
Evaluation of Nitrobenzyl Derivatives of Camptothecin as Anti-Cancer Agents and Potential Hypoxia Targeting Prodrugs

 and
and
Abstract
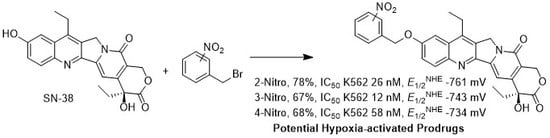
:
1. Introduction
2. Results
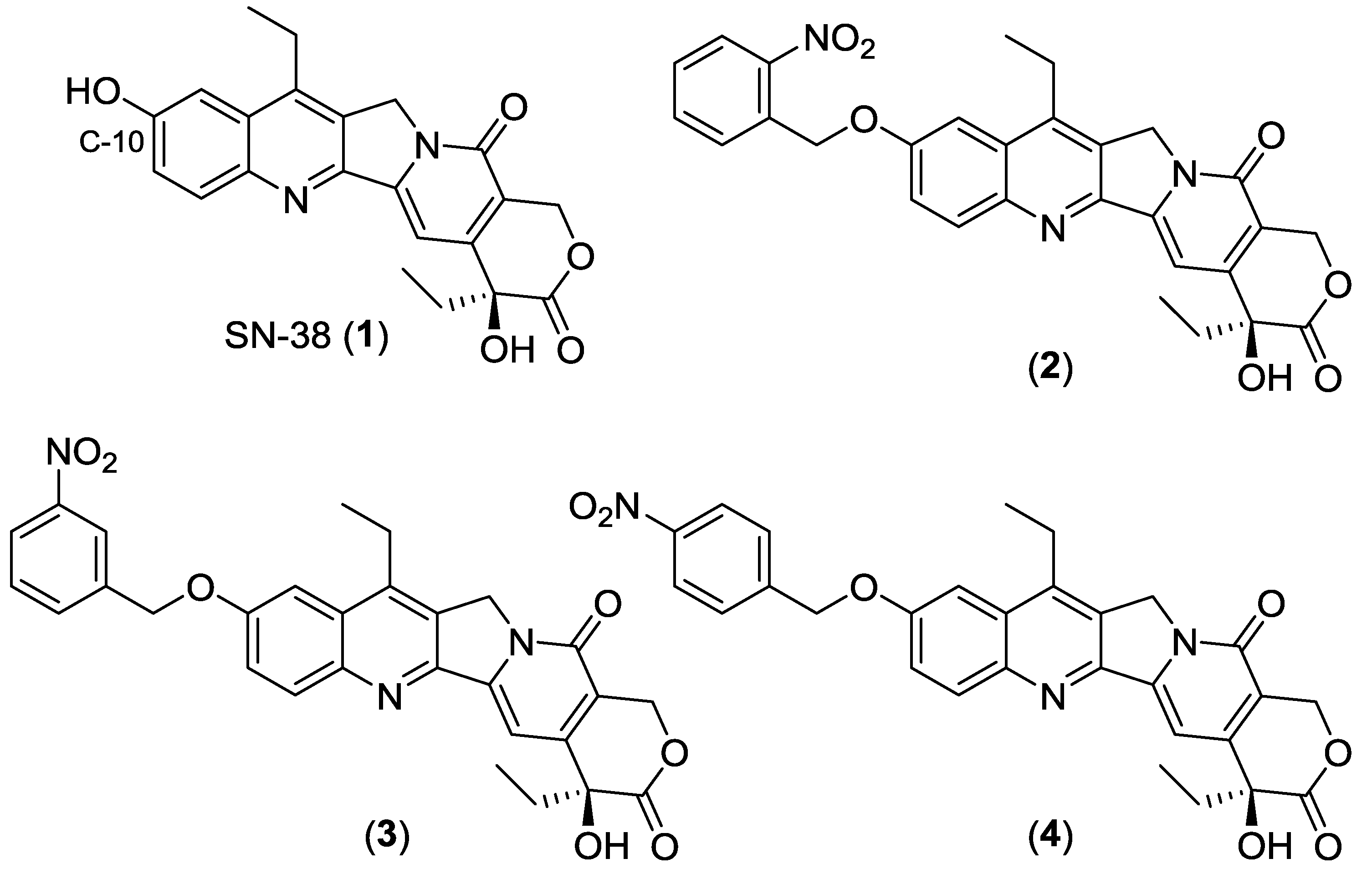
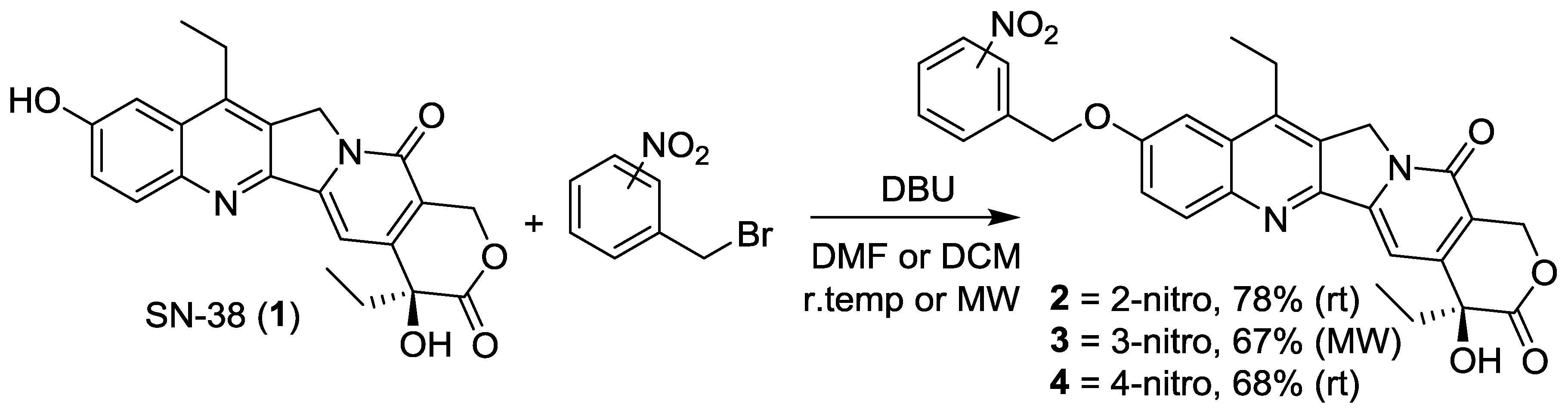
2.1. Synthesis of Analogs 2, 3 and 4
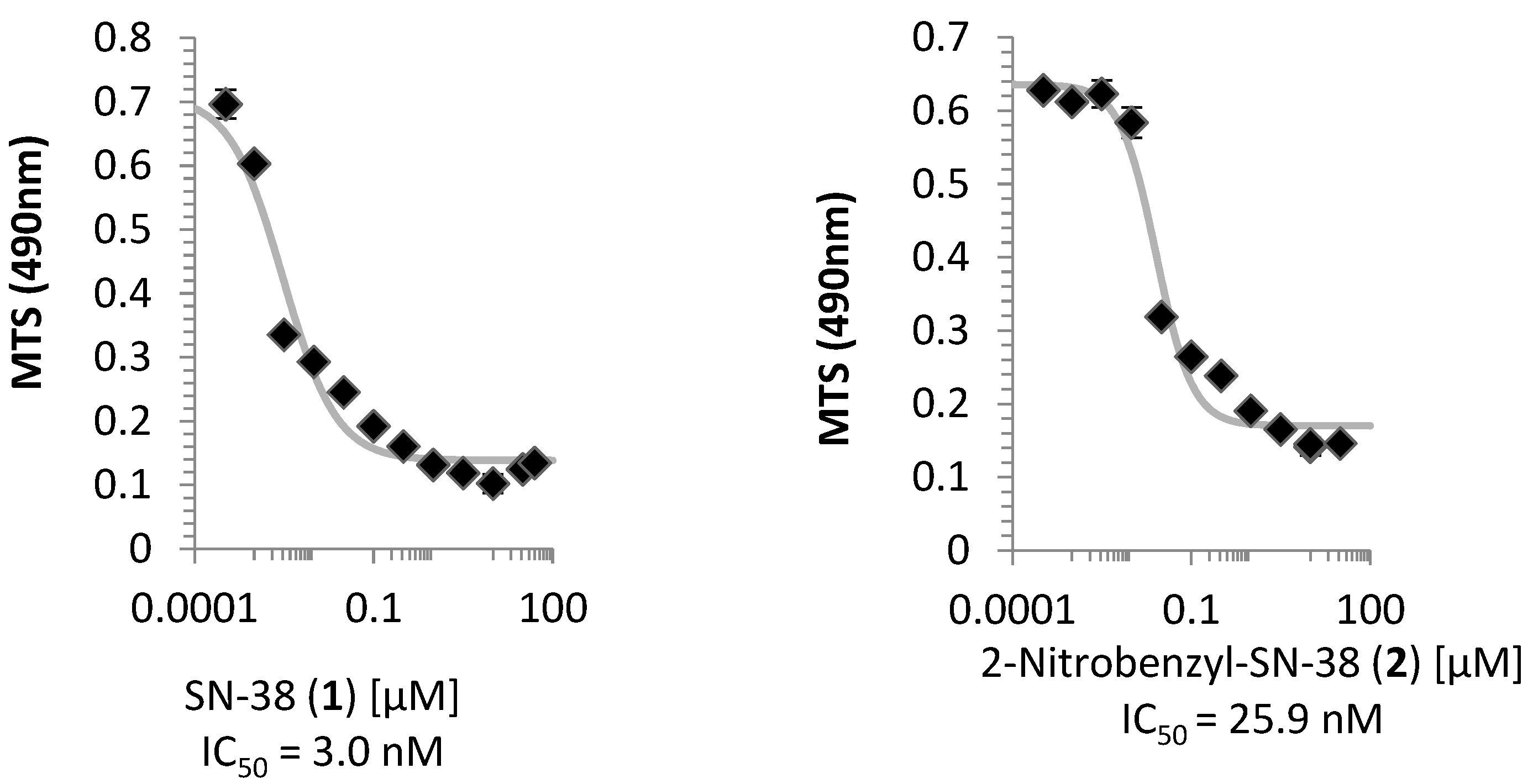
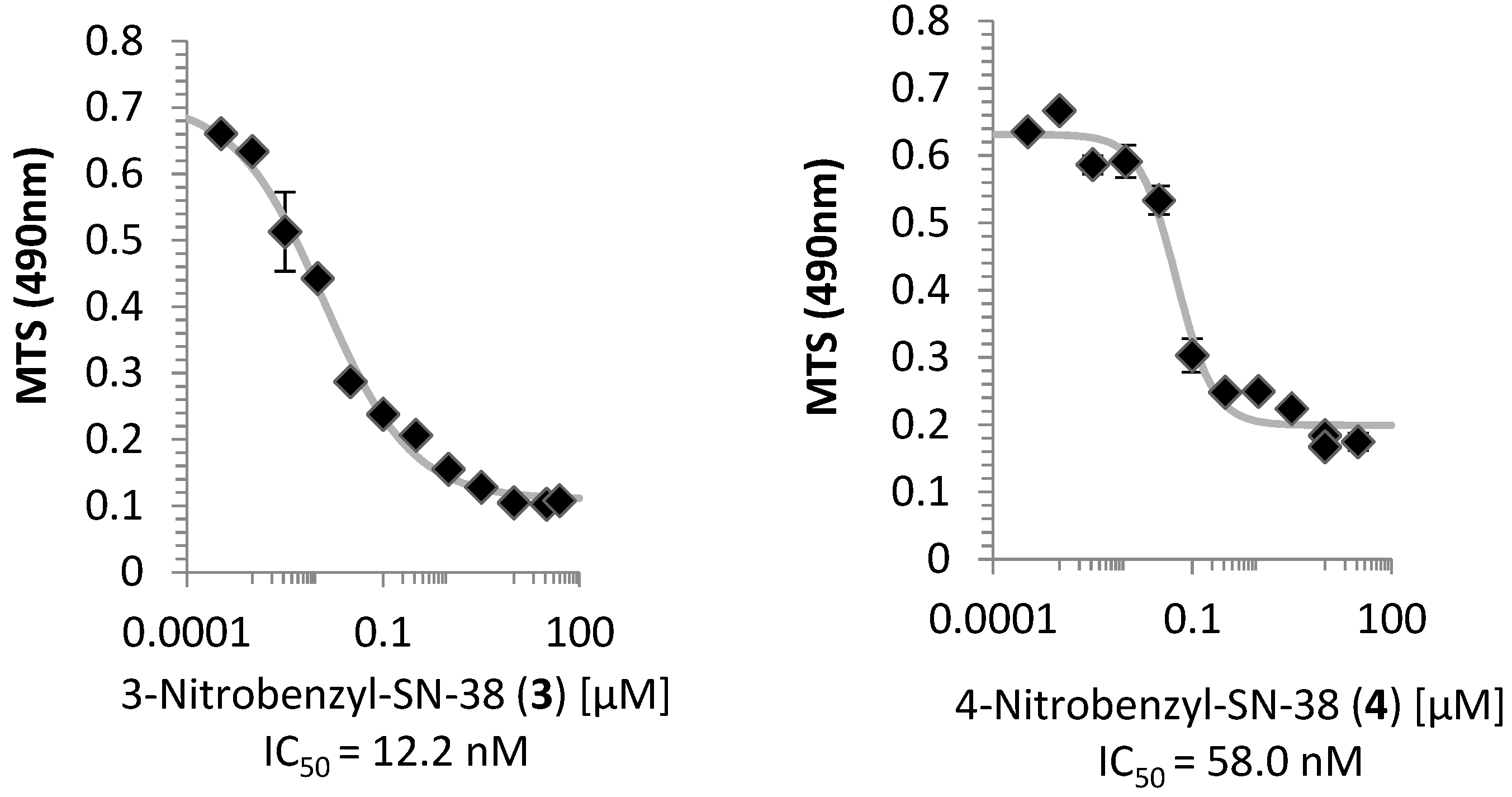
2.2. Cell Viability Assay
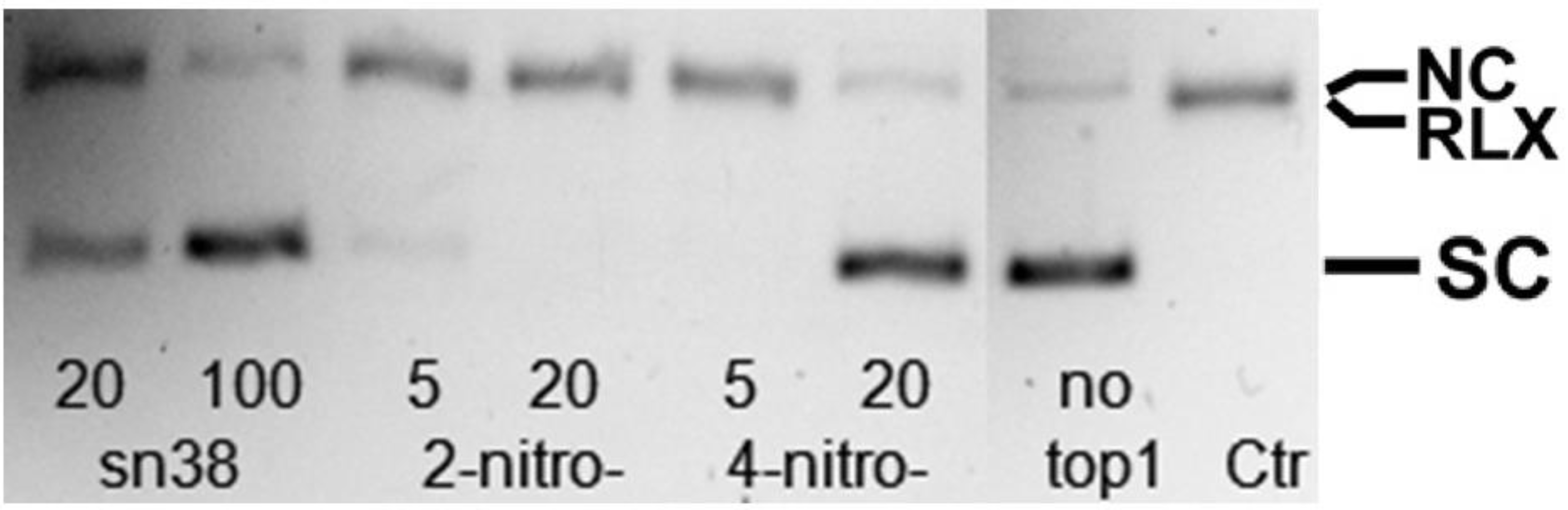
2.3. Topoisomerase I Inhibition Assay
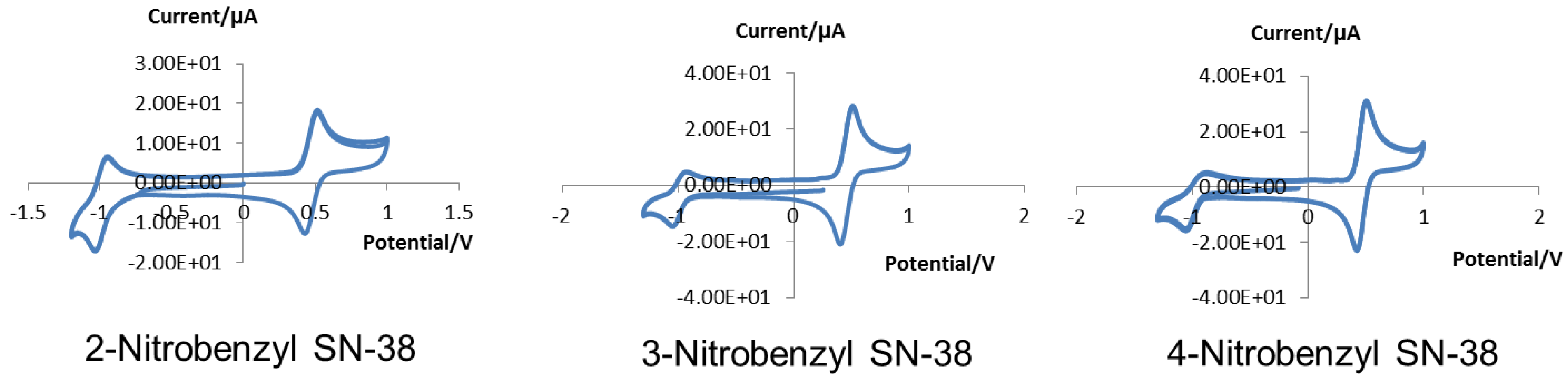
2.4. Cyclic Voltammetry
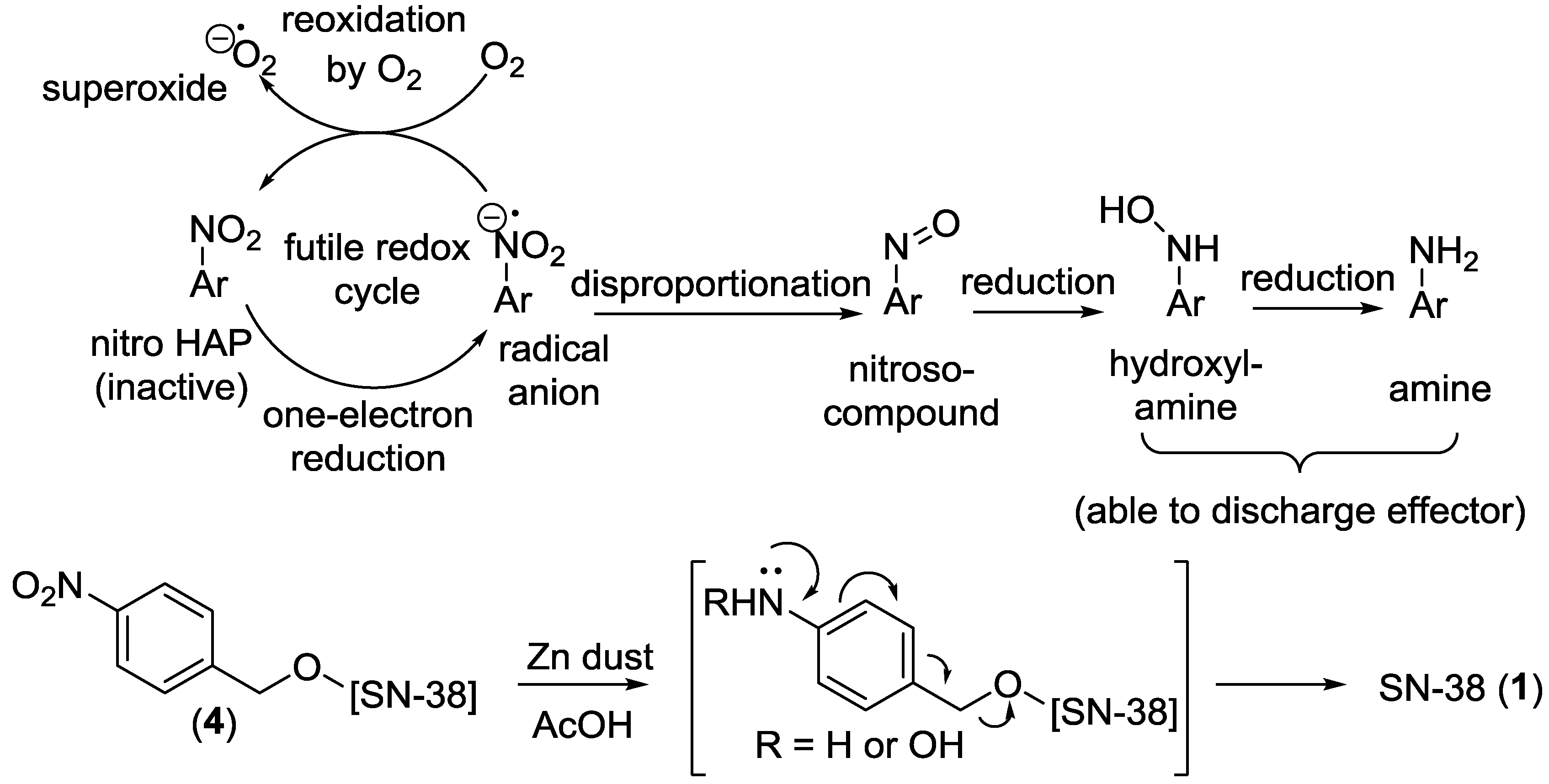
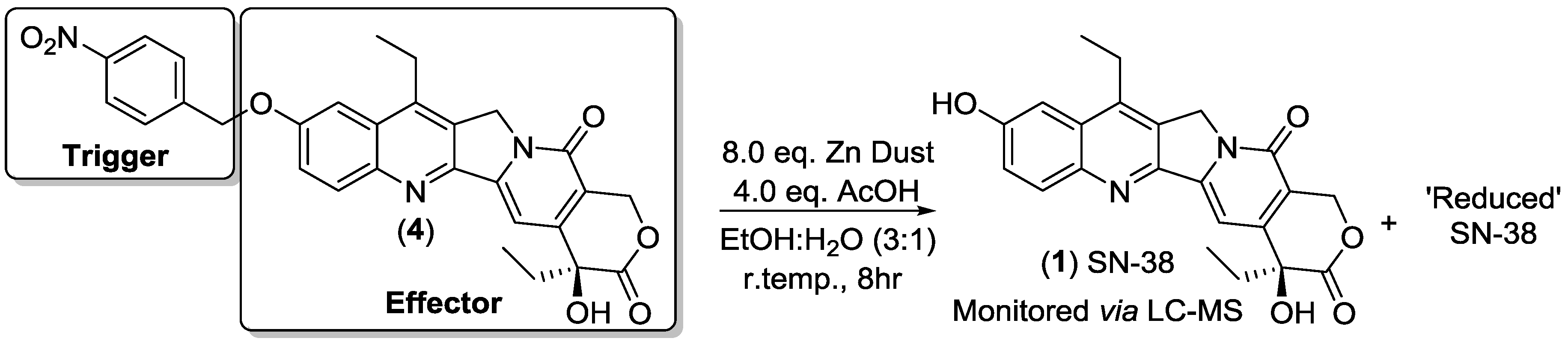
2.5. Chemical Reduction of Prodrug
3. Discussion
3.1. Cell Viability and Topoisomerase I Inhibition Assay
3.2. Cyclic Voltammetry
3.3. Reduction of Prodrug
4. Materials and Methods
4.1. General Information
4.2. Synthesis and Characterization of 2-Nitrobenzyl-C10-SN-38 (2)
4.3. Synthesis and Characterization of 3-Nitrobenzyl-C10-SN-38 (3)
4.4. Synthesis and Characterization of 4-Nitrobenzyl-C10-SN-38 (4)
4.5. Topoisomerase I Inhibitory Assay
4.6. Cell Viability Assay
4.7. Reduction of Prodrug 4-Nitrobenzyl-C10-SN-38 (4)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DBU | Diazabicyclo[5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DMSO | Dimethyl Sulfoxide |
| FMN | Flavin Mononucleotide |
| iNOS | Inducible Nitric Oxide Synthase |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MTRR | Methionine Synthase Reductase |
