the addition of TBSOTf (35 μL, 0.17 mmol, 2.5 equiv). The resulting mixture was warmed to room temperature (rt) naturally, stirred for 6 h, and then quenched with water (2.0 mL). The mixture was then stirred for 10 min, followed by an extraction with EtOAc (10.0 mL) three times, and the EtOAc layer was dried over anhydrous Na2SO4 and subsequently concentrated. The residue was further purified by semi-preparative HPLC (85% MeOH in H2O, 3 mL/min, a HITACHI Chromaster system equipped with a DAD detector, an Agilent ZORBAX SB-C18 column, 150 mm × 9.4 mm i.d., 5 μm) to yield compound 2a (6.8 mg, tR = 19.8 min) and 3a (4.2 mg, tR = 15.8 min) as white solids. Compound 2a: 1H-NMR (400 MHz, CDCl3): δH 7.17 (1H, s, H-5), 6.18 (1H, s, H-3), 3.91 (3H, s, Me-15), 3.24 (1H, m, H-11), 2.29 (3H, s, Me-14), 1.30 (6H, d, J = 6.8 Hz, Me-12, 13), 1.03 (9H, s, Me-19, 20, 21), 0.24 (6H, s, Me-16, 17), 13C-NMR (100 MHz, CDCl3): δC 162.5 (C-4), 161.7 (C-2), 150.1 (C-7), 146.9 (C-10), 138.3 (C-8), 126.5 (C-6), 119.5 (C-5), 113.4 (C-9), 108.5 (C-3), 61.1 (C-15), 28.7 (C-11), 26.1 × 3 (C-19, 20, 21), 22.1 × 2 (C-12, 13), 19.0 (C-18), 17.7 (C-14), −4.0 × 2 (C-16, 17). Compound 3a: 1H-NMR (400 MHz, CDCl3): δH 7.07 (1H, s, H-5), 6.21 (1H, s, H-3), 3.82 (3H, s, Me-15), 3.24 (1H, m, H-11), 2.32 (3H, s, Me-14), 1.30 (6H, d, J = 6.8 Hz, Me-12, 13), 1.08 (9H, s, Me-19, 20, 21), 0.25 (6H, s, Me-16, 17), 13C-NMR (100 MHz, CDCl3): δC 162.2 (C-4), 161.4 (C-2), 152.5 (C-7), 145.3 (C-10), 137.2 (C-8), 127.8 (C-6), 117.0 (C-5), 115.4 (C-9), 109.8 (C-3), 60.2 (C-15), 28.7 (C-11), 25.9 × 3 (C-19, 20, 21), 22.1 × 2 (C-12, 13), 18.8 (C-18), 16.4 (C-14), −4.2 × 2 (C-16, 17).
3.5.2. Desilication of Compound 2a
To a solution of compound 2a (6.8 mg, 0.0188 mmol) in dry THF (0.1 mL), TBAF (1 M in THF, 19 μL, 0.0197 mmol, 1.05 equiv) was added at 0 °C. The resulting mixture was stirred at 0 °C for 5 min and then quenched by adding 1.0 mL of the saturated ammonium chloride aqueous solution. The resulting mixture was then extracted by EtOAc (5.0 mL) three times and the combined organic extracts were dried over anhydrous Na2SO4 and were then concentrated. The residue was purified by using flash column chromatography on the silica gel (200–300 mush, 1.0 × 3.0 cm, petroleum ether/EtOAc 4:1), which yielded compound 2 (4.5 mg, 96.6% yield) as a white solid. Compound 2: UV (MeOH) λmax (log ε): 206 (3.81) nm, 218 (3.52) nm, 250 (2.78) nm, 330 (3.29) nm; IR (KBr) νmax 3548, 3466, 3169, 2942, 1668, 1602, 1460, 1404, 1229, 1094, 1024, 925, 856; 1H-NMR (600 MHz, CDCl3): δH 7.17 (1H, s, H-5), 6.16 (1H, s, H-3), 4.08 (3H, s, Me-15), 3.25 (1H, m, H-11), 2.31 (3H, s, Me-14), 1.30 (3H, d, J = 6.8 Hz, Me-12), 1.30 (3H, d, J = 6.8 Hz, Me-13); 13C-NMR (150 MHz, CDCl3): δC 163.0 (C-4), 161.5 (C-2), 150.0 (C-7), 145.5 (C-10), 133.6 (C-8), 121.1 (C-6), 120.0 (C-5), 112.2 (C-9), 107.8 (C-3), 61.9 (C-15), 28.7 (C-11), 22.1 (C-12), 22.1 (C-13), 15.9 (C-14), HR-ESI-MS: m/z 247.0975[M − H]−, calcd. 247.0976 for C14H15O4.
