Hexanoic, Octanoic and Decanoic Acids Promote Basal and Insulin-Induced Phosphorylation of the Akt-mTOR Axis and a Balanced Lipid Metabolism in the HepG2 Hepatoma Cell Line


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Chemicals and Reagents
2.2. Fatty Acid Conjugation to Bovine Serum Albumin
2.3. Cell Culture
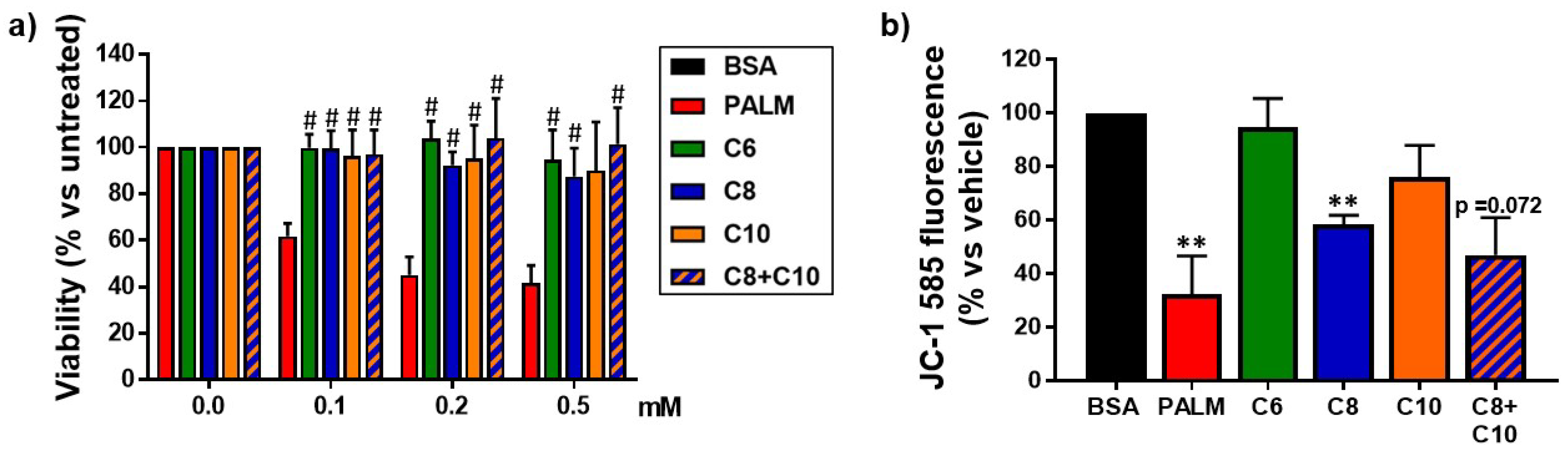
2.4. MTT Cell Viability Assay
2.5. Mitochondrial Membrane Potential Measurement
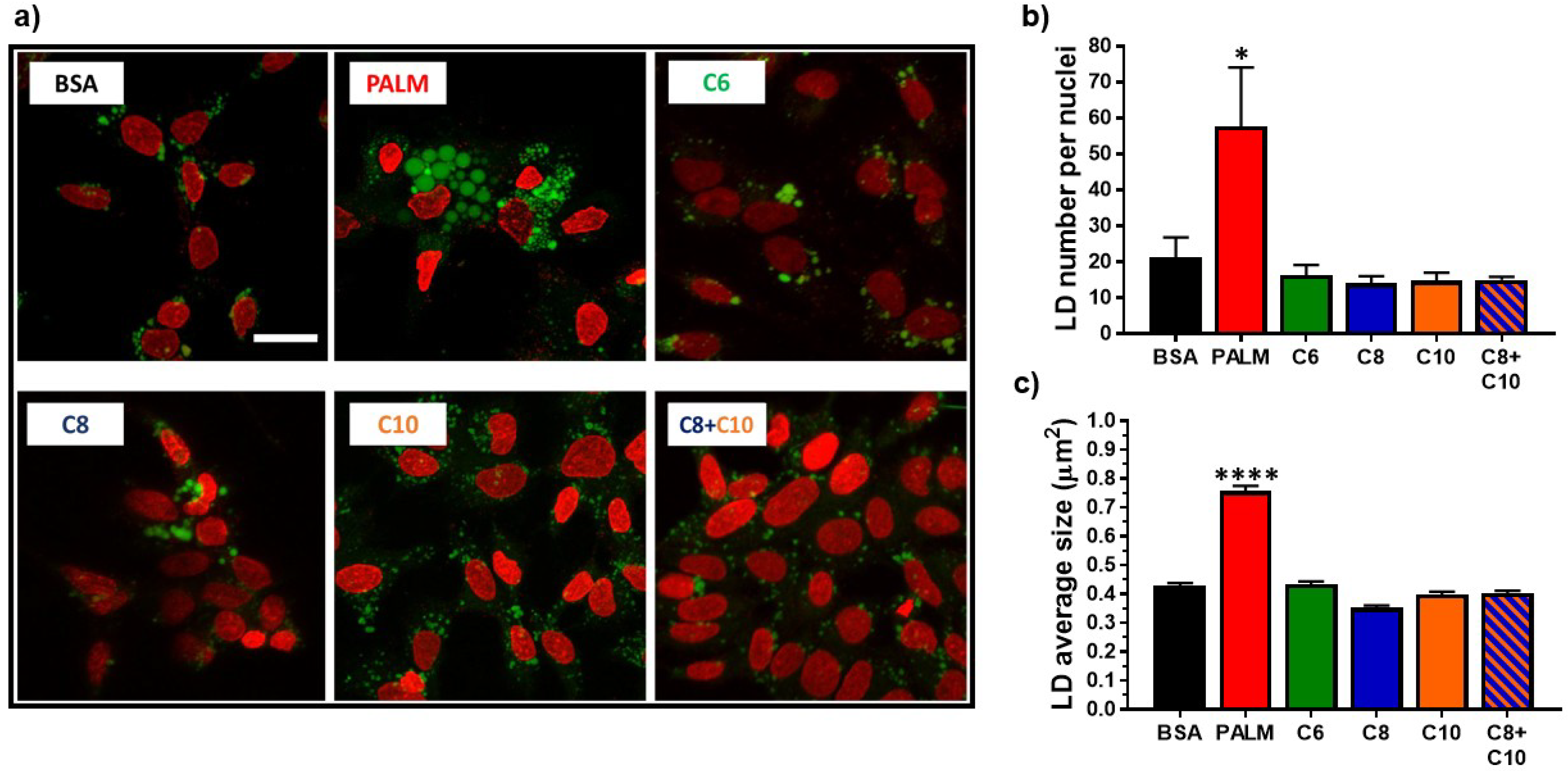
2.6. Lipid Droplet Staining
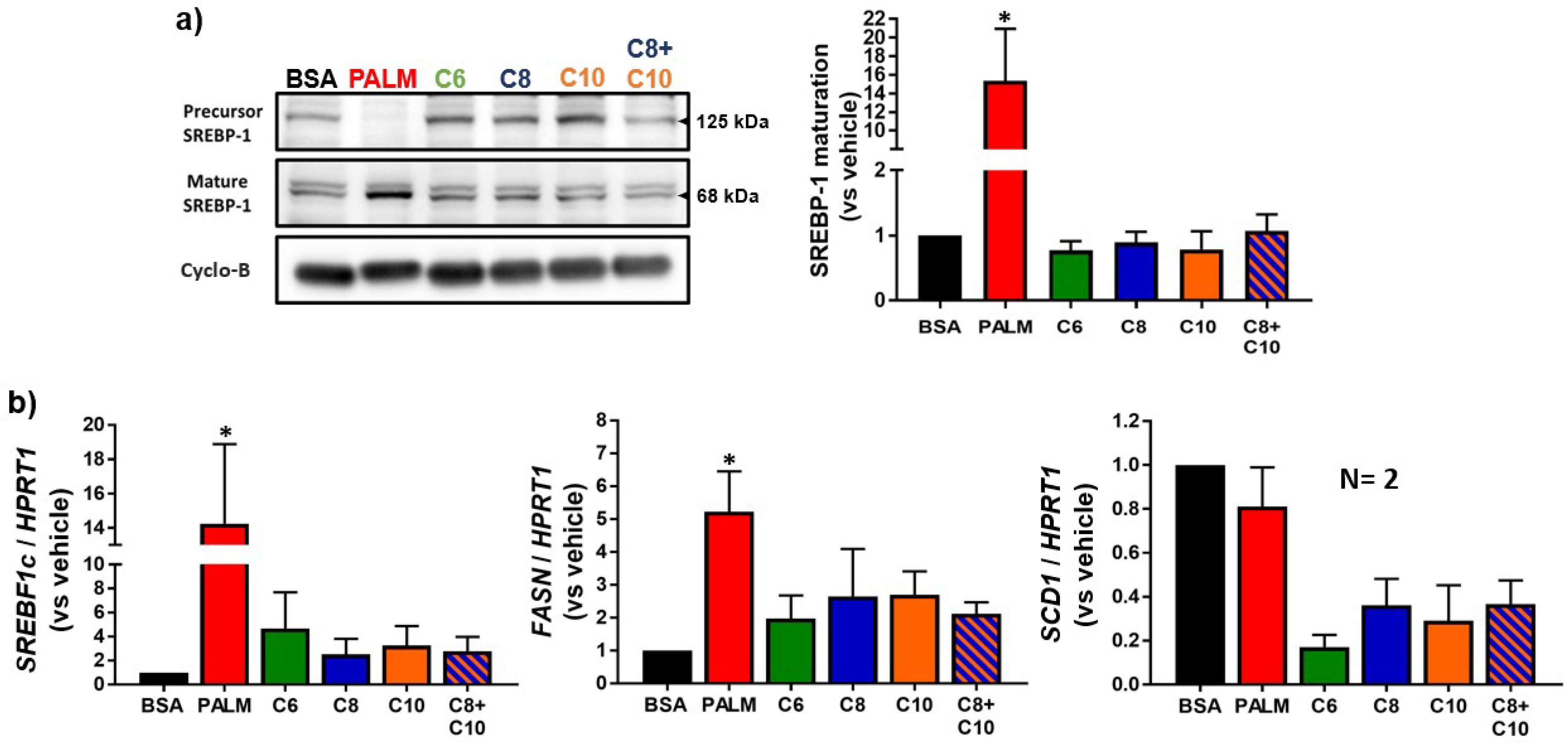
2.7. Western Blot Analysis
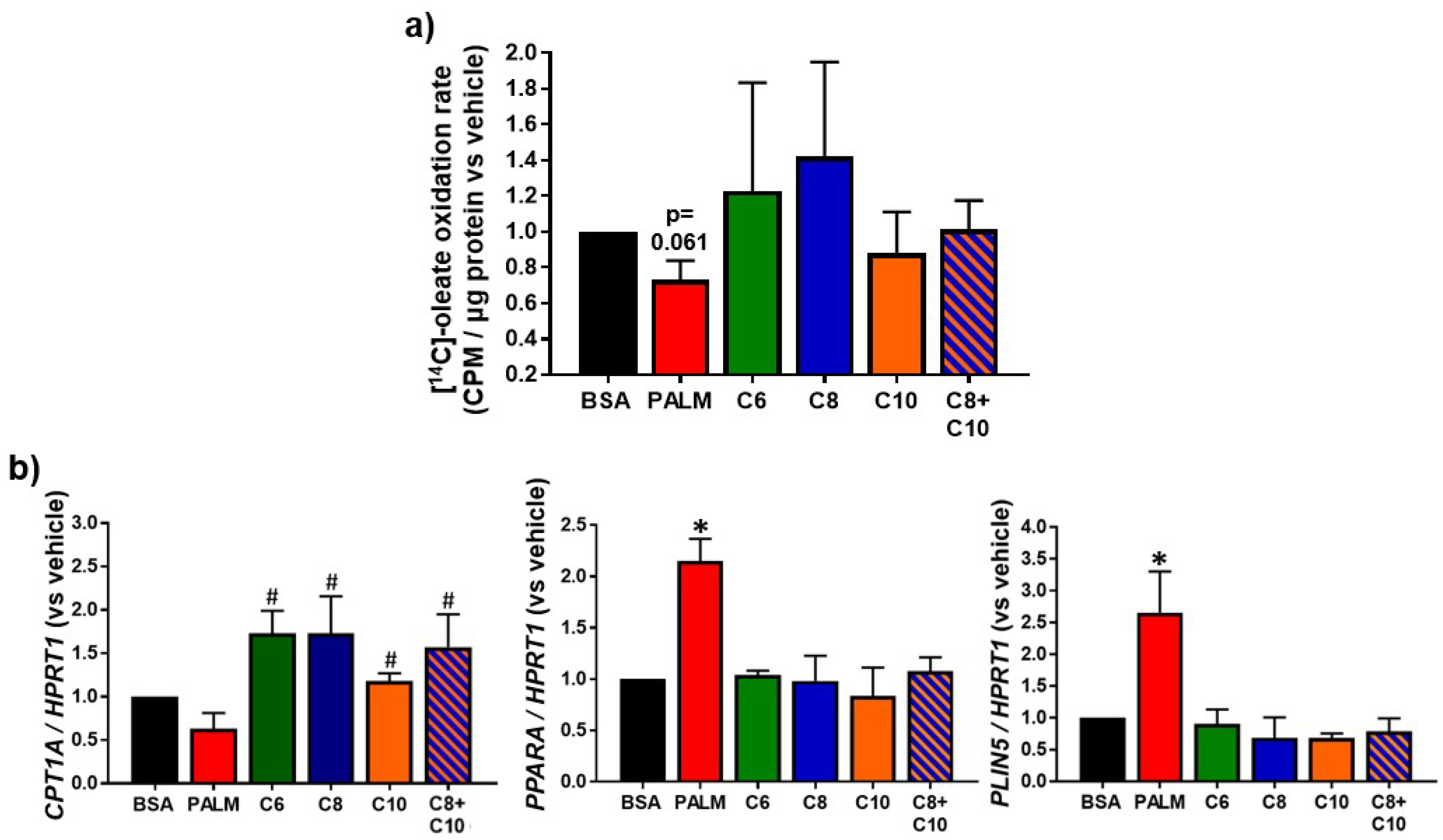
2.8. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) Analysis
2.9. Oleate Oxidation Assay
2.10. Glucose Uptake Assay
2.11. Statistical Analysis
3. Results
3.1. Medium Chain Fatty Acids Are Not Cytotoxic but Induce Differential Effects on Mitochondria
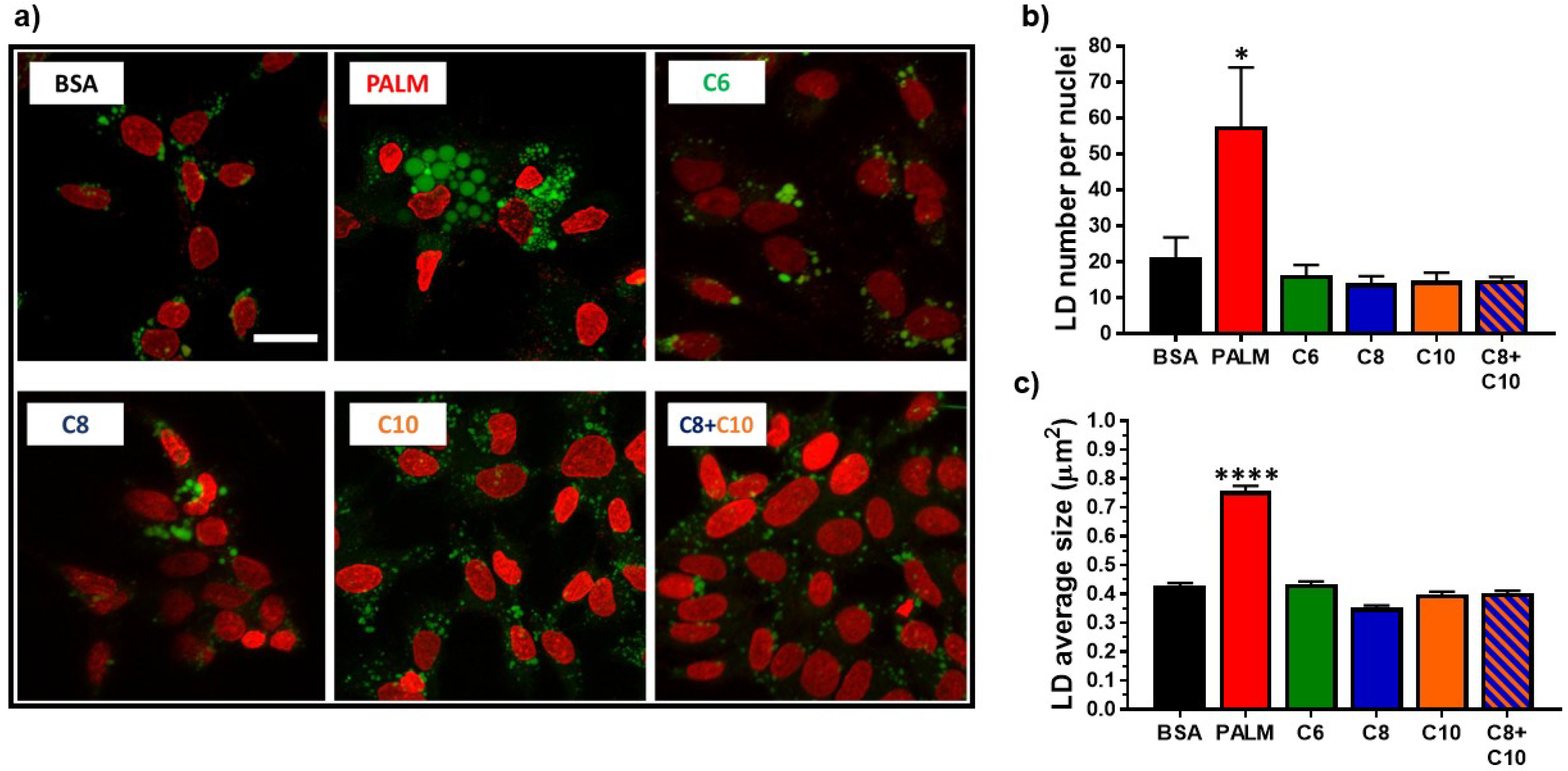
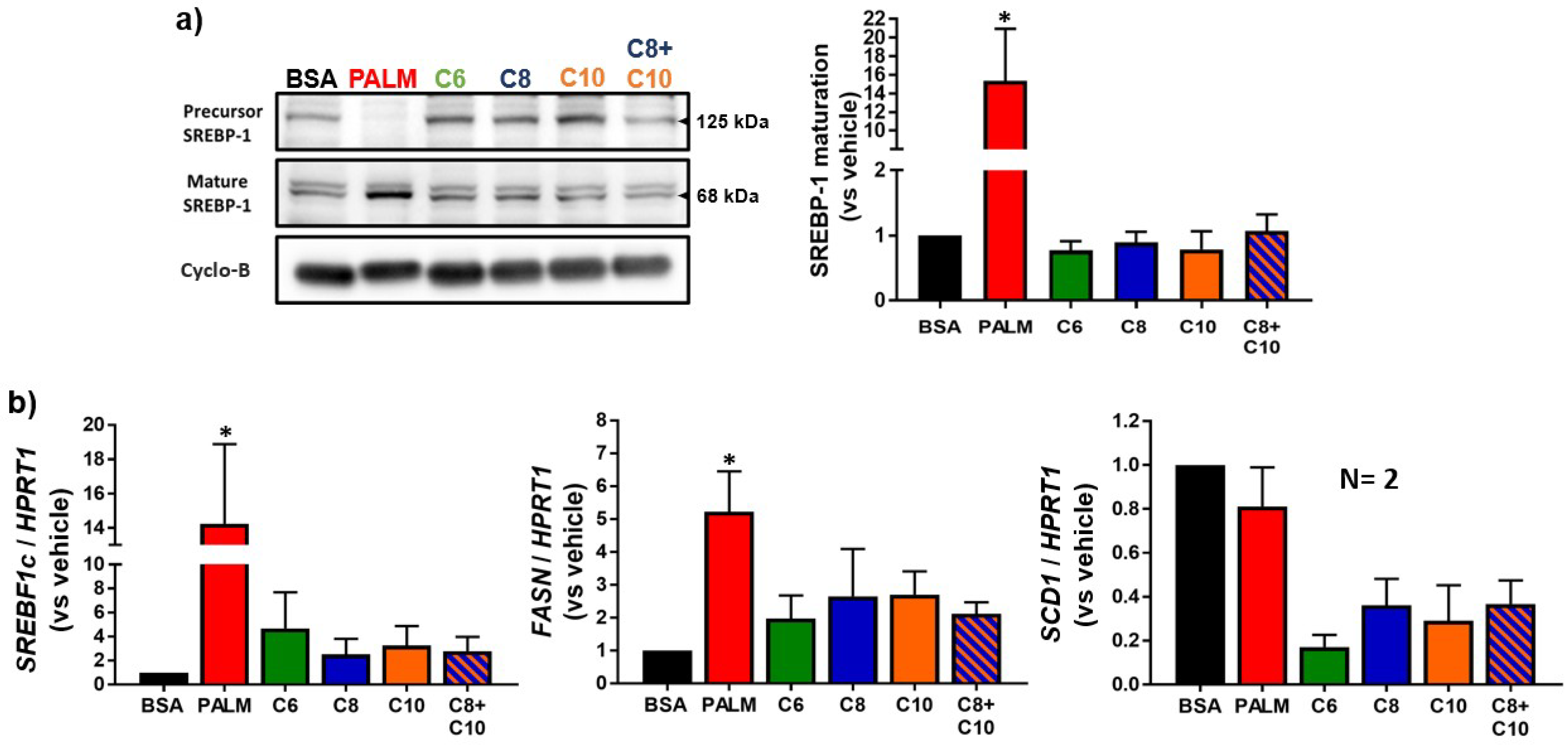
3.2. Medium Chain Fatty Acids Do Not Trigger Intracellular Lipid Storage or De Novo Lipogenesis
3.3. Medium Chain Fatty Acids Sustain Lipid Catabolism
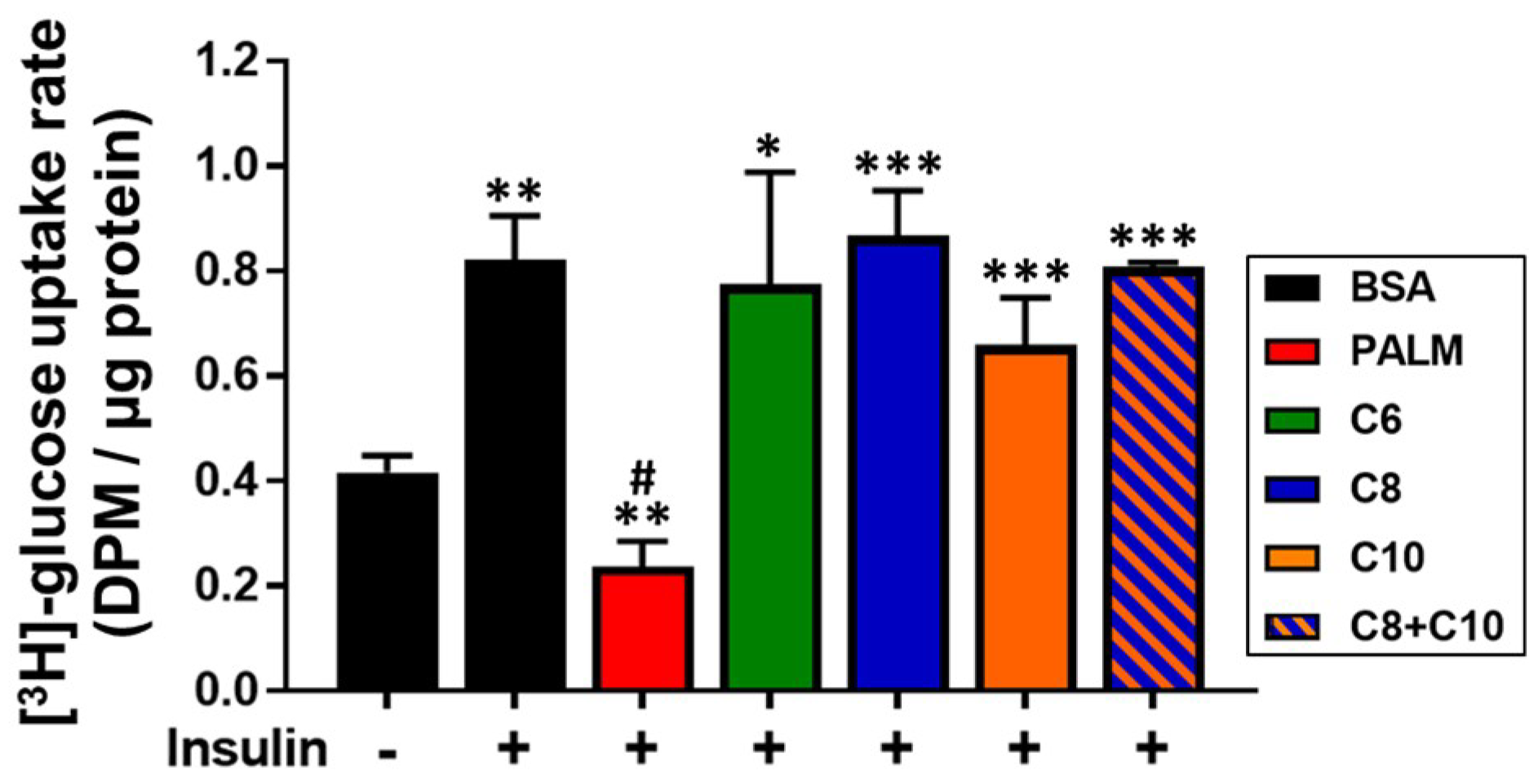
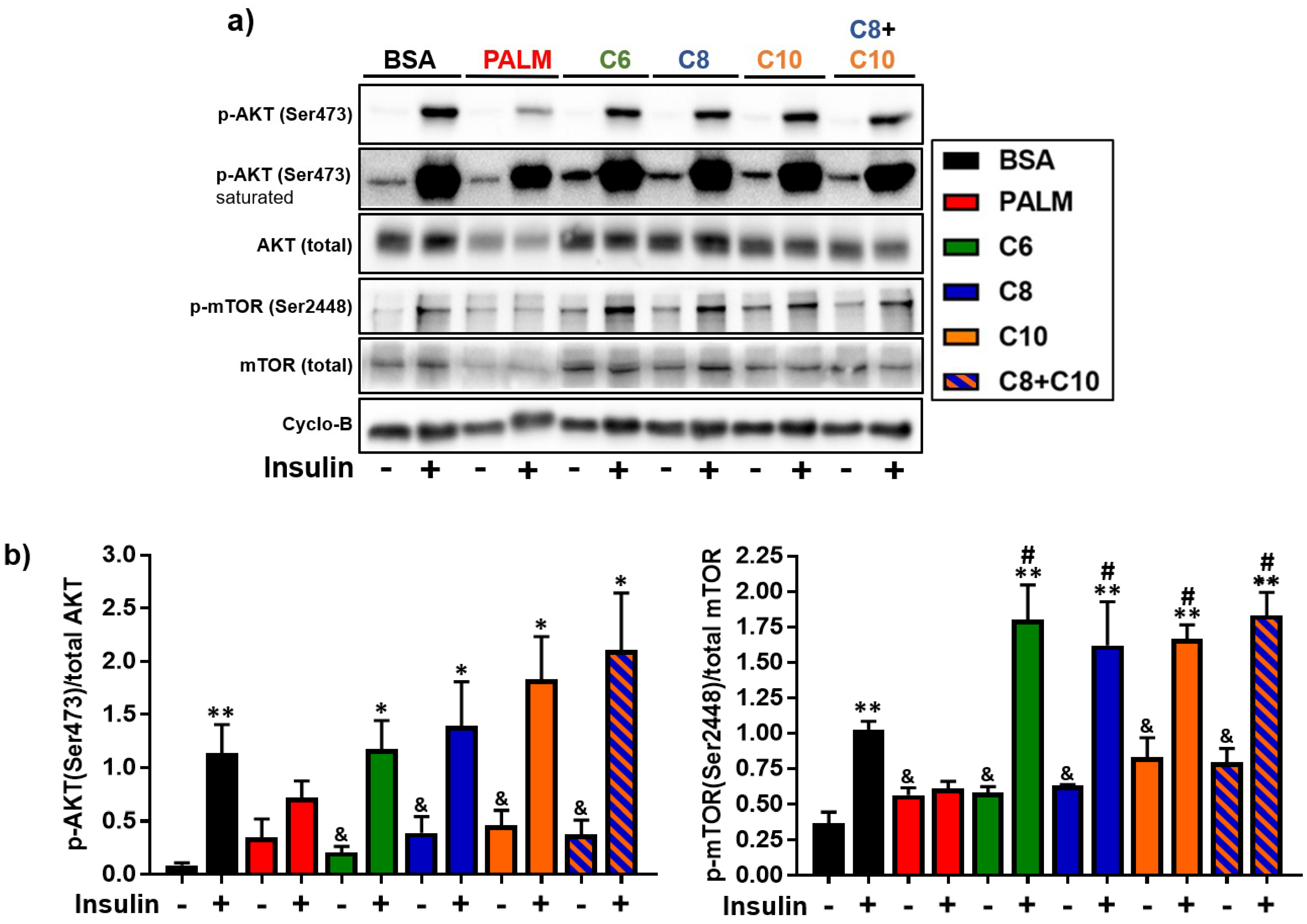
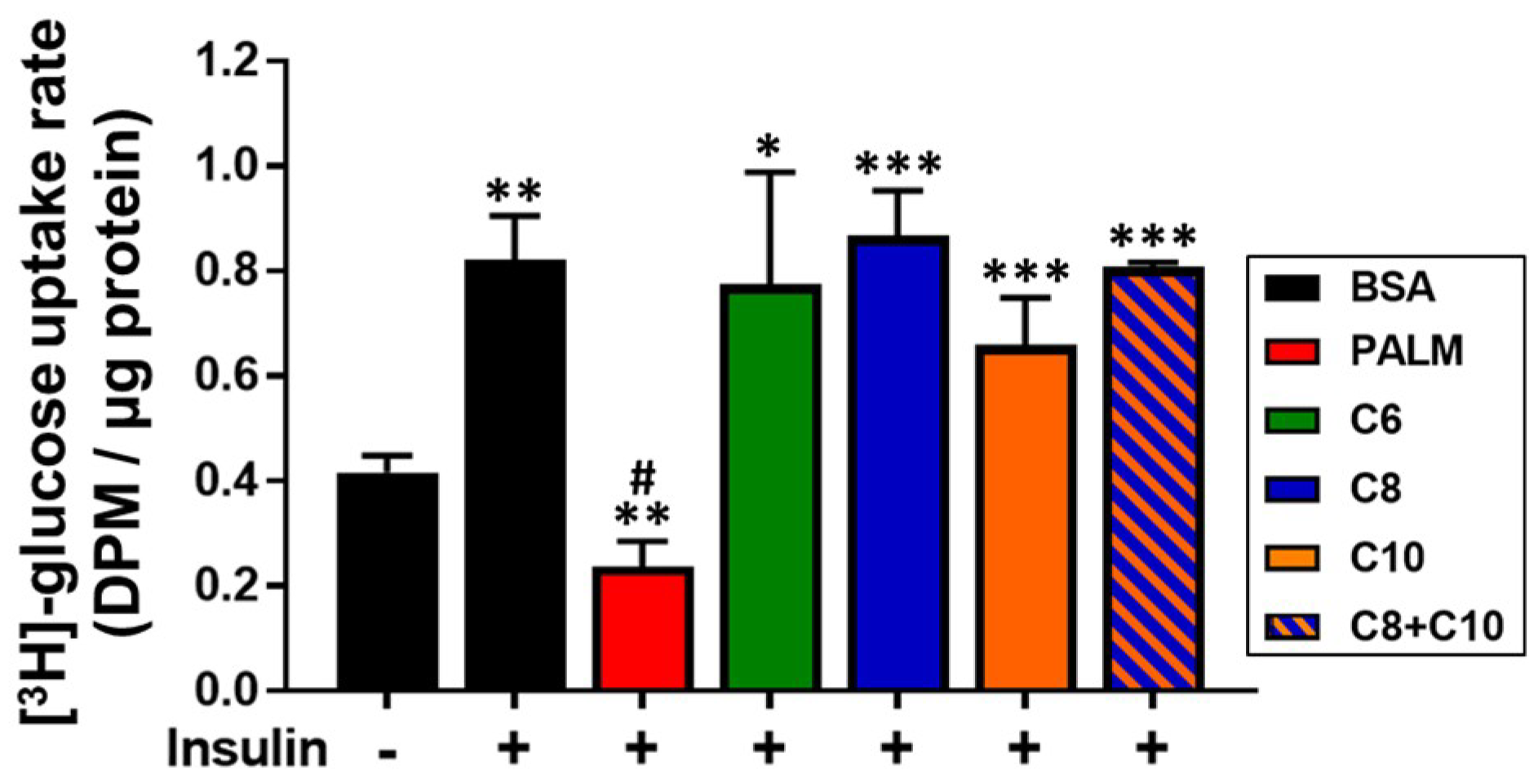
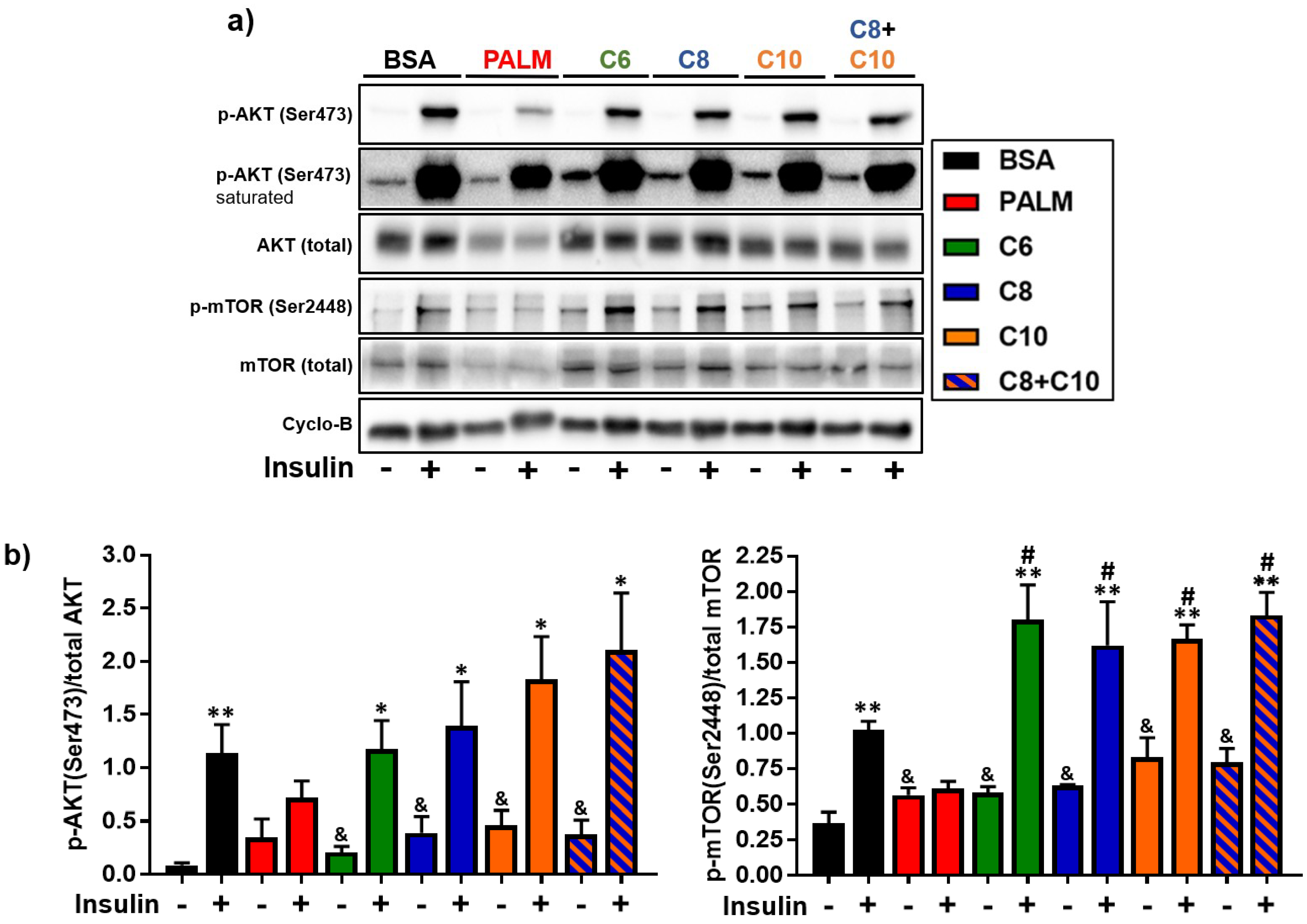
3.4. Medium Chain Fatty Acids Maintain Insulin Sensitivity and Foster Akt-mTOR Axis Activation
4. Discussion
4.1. Differential Effects of MCFA on Mitochondria
4.2. Lipid Catabolism in MCFA-Treated Hepatocytes
4.3. Akt-mTOR Phosphorylation in MCFA-Treated Hepatocytes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ofosu, A.; Ramai, D.; Reddy, M. Non-alcoholic fatty liver disease: Controlling an emerging epidemic, challenges, and future directions. Ann. Gastroenterol. 2018, 31, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Dumitrascu, D.L.; Neuman, M.G. Non-alcoholic fatty liver disease: An update on diagnosis. Clujul Med. (1957) 2018, 91, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Al-Dayyat, H.M.; Rayyan, Y.M.; Tayyem, R.F. Non-alcoholic fatty liver disease and associated dietary and lifestyle risk factors. Diabetes Metab. Syndr. 2018, 12, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Lounis, M.A.; Rial, S.; Ntambi, J.M.; Mounier, C. Role of lipogenesis and lipid desaturases in non-alcoholic fatty liver disease. In Hepatic de Novo Lipogenesis and Regulation of Metabolism; Ntambi, J.M., Ed.; Springer: Madison, WI, USA, 2015; pp. 143–164. [Google Scholar]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. Molecular pathways in non-alcoholic fatty liver disease. Clin. Exp. Gastroenter. 2014, 7, 221–239. [Google Scholar]
