
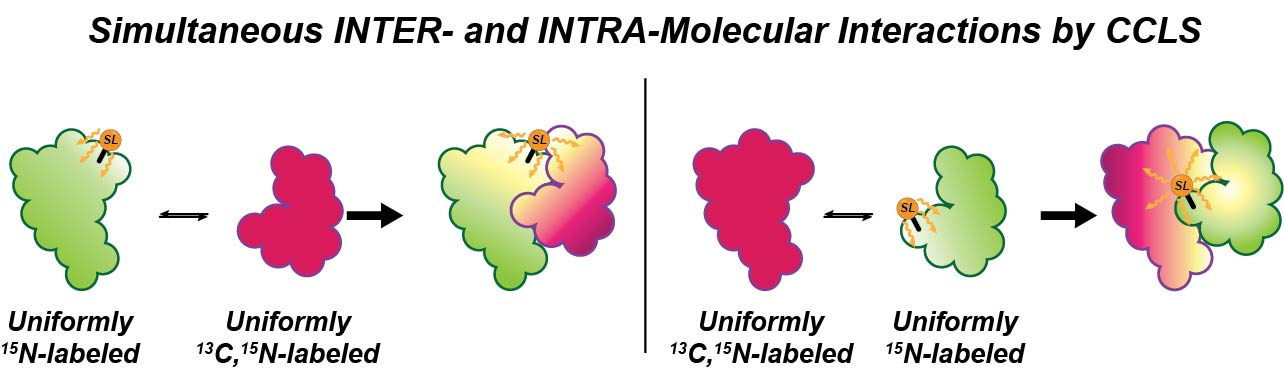
Probing Protein-Protein Interactions Using Asymmetric Labeling and Carbonyl-Carbon Selective Heteronuclear NMR Spectroscopy

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
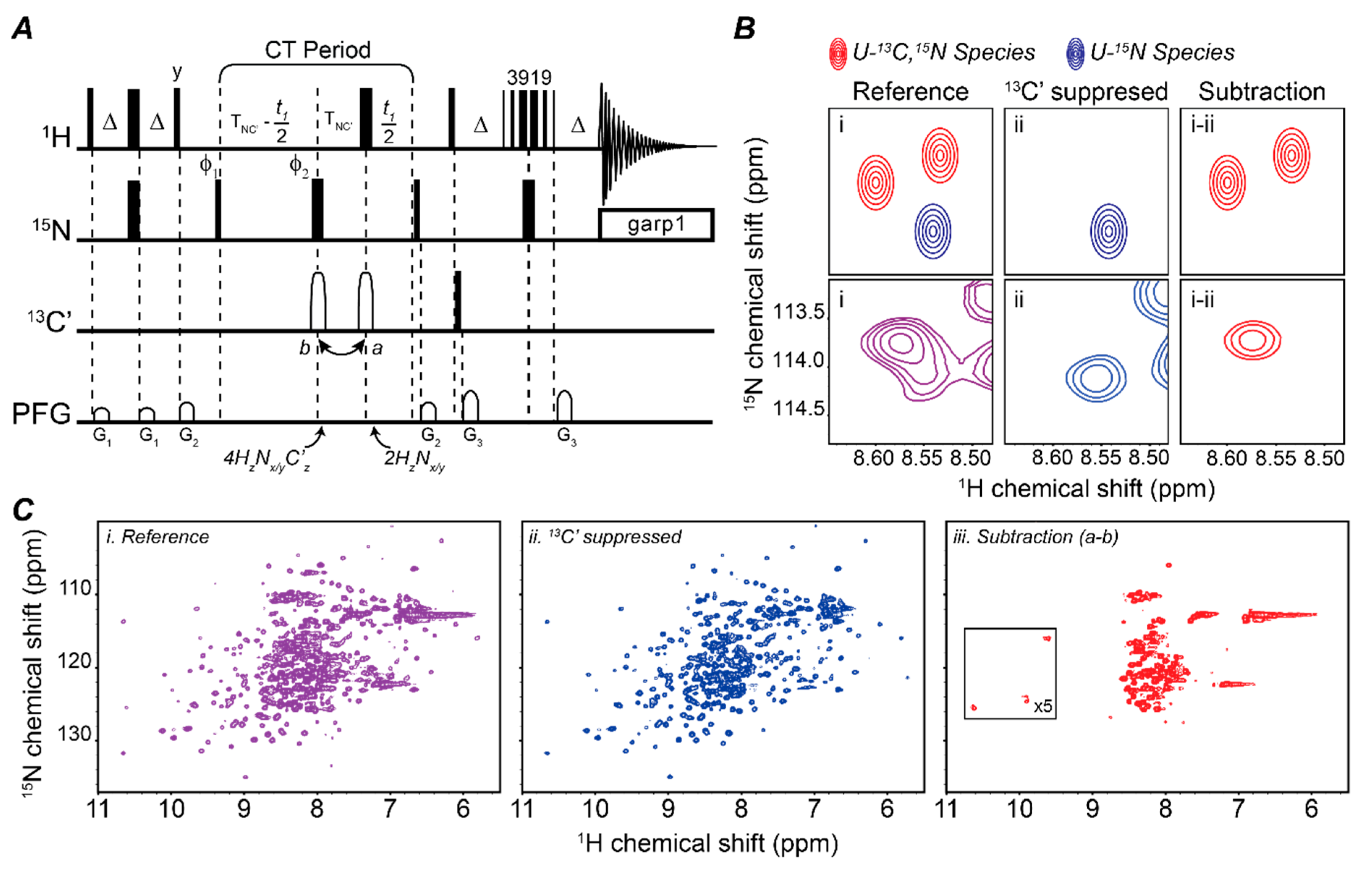
2.1. Mapping Two Binding Partners Fingerprints Simultaneously
2.2. Fingerprinting Three Binding Partners Using One Sample
2.3. Measuring Residual Dipolar Coupling (RDC) of Complexes Using One Sample
2.4. Measuring Long-Range Distances and Transient Complexes Using CCLS for Paramagnetic Relaxation Enhancements (PRE)
2.5. Improving Sensitivity with the G5 Pulse
3. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Marintchev, A.; Frueh, D.; Wagner, G. NMR methods for studying protein–protein interactions involved in translation initiation. In Methods in Enzymology; Lorsch, J., Ed.; Academic Press: Cambridge, MA, USA, 2007; pp. 283–331. [Google Scholar]
- Tord, B.; Sara, L.; Peter, J. Methods for the detection and analysis of protein–protein interactions. Proteomics 2007, 7, 2833–2842. [Google Scholar]
- Ngounou Wetie, A.G.; Izabela, S.; Alisa, G.W.; Urmi, R.; Joseph, A.L.; Costel, C.D. Investigation of stable and transient protein-protein interactions: Past, present and future. Proteomics 2013, 13, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Barile, E.; Pellecchia, M. NMR-Based Approaches for the Identification and Optimization of Inhibitors of Protein–Protein Interactions. Chem. Rev. 2014, 114, 4749–4763. [Google Scholar] [CrossRef] [PubMed]
- Zuiderweg, E.R.P. Mapping protein−protein interactions in solution by NMR Spectroscopy. Biochemistry 2002, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Shimada, I. An NMR Method to study protein–protein interactions. In Integrin and Cell. Adhesion Molecules: Methods and Protocols; Shimaoka, M., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 129–137. [Google Scholar]
- Ishima, R.; Torchia, D.A. Protein dynamics from NMR. Nat. Struct. Biol. 2000, 7, 740. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.E. Protein dynamics from NMR. Biochem. Cell Biol. 1998, 76, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kempf, J.G.; Loria, J.P. Protein dynamics from solution NMR. Cell Biochem. Biophys. 2002, 37, 187–211. [Google Scholar] [CrossRef]
- Olivieri, C. Simultaneous detection of intra- and inter-molecular paramagnetic relaxation enhancements in protein complexes. J. Biomol. NMR 2018, 70, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Wang, V.S.; Radoicic, J.; De Angelis, A.A.; Berkamp, S.; Opella, S.J. Paramagnetic relaxation enhancement of membrane proteins by incorporation of the metal-chelating unnatural amino acid 2-amino-3-(8-hydroxyquinolin-3-yl)propanoic acid (HQA). J. Biomol. NMR 2015, 61, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Schwieters, C.D.; Tang, C. Theory and practice of using solvent paramagnetic relaxation enhancement to characterize protein conformational dynamics. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Riek, R.; Wider, G.; Wüthrich, K. Transverse Relaxation-Optimized Spectroscopy (TROSY) for NMR Studies of Aromatic Spin Systems in 13C-Labeled Proteins. J. Am. Chem. Soc. 1998, 120, 6394–6400. [Google Scholar] [CrossRef]