3.5.3. Desilication of Compound 3a
To a solution of compound 3a (4.2 mg, 0.0116 mmol) in dry THF (0.1 mL), TBAF (1 M in THF, 12 μL, 0.0121 mmol, 1.05 equiv) was added at 0 °C. The resulting mixture was stirred at 0 °C for 5 min and then quenched by adding 1.0 mL of saturated ammonium chloride aqueous solution. The resulting mixture was then extracted by EtOAc (5.0 mL) three times and the combined organic extracts were dried over anhydrous Na2SO4 and concentrated. The residue was purified by flash column chromatography on the silica gel (200–300 mush, 1.0 × 3.0 cm, petroleum ether/EtOAc 4:1), which yielded compound 3 (2.8 mg, 97.4% yield) as a white solid. Compound 3: 1H-NMR (600 MHz, CDCl3): δH 7.01 (1H, s, H-5), 6.22 (1H, s, H-3), 3.95 (3H, s, Me-15), 3.25 (1H, m, H-11), 2.32 (3H, s, Me-14), 1.31 (6H, d, J = 6.8 Hz, Me-12, 13); 13C-NMR (150 MHz, CDCl3): δC 163.0 (C-4), 161.1 (C-2), 147.8 (C-7), 141.3 (C-10), 136.7 (C-8), 127.5 (C-6), 115.5 (C-5), 114.5 (C-9), 109.2 (C-3), 60.5 (C-15), 28.7 (C-11), 21.9 (C-12 & C-13), and 16.3 (C-14).
3.6. MTT and CCK-8 Assay
Human HepG2, HeLa, U251, HOS, MG63, U2OS, MB231, and SKBR-3 cells were obtained from the Cell Library of Committee on Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured at 37 °C, 5% CO2 in the Dulbecco’s Modified Eagle’s medium (DMEM), Minimum Eagle’s medium (MEM), or the Roswell Park Memorial Institute (RPMI) medium containing 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin.
Compounds 1–8 were dissolved in DMSO and diluted with DMEM medium (containing 1% FBS and 100 U/mL penicillin/streptomycin) for certain concentrations (1.5625 μM, 3.125 μM, 6.25 μM, 12.5 μM, 25 μM, 50 μM, and 100 μM). The concentration of DMSO in the final solutions was no more than 0.1%. Human HepG2, HeLa, U251, HOS, MG63, U2OS, MB231, and SKBR-3 cells were seeded in 96-well micro-titer plates (100 μL, 1 × 104 cells/well). When the cells grew to certain concentrations (70%–80% of the well), the medium was removed and the diluted compounds (200 μL) were added to each well. Blank (only medium) and control (cells with DMEM medium) group were set to calculate the cell viability and Taxol was used as a positive control. After 48 h, the 96-well micro-titer plates were taken out from the incubator and the medium was removed. When using the MTT assay, DMEM medium (200 μL) should be first added into the well and then followed by MTT (20 μL, 5 mg/mL dissolved in PBS). After culturing for 4 h, the 96-well micro-titer plates were taken out from the incubator and the medium was removed and then 150 μL of DMSO was added into the well. The absorbance was measured by a microplate reader (Bio-Rad, America) at 560 nm. When using CCK-8 assay, DMEM medium (100 μL) should be first added into the well and then CCK-8 (Dojindo, Kumamoto, Japan) followed. After culturing for 2.5 h, the 96-well micro-titer plates were taken out from the incubator and the absorbance was measured by a microplate reader at 450 nm. The cell viability = (Lab group − Blank group)/(Control group − Blank group) and the IC50 value was calculated by the software GraphPad Prism 7.0 with the cell viability value.