- Engin, A.B. What is lipotoxicity? In Advances in Experimental Medicine and Biology; Springer Nature: Berlin, Germany, 2017; pp. 197–220. [Google Scholar]
- Tamura, S.; Shimomura, I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1139–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating tnf-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, L.; Liu, X.; Luo, R.; Liao, G.; Li, L.; Liu, J.; Cheng, J.; Lu, Y.; Chen, Y. Oleic acid protects saturated fatty acid mediated lipotoxicity in hepatocytes and rat of non-alcoholic steatohepatitis. Life Sci. 2018, 203, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, P.; Perez-Jimenez, F.; Lopez-Miranda, J. N-3 pufa and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lai, Z.W.; Christiano, R.; Gazos-Lopes, F.; Walther, T.C.; Farese, R.V. Global analyses of selective insulin resistance in hepatocytes caused by palmitate lipotoxicity. Mol. Cell. Proteom. MCP 2018, 17, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhang, W.; Wan, C.; Xu, G.; Nie, X.; Zhu, X.; Xia, N.; Zhao, Y.; Wang, S.; Cui, S.; et al. Tram1 protect HepG2 cells from palmitate induced insulin resistance through er stress-jnk pathway. Biochem. Biophys. Res. Commun. 2015, 457, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The effects of palmitate on hepatic insulin resistance are mediated by nadph oxidase 3-derived reactive oxygen species through jnk and p38mapk pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.-Q.; Li, X.-Y.; Wang, L.; Feng, Z.-L.; Li, X.-F.; Wen, Y.-F.; Han, J.-X. Palmitate induces fat accumulation by activating c/ebpβ-mediated g0s2 expression in HepG2 cells. World J. Gastroenterol. 2017, 23, 7705–7715. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, H.; Qi, X.; Wang, Y.; Xu, C.; Liu, G.; Wen, G.; Liu, J. Zinc alpha2 glycoprotein alleviates palmitic acid-induced intracellular lipid accumulation in hepatocytes. Mol. Cell. Endocrinol. 2017, 439, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Kaplowitz, N. Mitochondria in Liver Disease; CRC Press: Boca Raton, FL, USA, 2015; pp. 365–382. [Google Scholar]
- Wang, S.; Zhao, Y.; Xia, N.; Zhang, W.; Tang, Z.; Wang, C.; Zhu, X.; Cui, S. Kpnbeta1 promotes palmitate-induced insulin resistance via nf-kappab signaling in hepatocytes. J. Physiol. Biochem. 2015, 71, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, X. Emerging role of jnk in insulin resistance. Curr. Diabetes Rev. 2013, 9, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Capeau, J. Insulin signaling: Mechanisms altered in insulin resistance. Med. Sci. 2003, 19, 834–839. [Google Scholar]
