Efficacy of Prosopilosidine from Prosopis glandulosa var. glandulosa against Cryptococcus neoformans Infection in a Murine Model

 ,
, 
Abstract
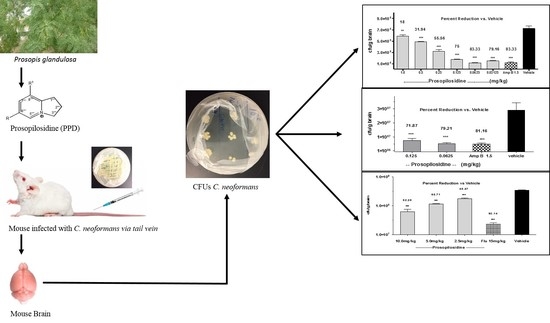
:
1. Introduction
2. Results and Discussion
2.1. In Vitro Toxicity of 1–6 against HepG2 Cells
2.2. In Vivo Anti-Cryptococcal Activity of PPD
2.3. Bid Treatment with PPD
2.4. Oral Administration of PPD
3. Materials and Methods
3.1. Compounds and Chemicals
3.2. Animals
3.3. Inoculum
3.4. In Vitro Cytotoxicity Assay
3.5. Maximum Tolerated Dose
3.6. Experimental Design
3.7. In Vivo Clinical Chemistry of PPD
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Meara, T.R.; Alspaugh, J.A. The Cryptococcus neoformans Capsule: A Sword and a Shield. Clin. Microbiol. Rev. 2012, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Alspaugh, J.A. Virulence Mechanisms and Cryptococcus neoformans pathogenesis. Fungal Genet. Biol. 2015, 78, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.F.; Valencia-Rey, P.A.; Davis, W.B. Pulmonary Cryptococcosis in the Immunocompetent Patient-Many Questions, Some Answers. Open Forum Infect. Dis. 2016, 3, ofw167. [Google Scholar] [CrossRef] [PubMed]
- Lui, G.; Lee, N.; Ip, M.; Choi, K.W.; Tso, Y.K.; Lam, E.; Chau, S.; Lai, R.; Cockram, C.S. Cryptococcosis in apparently immunocompetent patients. QJM 2006, 99, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabiiti, W.; May, R.C. Mechanisms of infection by the human fungal pathogen Cryptococcus neoformans. Future Microbiol. 2012, 7, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- Currie, B.P.; Casadevall, A. Estimation of the prevalence of cryptococcal infection among HIV infected individuals in New York city. Clin. Infect. Dis. 1994, 19, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Chayakulkeeree, M.; Perfect, J.R. Cryptococcosis. Infect. Dis. Clin. N. Am. 2006, 20, 507–544. [Google Scholar] [CrossRef] [PubMed]
- McKenney, J.; Smith, R.M.; Chiller, T.M.; Detels, R.; French, A.; Margolick, J.; Klausner, J.D. Prevalence and correlates of cryptococcal antigen positivity among AIDS patients—United States, 1986–2012. Morb. Mortal. Wkly. Rep. 2014, 63, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.A.; Phelan, M.; Rimland, D.; Graviss, E.; Hamill, R.; Brandt, M.E.; Gardner, T.; Sattah, M.; de Leon, G.P.; Baughman, W.; et al. The changing epidemiology of cryptococcosis: An update from population-based active surveillance in 2 large metropolitan areas, 1992–2000. Clin. Infect. Dis. 2003, 36, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.E.; Hanson, D.; Dworkin, M.S.; Frederick, T.; Bertolli, J.; Lindegren, M.L.; Holmberg, S.; Jones, J.L. Epidemiology of human immunodeficiency virus-associated opportunistic infections in the United States in the era of highly active antiretroviral therapy. Clin. Infect. Dis. 2000, 30, S5–S14. [Google Scholar] [CrossRef] [PubMed]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global Burden of Disease of HIV-Associated Cryptococcal Meningitis: An Updated Analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef]
- Sanglard, D. Emerging Threats in Antifungal-Resistant Fungal Pathogens. Front. Med. Lausanne 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Onyewu, C.; Yoell, H.J.; Ali, R.Y.; Vilgalys, R.J.; Mitchell, T.G. Dynamic and Heterogeneous Mutations to fluconazole resistance in Cryptococcus neoformans. Antimicrob. Agent. Chemother. 2001, 45, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Yamazumi, T.; Pfaller, M.A.; Messer, S.A.; Houston, A.K.; Boyken, L.; Hollis, R.J.; Furuta, I.; Jones, R.N. Characterization of heteroresistance to fluconazole among clinical Isolates of Cryptococcus neoformans. J. Clin. Microbiol. 2003, 41, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Bicanic, T.; Harrison, T.; Niepieklo, A.; Dyakopu, N.; Meintjes, G. Symptomatic relapse of HIV-associated cryptococcal meningitis after initial fluconazole monotherapy: The role of fluconazole resistance and immune reconstitution. Clin. Infect. Dis. 2006, 43, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Seilmaier, M.; Hecht, A.; Guggemos, W.; Rüdisser, K. Cryptococcal Meningoencephalitis Related to HIV Infection with Resistance to Fluconazole, Relapse, and IRIS. Med. Klin. Munich 2009, 104, 58–62. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Rapid Advice: Diagnosis, Prevention and Management of Cryptococcal Disease in HIV-Infected Adults, Adolescents and Children; World Health Organization: Geneva, Switzerland, 2011; Available online: http://www.who.int/hiv/pub/cryptococcal_disease2011/en/ (accessed on 25 January 2018).