- Tugarinov, V.; Hwang, P.M.; Kay, L.E. Nuclear magnetic resonance spectroscopy of high.-molecular-weight proteins. Annu. Rev. Biochem. 2004, 73, 107–146. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.H.; Zhang, X.; Gehring, K.; Kay, L.E. Solution NMR studies of a 42 kda escherichia coli maltose binding protein/β-cyclodextrin complex.: Chemical shift assignments and analysis. J. Am. Chem. Soc. 1998, 120, 11738–11748. [Google Scholar] [CrossRef]
- Walters, K.J.; Ferentz, A.E.; Hare, B.J.; Hidalgo, P.; Jasanoff, A.; Matsuo, H.; Wagner, G. Characterizing protein-protein complexes and oligomers by nuclear magnetic resonance spectroscopy. In Methods in Enzymology; James, T.L., Dötsch, V., Schmitz, U., Eds.; Academic Press: Cambridge, MA, USA, 2001; pp. 238–258. [Google Scholar]
- Göbl, C.; Madl, T.; Simon, B.; Sattler, M. NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog. Nuclear Mag. Res. Spect. 2014, 80, 26–63. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Predicting Protein-Protein Interactions from the Molecular to the Proteome Level. Chem. Rev. 2016, 116, 4884–4909. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Wang, J.; Sun, F. Protein-protein interactions among signaling pathways may become new therapeutic targets in liver cancer. Oncol. Rep. 2016, 35, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Braun, P.; Gingras, A.C. History of protein-protein interactions: From egg-white to complex networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Guerois, R. Evolution of protein interactions: From interactomes to interfaces. Arch. Biochem. Biophys. 2014, 554, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Lage, K. Protein-protein interactions and genetic diseases: The interactome. Biochim. Biophys. Acta 2014, 1842, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kim, Y.C.; Mittal, J. Protein-protein interactions in a crowded environment. Biophys. Rev. 2013, 5, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Vuister, G.W.; Wang, A.C.; Bax, A. Measurement of three-bond nitrogen-carbon J. couplings in proteins uniformly enriched in nitrogen-15 and carbon-13. J. Am. Chem. Soc. 1993, 115, 5334–5335. [Google Scholar] [CrossRef]
- Montelione, G.T.; Winkler, M.E.; Rauenbuehler, P.; Wagner, G. Accurate measurements of long-range heteronuclear coupling constants from homonuclear 2D NMR spectra of isotope-enriched proteins. J. Magn. Reson. 1989, 82, 198–204. [Google Scholar] [CrossRef]
- Wider, G.; Neri, D.; Otting, G.; Wüjthrich, K. A heteronuclear three-dimensional NMR experiment for measurements of small heteronuclear coupling constants in biological macromolecules. J. Magn. Reson. 1989, 85, 426–431. [Google Scholar] [CrossRef]