- Babayan, V.K. Medium chain triglycerides and structured lipids. Lipids 1987, 22, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.C.; Babayan, V.K. Medium-chain triglycerides: An update. Am. J. Clin. Nutr. 1982, 36, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Eyres, L.; Eyres, M.F.; Chisholm, A.; Brown, R.C. Coconut oil consumption and cardiovascular risk factors in humans. Nutr. Rev. 2016, 74, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papamandjaris, A.A.; MacDougall, D.E.; Jones, P.J. Medium chain fatty acid metabolism and energy expenditure: Obesity treatment implications. Life Sci. 1998, 62, 1203–1215. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Rial, S.A.; Karelis, A.D.; Bergeron, K.F.; Mounier, C. Gut microbiota and metabolic health: The potential beneficial effects of a medium chain triglyceride diet in obese individuals. Nutrients 2016, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- St-Onge, M.P.; Mayrsohn, B.; O’Keeffe, M.; Kissileff, H.R.; Choudhury, A.R.; Laferrere, B. Impact of medium and long chain triglycerides consumption on appetite and food intake in overweight men. Eur. J. Clin. Nutr. 2014, 68, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaton, T.B.; Welle, S.L.; Warenko, M.K.; Campbell, R.G. Thermic effect of medium-chain and long-chain triglycerides in man. Am. J. Clin. Nutr. 1986, 44, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Scalfi, L.; Coltorti, A.; Contaldo, F. Postprandial thermogenesis in lean and obese subjects after meals supplemented with medium-chain and long-chain triglycerides. Am. J. Clin. Nutr. 1991, 53, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.O.; Peters, J.C.; Yang, D.; Sharp, T.; Kaler, M.; Abumrad, N.N.; Greene, H.L. Thermogenesis in humans during overfeeding with medium-chain triglycerides. Metabolism 1989, 38, 641–648. [Google Scholar] [CrossRef]
- Dulloo, A.G.; Fathi, M.; Mensi, N.; Girardier, L. Twenty-four-hour energy expenditure and urinary catecholamines of humans consuming low-to-moderate amounts of medium-chain triglycerides: A dose-response study in a human respiratory chamber. Eur. J. Clin. Nutr. 1996, 50, 152–158. [Google Scholar] [PubMed]
- St-Onge, M.P.; Jones, P.J. Greater rise in fat oxidation with medium-chain triglyceride consumption relative to long-chain triglyceride is associated with lower initial body weight and greater loss of subcutaneous adipose tissue. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 1565–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Onge, M.P.; Ross, R.; Parsons, W.D.; Jones, P.J. Medium-chain triglycerides increase energy expenditure and decrease adiposity in overweight men. Obes. Res. 2003, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Akpa, M.M.; Point, F.; Sawadogo, S.; Radenne, A.; Mounier, C. Inhibition of insulin and t3-induced fatty acid synthase by