3.7. Immunoregulation Activity
THP-1 cells were obtained from the Harbin medical university and was cultured in RPMI medium. The cell was seeded in 6-well plate (2 mL, 2 × 106 cell/well) and starved for 12 h. Lipopolysaccharide (LPS, Sigma, St. Louis, MI, USA) (2 mg/mL in PBS) was then added in the well to stimulate the cell. One hour later, each compound was dissolved in DMSO at a concentration of 20 μM. It was added in the well and cultivated for 24 h. The cell was then collected. Following the manufacturer’s instructions, the Trizol reagent (Invitrogen, Carlsbad, CA, USA) was used to isolate the total RNA of THP-1 cell. The extracted total RNA was dissolved in RNA enzyme-free water and added into a 100 μL reaction mixture for reverse transcription into complementary DNA (cDNA). The extracted RNA solution contained 8 μg, 8 μL of 50 pmol/μL Oligo d(T)18 and the volume was brought up to 46 μL with RNA enzyme-free water, incubated at 70 °C for 5 min, and then 4 °C for 5 min. Afterward, 20 μL of 2.5 μmol/mL dNTP, 20 μL of 5× RT buffer, 8 μL of dTT, 2 μL of RNA inhibitor, and 4 μL of M-MLV were added and the mixture was incubated at 42 °C for 3 h. The 100 μL mixture was stored at −20 °C for qualitative PCR (qPCR). Detected via qPCR, the gene expression levels were identified with a LightCycler® 480 System (Roche, Basel, Switzerland) using the TransStart® Tip Green qPCR SuperMix (TRANSGEN BIOTECH, Beijing, China). The cDNA was added into a 20 μL reaction mixture: 10 μL of 2× TransStart® Tip Green qPCR SuperMix, 2 μL of cDNA Template (100–200 pg), 0.8 μL of primer mixture (10 μM), 0.4 μL of Passive Reference Dye (50×), and 6.8 μL of ddH2O. The PCR procedure includes a temperature of 94 °C and a time period of 30 s followed by 40 cycles at 94 °C for 5 s, 57 °C for 15 s, and 72 °C for 10 s. The β-actin gene was used a control to quantify other genes. The results were calculated using 2−ΔΔCt where ΔΔCt = (Ct Target − Ct β-actin)Lab − (Ct Target − Ct β-actin) Control. Primers of the target gene TNF-α, IL-1β, and β-actin were designed by Primer 5.0 software following the published gene sequence in GenBank: TNF-α Forward: CAGCAAGGGACAGCAGAGG, Reverse: AGTATGTGAGAGGAAGAGAACC; IL-1β Forward: TGATGGCTTATTACAGTGGCAATG, Reverse: TGATGGCTTATTACAGTGGCAATG; β-actin Forward: ATCGGCAATGAGCGGTTCC, Reverse: ATCGGCAATGAGCGGTTCC.
4. Conclusions
In this work, two new phenolic derivatives dryofragone (1) and dryofracoumarin B (2) were isolated from Dryopteris fragrans by cytotoxicity-guided tracking. Two coumarin isomers dryofracoumarin B (2) and dryofracoumarin A (3) were separated by a silicon etherification-desilication procedure. Compounds (4) and (6) were first reported in this plant. The cytotoxicity and immuno-regulation activity were examined among the eight compounds and the relationship between cytotoxicity and immuno-regulation activity revealed that compounds may activate the LPS signaling to regulate the growth of tumor cells through immuno-regulation. This relation needs further study.
Supplementary Materials
The following are available online. Supplementary materials included Figures S1 and S2: Two round screening of cytotoxicity with MTT or CCK-8 assay, Figures S3–S27: Spectrum and Spectroscopy data of compounds 1–3, 2a and 3a, and NMR spectral data of compounds 4–8.
Author Contributions
S.-X.H. and Y.C. handled the conceptualization. T.Z. managed the data curation. T.Z. performed a formal analysis. S.-X.H. and Y.C. handled the funding acquisition. T.Z., L.W., D.-H.D., and Y.-H.Z. were responsible for the methodology. L.W. handled the software. S.-X.H. and Y.C. supervised the project. T.Z. wrote the original draft. Writing—review & editing, L.W. took part in reviewing and editing the manuscript.