- Day, J.N.; Chau, T.T.; Wolbers, M.; Mai, P.P.; Dung, N.T.; Mai, N.H.; Phu, N.H.; Nghia, H.D.; Phong, N.D.; Thai, C.Q.; et al. Combination antifungal therapy for cryptococcal meningitis. N. Engl. J. Med. 2013, 368, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.C.; Palacios, D.S.; Dailey, I.; Endo, M.M.; Uno, B.E.; Wilcock, B.C.; Burke, M.D. Amphotericin primarily kills yeast by simply binding ergosterol. Proc. Natl. Acad. Sci. USA 2012, 109, 2234–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, A.; Scholer, H.J. Mode of action of 5-fluorocytosine and mechanisms of resistance. Chemotherapy 1975, 21, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Diasio, R.B.; Bennett, J.E.; Myers, C.E. Mode of action of 5-fluorocytosine. Biochem. Pharmacol. 1978, 27, 703–707. [Google Scholar] [CrossRef]
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Bicanic, T.; Meintjes, G.; Wood, R.; Hayes, M.; Rebe, K.; Bekker, L.G.; Harrison, T. Fungal burden, early fungicidal activity, and outcome in cryptococcal meningitis in antiretroviral-naive or antiretroviral-experienced patients treated with amphotericin B or fluconazole. Clin. Infect. Dis. 2007, 45, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Rajasingham, R.; Rolfes, M.A.; Birkenkamp, K.E.; Meya, D.B.; Boulware, D.R. Cryptococcal meningitis treatment strategies in resource-limited settings: A cost-effectiveness analysis. PLoS Med. 2012, 9, e1001316. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, C.A.; Frame, P.T. Bone marrow toxicity associated with 5-fluorocytosine therapy. Antimicrob. Agents Chemother. 1977, 11, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Bicanic, T.; Bottomley, C.; Loyse, A.; Brouwer, A.E.; Muzoora, C.; Taseera, K.; Jackson, A.; Phulusa, J.; Hosseinipour, M.C.; van der Horst, C.; et al. Toxicity of Amphotericin B Deoxycholate-Based Induction Therapy in Patients with HIV-Associated Cryptococcal Meningitis. Antimicrob. Agents Chemother. 2015, 59, 7224–7231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falci, D.R.; da Rosa, F.B.; Pasqualotto, A.C. Hematological toxicities associated with amphotericin B formulations. Leuk. Lymphoma 2015, 56, 2889–2894. [Google Scholar] [CrossRef] [PubMed]
- Bukhart, A.A. Monograph of the genus Prosopis. J. Arnold Arbor. 1976, 57, 450–525. [Google Scholar]
- Hilu, Y.W.; Boyd, S.; Felker, P. Morphological diversity and toaxonomy of California mesquites (Prosopis, Lepminosae). Madrono 1982, 29, 237–254. [Google Scholar]
- Snider, B.B.; Neubert, B.J. Syntheses of Ficuseptine, Juliprosine, and Juliprosopine by Biomimetic Intramolecular Chichibabin Pyridine Syntheses. Org. Lett. 2005, 7, 2715–2718. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ahmad, V.; Khalid, M.S.; Ansari, F.A.; Khan, K.A. Study on the Antifungal efficacy of Juliflorine and a Benzene-insoluble alkaloidal fraction of Prosopis juliflora. Philipp. J. Sci. 1997, 126, 175–182. [Google Scholar]