- Kurz, M.; Schmieder, P.; Kessler, H. HETLOC, an efficient method for determining heteronuclear long-range couplings with heteronuclei in natural abundance. Angew. Chem. Int. Ed. 1991, 30, 1329–1331. [Google Scholar] [CrossRef]
- Edison, A.S.; Westler, W.M.; Markley, J.L. Elucidation of amino acid spin systems in proteins and determination of heteronuclear coupling constants by carbon-proton-proton three-dimensional NMR. J. Magn. Reson. 1991, 92, 434–438. [Google Scholar] [CrossRef]
- Xu, R.X.; Olejniczak, E.T.; Fesik, S.W. Stereospecific assignments and χ1 rotamers for FKBP when bound to ascomycin from 3JHα,Hβ and 3JN,Hβ coupling constants. FEBS Lett. 1992, 305, 137–143. [Google Scholar] [CrossRef]
- Sattler, M.; Schwalbe, H.; Griesinger, C. Stereospecific assignment of leucine methyl groups with carbon-13 in natural abundance or with random 13C labeling. J. Am. Chem. Soc. 1992, 114, 1126–1127. [Google Scholar] [CrossRef]
- Tonelli, M.; Masterson, L.R.; Hallenga, K.; Veglia, G.; Markley, J.L. Carbonyl carbon label selective (CCLS) (1)H–(15)N HSQC experiment for improved detection of backbone (13)C–(15)N cross peaks in larger proteins. J. Biomol. NMR 2007, 39, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Yang, J.; Wu, J.; Haste, N.M.; Radzio-Andzelm, E.; Anand, G. PKA: A portrait of protein kinase dynamics. BBA 2004, 1697, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kim, C.; Vigil, D.; Haste, N.M.; Yang, J.; Wu, J.; Anand, G.S. Dynamics of signaling by PKA. BBA 2005, 1754, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Langer, T.; Vogtherr, M.; Elshorst, B.; Betz, M.; Schieborr, U.; Saxena, K.; Schwalbe, H. NMR backbone assignment of a protein kinase catalytic domain by a combination of several approaches: Application to the catalytic subunit of camp-dependent protein kinase. ChemBioChem 2004, 5, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Langer, T.; Sreeramulu, S.; Vogtherr, M.; Elshorst, B.; Betz, M.; Schieborr, U.; Schwalbe, H. Folding and activity of cAMP-dependent protein kinase mutants. FEBS Lett. 2005, 579, 4049–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogtherr, M.; Saxena, K.; Grimme, S.; Betz, M.; Schieborr, U.; Pescatore, B.; Schwalbe, H. NMR backbone assignment of the mitogen-activated protein (MAP) kinase p38. J. Biomol. NMR 2005, 32, 175. [Google Scholar] [CrossRef] [PubMed]
- Vogtherr, M.; Saxena, K.; Hoelder, S.; Grimme, S.; Betz, M.; Schieborr, U.; Wendt, K.U. NMR Characterization of Kinase p38 Dynamics in Free and Ligand-Bound Forms. Angew. Chem. Int. Ed. 2006, 45, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Masterson, L.R.; Shi, L.; Tonelli, M.; Mascioni, A.; Mueller, M.M.; Veglia, G. Backbone NMR resonance assignment of the catalytic subunit of cAMP-dependent protein kinase A in complex with AMP-PNP. Biomol. NMR Assign. 2009, 3, 115–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masterson, L.R.; Shi, L.; Metcalfe, E.; Gao, J.; Taylor, S.S.; Veglia, G. Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 6969–6974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.K.; McDonald, L.R.; Cembran, A.; Kim, J.; Masterson, L.R.; McClendon, C.L.; Veglia, G. Synchronous opening and closing motions are essential for camp-dependent protein kinase a signaling. Structure 2014, 22, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ahuja, L.G.; Chao, F.A.; Xia, Y.; McClendon, C.L.; Kornev, A.P.; Veglia, G. A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci. Adv. 2017, 3, e1600663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, G.D.; Dewey, W.L. Protein kinase inhibitor peptide (PKI): A family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides 2006, 40, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Abeygunawardana, C.; Johnson, M.O.; Vanzijl, P.C.M. Improved Sensitivity of HSQC Spectra of Exchanging Protons at Short Interscan Delays Using a New Fast HSQC (FHSQC) Detection Scheme That Avoids Water Saturation. J. Magn. Reson. Ser. B 1995, 108, 94–98. [Google Scholar] [CrossRef]
- Masterson, L.R.; Tonelli, M.; Markley, J.L.; Veglia, G. Simultaneous detection and deconvolution of congested NMR spectra containing three isotopically labeled species. J. Am. Chem. Soc. 2008, 130, 7818–7819. [Google Scholar] [CrossRef] [PubMed]
- Wirmer, J.; Schwalbe, H. Angular dependence of 1J(Ni,Cαi) and 2J(Ni,Cαi−1) coupling constants measured in J-modulated HSQCs. J. Biomol. NMR 2002, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Traaseth, N.J.; Verardi, R.; Veglia, G. Asymmetric methyl group labeling as a probe of membrane protein homo-oligomers by NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 2400–2401. [Google Scholar] [CrossRef] [PubMed]
- Verardi, R.; Traaseth, N.J.; Masterson, L.R.; Vostrikov, V.V.; Veglia, G. Isotope labeling for solution and solid-state NMR spectroscopy of membrane proteins. Adv. Exp. Med. Biol. 2012, 992, 35–62. [Google Scholar] [PubMed]
- Atreya, H.S. Isotope Labeling in Biomolecular NMR; Springer: Dordrecht, The Netherlands; New York, NY, USA, 2012; p. 219. [Google Scholar]
- Anglister, J.; Srivastava, G.; Naider, F. Detection of intermolecular NOE interactions in large protein complexes. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Anglister, J.; Levy, R.; Scherf, T. Interactions of antibody aromatic residues with a peptide of cholera toxin observed by two-dimensional transferred nuclear Overhauser effect difference spectroscopy. Biochemistry 1989, 28, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Scherf, T.; Anglister, J. A T1 rho-filtered two-dimensional transferred NOE spectrum for studying antibody interactions with peptide antigens. Biophys. J. 1993, 64, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.F.; Vallurupalli, P.; Lundström, P.; Neudecker, P.; Kay, L.E. Probing chemical shifts of invisible states of proteins with relaxation dispersion NMR spectroscopy: How well can we do? J. Am. Chem. Soc. 2008, 130, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- Lundström, P.; Teilum, K.; Carstensen, T.; Bezsonova, I.; Wiesner, S.; Hansen, D.F.; Tomasz, L.R.; Mikael, A.; Kay, L. EFractional 13C enrichment of isolated carbons using [1-13C]- or [2-13C]-glucose facilitates the accurate measurement of dynamics at backbone Cα and side-chain methyl positions in proteins. J. Biomol. NMR 2007, 38, 199–212. [Google Scholar] [CrossRef] [PubMed]