| NDOR | NADPH Dependent Diflavin Oxidoreductase 1 |
| NHE | Normal Hydrogen Electrode |
| POR | NADPH:cytochrome P450 Oxidoreductase |
| SCE | Saturated Calomel Electrode |
| 1H-NMR | Proton Nuclear Magnetic Resonance |
| 13C-NMR | Carbon Nuclear Magnetic Resonance |
References
- Pizzolato, J.F.; Saltz, L.B. The camptothecins. Lancet 2003, 361, 2235–2242. [Google Scholar] [CrossRef]
- Höckel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors: A Review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Höckel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schäffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Cater, D.B.; Phillips, A.F. Measurement of electrode potentials in living and dead tissues. Nature 1954, 174, 121–123. [Google Scholar] [CrossRef]
- Jiang, J.; Auchinvole, C.; Fisher, K.; Campbell, C.J. Quantitative measurement of redox potential in hypoxic cells using SERS nanosensors. Nanoscale 2014, 6, 12104–12110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy. Int. J. Radiat. Oncol. Boil. Phys. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Wilson, W.R.; Hay, M.P. Recent developments in the design of bioreductive drugs. Br. J. Cancer Suppl. 1996, 27, S32–S38. [Google Scholar] [PubMed]
- Sum, B.G.; Denny, W.A.; Wilson, W.R. Nitro reduction as an electronic switch for bioreductive drug activation. Oncol. Res. 1997, 9, 357–369. [Google Scholar]