Acknowledgments
This work received no external funding and was financially supported by the National Natural Science Foundation of China (No. 81522044 to S.-X.H.), the Applied Basic Research Foundation of Yunnan Province (No. 2013HA022 to S.-X.H.), Foundation from Chinese Academy of Sciences (QYZDB-SSW-SMC051 to S.-X.H.), and the National Natural Science Foundation of China (No. 31570189 to Y.C.). We appreciate the help of Yunzhou Wu and Wenfei Wang in this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kuang, H.; Zhang, Y.; Li, G.; Zeng, W.; Wang, H.; Song, Q. A new phenolic glycoside from the aerial parts of Dryopteris fragrans. Fitoterapia 2008, 79, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Patama, T.T.; Widen, C.J. Phloroglucinol derivatives from Dryopteris fuscoatra and D. hawaiiensis. Phytochemistry 1991, 30, 3305–3310. [Google Scholar] [CrossRef]
- Zhao, D.D.; Zhao, Q.S.; Liu, L.; Chen, Z.Q.; Zeng, W.M.; Lei, H.; Zhang, Y.L. Compounds from Dryopteris fragrans (L.) Schott with cytotoxic activity. Molecules 2014, 19, 3345–3355. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Zeng, W.M.; Li, G.Y.; Liu, G.Q.; Zhao, D.D.; Wang, J.; Zhang, Y.L. Characterization of a new sesquiterpene and antifungal activities of chemical constituents from Dryopteris fragrans (L.) Schott. Molecules 2013, 19, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Muranaka, T.; Mori, K.; Jin, Z.X.; Tokuda, H.; Nishino, H.; Yoshida, T. Ichthyotoxic phloroglucinol derivatives from Dryopteris fragrans and their anti-tumor promoting activity. Chem. Pharm. Bull. 2000, 31, 1190–1195. [Google Scholar] [CrossRef]
- Peng, B.; Bai, R.F.; Li, P.; Han, X.Y.; Wang, H.; Zhu, C.C.; Zeng, Z.P.; Chai, X.Y. Two new glycosides from Dryopteris fragrans with anti-inflammatory activities. J. Asian Nat. Prod. Res. 2016, 18, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Wang, W.; Luo, M.; Li, C.Y.; Zu, Y.G.; Mu, P.S.; Fu, Y.J. Solvent-free microwave extraction of essential oil from Dryopteris fragrans and evaluation of antioxidant activity. Food Chem. 2012, 133, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, M.; Zu, Y.; Fu, Y.; Gu, C.; Wang, W.; Yao, L.; Efferth, T. Dryofragin, a phloroglucinol derivative, induces apoptosis in human breast cancer MCF-7 cells through ROS-mediated mitochondrial pathway. Chem. Biol. Interact. 2012, 199, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Jiang, C.; Hua, X.; Wang, T.; Chai, Y. Cell cycle arrest and apoptosis induced by aspidin PB through the p53/p21 and mitochondria-dependent pathways in human osteosarcoma cells. Anti Cancer Drugs 2015, 26, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wan, D.; Song, W. Dryofragin inhibits the migration and invasion of human osteosarcoma U2OS cells by suppressing MMP-2/9 and elevating TIMP-1/2 through PI3K/AKT and p38 MAPK signaling pathways. Anti Cancer Drugs 2016, 27, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Gao, C.; Luo, M.; Wang, W.; Gu, C.; Zu, Y.; Li, J.; Efferth, T.; Fu, Y. Aspidin PB, a phloroglucinol derivative, induces apoptosis in human hepatocarcinoma HepG2 cells by modulating PI3K/Akt/GSK3β pathway. Chem. Biol. Interact. 2013, 201, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Mu, F.; Li, C.; Wang, W.; Luo, M.; Fu, Y.; Zu, Y. Aspidin BB, a phloroglucinol derivative, induces cell cycle arrest and apoptosis in human ovarian HO-8910 cells. Chem. Biol. Interact. 2013, 204, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.C.; Zhao, D.D.; Liu, Z.D.; Jiang, S.; Zhang, Y.L. A new human cancer cell proliferation inhibition sesquiterpene, dryofraterpene A, from medicinal plant Dryopteris fragrans (L.) Schott. Molecules 2017, 22, 180. [Google Scholar] [CrossRef] [PubMed]