- LeMaster, D.M.; Kushlan, D.M. Dynamical mapping of E. coli thioredoxin via 13C NMR relaxation analysis. J. Am. Chem. Soc. 1996, 118, 9255–9264. [Google Scholar] [CrossRef]
- Takeuchi, K.; Ng, E.; Malia, T.J.; Wagner, G. 1-13C amino acid selective labeling in a 2H15N background for NMR studies of large proteins. J. Biomol. NMR 2007, 38, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Sun, Z.-Y.J.; Wagner, G. Alternate 13C-12C labeling for complete mainchain resonance assignments using Cα direct-detection with applicability toward fast relaxing protein systems. J. Am. Chem. Soc. 2008, 130, 17210–17211. [Google Scholar] [CrossRef] [PubMed]
- Prestegard, J.H.; Bougault, C.M.; Kishore, A.I. Residual dipolar couplings in structure determination of biomolecules. Chem. Rev. 2004, 104, 3519–3540. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, R.S.; Tjandra, N. Residual dipolar couplings in NMR structure analysis. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 387–413. [Google Scholar] [CrossRef] [PubMed]
- Bax, A.; Grishaev, A. Weak alignment NMR: A hawk-eyed view of biomolecular structure. Curr. Opin. Struct. Biol. 2005, 15, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Tolman, J.R.; Ruan, K. NMR residual dipolar couplings as probes of biomolecular dynamics. Chem. Rev. 2006, 106, 1720–1736. [Google Scholar] [CrossRef] [PubMed]
- Ottiger, M.; Delaglio, F.; Bax, A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 1998, 131, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Gronenborn, A.M. Sensitivity-enhanced 2D IPAP, TROSY-anti-TROSY, and E.COSY experiments: Alternatives for measuring dipolar 15N–1HN couplings. J. Magn. Reson. 2003, 163, 208–214. [Google Scholar] [CrossRef]
- Tonelli, M.; Masterson, L.R.; Cornilescu, G.; Markley, J.L.; Veglia, G. One-sample approach to determine the relative orientations of proteins in ternary and binary complexes from residual dipolar coupling measurements. J. Am. Chem. Soc. 2009, 131, 14138–14139. [Google Scholar] [CrossRef] [PubMed]
- Sharff, A.J.; Rodseth, L.E.; Spurlino, J.C.; Quiocho, F.A. Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry 1992, 31, 10657–10663. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, S.; Bugg, C.E.; Cook, W.J. Structure of ubiquitin refined at 1.8 Åresolution. J. Mol. Biol. 1987, 194, 531–544. [Google Scholar] [CrossRef]
- Clore, G.M.; Iwahara, J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009, 109, 4108–4139. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I. Relaxation processes in a system of two spins. Phys. Rev. 1955, 99, 559–565. [Google Scholar] [CrossRef]