- Wardman, P. Electron transfer and oxidative stress as key factors in the design of drugs selectively active in hypoxia. Curr. Med. Chem. 2001, 8, 739–761. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Miller, G.H.; Tranmer, G.K. Hypoxia activated prodrugs: Factors influencing design and development. Curr. Med. Chem. 2015, 22, 4313–4325. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Sheng, G.; Wu, X.; Cai, X.; Zhang, W. Cooperation of a Reductant and an Oxidant in One Pot To Synthesize Amides from Nitroarenes and Aldehydes. Synthesis 2015, 47, 949–954. [Google Scholar]
- Porzelle, A.; Woodrow, M.D.; Tomkinson, N.C.O. Facile Procedure for the Synthesis of N-Aryl-N-hydroxy Carbamates. Synlett 2009, 2009, 798–802. [Google Scholar]
- Kelly, S.M.; Lipshutz, B.H. Chemoselective Reductions of Nitroaromatics in Water at Room Temperature. Org. Lett. 2014, 16, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Wilson, W.R. Considerations for the Design of Nitrophenyl Mustards as Agents with Selective Toxicity for Hypoxic Tumor Cells. J. Med. Chem. 1986, 29, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Kassel, M.A.; Feinberg, B.A.; Corbett, M.D.; McClelland, R.A. Reduction potentials in relation to physiological activities of benzenoid and heterocyclic nitroso compounds: Comparison with the nitro precursors. Bioorg. Chem. 1990, 18, 265–275. [Google Scholar] [CrossRef]