- Samoylenko, V.; Ashfaq, M.K.; Jacob, M.R.; Tekwani, B.L.; Khan, S.I.; Manly, S.P.; Joshi, V.C.; Walker, L.A.; Muhammad, I. Indolidine, Antiinfective and Antiparasitic Compounds from Prosopis glandulosa var. glandulosa. J. Nat. Prod. 2009, 72, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Primer on BELLE. In Biological Effects of Low Level Exposures: Dose-Response Relationships; Calabrese, E.J., Ed.; CRC/Lewis Publishers: Boca Raton, FL, USA, 1994; pp. 27–42. [Google Scholar]
- Calabrese, E.J.; Baldwin, L.A. Hormesis as a biological hypothesis. Environ. Health Perspect. 1998, 106, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Klaassen, C.D. Perfluorocarboxylic Acids Induce Cytochrome P450 Enzymes in Mouse Liver through Activation of PPAR-α and CAR Transcription Factors. Toxicol. Sci. 2008, 106, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of compounds 1–6 mentioned in this manuscript are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose of 1 | Mean Body Weight of Mice on Day | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Vehicle | 24.40 ± 1.36 | 24.58 ± 1.4 | 24.37 ± 1.37 | 24.47 ± 1.41 | 24.55 ± 1.39 | 25.28 ± 0.88 |
| 1.0 mg/kg | 24.83± 1.79 | 24.79 ± 1.81 | 23.66 ± 1.02 | 23.81 ± 0.95 | 24.28 ± 0.96 | 24.39 ± 0.93 |
| 2.5 mg/kg | 24.8 ± 0.58 | 25.01 ± 0.68 | 25.1 ± 0.62 | 24.89 ± 0.37 | 24.85 ± 0.29 | 24.94 ± 0.28 |
| 5.0 mg/kg | 23.55 ± 0.77 | - | - | - | - | - |
| 10 mg/kg | 25.12 ± 0.66 | - | - | - | - | - |
| Parameters | Vehicle | Prosopilosidine (20 mg/kg) |
|---|---|---|
| Albumin (g/dL) | 3.5 ± 0 | 3.1 ± 0 |
| Alanine transaminase (ALT) U/L | 35.0 ± 0 | 54.5 ± 26.5 |
| Total bilirubin (mg/dL) | 0.3 ± 0 | 0.2 ± 0 |
| Blood urea nitrogen (BUN) mg/dL | 21.0 ± 1 | 17.5 ± 0.5 |
| Creatinine (CRE) mg/dL | 0.245 ± 0.05 | 0.3 ± 0 |
| BUN/CRE | 89.3 ± 15.97 | 58.33 ± 1.67 |
| Calcium (mg/dL) | 10.65 ± 0.05 | 10.0 ± 0.3 |
| Phosphate (mg/dL) | 7.3 ± 0.1 | 9.5 ± 0 |
| Glucose (mg/dL) | 70.0 ± 9 | 145.5 ± 29.5 |
| Na+ (mmol/L) | 156.5 ± 2.5 | 150.5 ± 0.5 |
| K+ (mmol/L) | 8.4 ± 0.2 | 5.45 ± 0.25 |
| Total protein (g/dL) | 5.4 ± 0 | 5.0 ± 0 |
| Globulin (g/dL) | 1.95 ± 0.05 | 1.9 ± 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashfaq, M.K.; Abdel-Bakky, M.S.; Tahir Maqbool, M.; Samoylenko, V.; Abdur Rahman, A.; Muhammad, I. Efficacy of Prosopilosidine from Prosopis glandulosa var. glandulosa against Cryptococcus neoformans Infection in a Murine Model. Molecules 2018, 23, 1674. https://doi.org/10.3390/molecules23071674
Ashfaq MK, Abdel-Bakky MS, Tahir Maqbool M, Samoylenko V, Abdur Rahman A, Muhammad I. Efficacy of Prosopilosidine from Prosopis glandulosa var. glandulosa against Cryptococcus neoformans Infection in a Murine Model. Molecules. 2018; 23(7):1674. https://doi.org/10.3390/molecules23071674
Chicago/Turabian StyleAshfaq, Mohammad K., Mohamed Sadek Abdel-Bakky, Mir Tahir Maqbool, Volodymyr Samoylenko, Aziz Abdur Rahman, and Ilias Muhammad. 2018. "Efficacy of Prosopilosidine from Prosopis glandulosa var. glandulosa against Cryptococcus neoformans Infection in a Murine Model" Molecules 23, no. 7: 1674. https://doi.org/10.3390/molecules23071674