hexanoate. Lipids 2010, 45, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Strable, M.S.; Ntambi, J.M. Genetic control of de novo lipogenesis: Role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fu, J.; Li, L.; Gong, D.; Wen, X.; Yu, P.; Zeng, Z. Medium-chain fatty acid reduces lipid accumulation by regulating expression of lipid-sensing genes in human liver cells with steatosis. Int. J. Food Sci. Nutr. 2016, 67, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, B.; Yu, P.; Wen, X.; Gong, D.; Zeng, Z. Medium and long chain fatty acids differentially modulate apoptosis and release of inflammatory cytokines in human liver cells. J. Food Sci. 2016, 81, H1546–H1552. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Zhu, W.; Xie, C.; Li, X.; Wu, J.; Liang, Z.; Xie, W.; Zhu, J.; Huang, C.; Zhu, M.; et al. Medium-chain triglyceride ameliorates insulin resistance and inflammation in high fat diet-induced obese mice. Eur. J. Nutr. 2016, 55, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.; Baumgardner, J.N.; Sharma, N.; Vantrease, J.; Ferguson, M.; Tong, Y.; Wu, X.; Cleves, M.A.; Badger, T.M. Medium chain triglycerides dose-dependently prevent liver pathology in a rat model of non-alcoholic fatty liver disease. Exp. Biol. Med. 2013, 238, 151–162. [Google Scholar] [CrossRef] [PubMed]
- De Vogel-van den Bosch, J.; van den Berg, S.A.; Bijland, S.; Voshol, P.J.; Havekes, L.M.; Romijn, H.A.; Hoeks, J.; van Beurden, D.; Hesselink, M.K.; Schrauwen, P.; et al. High-fat diets rich in medium- versus long-chain fatty acids induce distinct patterns of tissue specific insulin resistance. J. Nutr. Biol. 2011, 22, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Pujol, J.B.; Christinat, N.; Ratinaud, Y.; Savoia, C.; Mitchell, S.E.; Dioum, E.H.M. Coordination of gpr40 and ketogenesis signaling by medium chain fatty acids regulates beta cell function. Nutrients 2018, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Chamma, C.M.; Bargut, T.C.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. A rich medium-chain triacylglycerol diet benefits adiposity but has adverse effects on the markers of hepatic lipogenesis and beta-oxidation. Food Funct. 2017, 8, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Senthilraja, P.; Kathiresan, K. In vitro cytotoxicity mtt assay in vero, HepG2 and mcf -7 cell lines study of marine yeast. J. Appl. Pharm. Sci. 2015, 5, 80–84. [Google Scholar] [CrossRef]