- Wardman, P. Some reactions and properties of nitro radical-anions important in biology and medicine. Environ. Health Perspect. 1985, 64, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, A.J.; Williams, M.; Jenkins, T.C. Calculation of one-electron reduction potentials for nitroheterocyclic hypoxia-selective agents. J. Chem. Soc.-Faraday Trans. 1996, 92, 763–768. [Google Scholar] [CrossRef]
- Mason, R.P.; Holtzman, J.L. The role of catalytic superoxide formation in the O2 inhibition of nitroreductase. Biochem. Biophys. Res. Commun. 1975, 67, 1267–1274. [Google Scholar] [CrossRef]
- Denny, W.A.; Wilson, W.R. Bioreducible mustards: a paradigm for hypoxia-selective prodrugs of diffusible cytotoxins (HPDCs). Cancer Metastasis Rev. 1993, 12, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Allalunis, M.J.; Chapman, J.D.; Turner, A.R. Identification of a hypoxic population of bone marrow cells. Int. J. Radiat. Oncol. Boil. Phys. 1983, 9, 227–232. [Google Scholar] [CrossRef]
- Nombela-Arrieta, C.; Pivarnik, G.; Winkel, B.; Canty, K.J.; Harley, B.; Mahoney, J.E.; Park, S.Y.; Lu, J.; Protopopov, A.; Silberstein, L.E. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell Boil. 2013, 15, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Mauch, P.; Vergilio, J.A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parliament, M.B.; Franko, A.J.; Wiebe, L.I. Nitroimidazole adducts as markers for tissue hypoxia: Mechanistic studies in aerobic normal tissues and tumour cells. Br. J. Cancer 1992, 66, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.E.; Wilson, W.R. Hypoxia-dependent retinal toxicity of bioreductive anticancer prodrugs in mice. Toxicol. Appl. Pharmacol. 