- Elisia, I.; Popovich, D.G.; Hu, C.; Kitts, D.D. Evaluation of viability assays for anthocyanins in cultured cells. Phytochem. Anal. 2008, 19, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Gong, Y.; Zhu, Y.; Li, A.; Dong, N.; Piao, Y.; Yuan, Y. The biological activity of H. pylori SlyD in vitro. Helicobacter 2013, 18, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.G.; Lee, D.Y.; Lee, J.W.; Lee, D.G.; Lee, Y.H.; Kim, S.Y.; Kim, S.H. Acyclic diterpenoids from the leaves of Capsicum annuum. J. Korean Soc. Appl. Biol. Chem. 2009, 52, 128–132. [Google Scholar] [CrossRef]
- Amzad, H.M. Synthesis of 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone. Bangladesh J. Sci. Ind. Res. 2001, 36, 50–54. [Google Scholar]
- Ayräs, P.; Lötjönen, S.; Widén, C.J. NMR spectroscopy of naturally occurring phloroglucinol derivatives. Planta Med. 1981, 42, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Lobo-Echeverri, T.; Rivero-Cruz, J.F.; Su, B.N.; Chai, H.B.; Cordell, G.A.; Pezzuto, J.M.; Swanson, S.M.; Soejarto, D.D.; Kinghorn, A.D. Constituents of the leaves and twigs of Calyptranthes pallens collected from an experimental plot in Southern Florida. J. Nat. Prod. 2005, 68, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Nisa, K.; Rakainsa, S.K.; Lallo, S.; Morita, H. New phloroglucinol derivatives from Indonesian Baeckea frutescens. Tetrahedron 2017, 73, 1177–1181. [Google Scholar] [CrossRef]
- Yang, L.; Guo, H.; Li, Y.; Meng, X.; Yan, L.; Dan, Z.; Wu, S.; Zhou, H.; Peng, L.; Xie, Q.; et al. Oleoylethanolamide exerts anti-inflammatory effects on LPS-induced THP-1 cells by enhancing PPARα signaling and inhibiting the NF-κB and ERK1/2/AP-1/STAT3 pathways. Sci. Rep. 2016, 6, 34611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds 1–8 are available from the authors. |
Figure 1.
Dryopteris fragrans plant.

Figure 2.
Structures of compounds 1–8.

Figure 3.
Key HMBC correlations of 1 and 2.

Figure 4.
Silicon etherification involved the isolation of 2 and 3.

Figure 5.
The influence of eight compounds on immuno-regulation factors over a period of 24 h. (A) for the factor TNF-α and (B) for the factor IL-1β. The “*” indicates that there were significant differences (p < 0.05) between other compounds and the BLANK group amd the “#” indicates that there were significant differences (p < 0.05) between other compounds and the BLANK + LPS group. Each value represented the means ± SD of three independent experiments.
Figure 5.
The influence of eight compounds on immuno-regulation factors over a period of 24 h. (A) for the factor TNF-α and (B) for the factor IL-1β. The “*” indicates that there were significant differences (p < 0.05) between other compounds and the BLANK group amd the “#” indicates that there were significant differences (p < 0.05) between other compounds and the BLANK + LPS group. Each value represented the means ± SD of three independent experiments.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
NMR data for Compound 1 (TMS as the internal standard, δ in ppm, J in Hz) a.
| No. | δH | δC | No. | δH | δC |
|---|---|---|---|---|---|
| 1 | 196.32 (C) | 8a | 2.99 (1H, m) | 41.04 (CH2) | |
| 2 | 75.37 (C) | 8b | 2.92 (1H, m) | ||
| 3 | 176.04 (C) | 9 | 1.69 (2H, m) | 18.71 (CH2) | |
| 4 | 5.37 (1H, s) | 94.49 (CH) | 10 | 1.01 (3H, t, J = 7.4) | 14.07 (CH3) |
| 5 | 189.34 (C) | 11 | 1.54 (3H, s) | 30.20 (CH3) | |
| 6 | 104.49 (C) | 12 | 3.91 (3H, s) | 57.35 (CH3) | |
| 7 | 203.65 (C) |
a1H-NMR and 13C-NMR data were recorded in CDCl3 at 600 MHz and 150 MHz, respectively.
Table 2.