- Smiley, S.T.; Reers, M.; Mottola-Hartshorn, C.; Lin, M.; Chen, A.; Smith, T.W.; Steele, G.D.; Chen, L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a j-aggregate-forming lipophilic cation jc-1. Proc. Natl. Acad. Sci. USA 1991, 88, 3671–3675. [Google Scholar] [CrossRef] [PubMed]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. Jc-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Disease 2012, 3, e430. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Guo, W.; Huang, N.; Cai, J.; Xie, W.; Hamilton, J.A. Fatty acid transport and metabolism in HepG2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G528–534. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.; Petrov, P.; Bonet, M.L.; Ribot, J.; Palou, A. Induction of carnitine palmitoyl transferase 1 and fatty acid oxidation by retinoic acid in HepG2 cells. Int. J. Biochem. Cell Biol. 2012, 44, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Kikani, C.K.; Riojas, R.A.; Langlais, P.; Wang, L.; Ramos, F.J.; Fang, Q.; Christ-Roberts, C.Y.; Hong, J.Y.; Kim, R.Y.; et al. Appl1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 2006, 8, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Lee, J.H.; Park, C.E.; Kim, M.-J.; Min, B.-I.; Bae, H.; Choe, W.; Kang, I.; Kim, S.-S.; Ha, J. Inulin increases glucose transport in c2c12 myotubes and HepG2 cells via activation of amp-activated protein kinase and phosphatidylinositol 3-kinase pathways. J. Med. Food 2009, 12, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ni, Y.; Ma, X.; Zhao, A.; Bao, Y.; Liu, J.; Chen, T.; Xie, G.; Panee, J.; Su, M.; et al. A panel of free fatty acid ratios to predict the development of metabolic abnormalities in healthy obese individuals. Sci. Rep. 2016, 6, 28418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, S.; Abughazaleh, A.; Webel, S.; Jones, K. Circulating fatty acid profiles in response to three levels of dietary omega-3 fatty acid supplementation in horses. J. Anim. Sci. 2008, 86, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, I.; Solis-Munoz, P.; Fernandez-Moreira, D.; Munoz-Yague, T.; Solis-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis. Model. Mech. 2015, 8, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.-R.; Liu, J.; Plumeri, D.; Cao, Y.-B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.-Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res. 2011, 3, 284–291. [Google Scholar] [PubMed]
- Janani, C.; Ranjitha Kumari, B.D. Ppar gamma gene—A review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernandez-Fernandez, C.; Mourino-Bayolo, D. Mitochondrial beta-oxidation of saturated fatty acids in humans. Mitochondrion 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.-W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalen, K.T.; Dahl, T.; Holter, E.; Arntsen, B.; Londos, C.; Sztalryd, C.; Nebb, H.I. LSDP5 is a pat protein specifically expressed in fatty acid oxidizing tissues. Biochim. Biophys. Acta 2007, 1771, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Maeda, A.; Tani, S.; Akagawa, M. Palmitate induces insulin resistance in human HepG2 hepatocytes by enhancing ubiquitination and proteasomal degradation of key insulin signaling molecules. Arch. Biochem. Biophys. 