2000, 163, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.M.; Schrlau, A.E.; Chalian, A.A.; Zhang, P.; Koch, C.J. Oxygen levels in normal and previously irradiated human skin as assessed by EF5 binding. J. Investig. Dermatol. 2006, 126, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.; Von Eschwege, K.G.; Conradie, J. Reduction potentials of para-substituted nitrobenzenes-an infrared, nuclear magnetic resonance, and density functional theory study. J. Phys. Org. Chem. 2012, 25, 58–68. [Google Scholar] [CrossRef]
- Wardman, P. Reduction Potentials of One Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef]
- Meng, F.; Evans, J.W.; Bhupathi, D.; Banica, M.; Lan, L.; Lorente, G.; Duan, J.X.; Cai, X.; Mowday, A.M.; Guise, C.P.; et al. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol. Cancer Ther. 2012, 11, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Penketh, P.G.; Baumann, R.P.; Shyam, K.; Williamson, H.S.; Ishiguro, K.; Zhu, R.; Eriksson, E.S.E.; Eriksson, L.A.; Sartorelli, A.C. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl]hydrazine (KS119): A Cytotoxic Prodrug with Two Stable Conformations Differing in Biological and Physical Properties. Chem. Boil. Drug Des. 2011, 78, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Gu, Y.; Pruijn, F.B.; Smaill, J.B.; Patterson, A.V.; Guise, C.P.; Wilson, W.R. Zinc finger nuclease knock-out of NADPH:cytochrome P450 oxidoreductase (POR) in human tumor cell lines demonstrates that hypoxia-activated prodrugs differ in POR dependence? J. Boil. Chem. 2013, 288, 37138–37153. [Google Scholar] [CrossRef] [PubMed]
- Guise, C.P.; Abbattista, M.R.; Tipparaju, S.R.; Lambie, N.K.; Su, J.; Li, D.; Wilson, W.R.; Dachs, G.U.; Patterson, A.V. Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol. Pharmacol. 2012, 81, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.J.I.; Garner, A.P.; Powell, D.; Sibbald, J.; Sales, M.; Pratt, N.; Smith, T.; Tew, D.G.; Wolf, C.R. Cloning and characterization of a novel human dual flavin reductase. J. Boil. Chem. 2000, 275, 1471–1478. [Google Scholar] [CrossRef]
- Garner, A.P.; Paine, M.J.I.; Rodriguez-Crespo, I.; Chinje, E.C.; De Montellano, P.O.; Stratford, I.J.; Tew, D.G.; Wolf, C.R. Nitric oxide synthases catalyze the activation of redox cycling and bioreductive anticancer agents. Cancer Res. 1999, 59, 1929–1934. [Google Scholar] [PubMed]
- Aigrain, L.; Fatemi, F.; Frances, O.; Lescop, E.; Truan, G. Dynamic control of electron transfers in diflavin reductases. Int. J. Mol. Sci. 2012, 13, 15012–15041. [Google Scholar] [CrossRef] [PubMed]