NMR data for compound 2 and 3 (TMS as the internal standard, δ in ppm, J in Hz) a.
| No. | 2 | 3 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | ||||
| 2 | 161.5 (C) | 161.1 (C) | ||
| 3 | 6.16 (1H, s) | 107.8 (CH) | 6.22 (1H, s) | 109.2 (CH) |
| 4 | 163.0 (C) | 163.0 (C) | ||
| 5 | 7.17 (1H, s) | 120.0 (CH) | 7.01 (1H, s) | 115.5 (CH) |
| 6 | 121.1 (C) | 127.5 (C) | ||
| 7 | 150.0 (C) | 147.8 (C) | ||
| 8 | 133.6 (C) | 136.7 (C) | ||
| 9 | 112.2 (C) | 114.5 (C) | ||
| 10 | 145.5 (C) | 141.3 (C) | ||
| 11 | 3.25 (1H, m) | 28.7 (CH) | 3.25 (1H, m) | 28.7 (CH) |
| 12 | 1.30 (3H, d, J = 6.8) | 22.1 (CH3) | 1.31 (3H, d, J = 6.8) | 21.9 (CH3) |
| 13 | 1.30 (3H, d, J = 6.8) | 22.1 (CH3) | 1.31 (3H, d, J = 6.8) | 21.9 (CH3) |
| 14 | 2.31 (3H, s) | 15.9 (CH3) | 2.32 (3H, s) | 16.3 (CH3) |
| 15 | 4.08 (3H, s) | 61.9 (OCH3) | 3.95 (3H, s) | 60.5 (OCH3) |
a1H-NMR and 13C-NMR data were recorded in CDCl3 at 600 MHz and 150 MHz, respectively.
Table 3.
IC50 values (μM) of cytotoxicity for eight compounds against nine human cancer cell lines.
| Compound | HepG2 | A549 | HeLa | U251 | HOS | MG63 | U2OS | MB231 | SKBR-3 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | - | 45.86 ± 1.64 | 25.37 ± 2.62 | 46.13 ± 1.90 | - | - | - | - | - |
| 2 | 45.52 ± 3.21 | 47.70 ± 2.43 | 15.12 ± 4.01 | 46.14 ± 2.40 | - | - | - | 25.59 ± 2.30 | - |
| 3 | 48.39 ± 2.15 | 38.01 ± 3.56 | - | - | - | - | - | 38.09 ± 2.40 | - |
| 4 | 38.13 ± 1.03 | 37.41 ± 1.24 | 1.24 ± 0.08 | - | - | - | 47.56 ± 2.23 | 41.95 ± 2.35 | - |
| 5 | - | 49.74 ± 3.35 | - | - | - | - | - | - | - |
| 6 | - | 47.42 ± 2.25 | - | - | - | - | - | - | |
| 7 | - | 40.03 ± 0.98 | - | - | - | - | - | - | |
| 8 | 18.02 ± 0.89 | - | 17.76 ± 3.43 | 41.21 ± 1.35 | 43.67 ± 2.52 | 22.76 ± 2.65 | 36.36 ± 1.32 | 39.61 ± 1.50 | 33.40 ± 1.50 |
| Taxol a | 5.32 ± 0.12 | 3.46 ± 0.23 | 0.17 ± 0.02 | 5.02 ± 0.21 | 3.71 ± 0.33 | 5.86 ± 0.24 | 1.01 ± 0.03 | 6.23 ± 0.36 | 3.12 ± 0.25 |
Note: IC50 values represented the means ± SD of six independent experiments and “-” means the IC50 value is above 50 μM. a Taxol was used as a positive control.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, T.; Wang, L.; Duan, D.-H.; Zhang, Y.-H.; Huang, S.-X.; Chang, Y. Cytotoxicity-Guided Isolation of Two New Phenolic Derivatives from Dryopteris fragrans (L.) Schott. Molecules 2018, 23, 1652. https://doi.org/10.3390/molecules23071652
AMA Style
Zhang T, Wang L, Duan D-H, Zhang Y-H, Huang S-X, Chang Y. Cytotoxicity-Guided Isolation of Two New Phenolic Derivatives from Dryopteris fragrans (L.) Schott. Molecules. 2018; 23(7):1652. https://doi.org/10.3390/molecules23071652
Chicago/Turabian StyleZhang, Tong, Li Wang, De-Hua Duan, Yi-Hao Zhang, Sheng-Xiong Huang, and Ying Chang. 2018. "Cytotoxicity-Guided Isolation of Two New Phenolic Derivatives from Dryopteris fragrans (L.) Schott" Molecules 23, no. 7: 1652. https://doi.org/10.3390/molecules23071652