2015, 566, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kerimi, A.; Jailani, F.; Williamson, G. Modulation of cellular glucose metabolism in human HepG2 cells by combinations of structurally related flavonoids. Mol. Nutr. Food Res. 2015, 59, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Wei, D.; Mo, C.; Zhang, J.; Wang, X.; Han, X.; Wang, Z.; Xiao, H. Saturated fatty acid palmitate-induced insulin resistance is accompanied with myotube loss and the impaired expression of health benefit myokine genes in c2c12 myotubes. Lipids Health Disease 2013, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Deng, J.; Guo, Y.; Niu, Y.; Yuan, F.; Yu, J.; Chen, S.; Guo, F. BTG1 ameliorates liver steatosis by decreasing stearoyl-coa desaturase 1 (SCD1) abundance and altering hepatic lipid metabolism. Sci. Signal. 2016, 9, ra50. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Lei, T.; Wang, T.; Corkey, B.E.; Han, J. Octanoate inhibits triglyceride synthesis in 3T3-L1 and human adipocytes. J. Nutr. 2003, 133, 2512–2518. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Xu, Y.; Xie, J.; Sun, C.; Zheng, X.; Chen, W. Systematic evaluation of phenolic compounds and protective capacity of a new mulberry cultivar J33 against palmitic acid-induced lipotoxicity using a simulated digestion method. Food Chem. 2018, 258, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Zhou, R. Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 2014, 3, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, L.; Fu, J.; Yu, P.; Gong, D.; Zeng, C.; Zeng, Z. Effects of long-chain and medium-chain fatty acids on apoptosis and oxidative stress in human liver cells with steatosis. J. Food Sci. 2016, 81, H794–H800. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Zhang, X.; Singleton, T.P.; Kiechle, F.L. Mitochondrial membrane potential change induced by hoechst 33342 in myelogenous leukemia cell line hl-60. Ann. Clin. Lab. Sci. 2004, 34, 458–466. [Google Scholar] [PubMed]
- Zhang, H.; Li, Y.; Hou, X.; Zhang, L.; Wang, T. Medium-chain tag improve energy metabolism and mitochondrial biogenesis in the liver of intra-uterine growth-retarded and normal-birth-weight weanling piglets. Br. J. Nutr. 2016, 115, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Q.; Liu, Y.H.; Zhang, X.S.; Wang, J.; Yu, X.M.; Zhang, R.X.; Xue, C.; Yang, X.Y.; Xue, C.Y. Medium-chain triglyceride activated brown adipose tissue and induced reduction of fat mass in