- Finn Robert, D.; Basran, J.; Roitel, O.; Wolf, C.R.; Munro Andrew, W.; Paine Mark, J.I.; Scrutton Nigel, S. Determination of the redox potentials and electron transfer properties of the FAD- and FMN-binding domains of the human oxidoreductase NR1. Eur. J. Biochem. 2003, 270, 1164–1175. [Google Scholar] [CrossRef] [Green Version]
- Munro, A.W.; Noble, M.A.; Robledo, L.; Daff, S.N.; Chapman, S.K. Determination of the redox properties of human NADPH-cytochrome P450 reductase. Biochemistry 2001, 40, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.T.; Smith, S.M.E.; Weinberg, J.B.; Montgomery, H.J.; Newman, E.; Guillemette, J.G.; Ghosh, D.K.; Roman, L.J.; Martasek, P.; Salerno, J.C. Thermodynamics of Oxidation-Reduction Reactions in Mammalian Nitric-oxide Synthase Isoforms. J. Boil. Chem. 2004, 279, 18759–18766. [Google Scholar] [CrossRef] [PubMed]
- Wolthers, K.R.; Basran, J.; Munro, A.W.; Scrutton, N.S. Molecular dissection of human methionine synthase reductase: Determination of the flavin redox potentials in full-length enzyme and isolated flavin-binding domains. Biochemistry 2003, 42, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Wu, X.; Yalowich, J.C.; Hasinoff, B.B. A three-dimensional quantitative structure-activity analysis of a new class of bisphenol topoisomerase IIα inhibitors. Mol. Pharmacol. 2008, 73, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Hasinoff, B.B.; Creighton, A.M.; Kozlowska, H.; Thampatty, P.; Allan, W.P.; Yalowich, J.C. Mitindomide is a catalytic inhibitor of DNA topoisomerase II that acts at the bisdioxopiperazine binding site. Mol. Pharmacol. 1997, 52, 839–845. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EpaAg/mV | ipa/µA | EpcAg/mV | ipc/µA | EpaAg(Fc)/mV | EpcAg(Fc)/mV | |