- Iwahara, J.; Tang, C.; Clore, G.M. Practical aspects of (1)h transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 2007, 184, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Janowska, M.K.; Baum, J. Intermolecular paramagnetic relaxation enhancement (pre) studies of transient complexes in intrinsically disordered proteins. Methods Mol. Biol. 2016, 1345, 45–53. [Google Scholar] [PubMed]
- Newby, F.N.; De Simone, A.; Yagi-Utsumi, M.; Salvatella, X.; Dobson, C.M.; Vendruscolo, M. Structure-free validation of residual dipolar coupling and paramagnetic relaxation enhancement measurements of disordered proteins. Biochemistry 2015, 54, 6876–6886. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D. Distance information for disordered proteins from NMR and ESR measurements using paramagnetic spin labels. Methods Mol. Biol. 2012, 895, 127–138. [Google Scholar] [PubMed]
- Mainz, A.; Religa, T.L.; Sprangers, R.; Linser, R.; Kay, L.E.; Reif, B. NMR spectroscopy of soluble protein complexes at one mega-dalton and beyond. Angew. Chem. Int. Ed. 2013, 52, 8746–8751. [Google Scholar] [CrossRef] [PubMed]
- Tzakos, A.G.; Grace, C.R.; Lukavsky, P.J.; Riek, R. NMR techniques for very large proteins and rnas in solution. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 319–342. [Google Scholar] [CrossRef] [PubMed]
- Manu, V.S.; Veglia, G. Genetic algorithm optimized triply compensated pulses in NMR spectroscopy. J. Magn. Reson. 2015, 260, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Rossi, P.; Subrahmanian, M.V.; Huang, C.; Saleh, T.; Olivieri, C.; Veglia, G. Enhancing the sensitivity of multidimensional NMR experiments by using triply-compensated pi pulses. J. Biomol. NMR 2017, 69, 237–243. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsen, E.K.; Olivieri, C.; Walker, C.; V.S., M.; Gao, J.; Bernlohr, D.A.; Tonelli, M.; Markley, J.L.; Veglia, G. Probing Protein-Protein Interactions Using Asymmetric Labeling and Carbonyl-Carbon Selective Heteronuclear NMR Spectroscopy. Molecules 2018, 23, 1937. https://doi.org/10.3390/molecules23081937
Larsen EK, Olivieri C, Walker C, V.S. M, Gao J, Bernlohr DA, Tonelli M, Markley JL, Veglia G. Probing Protein-Protein Interactions Using Asymmetric Labeling and Carbonyl-Carbon Selective Heteronuclear NMR Spectroscopy. Molecules. 2018; 23(8):1937. https://doi.org/10.3390/molecules23081937
Chicago/Turabian StyleLarsen, Erik K., Cristina Olivieri, Caitlin Walker, Manu V.S., Jiali Gao, David A. Bernlohr, Marco Tonelli, John L. Markley, and Gianluigi Veglia. 2018. "Probing Protein-Protein Interactions Using Asymmetric Labeling and Carbonyl-Carbon Selective Heteronuclear NMR Spectroscopy" Molecules 23, no. 8: 1937. https://doi.org/10.3390/molecules23081937
APA StyleLarsen, E. K., Olivieri, C., Walker, C., V.S., M., Gao, J., Bernlohr, D. A., Tonelli, M., Markley, J. L., & Veglia, G. (2018). Probing Protein-Protein Interactions Using Asymmetric Labeling and Carbonyl-Carbon Selective Heteronuclear NMR Spectroscopy. Molecules, 23(8), 1937. https://doi.org/10.3390/molecules23081937