C57BL/6J mice fed high-fat diet. Biomed. Environ. Sci. 2015, 28, 97–104. [Google Scholar] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-Dominguez, M.; Mir, J.F.; Fucho, R.; Weber, M.; Serra, D.; Herrero, L. Fatty acid metabolism and the basis of brown adipose tissue function. Adipocyte 2016, 5, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.C.; Carpenter, C.A.; Saggerson, E.D. Intertissue differences in the hysteretic behaviour of carnitine palmitoyltransferase in the presence of malonyl-coa. Biochem. J. 1986, 237, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Borén, J.; Dulloo, A.G. De novo lipogenesis in metabolic homeostasis: More friend than foe? Mol. Metab. 2015, 4, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Khabbush, A.; Orford, M.; Tsai, Y.C.; Rutherford, T.; O’Donnell, M.; Eaton, S.; Heales, S.J.R. Neuronal decanoic acid oxidation is markedly lower than that of octanoic acid: A mechanistic insight into the medium-chain triglyceride ketogenic diet. Epilepsia 2017, 58, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of pparα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, Y.; Jacoby, J.J.; Jiang, Y.; Zhang, Y.; Yu, L.L. Effects of medium- and long-chain triacylglycerols on lipid metabolism and gut microbiota composition in C57BL/6J mice. J. Agric. Food Chem. 2017, 65, 6599–6607. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. “New” hepatic fat activates pparalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005, 1, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Pineda Torra, I.; Jamshidi, Y.; Flavell, D.M.; Fruchart, J.C.; Staels, B. Characterization of the human pparalpha promoter: Identification of a functional nuclear receptor response element. Mol. Endocrinol. 2002, 16, 1013–1028. [Google Scholar] [PubMed]
- Titchenell, P.M.; Quinn, W.J.; Lu, M.; Chu, Q.; Lu, W.; Li, C.; Chen, H.; Monks, B.R.; Chen, J.; Rabinowitz, J.D.; et al. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 2016, 23, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Hong, T.; Wen, G.B.; Han, J.; Zuo, D.; Liu, Z.; Cao, W. Increased basal level of Akt-dependent insulin signaling may be responsible for the development of insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E898–906. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, Y.; Liu, Y.; Ping, J.; Shou, Q.; Chen, F.; Ruo, R. Metformin improves hepatic IRS2/PI3k/AKT signaling in insulin-resistant rats of nash and cirrhosis. J. Endocrinol. 