| 2-Nitrobenzyl- (2) | −948 | 5.81 | −1033 | −9.86 | 427 | 514 |
| 3-Nitrobenzyl- (3) | −929 | 5.55 | −1043 | −10.14 | 405 | 509 |
| 4-Nitrobenzyl- (4) | −882 | 4.17 | −1055 | −11.97 | 427 | 504 |
| Cont’d | ipa/ipc | E1/2Ag(Fc)/mV | E1/2Ag/mV | E1/2Fc/mV | E1/2NHE/mV | |
| (2) | 0.589 | 471 | −991 | −1461 | −761 | |
| (3) | 0.548 | 457 | −986 | −1443 | −743 | |
| (4) | 0.348 | 466 | −969 | −1434 | −734 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, D.; Wu, X.; Hasinoff, B.B.; Herbert, D.E.; Tranmer, G.K. Evaluation of Nitrobenzyl Derivatives of Camptothecin as Anti-Cancer Agents and Potential Hypoxia Targeting Prodrugs. Molecules 2018, 23, 2041. https://doi.org/10.3390/molecules23082041
Liang D, Wu X, Hasinoff BB, Herbert DE, Tranmer GK. Evaluation of Nitrobenzyl Derivatives of Camptothecin as Anti-Cancer Agents and Potential Hypoxia Targeting Prodrugs. Molecules. 2018; 23(8):2041. https://doi.org/10.3390/molecules23082041
Chicago/Turabian StyleLiang, Dinghua, Xing Wu, Brian B. Hasinoff, David E. Herbert, and Geoffrey K. Tranmer. 2018. "Evaluation of Nitrobenzyl Derivatives of Camptothecin as Anti-Cancer Agents and Potential Hypoxia Targeting Prodrugs" Molecules 23, no. 8: 2041. https://doi.org/10.3390/molecules23082041