2016, 229, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, Y.; Jiang, Y.; Zhang, Z.; Sun, X.; Yu, L.L. Dietary intake of structured lipids with different contents of medium-chain fatty acids on obesity prevention in C57BL/6J mice. J. Food Sci. 2017, 82, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Wein, S.; Wolffram, S.; Schrezenmeir, J.; Gasperikova, D.; Klimes, I.; Sebokova, E. Medium-chain fatty acids ameliorate insulin resistance caused by high-fat diets in rats. Diabetes Metab. Res. 2009, 25, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. Mtor: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell. Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozengurt, E.; Soares, H.P.; Sinnet-Smith, J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: An unintended consequence leading to drug resistance. Mol. Cancer Ther. 2014, 13, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds C6, C8, C10 and palmitic acid are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Target | Manufacturer | Catalog Number | Concentration Used |
|---|---|---|---|
| SREBP-1 | Santa Cruz Biotechnology | #sc-8984 | 1:2000 |
| p-Akt (Ser473) | Cell Signaling Technology | #4060 | 1:1000 |
| p-mTOR (Ser2448) | Cell Signaling Technology | #2971 | 1:1000 |
| Akt (pan) | Cell Signaling Technology | #4691 | 1:1500 |
| mTOR | Cell Signaling Technology | #2972 | 1:1000 |
| Cyclophilin-B | Abcam | #ab16045 | 1:50,000 |
| Gene Target | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| SREBF1c | ACAGTGACTTCCCTGGCCTAT | GCATGGACGGGTACATCTTCAA |
| CPT1A | ATCAATCGGACTCTGGAAACGG | TCAGGGAGTAGCGCATGGT |
| PLIN5 | AAGGCCCTGAAGTGGGTTT | GCATGTGGTCTATCAGCTCCA |
| PPARA | CGGTGACTTATCCTGTGGTCC | CCGCAGATTCTACATTCGATGTT |
| FASN | AAGGACCTGTCTAGGTTTGATGC | TGGCTTCATAGGTGACTTCCA |
| SCD1 | TTCCTACCTGCAAGTTCTACACC | CCGAGCTTTGTAAGAGCGGT |
| HPRT1 | CCTGGCGTCGTGATTAGTGAT | AGACGTTCAGTCCTGTCCATAA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rial, S.A.; Ravaut, G.; Malaret, T.B.; Bergeron, K.-F.; Mounier, C. Hexanoic, Octanoic and Decanoic Acids Promote Basal and Insulin-Induced Phosphorylation of the Akt-mTOR Axis and a Balanced Lipid Metabolism in the HepG2 Hepatoma Cell Line. Molecules 2018, 23, 2315. https://doi.org/10.3390/molecules23092315
Rial SA, Ravaut G, Malaret TB, Bergeron K-F, Mounier C. Hexanoic, Octanoic and Decanoic Acids Promote Basal and Insulin-Induced Phosphorylation of the Akt-mTOR Axis and a Balanced Lipid Metabolism in the HepG2 Hepatoma Cell Line. Molecules. 2018; 23(9):2315. https://doi.org/10.3390/molecules23092315
Chicago/Turabian StyleRial, Sabri Ahmed, Gaetan Ravaut, Tommy B. Malaret, Karl-F. Bergeron, and Catherine Mounier. 2018. "Hexanoic, Octanoic and Decanoic Acids Promote Basal and Insulin-Induced Phosphorylation of the Akt-mTOR Axis and a Balanced Lipid Metabolism in the HepG2 Hepatoma Cell Line" Molecules 23, no. 9: 2315. https://doi.org/10.3390/molecules23092315